Back to Journals » Clinical Ophthalmology » Volume 16
Retinitis Pigmentosa: Burden of Disease and Current Unmet Needs
Authors Cross N ![]() , van Steen C
, van Steen C ![]() , Zegaoui Y, Satherley A, Angelillo L
, Zegaoui Y, Satherley A, Angelillo L
Received 11 March 2022
Accepted for publication 12 May 2022
Published 20 June 2022 Volume 2022:16 Pages 1993—2010
DOI https://doi.org/10.2147/OPTH.S365486
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Nancy Cross,1 Cécile van Steen,2 Yasmina Zegaoui,1 Andrew Satherley,1 Luigi Angelillo2
1Market Access, Lightning Health, London, England, UK; 2Market Access HTA & HEOR, EMEA, Santen GmbH, Munich, Bavaria, Germany
Correspondence: Yasmina Zegaoui, Market Access, Lightning Health, 8 Devonshire Square, London, EC2M 4PL, England, UK, Tel +447770918748, Email [email protected]
Abstract: Retinitis Pigmentosa (RP), a group of inherited retinal dystrophies characterised by progressive vision loss, is the leading cause of visual disability and blindness in subjects less than 60 years old. Currently incurable, therapy is aimed at restricting degeneration of vision, treating complications, and helping patients to cope with the psychosocial impact of their disease. Hence, RP is associated with a high burden of disease. This paper describes the current therapeutic landscape for RP and the disease burden for patients, caregivers, and society. A review of available data was conducted in three stages: (1) a literature search of publicly available information on all domains of RP; (2) a systematic literature review using Medline, Embase, the Cochrane Library and grey literature (GlobalData) on epidemiology and cost of RP; and (3) qualitative research with senior physicians treating RP patients in the EU4 and the UK to validate research findings from secondary sources. RP severely impacts the daily lives of over a million people worldwide. Progressive vision loss significantly affects the ability to perform basic daily tasks, to maintain employment, and maintain independence. Consequently, most patients will experience reduced quality of life, with a greater emotional and psychological impact than other conditions related to vision loss such as diabetic retinopathy or age-related macular degeneration. RP is also associated with a high level of carer burden, arising from psychological and financial stress. The therapeutic landscape for RP is limited, with few treatment options and minimal guidance for the diagnosis, treatment, and care of patients. A curative intervention, voretigene neparvovec (Luxturna®), only exists for 1– 6% of patients. Although disease management can be successful in developing coping strategies, most patients live with this chronic, progressive condition without interventions to change the disease course. Innovative new therapies can transform the therapeutic landscape, provided appropriate clinical guidance is forthcoming.
Keywords: treatment, burden of disease, retinitis pigmentosa, visual impairment, retinal dystrophy
Introduction
Retinitis Pigmentosa (RP), the leading cause of visual disability and blindness in subjects less than 60 years old,1 encompasses a group of inherited retinal dystrophies (IRDs) characterised by progressive vision loss.2–4 With no current treatments to stop disease progression or restore vision, RP is considered incurable and is associated with a high unmet need. Therapy is aimed at restricting degeneration of vision, treating complications, and helping patients to cope with the psychosocial impact of their disease.5
Patients present with night vision problems which progress to the loss of peripheral vision, whilst the central retina is initially relatively spared.2,4 Individuals with RP experience a lower quality of life than healthy individuals, associated with reduced mobility and challenges with activities of daily living, increased financial burden due to difficulties staying in work, reduced autonomy and often social isolation.1,6,7
In the context of the high unmet need associated with RP, this paper aims to describe the burden of the disease on patients, caregivers, and society.
Methods
This review was conducted in three stages: (1) a literature search on all domains of RP; (2) a systematic literature review regarding the epidemiology and cost of RP; and (3) primary research with senior physicians treating RP patients to validate and augment research findings from secondary sources.
Literature Search on All Domains of RP
A literature search of publicly available information was performed using PubMed (https://pubmed.ncbi.nlm.nih.gov/) and Google Scholar (https://scholar.google.com/). The search was limited to English language publications between the years 2000–2021, using the search terms or variations of the following terms: “retinitis pigmentosa”; “inherited retinal dystrophies”; “etiology”; “pathophysiology”; “epidemiology”; “prevalence”; “disease presentation”; “disease progression”; “patient pathway”; “diagnosis”; “therapeutic landscape”; “burden of disease”; “unmet need”. Information was also retrieved from the following databases: Orphanet (https://www.orpha.net/consor/cgi-bin/index.php), GlobalData (https://www.globaldata.com), Genetic Testing Registry (https://www.ncbi.nlm.nih.gov/gtr); government websites; independent professional ophthalmology bodies and the webpages of relevant organisations including independent professional ophthalmology organisations, and visual disability charities.
Systematic Literature Review (SLR) for Epidemiology and Cost of RP
A search strategy was developed, with the search terms or variations of the following terms used in Medline, Embase, and the Cochrane Library databases: “retinitis pigmentosa”, “epidemiology” or “comorbidity” or “incidence” or “prevalence”, “disease progression”, “cost” or “budget” or “expenditure” or “resource utilisation” or “economic” or “hospitalisation”. Further details on the search strategy are available in Supplementary Material.
The search strategy identified peer-reviewed publications that met the following eligibility criteria, using the Population, Intervention, Comparator, Outcomes, Study Design (PICOS) tool: P: all RP patients; I: all pharmacological therapies; C: active comparator, placebo/sham; O: epidemiology data for RP, cost of illness data for RP, cost of illness data of comorbidities of patients with RP; S: all, except case reports and letters to the editor. The geographical scope was limited to France, Germany, Italy, Spain and the UK. Countries were selected based on available data through literature review and online open sources, and the number of clinical experts in the field, as measured by the number of publications in RP (using the Expertscape database: https://expertscape.com/).
Primary Research with Senior Physicians
Semi-structured qualitative research was completed with senior physicians responsible for the treatment of RP at specialist treatment centres in France, Germany, Italy, Spain, and the UK. Key topics addressed included the current treatment landscape for RP, the patient pathway between initial presentation of symptoms, diagnosis, treatment and follow up, and key clinical, economic, and humanistic unmet needs for patients and their support networks.
Systematic Selection of Studies
Flowcharts for the study can be viewed in Figures 1 and 2.
