Back to Journals » Infection and Drug Resistance » Volume 16
Retrospective Study of the Epidemiology of Clostridioides difficile Infection in the Neurosurgery Department of a Tertiary Hospital in China
Authors Bi X, Zheng L, Yang Z, Lv T, Tong X, Chen Y ![]()
Received 21 November 2022
Accepted for publication 19 January 2023
Published 26 January 2023 Volume 2023:16 Pages 545—554
DOI https://doi.org/10.2147/IDR.S397544
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Héctor Mora-Montes
Xiajing Bi,1 Lisi Zheng,2 Zhi Yang,1 Tao Lv,2 Xiaofei Tong,1 Yunbo Chen2,3
1Department of Neurosurgery, The First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, People’s Republic of China; 2State Key Laboratory for Diagnosis and Treatment of Infectious Diseases, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, The First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, People’s Republic of China; 3Microbiology Laboratory, Jinan Microecological Biomedicine Shandong Laboratory, Jinan, People’s Republic of China
Correspondence: Xiaofei Tong; Yunbo Chen, The First Affiliated Hospital, School of Medicine, Zhejiang University, 79 Qingchun Road, Hangzhou, Zhejiang, 310003, People’s Republic of China, Tel/Fax +86 571 87236459, Email [email protected]; [email protected]
Background: Although the epidemiology of Clostridioides difficile is important, few studies examining transmission of C. difficile have been reported, especially in wards with low detection rates, such as neurosurgery departments.
Purpose: This retrospective study investigated the epidemiology of C. difficile infection in a neurosurgery department over a 24-month period, particularly examining the transmission of C. difficile using whole-genome sequencing (WGS).
Methods: Clostridioides difficile strains were isolated and identified from fecal samples of neurosurgical patients. Toxigenic strains were typed using multilocus sequence typing, PCR ribotyping and using capillary gel electrophoresis. WGS was used to characterize C. difficile ST-37/RT017 isolates, and comparative genomic analyses were performed to compare genomic differences between all ST-37 strains from other wards. The susceptibility to 8 antimicrobial agents was examined using the E-test.
Results: Comparative genomic analyses revealed that isolates obtained from neurosurgical patients clustered into two lineages. Only strains s11052403 and s10090304, respectively, isolated from a patient on the 8th floor of the neurosurgery ward and a patient on the 9th floor, were highly similar, exhibiting differences of only two single-nucleotide polymorphisms. All C. difficile ST-37/RT017 strains isolated from neurosurgical patients were resistant to multiple classes of antibiotics.
Conclusion: There is an urgent need to raise awareness of C. difficile infection, and epidemiologic surveillance is required to detect clustering and transmission of C. difficile cases in China. Strict disinfection of the environment is essential to reduce transmission of C. difficile and achieve effective infection control in the hospital setting.
Keywords: Clostridioides difficile infection, transmission, capillary gel electrophoresis, whole-genome sequencing
Introduction
Clostridioides difficile (C. difficile) is an anaerobic, gram-positive, spore-forming bacillus and important pathogen of antimicrobial-associated diarrhea and nosocomial diarrhea in humans.1 Clostridioides difficile infection (CDI) can progress from self-resolving diarrhea to colitis, life-threatening pseudomembranous colitis and toxic megacolon, and death.2,3 CDI is a toxin-mediated disease, and the major virulence factors associated with CDI include toxin A (TcdA, an enterotoxin) and toxin B (TcdB, a cytotoxin).4 Additionally, some strains exclusively produce a third toxin (binary toxin, CDT), which has been linked with the increasing severity of human infections.5
Since the emergence of hypervirulent variants of toxigenic C. difficile RT027, the morbidity and mortality of CDI have increased markedly, and this has been accompanied by large healthcare-associated outbreaks.3,6 CDI has aroused great concern worldwide due to its increasing incidence, recurrence, morbidity, and impact on healthcare spending. Current clinical practice guidelines for reducing the incidence of CDI cover a wide range of issues, including epidemiology, diagnosis, treatment, and infection control.2 Although sporadic cases and a small healthcare-associated outbreak of CDI caused by the hypervirulent RT027 strain have been reported in China, CDI is still an emerging problem.7 According to epidemiologic studies of C. difficile in China, the detection rate of CDI is similar to, or even slightly higher than, that in Western countries, equal to about 10%.7–10 Sequence types 35 (ST-35/RT012), ST-37/RT017, ST-54/RT012 and ST-3/RT001 are the predominant genotypes in China.10,11 These strains are primarily isolated from patients in intensive care units, geriatric wards, infection wards, hematology wards, and gastroenterology departments.8,9 Nevertheless, the epidemiology of C. difficile transmission in health-care settings in China remains poorly understood. Few epidemiologic studies have been reported regarding C. difficile in wards with low detection rates, such as neurosurgery departments.
For neurosurgical patients, prophylactic antibiotics are used to reduce the preoperative, intraoperative, and postoperative incidence of surgical site infections, respiratory tract infections, and bacteriuria.12–14 However, extended antimicrobial administration is associated with a clear increase in the risk of CDI.15 Currently, there is a paucity of data regarding CDI in neurosurgical patients in China. Thus, the main objective of this study was to retrospectively investigate the incidence of CDI over time and the epidemiology of C. difficile transmission in a neurosurgery department in China using whole-genome sequencing (WGS).
