Back to Journals » Clinical Ophthalmology » Volume 16
Current and Future Treatment of Retinitis Pigmentosa
Authors Cross N ![]() , van Steen C
, van Steen C ![]() , Zegaoui Y, Satherley A, Angelillo L
, Zegaoui Y, Satherley A, Angelillo L
Received 9 April 2022
Accepted for publication 14 July 2022
Published 31 August 2022 Volume 2022:16 Pages 2909—2921
DOI https://doi.org/10.2147/OPTH.S370032
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Nancy Cross,1 Cécile van Steen,2 Yasmina Zegaoui,1 Andrew Satherley,1 Luigi Angelillo2
1Market Access, Lightning Health, London, UK; 2Market Access, Health Technology Assessment & Health Economics and Outcome Research, Europe, the Middle East and Africa, Santen GmbH, Munich, Bavaria, Germany
Correspondence: Yasmina Zegaoui, Market Access, Lightning Health, 8 Devonshire Square, London, EC2M 4PL, UK, Tel +44 7770918748, Email [email protected]
Abstract: Retinitis Pigmentosa (RP) is a group of inherited retinal dystrophies (IRDs) characterised by progressive vision loss. Patients with RP experience a significant impact on daily activities, social interactions, and employment, reducing their quality of life. Frequent delays in referrals and no standard treatment for most patients also contribute to the high unmet need for RP. This paper aims to describe the evolving therapeutic landscape for RP including the rationale for advanced therapy medicinal products (ATMPs). A review of available data was conducted in three stages: (1) a search of publicly available literature; (2) qualitative research with physicians treating RP patients in France, Germany, Italy, Spain, and the UK; and (3) a review of leading candidates in the RP pipeline. Globally, there are currently over 100 drugs in development for RP; 50% of which are ATMPs. Amongst the 15 cell and gene therapies in late-stage development, 5 leading candidates have been selected to profile based on the development stage, drug target and geography: gene therapies AGN-151597, GS-030 and VMCO-1 and human stem cell therapies jCell and ReN-003. Hereditary retinal diseases are suitable for treatment with cell and gene therapies due to the accessibility of the retina and its immune privilege and compartmentalisation. Therapeutic approaches that aim to rescue photoreceptors (eg gene therapies) require that non-functional target cells are still present, whereas other therapies (eg cell therapies) are not reliant on the presence of viable photoreceptors. Gene therapies may be attractive as their fundamental goal is to restore vision; however, cell therapies will likely have a broader application and do not rely on genetic testing, which can delay treatment. Ensuring effective therapeutic options for RP patients across disease stages requires the continued diversification and advancement of the development pipeline, and sustained efforts to promote early patient identification and timely diagnosis.
Keywords: treatment, retinitis pigmentosa, retinal dystrophy, cell therapy, gene therapy, therapeutic landscape
Introduction
Retinitis Pigmentosa (RP) is a group of inherited retinal dystrophies characterised by progressive vision loss.1,2 RP is the most common inherited retinal dystrophy (IRD), with prevalence rates reported between 1 in 4000 and 1 in 3745,3,4 affecting over 1.5 million patients worldwide.2,5 Whilst RP can occur together with systemic disease, ie, syndromic RP, non-syndromic RP, involving vision loss alone, accounts for the majority of cases.2
RP is a progressive disease, with patients initially experiencing loss of night vision, followed by reduced visual acuity and a gradual narrowing of the visual field.2 By the age of 40, most patients are classified as legally blind.13 Worsening symptoms are associated with increased difficulty performing daily activities and reduced autonomy. This results in difficulties staying in work, higher levels of anxiety and depression, social isolation, and an overall reduced quality of life. As such, RP has significant effects across all domains of the patient’s life and there is a high burden on patients, families and caregivers, and society.1,6,7
With only one approved therapy for a small sub-population of patients, most patients are limited to the best supportive care, which is often inadequate to address these unmet needs.14 However, with recent advances in potentially curative cell and gene therapies, the evolving treatment landscape offers a promising future for patients with RP.
Methods
Literature Search
A literature search of publicly available information was performed using PubMed (https://pubmed.ncbi.nlm.nih.gov/) and Google Scholar (https://scholar.google.com/). The search was limited to English language publications between the years 2000–2021, using the search terms or variations of the following terms: “retinitis pigmentosa”; “inherited retinal dystrophies”; “epidemiology”; “prevalence”; “disease presentation”; “disease progression”; “patient pathway”; “diagnosis”; “burden of disease”; “unmet need”; “therapeutic landscape”; “treatment”; “advanced therapy medicinal products” or “ATMP”; “cell therapy”; “gene therapy”.