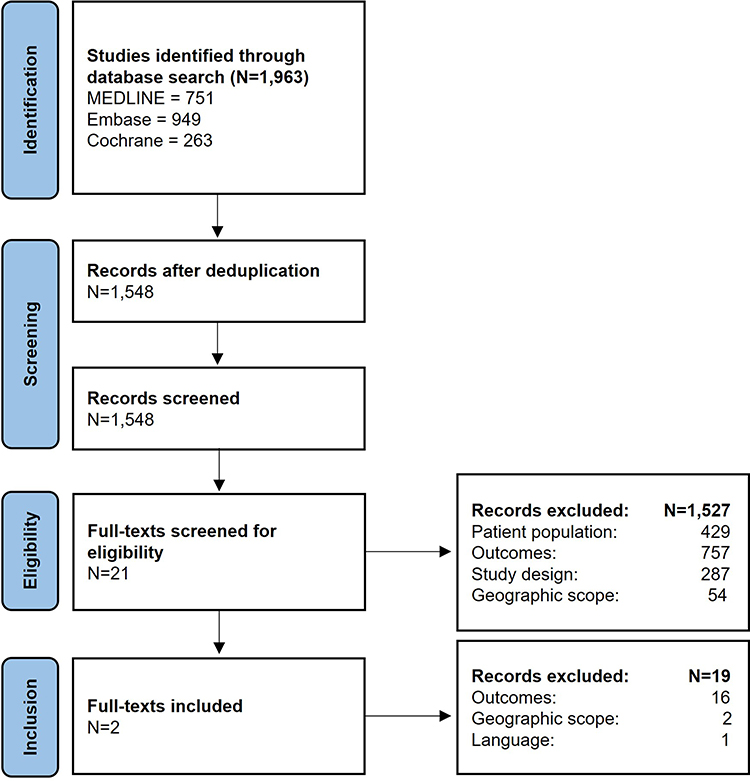 |
Figure 1 Identification of studies on the epidemiology of RP. Created using the official PRISMA 2020 flow diagram for new systematic reviews. Note: Adapted from Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ 2021;372:n71.59 For more information, visit: http://www.prisma-statement.org/. |
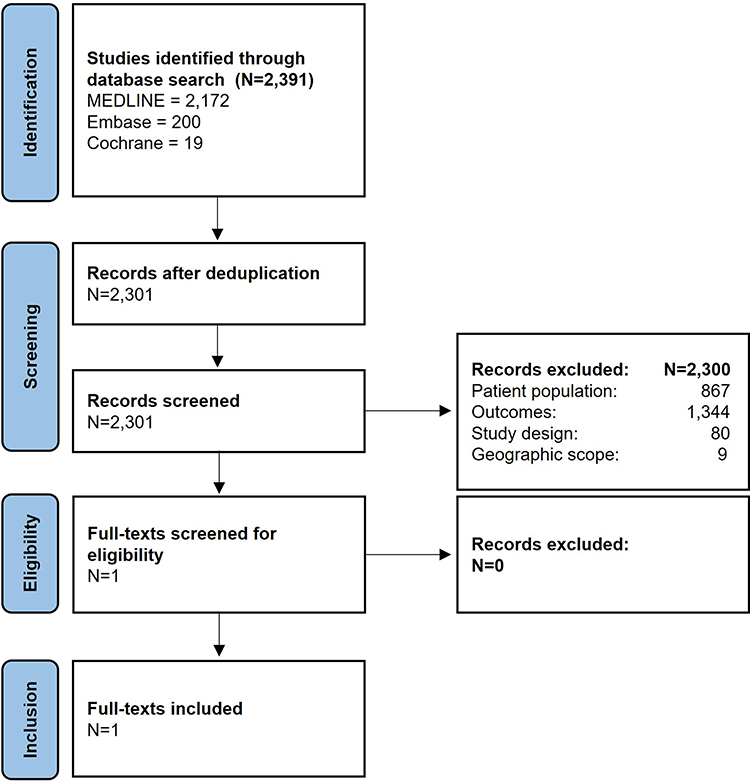 |
Figure 2 Identification of studies on the cost of RP. Created using the official PRISMA 2020 flow diagram for new systematic reviews. Note: Adapted from Page MJ, McKenzie JE, Bossuyt PM, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372:n71.59 For more information, visit: http://www.prisma-statement.org/. |
Etiology of Retinitis Pigmentosa
Over 100 genetic loci on 50 different genes have been implicated in patterns of inheritance and expression for RP3 although it is likely that there are many genes still to be identified.2 RP is generally inherited as mendelian traits, meaning that except for a small number of genes that can cause both dominant and recessive forms of RP, most genes are linked to only one form of inheritance.2
RP is generally categorized into syndromic and non-syndromic disease. Non-syndromic RP is the form of disease without systemic abnormalities, and accounts for 70–80% of all patients.3 When RP is associated with other, non-ocular syndromes and systemic disease, it is referred to as syndromic RP, accounting for 20–30% of all patients.3,4
Mutations in 80 different genes have been implicated in non-syndromic RP,4 and approximately 20% of cases are autosomal recessive, 10–20% are autosomal dominant (AD), and 10% are X-linked recessive.3 Figures from a recent study in Spain, representing the largest cohort of IRD patients reported worldwide, are in line with these estimates (see Figure 3).8 Whilst X-linked Retinitis Pigmentosa (XLRP) was previously thought to affect only males, it has been reported that female carriers can exhibit a range of phenotypes from asymptomatic to severe retinal disease.9 The remaining cases of RP are “sporadic” ie, there is no known inheritance pattern or molecular mechanism.3
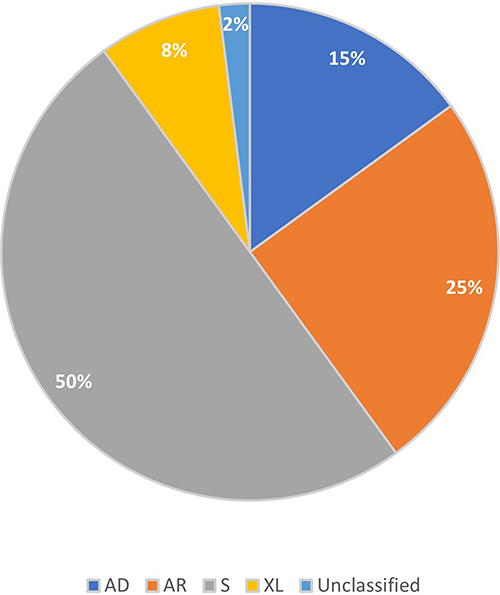 |
Figure 3 Classification of inheritance type of RP from a cohort of Spanish patients. Data from Perea- Romero et al.8 Abbreviations: AD, autosomal dominant; AR, autosomal recessive; S, sporadic; XL, X-linked. |
There have been some rare cases reported of di-genic diallelic inheritance caused by the simultaneous presence of a mutation in the RDS gene and ROM1 gene, RP caused by mitochondrial DNA mutations, uniparental isodisomy and incomplete penetrance.2,10
The unique loci and underlying genes identified for RP to date are outlined in Table 1, alongside the mapped loci and identified genes involved in each type of RP (RetNet, 2021; provided in the public domain by the University of Texas Houston Health Science Center).
 |
Table 1 Identified Genes and Loci Associated with Non-Syndromic RP |
Pathophysiology of Retinitis Pigmentosa
The genes associated with RP (see Table 1) play essential roles in the structure and function of photoreceptors responsible for vision.2,3 For example, mutations in Rhodopsin, the first component of the visual transduction pathway, encoded by the RHO gene (see Table 1), account for 30–40% of autosomal dominant RP cases.10
Typical disease presentation begins with night blindness, followed by progressive vision loss often resulting in loss of central vision later in the patient’s life. These visual symptoms reflect the degeneration of the two photoreceptor types. First, degeneration of rod photoreceptors which mediate achromatic vision at night or in low-light environments, and secondly degeneration of cone photoreceptors, responsible for high acuity central vision.12
The primary degeneration of rod photoreceptors, responsible for the initial presentation of night blindness, is caused by cell injury and death pathways. Mutations in the genes associated with RP can cause, for example, destabilization of the rod outer segment, increased vulnerability to oxygen toxicity, or interference with transduction pathways, resulting in degeneration of rod photoreceptors and progressive visual field loss.2–4
Rod degeneration can then contribute to the death of cone photoreceptors in several ways (as illustrated in Figure 4). For example, decreased cell density and increasing levels of oxygen at the outer retina trigger oxidative stress pathways; impaired cellular trafficking pathways result in reduced glucose transport into cone cells; and microglial activation, triggered by rod degeneration, stimulates a pro-inflammatory response that may contribute to the cell death.3,12 This sequence of cell death, with the secondary degeneration of cone photoreceptors, explains the central vision loss and dyschromatopsia (a deficiency in the perception of colors) that develops later in the disease.2,4
 |
Figure 4 Suggested mechanisms contributing to the death of cone photoreceptors in RP. Data from Narayan et al.12 |
The degeneration of rod and cone photoreceptors can be accelerated by phototoxic mechanisms, including mutations in retinol metabolism and acceleration of oxygen consumption, exacerbated by light exposure.3
The three clinical features of RP are vascular narrowing, the presence of bone spicule pigmentation, and optic nerve pallor.3,4 Narrowing of retinal vessels has been shown to correlate with a larger decline in visual acuity,13 although the exact cause for this is unknown.3 Pigmentation is caused by the detachment of Retinal Pigment Epithelium (RPE) cells which migrate to perivascular locations in the retina following photoreceptor degeneration, however the mechanisms behind the migration of RPE cells are not fully elucidated. Finally, optic nerve pallor is thought to result from the formation of glial cells which cover the optic disc, increase reflectivity, and produce the “waxy pallor” characteristic to RP.3,4
Epidemiology of Retinitis Pigmentosa
RP is the most common IRD, affecting more than 1.5 million patients worldwide,3,4 and is classified as a rare disease. Reported prevalence rates of IRDs vary across Europe, with under 1 in 7000 reported in Spain and approximately 1 in 3000–4000 reported in Denmark and Norway.8
Estimates for the worldwide prevalence of RP varying from 1 in 40004,12,14 to 1 in 3745.15 Estimates for the diagnosed prevalent patient population in the largest European countries range from 23,927 patients in Germany to 3617 in Spain (see Table 2),16 with non-syndromic RP accounting for 70–80% of all RP patients.3
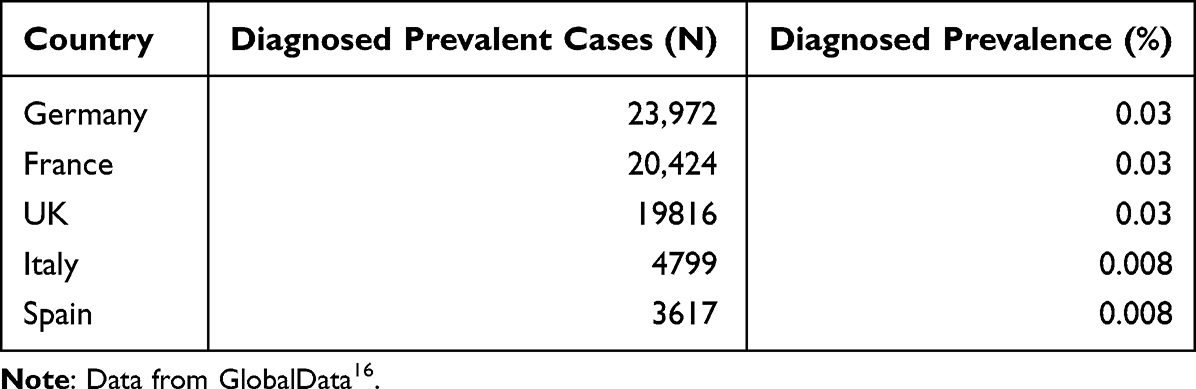 |
Table 2 Diagnosed Prevalent Cases of RP and Diagnosed Prevalence Percentage Across EU4 and UK Countries in 2021 (in Decreasing Prevalence) |
Disease Presentation and Progression
The initial symptoms of RP most commonly appear in young adulthood, with over three-quarters of patients developing symptoms by 30 years of age.3,4 However, due to delays in referral and diagnosis, diagnosis can often be delayed, with the average age of RP diagnosis reported to be 35.1 years (Figure 5).56
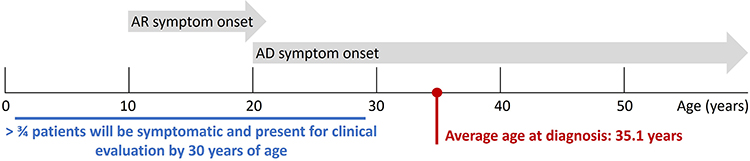 |