Materials and Methods
Collection of C. difficile Isolates
This retrospective study was conducted at the Department of Neurosurgery, First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, Zhejiang, China. Diarrhea was defined as three or more loose stools within 24 hours. Stool samples collected from neurosurgery department inpatients with diarrhea were submitted to the clinical microbiology laboratory for isolation of C. difficile. Inpatients with diarrhea, whose stool samples were positive for both C. difficile culture and toxin genes (tcdA and/or tcdB) and without evidence of other pathogens of diarrhea (including Shigella, nontyphoidal Salmonella, Campylobacter and Shiga toxin-producing Escherichia coli), were diagnosed as CDI.
Approximately 0.5 mL (0.5 g) of stool was mixed with 0.5 mL of 95% ethanol for 30 min at room temperature for spore selection before anaerobic isolation of C. difficile on selective cycloserine–cefoxitin–taurocholate agar (CCFA-TA; Oxoid) plates supplemented with 7% sheep blood incubated at 35°C for 48 h with anaerobic incubation (80% N2, 10% H2, 10% CO2). The C. difficile isolates were confirmed by matrix-assisted laser desorption ionization–time-of-flight mass spectrometry using a Bruker Daltonics Microflex LT system (Bruker Daltonik GmbH, Bremen, Germany).
Detection of Toxin Genes by Polymerase Chain Reaction (PCR)
Bacteria identified as C. difficile obtained after 48 h of anaerobic blood agar culture were suspended in distilled water in a microcentrifuge tube. Bacterial genomic DNA was extracted using the simplified alkaline lysis method. All isolated strains were tested for tcdA, tcdB, and binary toxin genes by PCR, as previously described.5,16
Multilocus Sequence Typing (MLST)
MLST of seven housekeeping genes (adk, atpA, dxr, glyA, recA, soda, and tpi) was used to genotype all toxigenic isolates, as previously described.17 Allele designations were obtained through the C. difficile PubMLST batch profile query page (http://pubmlst.org/organisms/clostridioides-difficile/) to determine the sequence type.
PCR Ribotyping
PCR ribotyping was performed using capillary gel electrophoresis as previously described.7 The 16S rRNA gene primers were labeled with carboxyfluorescein. PCR products were analyzed in the ABI 3100 genetic analyzer (Applied Biosystems, Foster City, CA, USA) with 36-cm capillary loaded with a POP4 gel (Applied Biosystems). A Size Standard-1200 bp TAMRA ladder (Chimerx, Milwaukee, WI, USA) was used as internal size marker for each sample. The peak Scanner software 1.0 (Applied Biosystems) was used to determine the size of each peak. The data obtained were submitted to the WEBRIBO database (https://webribo.ages.at/) for RT assignment.
Genome Sequencing, Assembly and Annotation
Sequencing-quality genomic DNA was extracted according to the manufacturer’s instructions using a DNeasy Blood and Tissue kit (Qiagen, Hilden, Germany) and quantitated using Qubit 2.0. WGS was performed on an Illumina HiSeq 2500 system (Illumina, San Diego, CA, USA). Before assembly, the quality control of raw sequenced reads was performed using FastQC v.0.11.5 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and adapter regions were trimmed using Trimmomatic v.0.40.18 Trimmed reads were assembled de novo using SPAdes v.3.6.19 Genome annotations were performed on the RAST server (http://rast.nmpdr.org) and with Prokka.20,21 Additionally, Roary v.3.13.0 was employed to estimate the size of the core and accessory genomes.22
SNP Calling, Phylogenetic and Comparative Genomic Analyses
Variant calls for SNP analysis were performed using Snippy (http://github.com/tseemann/snippy) with default parameters. The chromosome of C. difficile M68 (NC_017175.1) was set as the reference for all strains of C. difficile ST-37 in this study. The alignment file was filtered from variants with elevated densities of base substitutions as putative repetitive regions, mobile genetic elements (MGEs) and recombination events by Gubbins v.2.4.1, and used to calculate the pairwise cgSNP.23 Genome sequences were compared using BRIG software.24 The maximum likelihood trees based on core genome were constructed using MEGA X with 1000 bootstrap replicates and visualized using the Interactive Tree of Life (iToL) web server.25,26 Antimicrobial resistance genes were identified using the Comprehensive Antibiotic Resistance Database (CARD) Resistance Gene Identifier (RGI) software (https://card.mcmaster.ca/analyze/rgi).
Antimicrobial Susceptibility
Antimicrobial susceptibility testing was performed on Brucella agar plates containing 1 mg/L of vitamin K, 5 mg/L of hemin, and 5.0% sheep red blood cells with eight antimicrobial agents: metronidazole, vancomycin, clindamycin, erythromycin, linezolid, levofloxacin, moxifloxacin, and rifampicin. The minimum inhibitory concentrations (MICs) were read at the point at which the zone of complete inhibition intersected with the MIC scale according to the Clinical and Laboratory Standards Institute (CLSI) guidelines. The selected resistance breakpoints included 8 mg/L for erythromycin, clindamycin, and the fluoroquinolones, according to the CLSI interpretative categories approved for anaerobic bacteria. Breakpoint of 2 mg/L was selected for vancomycin and metronidazole according to the European Committee on Antimicrobial Susceptibility Testing (https://www.eucast.org). The breakpoint for linezolid was set at 4 mg/L. Since there was no susceptibility or resistance breakpoints determined for rifampicin against anaerobes, we designated isolates with MICs ≥32 μg/mL as resistance. The control isolate was C. difficile ATCC 700057.