Information was also retrieved from the GlobalData database (https://www.globaldata.com) and government websites.
Primary Research with Senior Physicians
Semi-structured qualitative research was completed with senior physicians responsible for the treatment of RP at specialist treatment centres in France, Germany, Italy, Spain, and the UK. Key topics addressed included the current treatment landscape for RP, the patient pathway between initial presentation of symptoms, diagnosis, treatment and follow-up, and key clinical, economic, and humanistic unmet needs for patients and their support networks.
Systematic Selection of Leading Candidates in Development for RP
The 19 drug candidates identified in Phase II or III development in the US, EU or globally were subject to further analysis to determine their eligibility for inclusion in this publication (Figure 1). For inclusion, therapies were required to have a broad application for the RP patient population to allow for better comparison between therapies. Commercialisation and distribution capabilities of the sponsors were analysed, resulting in the exclusion of one product under development by a university.
 |
Figure 1 Identification of leading candidates in RP. Created using the official PRISMA 2020 flow diagram for new systematic reviews. Notes: Adapted from: Page MJ, McKenzie JE, Bossuyt PM et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372:n71. doi:10.1136/bmj.n71.47 Copyright © 2021, BMJ Publishing Group Ltd. For more information, visit: http://www.prisma-statement.org/. |
The exclusion of the 11 products in Table 1 results in the identification of 8 candidates in the evolving treatment landscape of RP. As this review focuses on the rationale for cell and gene therapies in RP, further 3 products were excluded, leaving 5 key cell and gene therapies (outlined in Table 2).