Figure 5 RP diagnosis age based on the onset of symptoms, split by genotype. Data from these studies.3,56 Abbreviations: AR, autosomal recessive; AD, autosomal dominant. |
Both clinical presentation and the age of onset of symptoms varies widely, dependent on the genotype (Figure 5).3,4 For example, individuals with the autosomal recessive (AR) form of RP will become symptomatic in their early teenage years, whereas those with autosomal dominant (AD) RP may not experience symptoms until aged 20–30 years3 or in some cases, until aged over 50 years.2
As the pathophysiology of RP begins with the death of rod photoreceptors, patients initially present with night blindness and difficulties with dark adaptation. In some cases, RP patients can present with loss of mid-peripheral vision although this is not a common early symptom.4 As disease progresses, central cone degeneration leads to a decline in visual acuity, which typically becomes apparent in middle age.4 The central retina remains relatively preserved in the early stages of disease and patients only experience a narrowing field of vision as the disease progresses.3,4 In mid-stage disease, dyschromatopsia often becomes present and patients may become photophobic.2
Patients of all genotypes will likely experience tunnel vision in the advanced stages of the disease (Figure 6).3 Due to severe restrictions in the visual field, most patients are classified as legally blind by the age of 40,14 ie, 20/200 vision or worse in the better eye or visual field restriction to ≤20° diameter,6 and typically, patients lose central vision by the age of 60.14 As visual acuity worsens, patients experience a greater impact on their daily lives (Table 3). Although most patients will become legally blind, complete loss of vision is relatively uncommon.3
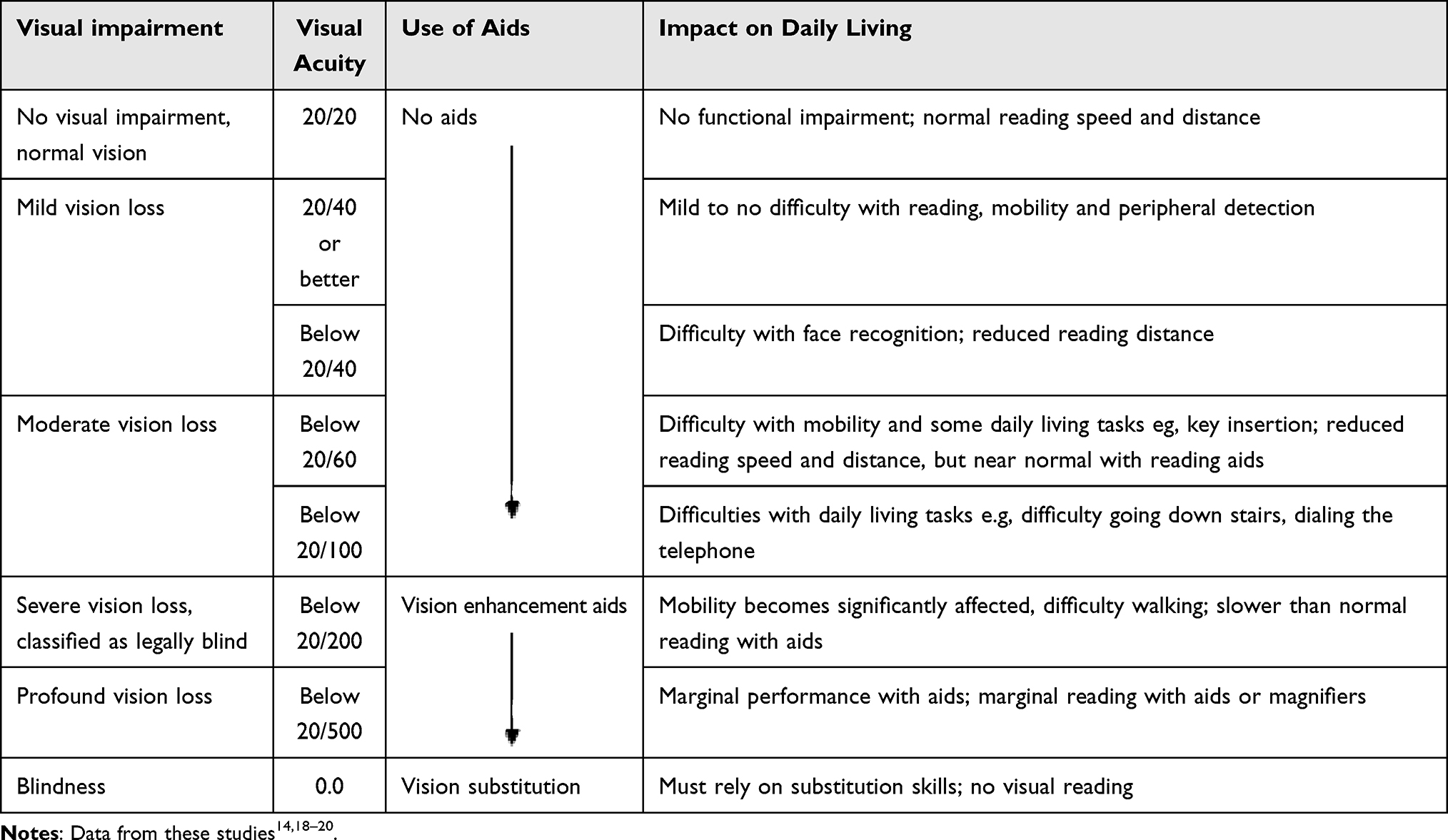 |
Table 3 Visual Acuity Related to Functionality and Daily Life |
 |
Figure 6 Progression of disease in patient with autosomal dominant RP. Top: representation of visual field; bottom: images from fundus photography; from left to right: progressing from normal visual field in a patient in teenage years (first column) to severely constricted visual field in a patient over 50 years (sixth column). Fundus images show the progression of vessel attenuation and retinal pigment epithelium atrophy, as well as sharp demarcation lines (arrow) between the healthy and affected retina. Note: Reprinted from Am J Ophthalmol, 140(5). Kozma P, Hughbanks-Wheaton DK, Locke KG, et al. Phenotypic characterization of a large family with RP10 autosomal-dominant retinitis pigmentosa: an Asp226Asn mutation in the IMPDH1 gene. 858–867. Copyright 2005, with permission from Elsevier.17 |
Although RP typically evolves over several decades, in extreme cases there can be a more rapid evolution over two decades.2 The most severe vision loss is expected for patients with XLRP3 who are four times more likely to have visual acuity ≤20/200 (legally blind), than those with autosomal dominant RP, and reach legally blind status on average by the age of 45 years.21
Patient Pathway
Initial Presentation of Disease
The initial presentation of RP typically involves vague visual disturbances including night blindness and difficulties with adapting to low light environments.3,4 For example, some patients find it difficult to drive at night, struggling to readjust to darkness after experiencing oncoming headlights.3
Symptoms may go relatively unnoticed in the early stages of disease as narrowing of the visual field is not initially obvious, and nighttime blindness can be hard to identify in artificially illuminated environments.3,4 Many patients, particularly children, are also able to compensate for peripheral visual loss that may develop4 using scanning eye movements to collect more information from their environment.22
As the disease progresses, patients start to experience symptoms characteristic of mid-stage disease. Night blindness is obvious, and patients may start to experience dyschromatopsia, photophobia and decreased visual acuity.2 At this stage, the disease has a substantial impact on the patient’s daily life, for example causing difficulties with driving at night, bumping into obstacles, and locating objects.22
Presentation of Patient to Primary Care
As the symptoms of RP become more advanced, patients are likely to consult an optician, non-specialist ophthalmologist, or their general practitioner (GP) (Figure 7).23,24 If a patient is aware of a family history of RP, they may be more likely to request an evaluation at an earlier stage or get a referral for genetic testing by either a specialist ophthalmologist or a geneticist.23
 |
Figure 7 Referral pathway for patients with RP. Data from Lightning Health. European ophthalmologist research; 2021.23 |
Assessment by an optician will typically include an examination of the retina using a slit lamp or an ophthalmoscope. If these tests reveal small, distinctive clumps of pigment around the retina, the optician may be able to detect RP.24,25 The healthcare provider may also perform a visual acuity determination, an examination of the central fundus25 and a field of vision test to evaluate changes to peripheral vision, although the latter is not offered routinely and may need to be requested by the patient if they are experiencing deficits in their peripheral vision.24
It is also possible that abnormalities may be detected during a routine eye test before the patient has reported any visual complaints.23
There is no clear set of signs and symptoms for RP as defined by scientific evidence, so diagnosis in the primary care setting is unlikely.26 If the screening physician has identified abnormalities or visual deficits characteristic of RP and/or the patient has a familial history of RP, they will usually be referred to an ophthalmologist to undergo further evaluation.26,27
Pediatricians should be particularly attentive to abnormalities such as ocular deviations. A child may be given glasses to correct for visual deficits, but if their vision does not improve, they will then be referred to an ophthalmologist.26 Pediatric patients with suspected RP experiencing poor balance and difficulties walking may also be referred to an ear, nose, and throat (ENT) specialist for further evaluation.26
Referral to Ophthalmologist: Timelines and Delays
The duration of time between initial patient presentation with symptoms of RP and referral to a specialist ophthalmologist can vary significantly between different settings across Europe. Often it can take 2–3 months to be referred to a specialist, depending on where the patient is initially assessed and the proximity or referral pathways to a specialist centre. However, in some cases specialist referral can be delayed by over a year following initial presentation, due to a range of contributing factors.23 For example, patients with early-stage disease often present with non-specific visual disturbances which may not be recognized by a physician (visual acuity is often affected later in the disease and abnormalities in the eye are often not clear in early-stage disease). Furthermore, patients in rural areas can face delays due to the geographical distance from a reference centre and a potential lack of referral pathways.23