Clinical Data
Clinical data were collected via an electronic medical record system to obtain basic information about patient age, hospitalization date, discharge date, inpatient floor, and sample collection date.
Data Analysis
SPSS version 23.0 for Windows (SPSS) was used for statistical analyses.
Results
Epidemiologic Analysis and Characterization of C. difficile Isolates
Of a total of 832 patients hospitalized in the neurosurgery department during the 24-month study period, six C. difficile isolates were identified from 19 patients suffering from diarrhea sent for C. difficile testing. Two strains were positive for both the tcdA and tcdB genes (A+B+), and these strains belonged to ST-35 and ST-14, respectively, whereas the remaining four strains were tcdA-negative/tcdB-positive (A–B+), and all four belonged to ST-37. Capillary electrophoresis mapping results showed that the four strains of C. difficile ST-37 isolated from the neurosurgery ward were almost indistinguishable (Figure 1B). The length of stay of all patients in the neurosurgery department is shown in Table 1.
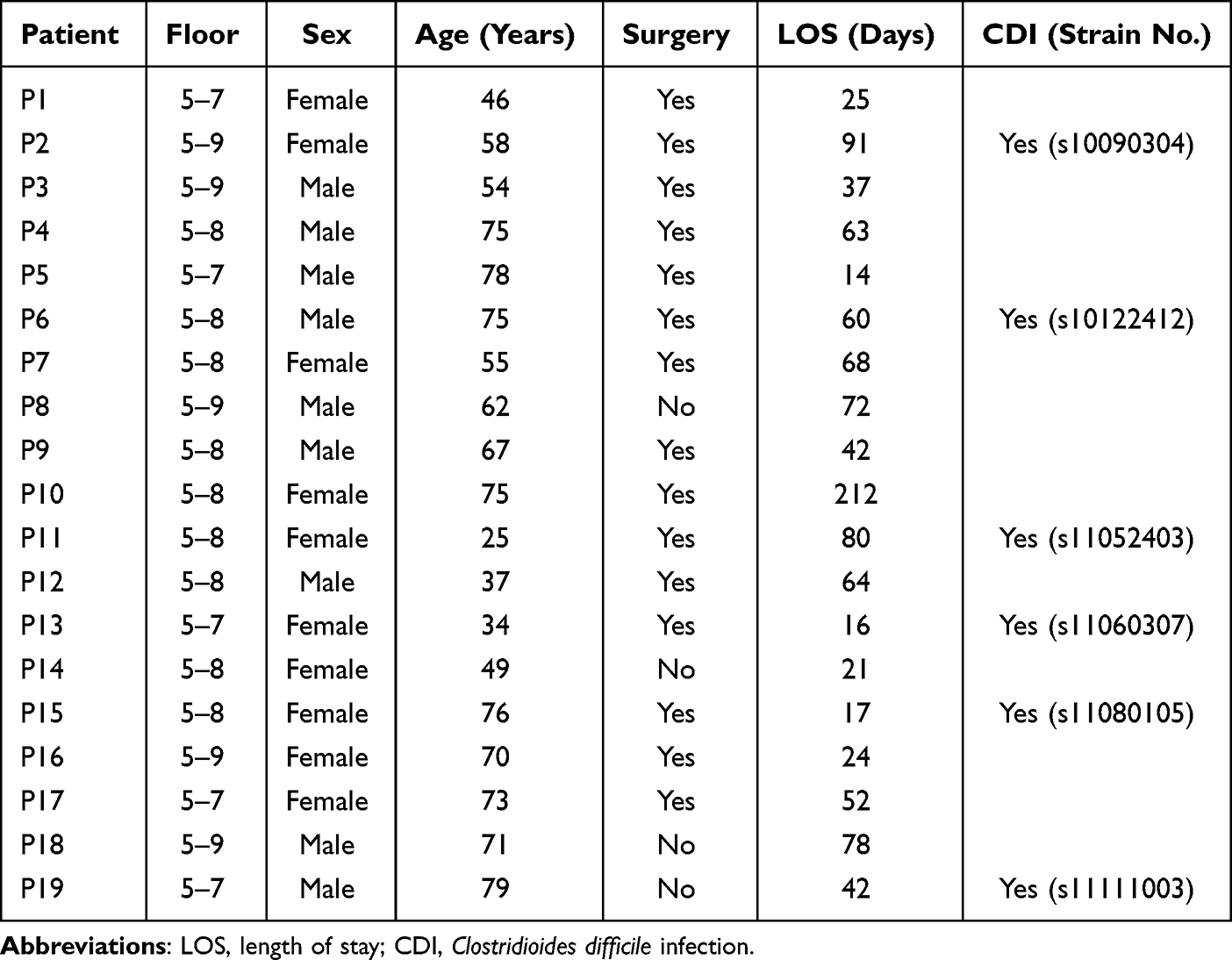 |
Table 1 Information Regarding the Patients Included in This Study |
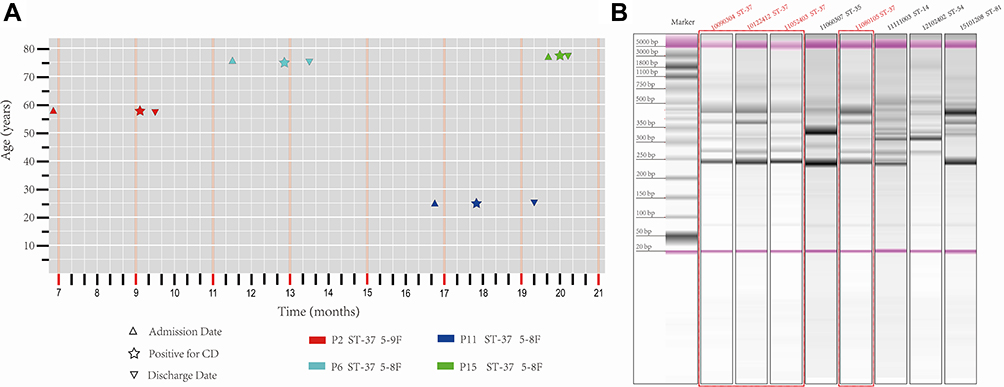 |
Figure 1 (A) Distribution of toxigenic C. difficile isolated from patients along with basic clinical data. Patient admission dates are marked by stars, the dates of becoming positive for CDI by triangles, and discharge dates by inverted triangles. (B) PCR ribotyping of toxigenic C. difficile isolated from the neurosurgery ward identified in the present study. Four isolates belong to ST-37/RT017 in this study are labeled with point brackets and in red font. |
Among the four ST-37 strains, C. difficile s10090304 was isolated from a stool sample from a 58-year-old female patient on the 9th floor of the neurosurgery ward (Figure 1A, P2). Strain s10122412 was isolated from a 75-year-old male patient on the 8th floor of the neurosurgery ward (Figure 1A, P6). Strain s11052403 was isolated from 25-year-old female patient on the 8th floor of the neurosurgery ward (Figure 1A, P11), and strain s11080105 was isolated from 76-year-old female patient on the 8th floor of the neurosurgery ward (Figure 1A, P15).