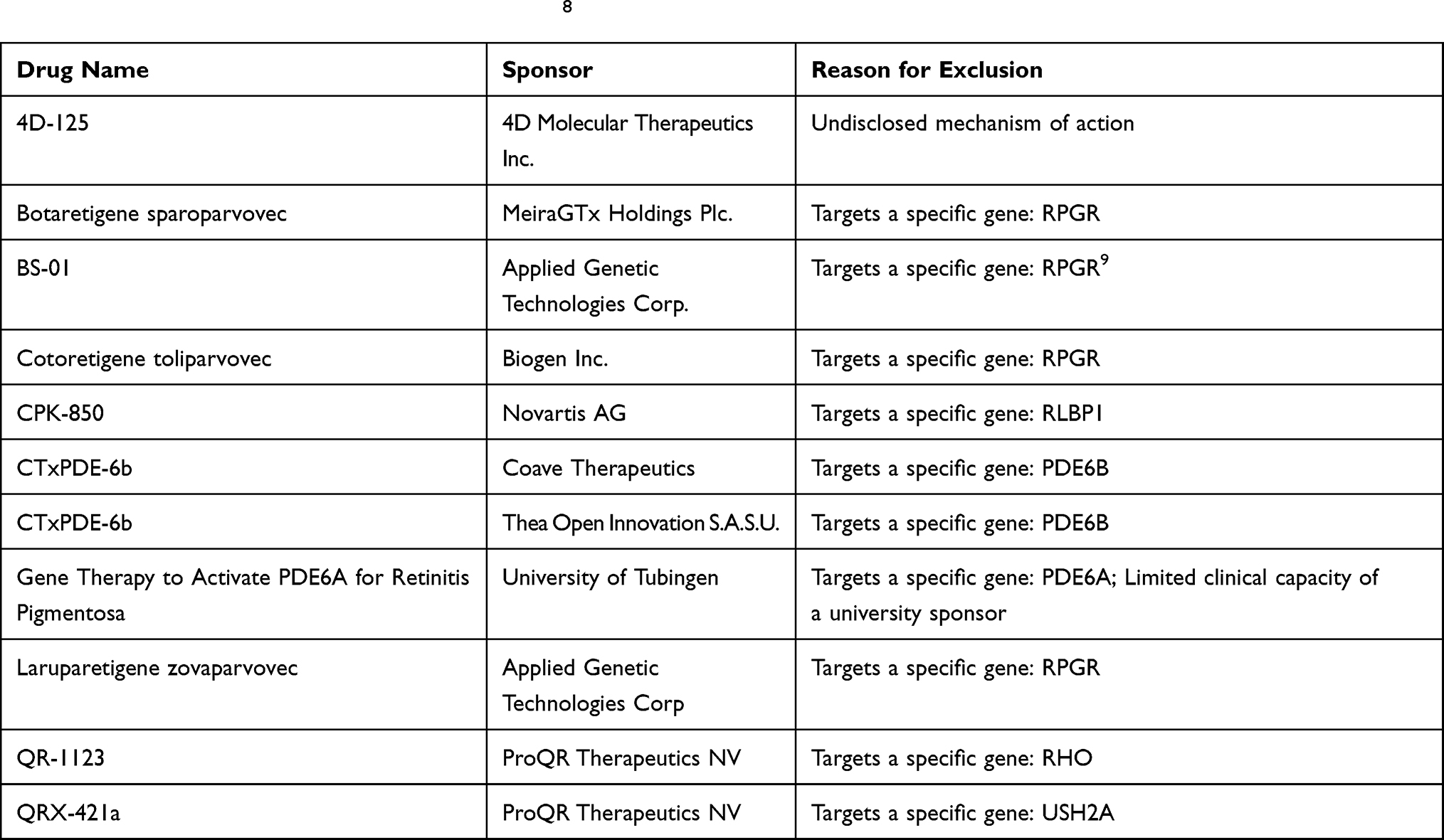 |
Table 1 Exclusion of Candidates in the RP pipeline8 |
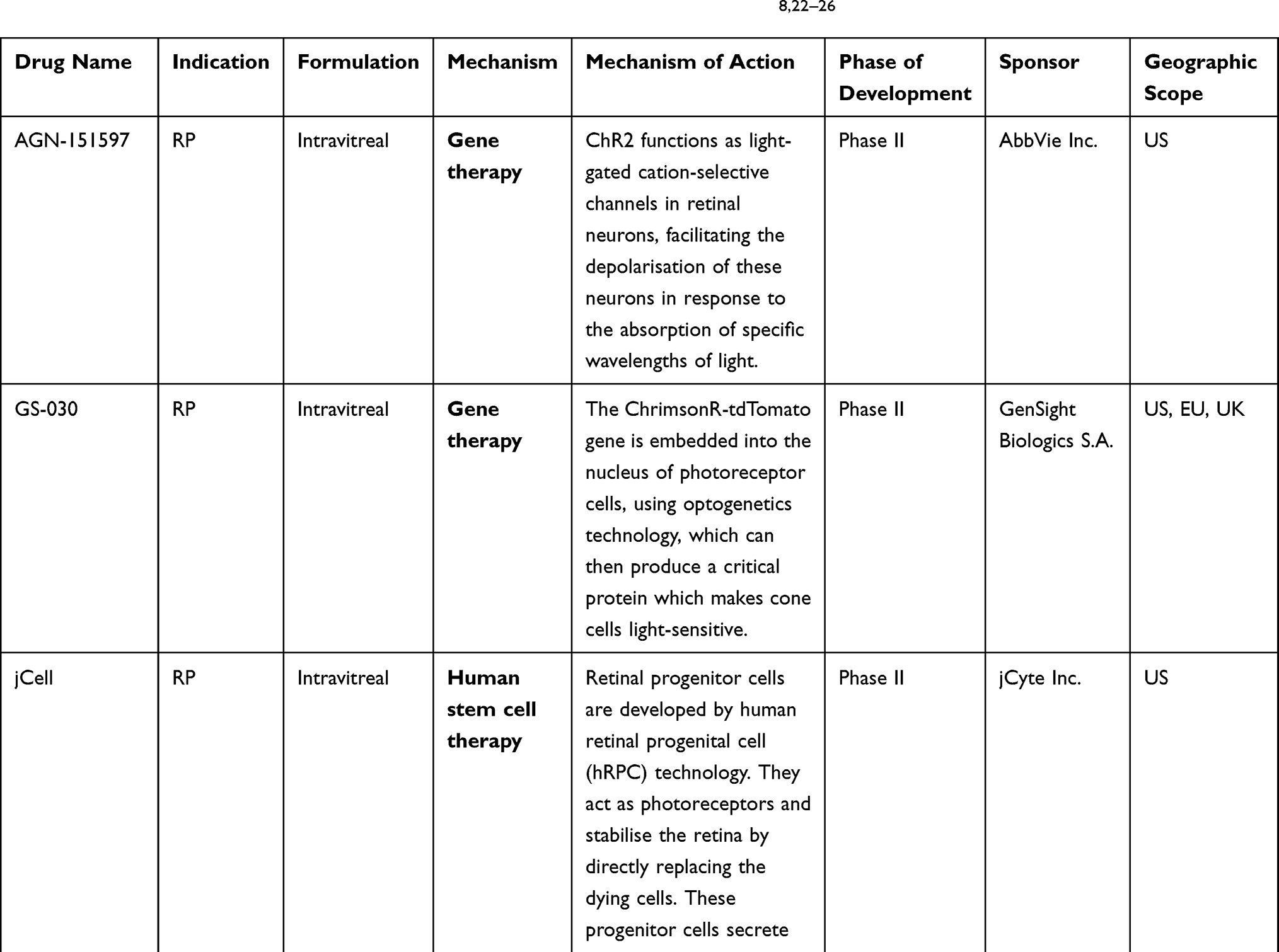 |
Table 2 Leading Cell and Gene Therapy Candidates in the Development for RP8,22–26 |
The Current Landscape for RP
Impact of Retinitis Pigmentosa on Patients
In the early stages of RP, patients will experience night-time blindness and difficulty adapting to low light environments. Mid-stage disease starts to have a significant effect on a patient’s quality of life, for example, they can have difficulty driving at night, start bumping into objects, and can have trouble locating objects.10,11
Due to frequent delays in referrals, patients are more likely to be diagnosed in the late-stage of the disease.12 By this stage of disease, visual field symptoms are progressively worse, and visual acuity is severely impaired.12 Late-stage disease is characterised by tunnel vision and severe restrictions in the visual field, and is associated with a high burden of disease.2 By the age of 40, most patients are classified as legally blind,13 defined as 20/200 vision or worse in the better eye or visual field restriction to ≤20° diameter,6 and typically patients lose central vision by the age of 60.13 This has significant effects on the patient’s quality of life, social interactions, and ability to work.
Diagnostic Challenges for Retinitis Pigmentosa
The initial presentation of the disease involves visual disturbances. Due to the vagueness of these symptoms, RP can go relatively unnoticed in the early stages of disease. The high genetic heterogeneity of RP can also lead to a wide range in the time of onset of symptoms and the type of symptoms that a patient first experiences.2,5 Consequently, there is no clear set of signs and symptoms for RP, which often leads to patients presenting to primary care in advanced disease stages.