It has also been reported that lack of awareness of RP amongst ophthalmologists as well as a lack of effective treatment options for the condition can reduce the impetus for specialist referral.23 Indeed, one report found that around 75% of patients with IRDs were frustrated with the lack of awareness and support for these conditions in the United Kingdom (UK).28
There are several factors that may lead to a reduced timeline for referral to a specialist ophthalmologist. For example, voretigene neparvovec (Luxturna®) is available in Germany, France, Italy, Spain and the UK for patients with confirmed biallelic RPE65 mutations,29–31 and referral timelines may be shortened if an RPE65 mutation is suspected and the patient may be eligible for treatment. Additionally, suspected cases of RP in children are usually prioritized to exclude the possibility of amblyopia, which, if not addressed quickly, could result in permanent deficits in vision. Rare disease networks have also increased awareness of RP to support earlier diagnosis and accelerated referral to specialist reference centres23 including the Alliance for Regenerative Medicine (ARM) and the European Organization for Rare Diseases (EURORDIS) which are dedicated to improving the lives of individuals with rare diseases across the world and Europe respectively.32,33 Additionally, the European Reference Network ERN-EYE, dedicated to rare eye diseases, consists of 44 healthcare providers across 20 countries in Europe. ERN-EYE aims to facilitate better access to accurate molecular diagnosis, reduction in the time to diagnosis and care, improved access to treatment, clinical trial involvement, and education.34
Despite these mitigating factors, a protracted period between initial presentation of symptoms and referral to a specialist centre for RP often means that when patients reach a formal diagnosis, they are likely to be in the advanced stage of disease, where visual field symptoms have become progressively worse and visual acuity is severely impaired.23
Patient Evaluation and Diagnosis
Guidelines for the diagnosis of RP are limited, and there is no real consensus on the techniques used in the evaluation of a patient. However, there are several key methods of investigation and evaluation commonly used to inform the diagnosis (Table 4).
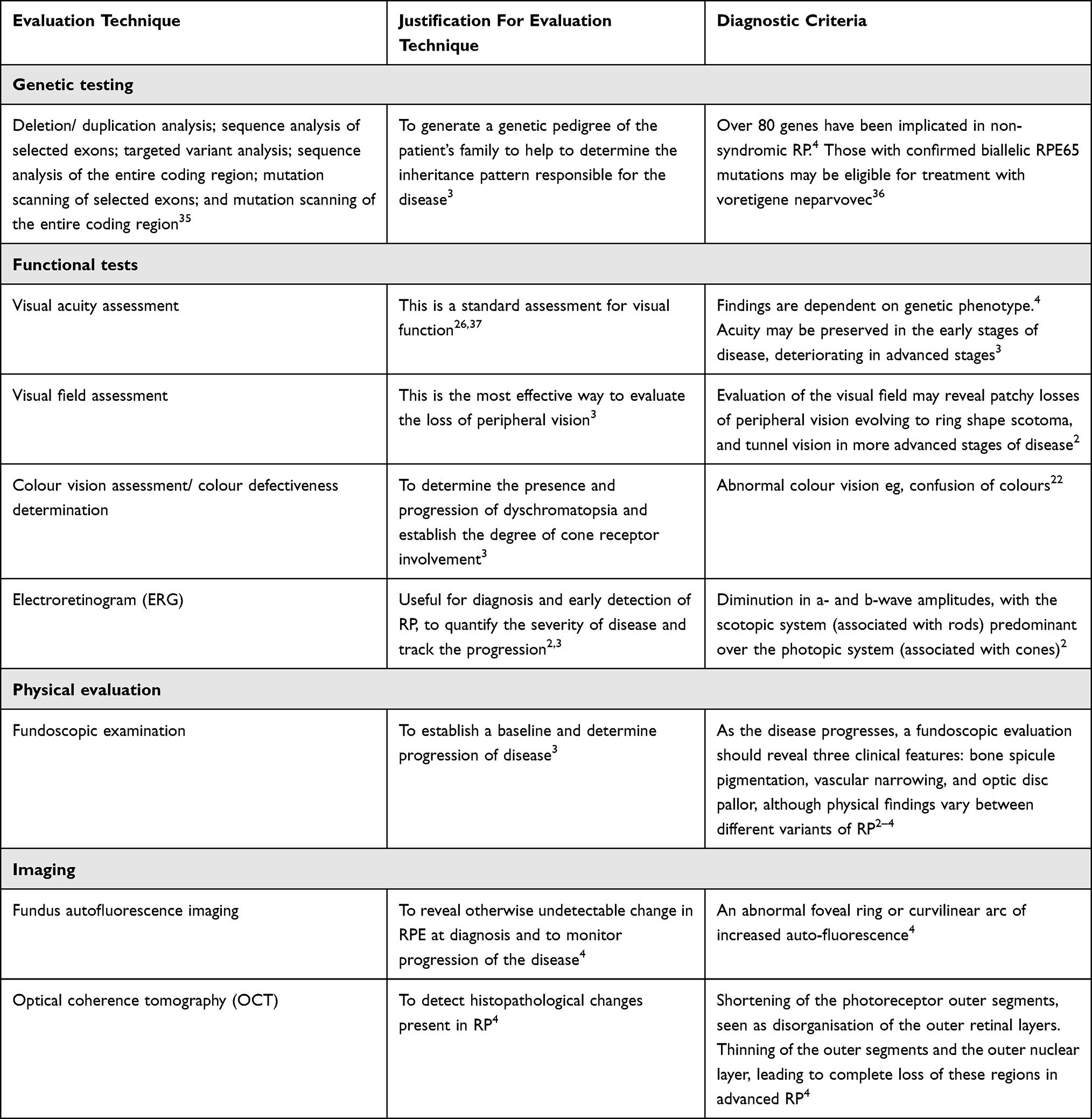 |
Table 4 Assessment Techniques Commonly Used to Inform Diagnosis of RP |
Given the high level of heterogeneity of RP and lack of universal access to genetic testing, genetic investigation can be challenging.4,26,38 Indeed, the genetic diagnosis rate has been previously reported to be as low as 50%.8 Therefore, many patients will never find out their sub-type of RP, affecting their understanding and acceptance of their disease and implications for their future, with a negative impact on quality of life.3 However, there have been significant advances in the methods used to identify genes,4 and in England it is expected that genetic testing will be offered in the future to all patients through the National Health Service (NHS).38
Evaluation of pediatric patients may differ from adult patients with RP, as young children may be unable to perform the functional tests applied to adults and the reliability of a visual field assessment in children under 7 years may be inadequate for diagnosis.39 Therefore, for young children and uncooperative adults, the use of an ERG, autofluorescence, and OCT is suggested.26 Leber congenital amaurosis (LCA), another IRD with a genetic and clinical overlap with RP, can also be diagnosed at a young age and therefore specific care is required to reach an accurate diagnosis in pediatric patients.4
The age limit for genetic testing is still under debate, as presymptomatic and/or predictive genetic testing at a young age can have a negative impact on the patient’s autonomy and quality of life.4,40
As part of the diagnostic process for RP, the psychological aspects of the condition must also be considered. Upon diagnosis, the patient may experience shock, fear, panic, disbelief, devastation, and a lack of confidence.7 Therefore, the diagnosing healthcare provider should offer support and education to the patient, including providing information on the chronic nature of RP, available treatments, and lifestyle adaptations to manage the disease. The ophthalmologist should also consider referral to the patient’s GP or a counsellor to support with the psychological aspects of disease management.22
Treatment and Management of Disease
Management of RP is generally coordinated by an ophthalmologist, although geneticists and pharmacists may also be consulted. A multi-disciplinary approach is required to achieve the best outcomes for the patient and to improve their quality of life.3,4,41
After diagnosis, the patient prognosis is relatively predictable,3 however, treatment pathways for RP are not well defined across Europe. Limited clinical guidance has been published in Spain as part of Clinical Practice Guidelines for Hereditary Retinal Dystrophies,26 in the UK as part of general guidance for healthcare providers,37 and in Germany by the Professional Association of Ophthalmologist on hereditary diseases of the retina, choroid or visual pathway.25
Although voretigene neparvovec is available for the 1–6% of RP patients with the RPE65 mutation,42 for most patients with RP, there is currently no cure or effective treatment to slow or stop disease progression.24
Traditionally, treatment has included vitamin A supplements to slow retinal deterioration, however, there is research demonstrating that this does not protect sight for patients with RP and can be detrimental to health in large doses.3,24
For example, adverse events resulting from excess Vitamin A include vomiting, diarrhea, headache due to cerebral edema, renal dysfunction, nerve dysfunctions including depression, and sometimes psychotic behaviour.26 Whilst Vitamin A toxicity in clinical practice is rare, this treatment option should be recommended with caution and patients taking Vitamin A should be evaluated regularly.24,26 Vitamin A toxicity is a particular concern for patients with the ABCA4 mutation, and therefore this mutation should be ruled out before Vitamin A is prescribed. Vitamin A is also unlikely to be prescribed to pediatric patients, pregnant women, patients with neuronal deficits, or patients with osteoporosis.26
Supportive care recommendations include sunlight protection, as light exposure can exacerbate phototoxic mechanisms leading to photoreceptor death. Therefore, patients are advised to avoid exposure to bright light and to wear sunglasses.3 There are several other interventions that could be implemented for low vision management as part of a multidisciplinary visual rehabilitation program. For example, magnification for poor high contrast visual acuity, field expanders for reduced visual field, night vision goggles for night blindness, and a cane for poor low contrast visual acuity.22