Similarly, six C. difficile isolates were typed as 3 different ribotypes: ST-37/RT017 (n = 4), ST-14/RT014 (n = 1) and ST-35/RT046 (n = 1).
Phylogenetic Analyses
The phylogenetic relationship between the four C. difficile ST-37/RT017 strains in the present study and all C. difficile ST-37/RT017 strains isolated from other departments in our hospital over the 24-month study period was examined. Bacterial DNA was extracted from a total of 19 strains, but due to substandard DNA quality, a library could not be constructed for C. difficile strains s11080105, which was isolated from a patient on the 8th floor of the neurosurgery ward. The phylogenetic tree revealed different group patterns. Strain s100122412, isolated from patient on the 8th floor of neurosurgery ward, clustered into an evolutionary branch with the strain isolated from the hematology ward. However, the remaining two strains isolated from patients in the neurosurgery ward clustered closely into the same evolutionary branch with the strain isolated from the hepatobiliary and pancreatic surgery ward (Figure 2). Another 16 non-duplicate toxigenic C. difficile strains belonging to ST-37/RT017 and isolated from diarrhea inpatients in other departments during the study period were combined to build the phylogenetic tree, as shown in Figure 2.
 |
Figure 2 Maximum likelihood phylogenetic tree of Clostridioides difficile ST-37/RT017 strains isolated from inpatients in our hospital during the 24-month study period, based on the core genome and grouped into different clades. Three isolates in this study are labeled with point brackets. A background color of Orange indicates that the strains were isolated from two patients on the 8th floor of the neurosurgery ward and green from a patient on the 9th floor. |
WGS Comparisons
Based on the phylogenetic tree, five closely related isolates were investigated to further define their genomic differences and relationship, and the paired-end DNA sequencing reads of these five strains were mapped to the reference genome. Two strains, s11052403 isolated from a patient on the 8th floor of the neurosurgery ward and s10090304 isolated from a patient on the 9th floor, were highly similar, differing only by two SNPs. However, the other strains differed from each other by more than three SNPs (Figure 3C).