Referrals to specialist geneticists and ophthalmologists can take between three and twelve months depending on the need for multiple consultations, proximity of the patient to a reference centre, patient age, and the stage of disease. This protracted period can mean that formal diagnoses are given to patients when they are in a more advanced disease stage, where visual field symptoms have become progressively worse.12
Current Landscape for the Management of Retinitis Pigmentosa
Currently, Luxturna® (voretigene neparvovec) is the only approved therapy for RP and is only authorised for the treatment of a small sub-population of patients that have the RPE65 mutation, which represents 0.3–1% of total RP cases.14 There is currently no standard treatment for patients without the RPE65 mutation, therefore most patients are limited to the best supportive care, including reliance on vitamin supplements, protection from sunlight, and visual aids.2 Comparison of seven clinical trials that measured visual acuity as a secondary outcome indicator showed that no significant difference was found in Early Treatment Diabetic Retinopathy Study (ETDRS) visual acuity with different doses of vitamin A and vitamin E in RP across treatment groups. Groups were either administered high-dose vitamin A, high-dose vitamin E, high-dose vitamin A and E, or trace amounts of both vitamins. This indicates that vitamin supplementation may have no beneficial effect in RP patients. Trials have also observed no significant difference between visual fields in control groups and groups supplemented with vitamin A.15 Consequently, for most patients with RP, there is currently no cure or effective treatment to slow or stop disease progression.11