Treatment may also be recommended to target complications associated with RP, the most common of which are cataracts and macular edema. Cataracts often develop during the mid-stage of disease and can cause further restrictions in sight by blurring the remaining central visual field. Surgery can be performed to break up the cataract by phacoemulsification before an artificial lens is implanted.2 Macular edema occurs frequently and is often chronic in patients with RP, although acute episodes may be treated with a Carbonic Anhydrase inhibitor such as acetazolamide sodium.2 Other complications include glaucoma and myopia, which must be monitored, and inflammatory reactions occurring in the vitreous, which can be treated with cryotherapy or laser treatment if large exudates develop in the peripheral retina.2
As there is no standard treatment for most patients with RP, management of the disease becomes the primary goal with the aim of developing psychosocial and behavioral coping strategies and maintaining independence for as long as possible.3,7 Education of the patient and family is important, and genetic counselling is a standard part of disease management3 to address the impact on the patient’s work, social life, environmental interactions, and overall mental wellbeing.6 Counsellors and clinical psychologists can also help patients accept their diagnosis, set realistic goals for disease management, and maintain a healthy lifestyle. Additionally, eye clinics may have a sight loss advisor or liaison officer to offer practical and emotional support to patients.24
Referral to support groups can allow patients to meet other individuals with RP, potentially influencing their coping mechanisms, functioning, and autonomy.7 As the disease progresses and patients lose their autonomy, it is also important that friends and family members are educated on how to recognize and deal with signs of depression.3
Throughout the course of disease, follow-up examinations are recommended to monitor progression.26,41 In line with German guidance, follow-up examinations should be performed by an ophthalmologist every 1–2 years after diagnosis and should be done close to the patient’s home.41 Follow-up examinations may include visual acuity assessment, ERG, and visual field testing.2
Recent advances in cell and gene therapies and electronic retinal implants have shown promise for vision restoration and preservation for RP patients.3 However, due to the current lack of curative treatments, patients with RP experience a high burden of disease, with a greater emotional and psychological impact than other conditions that result in loss of vision which are mostly treatable and have later onset.7
Burden of Disease
RP is the leading cause of visual disability and blindness in subjects less than 60 years old.1 This accounts for 25% to 30% of all visual disability cases,14 affecting over 1.5 million people worldwide.4 The condition impacts all aspects of a patient’s life including their mental health, independence and autonomy, ability to work, social life, and ability to perform activities of daily living (ADLs).5,6,38 As such, the overall burden of disease on patients and society is high.
The Burden of Disease Increases as the Disease Progresses
During early stages of disease, the impact on the patient is minimal. Early-onset symptoms such as mild night blindness2 can be associated with disorientation, confusion, and fear;38 however, even as the visual field starts to reduce, patients often do not report difficulties with ADLs.14
As the disease progresses to mid-stage, night blindness becomes more severe and patients can start to lose their peripheral vision.2 Reductions in visual acuity and visual field are correlated with perceived difficulty performing ADLs and are strong predictors of lower quality of life, anxiety, and depression.1,6 As visual disability becomes more advanced, the impact on job security, and recognition and compensation at work also becomes greater.1,21 Patients with end-stage disease experience significantly reduced autonomy due to loss of peripheral vision and subsequent tunnel vision.2
The progressive nature of RP means that patients must continually adapt to deteriorating vision.38 Adjusting to this progressive vision loss is a protracted process relying on the development of adaptive skills and is clearly linked with the emotional and psychological wellbeing of the patient.1,6
Physical Burden and Disability
Progressive loss of vision reduces the patient’s mobility and ability to perform common tasks1 such as reading, shopping, driving, and physical activities.7,21 Common examples of the physical burden include finding it challenging to walk at night or climb stairs in the dark, missing someone’s hand when handshaking and stepping on objects outside the field of vision.2 This reduction in mobility and coordination can increase a patient’s risk of injury.5
Patients with RP also commonly report sleep disturbances due to a lack of photic input (up to 76% of patients) and headaches. This in turn, can increase the risk of cardiovascular, metabolic, and psychiatric disorders.5
Difficulty driving at night is a common concern throughout the disease,2 with one study finding that 80% of RP patients do not meet legal requirements for driving in France.1 This has considerable implications for the patient’s social life and employment.6 Furthermore, difficulties with steering, lane positions, traffic-gap judgement, speed, blindside detection and collision avoidance, mean that individuals with RP are more likely to be involved in road accidents, affecting not only themselves, but presenting risks for other road users.43 Due to difficulties navigating the physical environment, many people with RP find it difficult to use public transport after they stop driving, further reducing their autonomy.7
Financial Burden for the Patient
RP can impact a patient’s career progression and ability to stay in work to a greater extent than other conditions that result in loss of vision eg, age-related macular degeneration. This is due to the early onset, potentially impacting vision for patients at school and during their formative years of career development.5–7
Studies show lower employment rates (61.49% vs 83.92%) and lower income levels by the age of 40 years for RP patient vs age paired controls without the disease. The employment rate for RP patients tends to decrease as the disease progresses, for example from 79.5% in the low vision group to 67.6% in the blind group in one study in France (Figure 8).1
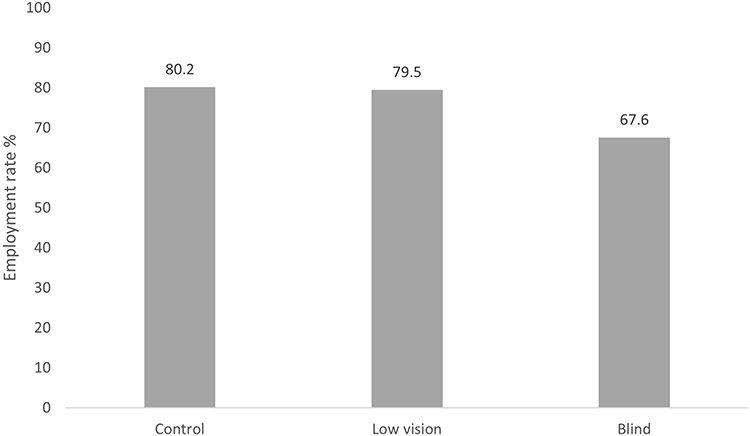 |
Figure 8 Employment rates across groups with different levels of visual impairment France. Data from Chaumet-Riffaud et al.1 |
Although patients may be granted visually disabled status or may be eligible for disability benefits (see Table 5), one study found that 50% of RP patients with low-vision were reluctant to request official recognition and financial support for their disability.1 This reluctance may stem from fears of discrimination or being treated differently in the workplace.7 In addition, the financial burden is increased by patient-borne costs associated with disease management, including travel to appointments, specialist equipment, home adaptations and domestic help.21 One study looking at out-of-pocket costs for patients with visual impairment in Australia, calculated the total personal costs for retinopathies at ASD$3318 per year, accounting for ASD$226 spent on medicinal products and equipment, ASD$167 on health and community services, ASD$1974 on informal care, and ASD$348 for other expenses.44