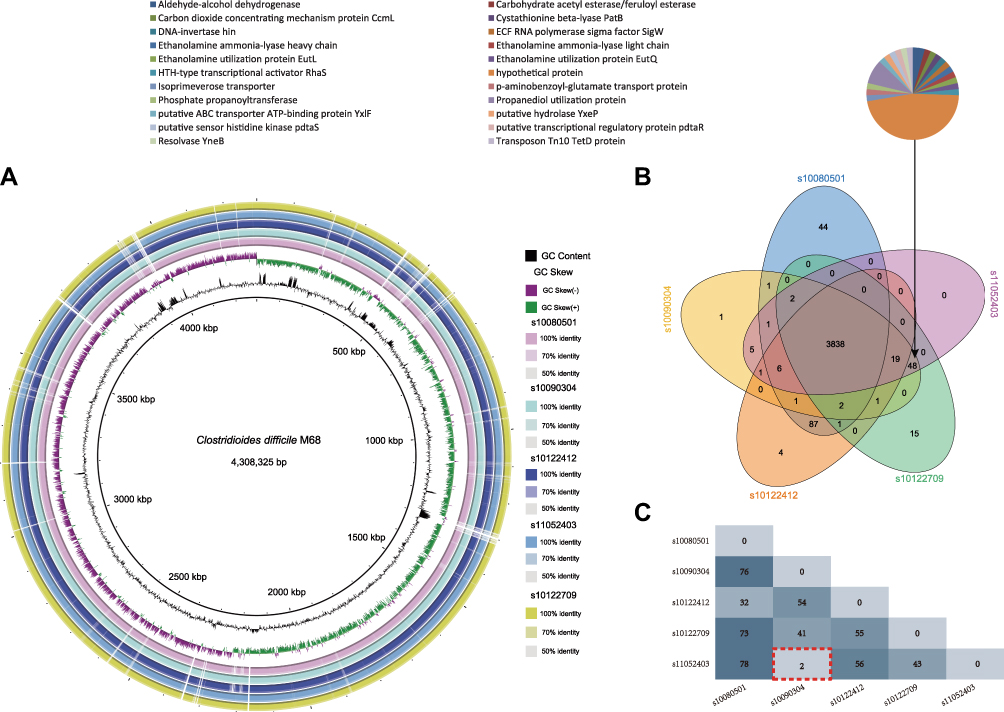 |
Figure 3 Comparative genomic analysis of five C. difficile ST-37/RT017 strains. (A) Whole-genome sequencing comparison of the strains. Circles from inside to outside indicate GC content of C. difficile 630, GC skew of strain C. difficile M68, and C. difficile stains s1008051, s10090304, s10122412, s11052403, and s10122709. Different BLAST identities are shown using different colors. (B) Venn diagram showing the number of unique, shared, or core genes between the five strains. Associated pie charts show the functional catalogues of the 48 predicted genes. (C) RAST annotation revealed several SNPs. Numbers in red boxes indicate the two SNPs that differed between C. difficile strain s10090304 and s11052403 in this study. Point brackets indicate the two different SNPs between the two isolates in this study. |
Comparisons of whole-genome sequences showed that the genomes of the five strains closely matched (Figure 3A). Comparative analyses identified a shared set of 3838 core genes without soft core genes and 238 different shell genes, as well as 44 genes unique to s10080501, 15 unique to s10122709, 4 unique to s10122412, 1 unique to s10090304, and none unique to s11052403. The annotations indicating the predicted functions of the proteins encoded by the different genes revealed that 22 proteins were encoded by only three strains, s11052403, s10090304, and s10122709, as shown in Figure 3B.
Antimicrobial Susceptibility
The antibiotic susceptibility patterns of C. difficile isolates are presented in Table 2. All toxigenic strains isolated from the neurosurgery department were sensitive to metronidazole, vancomycin, and linezolid. However, the four strains belonging to ST-37/RT017 were highly resistant to clindamycin, erythromycin, levofloxacin, moxifloxacin, and rifampicin. Among the four strains belonging to ST-37/RT017, three strains carrying ermB, tetM, AAC(6′)-Ie-APH(2″)-I and cdeA were distributed.
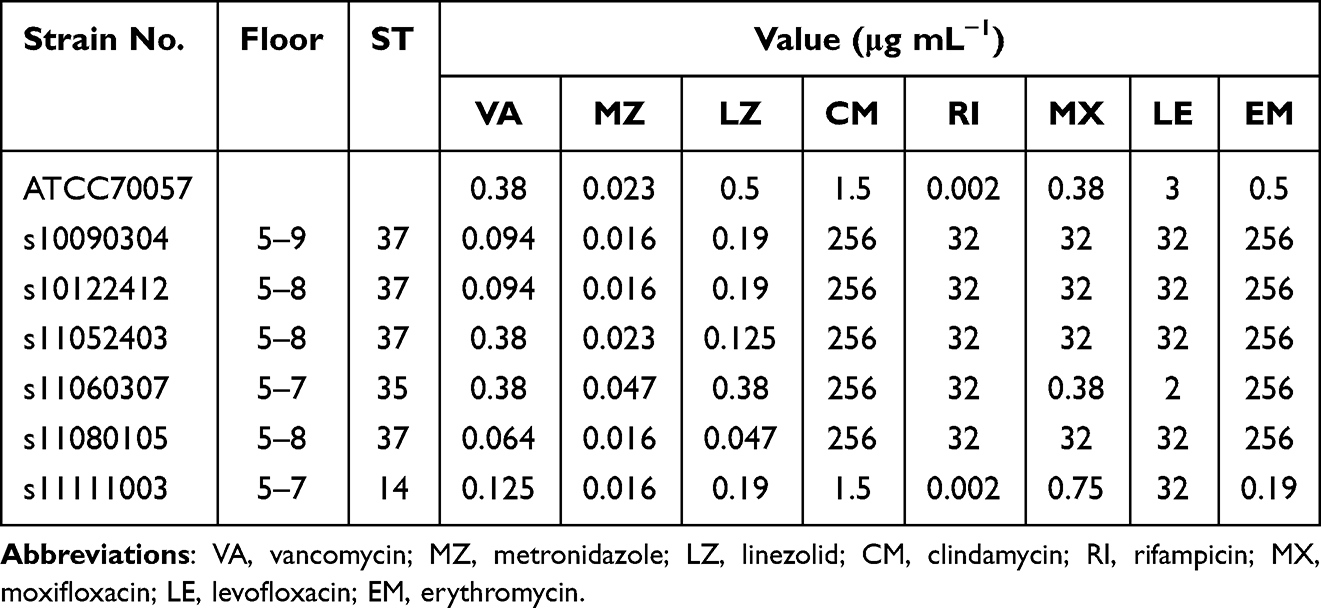 |
Table 2 Antimicrobial Susceptibility of the Six C. difficile Strains Isolated from the Neurosurgery Ward |
Discussion
CDI remains one of the most common healthcare-associated infections worldwide, with approximately 50% of CDI cases occurring during hospitalization.27 The molecular epidemiology of C. difficile is thus important, especially the epidemiology of C. difficile transmission in health-care settings.2 To our knowledge, this is the first study to investigate the epidemiology of C. difficile in a neurosurgery department.
Critically ill patients in some important wards, such as the neurosurgery department and intensive care unit (ICU), are particularly vulnerable to C. difficile.15 In this retrospective study, we found that the isolation rate of toxigenic C. difficile in neurosurgery patients with diarrhea was much higher than in patients from other departments in our previous studies.8 This phenomenon may be attributed to the use of prophylactic antibiotics,12,13 length of hospital stay, and older age,28,29 all of which are significantly associated with CDI.
Globally, C. difficile has emerged as an important cause of antimicrobial-associated diarrhea in humans, attributed to the production of two major toxins.6 However, not all toxigenic C. difficile strains produce both major toxins. As one of A–B+ toxin-types, C. difficile ST-37/RT017 could cause clinical symptoms as severe as other toxigenic strains, and has spread globally and furthermore been responsible for multiple outbreaks.30,31 In China, C. difficile ST-37/RT017 remains a major prevalent strain.7,8 In this study, genotyping by MLST identified three different STs. The hypervirulent strains C. difficile ST-1 and ST-11 were not detected. Among these strains, ST-37/RT017 was the dominant type in the neurosurgery department. Capillary electrophoresis ribotyping showed that the C. difficile ST-37/RT017 strains did not differ significantly from each other, but the phylogenetic tree revealed the presence of at least two C. difficile ST-37/RT017 clones in the neurosurgery department, and these clones were relatively independent of other ST-37/RT017 strains isolated in other departments during the same period. Although PCR-ribotyping is the gold-standard method for genotyping, it does not provide sufficient discriminatory power to distinguish related strains.32 Furthermore, it suggests that there was no epidemiologic transmission of C. difficile ST-37/RT017 strains between the departments in our hospital during this period. Comparing the same clones of C. difficile ST-37/RT017 strains isolated from the neurosurgery department, we found that there was no transmission of C. difficile ST-37/RT017 strains on the same floor, as the strains apparently derived from different clones. However, strains s11052403 and s10090304, which were isolated on different floors, derived from the same clone and were highly similar, differing by only two SNPs. According to a previous study in which 0–3 SNVs were identified between transmitted isolates obtained 124 to 364 days apart,33 there was clear epidemiologic transmission of C. difficile ST-37/RT017 strains between patients on different floors in the neurosurgery department. To the best of our knowledge, fecal-oral person-to-person transmission involving contaminated hands is the main route of CDI spread within inpatient health-care facilities.34 However, the hospitalization periods of the two affected patients did not overlap, and these patients did not share the same ward or even have direct contact based on the available clinical information. This strongly suggests that the epidemiologic transmission pathway of C. difficile ST-37/RT017 strains in the neurosurgery department was not via direct contact between patients. We speculate that environmental contamination may have played an important role in indirect transmission of the C. difficile ST-37/RT017 strains between the two patients, because available clinical information indicates they shared the same operating room.
A striking proportion of studies examining environmental sources of C. difficile in health-care institutions reported the importance of forward transmission mechanisms, including the transmission of spores by hands of health-care workers, as well as via contamination of bathrooms, toilets, hoppers, and janitorial equipment rooms.34–36 Notably, C. difficile spores can survive in the environment for months or years and can be found on multiple surfaces in health-care settings if surfaces are not adequately disinfected with the recommended working concentrations of chlorine-based cleaning agents.37,38 We found that all C. difficile ST-37/RT017 strains were resistant to numerous classes of antibiotics, with high MICs, including clindamycin, rifampicin, erythromycin, and fluoroquinolones, which could increase the risk of CDI.2 The acquisition of resistance to antimicrobial agents as well as enhanced virulence and survival properties may contribute to the dissemination of C. difficile in the environment.39 Undoubtedly, strict disinfection of clinical environments contaminated with C. difficile is essential to reduce transmission and achieve effective infection control in the hospital setting.40,41
Our study has some limitations. First, this was a retrospective study, and the extremely few samples were collected from neurosurgical patients over a 24-month period and therefore insufficient to accurately reflect the incidence of CDI. Determining the exact incidence rate will require further research. Second, though we thought to collect as much information as possible to observe the outcomes and risk factors associated with CDI, we could not search patient clinical information in detail because it had been archived. Third, we could not confirm the source of the C. difficile ST-37/RT017 strains because no environmental samples were collected.