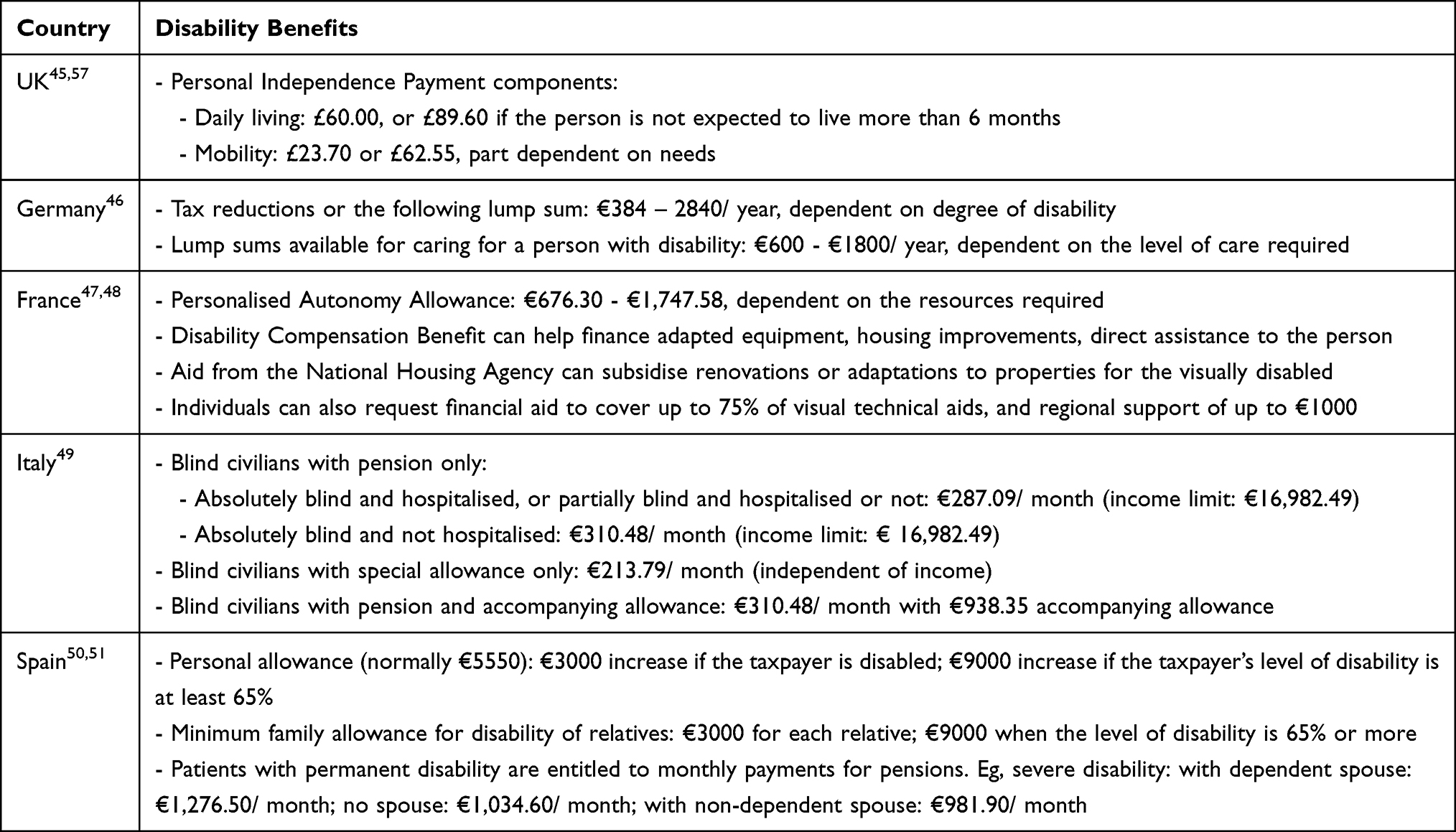 |
Table 5 Disability Benefits Available Across the EU4 and the UK in 2021 |
Psychological Burden and Impact on Patient Quality of Life
Adults living with RP are more likely to experience greater levels of psychological distress and depression and report lower levels of quality of life compared to adults living without the condition or with other health conditions.7 This is because RP normally has an earlier age of onset and is usually incurable.7
Patients often deal with perceived stigma due to their visual impairment, for example around their capabilities in social situations and the workplace, and their use of a cane or other visual aids. Minimising stigma is important in creating environments in which people with RP feel comfortable to adopt adaptive coping strategies.7
These factors have a significant impact on patient wellbeing, with a survey run by the charity Retina UK finding that 92% of RP patients considered that the disease affected their mental health.38
Patients’ mental health may be impacted by the presence of chronic visual hallucinations associated with vision loss, known as Charles Bonnet Syndrome. This has been reported in as many as 1 in 3 adults with advanced RP. These hallucinations are often not reported to healthcare professionals, in part due to the fear of experiencing stigma around psychiatric disorders, and can have severe negative emotional consequences for patients.53
Co-morbid mental health problems also contribute to increased treatment-related costs. Across multiple studies, it has been consistently shown that healthcare resource use is higher in patients with depression alongside a range of diseases compared to patients without co-morbidities.52
As patients experience greater difficulty performing ADLs as the disease progresses and visual impairment worsens, this is likely to contribute to patients feeling irritable, fatigued, disinterested, sad and potentially depressed.54 Additionally, financial burden, reduced job satisfaction and the need to frequently change jobs to suit abilities as the disease progresses all contribute to psychological stress and reduced quality of life.1,6,7 Worries may also manifest around the lack of protective laws in employment and the workplace culture, causing some people to conceal their condition due to fears of discrimination or employment termination.7
Individuals with RP also face considerable complications in maintaining their social life and social interactions. Night blindness can make dark places such as restaurants and theatres difficult to visit6 and other social activities and hobbies such as reading and playing sports may no longer be possible. This contributes to a sense of loss of identity and social isolation.7,21
Caregiver Burden and Burden on Families
RP can have significant emotional, physical, and financial impact on a patient’s family members, especially in cases of pediatric RP.38 Communication and interpersonal relationships are often disrupted, and patients often lose interest in spending time with extended family, leading to dissonance within families.6 Moreover, dependence on family members, especially as the disease progresses, can lead to psychological stress for those in a caregiving role and tensions between the patient and caregiver.6,7 Caregivers are also often forced to reduce their working hours to care for the patient and travel to medical appointments, which alongside having to pay for adaptive aids can contribute to financial stress.38
Due to the hereditary nature of the disease, family members may also experience feelings of guilt and blame. In particular, prospective parents may have concerns around reproductive risks and may fear societal judgement for choosing to have children despite these risks.6,37
Societal Burden
As RP progresses, patients become more dependent on others for daily tasks and are less able to contribute to society.7 For example, severe visual impairment is associated with lower employment rates (see Figure 8) and decreased productivity at work.1
People with RP are also more likely to use health services, compared to those without the condition,7 for example, due to other co-morbid ocular problems, headaches, nervous system disorders, and depression.5 Although the impact on healthcare resource use may be constrained as there are no approved treatments for most RP patients and the frequency and duration of inpatient hospital admissions are not significantly increased,5,21 a study in the United States found that individuals with RP had increased outpatient visits (on average 2.74 more visits annually) and a greater number of filled prescriptions (2.18 additional prescriptions filled annually).5,21
Reports show that RP is the largest contributor to the total costs of IRDs in the UK and the second largest in the United States (US), with an estimated total economic burden of £262.3 million and $3708.4 to $8790.6 million, respectively, in 2019. Wellbeing costs (the monetary value attributed to loss of disability-adjusted life years), loss of productivity, informal carer costs and healthcare system costs were key contributors to the overall economic burden.21
Burden on Pediatric Patients
As RP often develops in childhood or adolescence, when social interactions and interpersonal relationships are important in development, the social implications for an individual with RP can be extensive.6 An RP diagnosis can have an impact on development and success at school, and children may need considerable support to remain in mainstream education.38 Research also suggests that vision challenges at school that impact education can affect subsequent employment prospects and may increase the patient’s financial and psychological burden in later life.6
In Usher syndrome, where the retinal degeneration of RP is combined with bilateral sensorineural hearing loss, children experience significant burden. Depending on the subtype of Usher syndrome, children may derive from limited to no benefit from hearing aids and in Usher syndrome type 1 (Usher 1), many pediatric patients resort to using sign language to communicate. Children with Usher syndrome have also been reported to develop mental and behavioural disorders, including conduct disorder, schizophrenia, autism and learning difficulties. Dual sensory clinics are being established to promote accurate diagnosis of Usher syndrome in pediatric patients and provide supportive therapy for the disease, and associated mental health needs.58