Conclusions
Although C. difficile ST-37/RT017 strains are toxigenic and produce only the functional TcdB (A-B+), they can still cause serious outbreaks in hospitals, leading to severe disease.9 In mainland China, C. difficile ST-37/RT017 is one of the most prevalent phenotypes and exhibits a high rate of multidrug resistance.7,8,10 Further surveillance is absolutely necessary to detect clustering of cases and especially the epidemiologic transmission of C. difficile in China in order to avoid nosocomial outbreaks. Strict disinfection of environments contaminated with C. difficile is essential to reduce transmission and achieve effective infection control in the hospital setting. Moreover, it is crucial that clinical detecting for C. difficile should be conducted promptly in major hospitals in China, where CDI laboratory testing is lacking.
Data Availability
The genomic sequences of the 19 C. difficile isolates were deposited in GenBank under accession number PRJNA432876. The data of this study are available by contacting the corresponding author upon reasonable request.
Ethics Approval
This study was approved by the Ethics Committee of the First Affiliated Hospital, School of Medicine, Zhejiang University research ethics committee with an informed consent waiver (NO.2020-651). Due to the retrospective nature of the study, consent was waived. Therefore, we solemnly committed to cover patient data confidentiality and to comply with the Declaration of Helsinki.
Funding
This study was supported by Yunbo Chen, including National Nature Science Foundation of China (no. 82073609) and Research Project of Jinan Microecological Biomedicine Shandong Laboratory (JNL-2022037C).
Disclosure
All authors declare no competing interests in this study.
References
1. Sandhu BK, McBride SM. Clostridioides difficile. Trends Microbiol. 2018;26(12):1049–1050. doi:10.1016/j.tim.2018.09.004
2. McDonald LC, Gerding DN, Johnson S, et al. Clinical practice guidelines for Clostridium difficile infection in adults and children: 2017 update by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). Clin Infect Dis. 2018;66(7):987–994. doi:10.1093/cid/ciy149
3. Bauer MP, Notermans DW, van Benthem BH, et al. Clostridium difficile infection in Europe: a hospital-based survey. Lancet. 2011;377(9759):63–73. doi:10.1016/S0140-6736(10)61266-4
4. Smits WK, Lyras D, Lacy DB, Wilcox MH, Kuijper EJ. Clostridium difficile infection. Nat Rev Dis Primers. 2016;2(1):16020. doi:10.1038/nrdp.2016.20
5. Stubbs S, Rupnik M, Gibert M, Brazier J, Duerden B, Popoff M. Production of actin-specific ADP-ribosyltransferase (binary toxin) by strains of Clostridium difficile. FEMS Microbiol Lett. 2000;186(2):307–312. doi:10.1111/j.1574-6968.2000.tb09122.x
6. He M, Miyajima F, Roberts P, et al. Emergence and global spread of epidemic healthcare-associated Clostridium difficile. Nat Genet. 2013;45(1):109–113. doi:10.1038/ng.2478
7. Jin D, Luo Y, Huang C, et al. Molecular epidemiology of Clostridium difficile infection in hospitalized patients in Eastern China. J Clin Microbiol. 2017;55(3):801–810. doi:10.1128/JCM.01898-16
8. Chen YB, Gu SL, Wei ZQ, et al. Molecular epidemiology of Clostridium difficile in a tertiary hospital of China. J Med Microbiol. 2014;63(Pt 4):562–569. doi:10.1099/jmm.0.068668-0
9. Chia JH, Lai HC, Su LH, Kuo AJ, Wu TL. Molecular epidemiology of Clostridium difficile at a medical center in Taiwan: persistence of genetically clustering of A(-)B(+) isolates and increase of A(+)B(+) isolates. PLoS One. 2013;8(10):e75471. doi:10.1371/journal.pone.0075471
10. Chen YB, Gu SL, Shen P, et al. Molecular epidemiology and antimicrobial susceptibility of Clostridium difficile isolated from hospitals during a 4-year period in China. J Med Microbiol. 2018;67(1):52–59. doi:10.1099/jmm.0.000646
11. Zhao L, Luo Y, Bian Q, et al. High-level resistance of toxigenic Clostridioides difficile genotype to macrolide-lincosamide- streptogramin b in community acquired patients in Eastern China. Infect Drug Resist. 2020;13:171–181. doi:10.2147/idr.S238916
12. Han C, Song Q, Ren Y, Luo J, Jiang X, Hu D. Dose-response association of operative time and surgical site infection in neurosurgery patients: a systematic review and meta-analysis. Am J Infect Control. 2019;47(11):1393–1396. doi:10.1016/j.ajic.2019.05.025
13. Tsitsopoulos PP, Iosifidis E, Antachopoulos C, et al. A 5-year epidemiological study of nosocomial bloodstream infections in a neurosurgery department. Infect Control Hosp Epidemiol. 2010;31(4):414–417. doi:10.1086/651310
14. Sader E, Moore J, Cervantes-Arslanian AM. Neurosurgical infections. Semin Neurol. 2019;39(4):507–514. doi:10.1055/s-0039-1693107
15. Musa SA, Robertshaw H, Thomson SJ, Cowan ML, Rahman TM. Clostridium difficile-associated disease acquired in the neurocritical care unit. Neurocrit Care. 2010;13(1):87–92. doi:10.1007/s12028-010-9374-x
16. Kato Y, Tani T, Sotomaru Y, et al. Eight calves cloned from somatic cells of a single adult. Science. 1998;282(5396):2095–2098. doi:10.1126/science.282.5396.2095
17. Griffiths D, Fawley W, Kachrimanidou M, et al. Multilocus sequence typing of Clostridium difficile. J Clin Microbiol. 2010;48(3):770–778. doi:10.1128/JCM.01796-09
18. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi:10.1093/bioinformatics/btu170
19. Nurk S, Bankevich A, Antipov D, et al. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J Comput Biol. 2013;20(10):714–737. doi:10.1089/cmb.2013.0084
20. Aziz RK, Bartels D, Best AA, et al. The RAST server: rapid annotations using subsystems technology. BMC Genom. 2008;9(1):75. doi:10.1186/1471-2164-9-75
21. Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–2069. doi:10.1093/bioinformatics/btu153
22. Page AJ, Cummins CA, Hunt M, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015;31(22):3691–3693. doi:10.1093/bioinformatics/btv421
23. Croucher NJ, Page AJ, Connor TR, et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using gubbins. Nucleic Acids Res. 2015;43(3):e15. doi:10.1093/nar/gku1196
24. Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genom. 2011;12:402. doi:10.1186/1471-2164-12-402
25. Kumar S, Stecher G, Li M, Knyaz C, Tamura K, Battistuzzi FU. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35(6):1547–1549. doi:10.1093/molbev/msy096
26. Letunic I, Bork P. Interactive tree of life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 2011;39(WebServer issue):W475–W478. doi:10.1093/nar/gkr201
27. Khader K, Munoz-Price LS, Hanson R, et al. Transmission dynamics of Clostridioides difficile in 2 high-acuity hospital units. Clin Infect Dis. 2021;72(Suppl1):S1–S7. doi:10.1093/cid/ciaa1580
28. Drozdz M, Biesiada G, Piatek A, et al. Analysis of risk factors and outcomes of Clostridium difficile infection. Folia Med Cracov. 2018;58(4):105–116.
29. Ananthakrishnan AN. Clostridium difficile infection: epidemiology, risk factors and management. Nat Rev Gastroenterol Hepatol. 2011;8(1):17–26. doi:10.1038/nrgastro.2010.190
30. Imwattana K, Knight DR, Kullin B, et al. Clostridium difficile ribotype 017 - characterization, evolution and epidemiology of the dominant strain in Asia. Emerg Microbes Infect. 2019;8(1):796–807. doi:10.1080/22221751.2019.1621670
31. Xu X, Luo Y, Chen H, et al. Genomic evolution and virulence association of Clostridioides difficile sequence type 37 (ribotype 017) in China. Emerg Microbes Infect. 2021;10(1):1331–1345. doi:10.1080/22221751.2021.1943538
32. Seth-Smith HMB, Biggel M, Roloff T, et al. Transition from PCR-ribotyping to whole genome sequencing based typing of Clostridioides difficile. Front Cell Infect Microbiol. 2021;11:681518. doi:10.3389/fcimb.2021.681518
33. Eyre DW, Cule ML, Wilson DJ, et al. Diverse sources of C. difficile infection identified on whole-genome sequencing. N Engl J Med. 2013;369(13):1195–1205. doi:10.1056/NEJMoa1216064
34. Durovic A, Widmer AF, Tschudin-Sutter S. New insights into transmission of Clostridium difficile infection-narrative review. Clin Microbiol Infect. 2018;24(5):483–492. doi:10.1016/j.cmi.2018.01.027
35. Jia H, Du P, Yang H, et al. Nosocomial transmission of Clostridium difficile ribotype 027 in a Chinese hospital, 2012–2014, traced by whole genome sequencing. BMC Genom. 2016;17:405. doi:10.1186/s12864-016-2708-0
36. Asgary R, Snead JA, Wahid NA, Ro V, Halim M, Stribling JC. Risks and preventive strategies for Clostridioides difficile transmission to household or community contacts during transition in healthcare settings. Emerg Infect Dis. 2021;27(7):1776–1782. doi:10.3201/eid2707.200209
37. Jou J, Ebrahim J, Shofer FS, et al. Environmental transmission of Clostridium difficile: association between hospital room size and C. difficile infection. Infect Control Hosp Epidemiol. 2015;36(5):564–568. doi:10.1017/ice.2015.18
38. Freedberg DE, Salmasian H, Cohen B, Abrams JA, Larson EL. Receipt of antibiotics in hospitalized patients and risk for Clostridium difficile infection in subsequent patients who occupy the same bed. JAMA Intern Med. 2016;176(12):1801–1808. doi:10.1001/jamainternmed.2016.6193
39. Deneve C, Janoir C, Poilane I, Fantinato C, Collignon A. New trends in Clostridium difficile virulence and pathogenesis. Int J Antimicrob Agents. 2009;33(Suppl 1):S24–S28. doi:10.1016/S0924-8579(09)70012-3
40. Mayfield JL, Leet T, Miller J, Mundy LM. Environmental control to reduce transmission of Clostridium difficile. Clin Infect Dis. 2000;31(4):995–1000. doi:10.1086/318149
41. Apisarnthanarak A, Zack JE, Mayfield JL, et al. Effectiveness of environmental and infection control programs to reduce transmission of Clostridium difficile. Clin Infect Dis. 2004;39(4):601–602. doi:10.1086/422523
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Genomic Epidemiology of Clostridioides difficile ST81 in Multiple Hospitals in China
Xia X, Lv T, Zheng L, Zhao Y, Shen P, Zhu D, Chen Y
Infection and Drug Resistance 2024, 17:5535-5544
Published Date: 11 December 2024