Healthcare utilization and costs may be higher for pediatric patients with RP compared to older individuals with the disease. Data from the US indicates that total healthcare costs vary with age, with children under 12 years having the highest incremental costs. Additionally, for younger patients with RP and associated congenital comorbidities such as hearing impairments, the need for primary and specialty care to manage these chronic conditions may contribute to higher healthcare costs. For example, total annual healthcare costs for children aged <12 years were $11,072, compared to $9909 for those aged 55 to 64 years, $6871 for those aged 45–54, and $6019 for those aged 86 years and over.5
Conclusion
RP severely impacts the daily lives of over a million people across the world.4 Progressive vision loss significantly affects an individual’s ability to perform basic daily tasks, to maintain employment, to take part in social activities, and maintain their independence. Consequently, the burden of disease for RP patients is high, and most patients will experience dramatically reduced quality of life, with a greater emotional and psychological impact than other conditions that result in loss of vision such as diabetic retinopathy or age-related macular degeneration.6,7
RP is also associated with a high level of carer burden, arising from psychological and financial stress, and a wider societal burden, due to reduced productivity and increased healthcare resource use for these patients.1,5,6,38
For most RP patients today, there is no curative intervention available, with only 1–6% of patients eligible for treatment with voretigene neparvovec.42,55 Therefore, although disease management can be successful in developing coping strategies and encouraging acceptance of RP, most patients have to live with this chronic, progressive disease without any interventions to change the course of the disease.7
The therapeutic landscape for RP is currently limited, with few treatment options and minimal guidance for the diagnosis, treatment, and care of patients. New and innovative treatment strategies for RP have the potential to transform the therapeutic landscape for this patient group, however, to address the high burden of disease across Europe will require the development of appropriate clinical guidance and pathways for timely referral, diagnosis, and access to treatment.
Abbreviations
AD, autosomal dominant; ADL, activities of daily living; AR, autosomal recessive; ARM, Alliance for Regenerative Medicine; CDSR, Cochrane Database of Systematic Reviews; CENTRAL, Cochrane Central Register of Controlled Trials; DARE, Database of Abstracts of Reviews of Effects; ENT, ear, nose and throat; ERG, electroretinogram; ERN, European Reference Network; EU, European Union; EURORDIS, European Organisation for Rare Diseases; GP, general practitioner; HTA, health technology assessment; IRD, inherited retinal dystrophy; LCA, leber congenital amaurosis; NHS, National Health Service; NHSEED, NHS Economic Evaluation Database; OCT, optical coherence tomography; PICOS, P population; I: intervention; C: comparator; O: outcomes; S: study design; RP, retinitis pigmentosa; RPE, retinal pigment epithelium; PRISMA, Preferred Reporting Items for Systematic Reviews; S, sporadic; SLR, systematic literature review; UK, United Kingdom; US, United States; XL, X-linked; XLRP, X-linked retinitis pigmentosa.
Funding
This research was funded by Santen GmbH, Munich, Germany.
Disclosure
Cécile van Steen and Luigi Angelillo are employees of Santen. The views expressed in this article are the authors’ own and do not necessarily reflect the views of the company. The authors report no other conflicts of interest in this work.
References
1. Chaumet-Riffaud AE, Chaumet Riffaud P, Cariou A, et al. Impact of retinitis pigmentosa on quality of life, mental health, and employment among young adults. Am J Ophthalmol. 2017;177:169–174. doi:10.1016/j.ajo.2017.02.016
2. Hamel C. Retinitis pigmentosa. Orphanet J Rare Dis. 2006;1(1):40. doi:10.1186/1750-1172-1-40
3. O’Neal TB, Luther EE. Retinitis Pigmentosa. StatPearls Publishing; 2021.
4. Verbakel SK, van Huet RAC, Boon CJF, et al. Non-syndromic retinitis pigmentosa. Prog Retin Eye Res. 2018;66:157–186. doi:10.1016/j.preteyeres.2018.03.005
5. Frick KD, Roebuck MC, Feldstein JI, McCarty CA, Grover LL. Health services utilization and cost of retinitis pigmentosa. Arch Ophthalmol. 2012;130(5):629–634. doi:10.1001/archophthalmol.2011.2820
6. Jangra D, Ganesh A, Thackray R, et al. Psychosocial adjustment to visual loss in patients with retinitis pigmentosa. Ophthalmic Genet. 2007;28(1):25–30. doi:10.1080/13816810701201930
7. Garip G, Kamal A. Systematic review and meta-synthesis of coping with retinitis pigmentosa: implications for improving quality of life. BMC Ophthalmol. 2019;19(1):181. doi:10.1186/s12886-019-1169-z
8. Perea-Romero I, Gordo G, Iancu IF, et al. Genetic landscape of 6089 inherited retinal dystrophies affected cases in Spain and their therapeutic and extended epidemiological implications. Sci Rep. 2021;11(1):1526. doi:10.1038/s41598-021-81093-y
9. Churchill JD, Bowne SJ, Sullivan LS, et al. Mutations in the X-linked retinitis pigmentosa genes RPGR and RP2 found in 8.5% of families with a provisional diagnosis of autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2013;54(2):1411–1416. doi:10.1167/iovs.12-11541
10. Ferrari S, Iorio ED, Barbaro V, et al. Retinitis pigmentosa: genes and disease mechanisms. Curr Genomics. 2011;12(4):238–249. doi:10.2174/138920211795860107
11. RetNet. Summaries of genes and loci causing retinal diseases. Available from: https://sph.uth.edu/retnet/sum-dis.htm#C-complex.
12. Narayan DS, Wood JPM, Chidlow G, Casson RJ. A review of the mechanisms of cone degeneration in retinitis pigmentosa. Acta Ophthalmol. 2016;94(8):748–754. doi:10.1111/aos.13141
13. Nakagawa S, Oishi A, Ogino K, Majiyama Y, Kurimoto M, Yoshimura N. Association of retinal vessel attenuation with visual function in eyes with retinitis pigmentosa. Clin Ophthalmol. 2014;8:1487–1493. doi:10.2147/OPTH.S66326
14. Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368(9549):1795–1809. doi:10.1016/S0140-6736(06)69740-7
15. Orphanet. Prevalence and incidence of rare diseases: bibliographic data. Available from: https://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_decreasing_prevalence_or_cases.pdf.
16. GlobalData. Epidemiology and market size retinitis pigmentosa. Available from: https://www.globaldata.com/.
17. Kozma P, Hughbanks-Wheaton DK, Locke KG, et al. Phenotypic characterization of a large family with RP10 autosomal-dominant retinitis pigmentosa: an Asp226Asn mutation in the IMPDH1 gene. Am J Ophthalmol. 2005;140(5):858–867. doi:10.1016/j.ajo.2005.05.027
18. West SK, Rubin GS, Broman AT, Muñoz B, Bandeen-Roche K, Turano K. How does visual impairment affect performance on tasks of everyday life? The SEE Project. Salisbury Eye Evaluation . Arch Ophthalmol. 2002;120(6):774–780.
19. Kliegman RM, Lye PS, Bordini BJ, Toth H, Basel D. Paediatric Symptom-Based Diagnosis.
20. International Council of Ophthalmology. Visual standards: aspects and ranges of vision loss with emphasis on population surveys. Available from: https://lists.w3.org/Archives/Public/public-low-vision-comments/2016AprJun/att-0003/Colenbrander_Visual_Standards_Aspects_and_ranges_of_vision_loss_with_emp….pdf.
21. Chivers M, Li N, Pan F, Wieffer H, Slowik R, Leartsakulpanitch J. The burden of X-linked retinitis pigmentosa on patients and society: a narrative literature review. Clinicoecon Outcomes Res. 2021;13:565–572. doi:10.2147/CEOR.S297287
22. Herse P. Retinitis pigmentosa: visual function and multidisciplinary management. Clin Exp Optom. 2005;88(5):335–350. doi:10.1111/j.1444-0938.2005.tb06717.x
23. European Physician Research. Lightning health European ophthalmologist research; 2021.
24. RNIB. Understanding retinitis pigmentosa and other inherited retinal dystrophies. Available from: https://www.rnib.org.uk.
25. BVA. Erbliche Netzhaut-, Aderhaut- und Sehbahnerkrankungen. [Hereditary Diseases of theRetina, Choroid and Visual Tract]. Available from: https://www.awmf.org/uploads/tx_szleitlinien/045-023l_S1_Erbliche-Netzhaut-Aderhaut-Sehbahn-Erkrankungen_2021-09_01.pdf.
26. Ministero de Sanidad, Servicos Sociales, e Igualidad. Guía de Práctica Clínica para las Distrofias Hereditarias de Retina. [Clinical Practice Guideline for Hereditary Retinal Dystrophies]. Available from: https://www.researchgate.net/publication/346365712_Guia_de_Practica_Clinica_para_las_Distrofias_Hereditarias_de_Retina/link/60d3b26d458515ae7da76969/download.
27. NHS. Referral guideline ophthalmology overview. Available from: https://www.oxfordshireccg.nhs.uk/professional-resources/documents/clinical-guidelines/ophthalmology/Optom%20referral%20guidelines%20Ophthalmology.pdf.
28. Deloitte. The socioeconomic impact of inherited retinal dystrophies. Available from: https://www2.deloitte.com/au/en/pages/economics/articles/socioeconomic-impact-inherited-retinal-dystrophies.html.
29. Ronco V, Dilecce M, Lanati E, Canonico PL, Jommi C. Price and reimbursement of advanced therapeutic medicinal products in Europe: are assessment and appraisal diverging from expert recommendations? J Pharm Policy Pract. 2021;14(1):30. doi:10.1186/s40545-021-00311-0
30. AEMPS. Informe de Posicionamiento Terapéutico de voretigén neparvovec (Luxturna®) para distrofia retiniana asociada a la mutación RPE65 bialélica [Report on the therapeutic positioning of voretigene neparvovec (Luxturna®) for retinal dystrophyassociated with the biallelic RPE65 mutation]; 2021. Available from: https://www.aemps.gob.es/informa/informes-de-posicionamiento-terapeutico/informe-de-posicionamiento-terapeutico-de-voretigen-neparvovec-luxturna-para-distrofia-retiniana-asociada-a-la-mutacion-rpe65-bialelica/?lang=en.
31. Agenzia Italiana del Farmaco. Riclassificazione del medicinale per uso umano Luxturna ai sensi dell’art. 8, comma 10, della legge 24 dicembre 1993, n. 537. [Reclassification of the medicinal product for humanuse «Luxturna» pursuant to art. 8, paragraph 10, of the law of 24 December 1993, n. 537]. (Determina DG/1344/2020). (20A07274); 2020. Available from: https://www.gazzettaufficiale.it/atto/vediMenuHTML?atto.dataPubblicazioneGazzetta=2021-01-09&atto.codiceRedazionale=20A07274&tipoSerie=serie_generale&tipoVigenza=originario.
32. Alliance for regenerative medicine [homepage on the Internet]. Available from: https://alliancerm.org/.
33. EURORDIS. About EURORDIS. Available from: https://www.eurordis.org/about-eurordis. Accessed July, 2021.
34. European reference networks. Eye diseases (ERN-EYE). Available from: https://www.ern-eye.eu/network.
35. Genetic Testing Registry. Retinitis pigmentosa. Available from: https://www.ncbi.nlm.nih.gov/gtr/conditions/C0035334/.
36. EMC. Luxturna concentrate and solvent for solution for injection. Available from: https://www.medicines.org.uk/emc/product/9856/smpc#gref.
37. Knott L. Retinitis Pigmentosa. Patient. Available from: https://patient.info/doctor/retinitis-pigmentosa#nav-2.
38. NICE. Voretigene neparvovec for treating inherited retinal dystrophies caused by RPE65 gene mutations; 2019. Available from: https://www.nice.org.uk/guidance/hst11.
39. American Academy of Ophthalmology. Recommendations on clinical assessment of patients with inherited retinal degenerations; 2016. Available from: https://www.aao.org/clinical-statement/recommendations-on-clinical-assessment-of-patients.
40. Garrett JR, Lantos JD, Biesecker LG, et al. Rethinking the “open future” argument against predictive genetic testing of children. Genet Med. 2019;21(10):2190–2198. doi:10.1038/s41436-019-0483-4
41. G-BA. Supporting reasons to not include the diagnostics and care of patients with hereditary retinal degenerations in the catalog according to §116b Abs. 3 and 4 SGB V. s.l. Available from: https://www.g-ba.de/downloads/40-268-5204/2009-09-17-116b-Netzhaut_BMG.pdf.
42. Kang C, Scott LJ. Voretigene neparvovec: a review in RPE65 mutation-associated inherited retinal dystrophy. Mol Diagn Ther. 2020;24(4):487–495. doi:10.1007/s40291-020-00475-6
43. Patterson G, Howard C, Hepworth L, Rowe F. The impact of visual field loss on driving skills: a systematic narrative review. Br Ir Orthopt J. 2019;15(1):53. doi:10.22599/bioj.129
44. Wong EYH, Chou S-L, Lamoureux EL, Keeffe JE. Personal costs of visual impairment by different eye diseases and severity of visual loss. Ophthalmic Epidemiol. 2008;15(5):339–344. doi:10.1080/09286580802227394
45. Gov.UK. Personal Independence Payment (PIP). Available from: https://www.gov.uk/pip.
46. Bundesministerium fer Justiz. Einkommensteuergesetz (EStG) §33b Pauschbeträge für Menschen mit Behinderungen, Hinterbliebene und Pflegepersonen. [IncomeTax Act (EStG) § 33b lump sums for people with disabilities, surviving dependents and carers]. Available from: https://www.gesetze-im-internet.de/estg/__33b.html.
47. République Française. Vivre à domicile avec un handicap visual. [Living at home with a visual impairment 48 - Recognition of visual impairment and financial aid]. Available from: https://www.pour-les-personnes-agees.gouv.fr/vivre-a-domicile/vivre-a-domicile-avec-une-maladie-ou-un-handicap/vivre-domicile-avec-un-handicap-visuel#anchor6.
48. ORCAM. Reconnaissance du handicap visual et aides financières. Available from: https://www.orcam.com/fr/blog/reconnaissance-handicap-visuel/.
49. INPS. Risultati per tabelle. [Results by tables]. Available from: https://www.inps.it/Search122/ricercaNew.aspx?sTrova=tabelle&sDate=&sCategoria=Notizia.
50. PWC. Spain – individual deductions. Worldwide tax summaries. Available from: https://taxsummaries.pwc.com/spain/individual/deductions.
51. SeguridadSocial. Minimum Amounts. Available from: https://www.seg-social.es/wps/portal/wss/internet/Pensionistas/Revalorizacion/30434.
52. McDaid D, Park A-L. Counting all the costs: the economic impact of comorbidity. Karger. 2015;179:23–32.
53. O’Hare F, Bentley SA, Wu Z, Guymer RH, Luu CD, Ayton LN. Charles Bonnet syndrome in advanced retinitis pigmentosa. Ophthalmology. 2015;122(9):1951–1953. doi:10.1016/j.ophtha.2015.03.006
54. Mojon-Azzi SM, Sourza-Poza A, Mojon DS. Impact of low vision on well-being in 10 European countries. Ophthalmologica. 2008;222(3):205–212. doi:10.1159/000126085
55. The World Bank. Population estimates and projections. Available from: https://databank.worldbank.org/source/population-estimates-and-projections#.
56. Tsujikawa M, Wada Y, Sukegawa M. Age at onset curves of retinitis pigmentosa. Arch Ophthalmol. 2008;126(3):337–340. doi:10.1001/archopht.126.3.337
57. RNIB. Benefits rates and payments. Available from: https://www.rnib.org.uk/information-everyday-living-benefits-and-concessions/benefits-rates-and-payments.
58. Toms M, Pagarkar W, Moosajee M. Usher syndrome: clinical features, molecular genetics and advancing therapeutics. Therapeut Adv Ophthalmol. 2020;12:2515841420952194.
59. Page MJ, McKenzie JE, Bossuyt PM, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372:n71. doi:10.1136/bmj.n71
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Current and Future Treatment of Retinitis Pigmentosa
Cross N, van Steen C, Zegaoui Y, Satherley A, Angelillo L
Clinical Ophthalmology 2022, 16:2909-2921
Published Date: 31 August 2022