The Evolving Treatment Landscape for RP
The RP Pipeline - Cell and Gene Therapies in Clinical Development for the Treatment of RP
Globally, there are currently 131 drugs reported in all stages of clinical development for RP, including 80 drugs in preclinical development and 30 drugs in late-stage clinical development (24 in phase II and 6 in Phase III trials) (Figure 2A).16
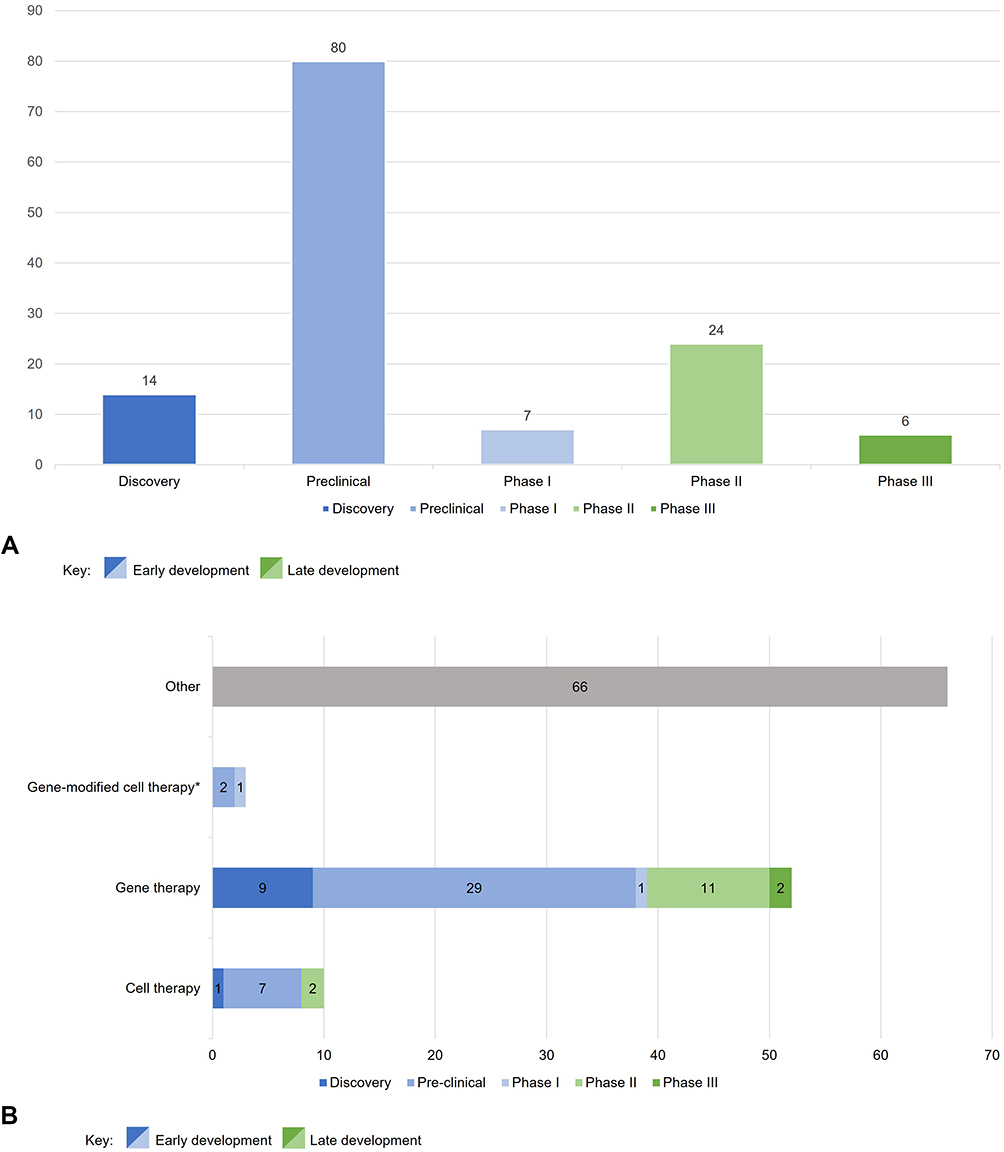 |
Figure 2 (A) RP pipeline by development phase. (B) Drug types across each stage of the clinical development pipeline for RP. Notes: Data from GlobalData.8 *Gene-modified cell therapies use gene transfer and cell-based technologies together to facilitate appropriate gene expression using cell carriers.17 |
From the drugs currently under development for the treatment of RP, several different types can be highlighted: cell therapies, gene therapies, gene-modified cell therapies, oligonucleotides (eg oligonucleotides, antisense oligonucleotides, and antisense ribonucleic acid interference (RNAi) oligonucleotides), antibodies, peptides, recombinant proteins and peptides, synthetic peptides, and small molecules. However, of the 131 drugs in clinical development for RP, around 50% are considered to be ATMPs, ie, cell and gene therapies. Of the 65 ATMPs in development, 10 are cell therapies (15.4%), 52 are gene therapies (80%), and 3 are gene-modified cell therapies (4.6%)8 (Figure 2B).
Most cell and gene therapy candidates are in the early stages of development. Currently, there are 2 cell therapies and 11 gene therapies in phase II development, with only 2 gene therapies in phase III development (Figure 2B).8
Gene therapy targets are generally diverse, although several targets are common across more than one drug. These include RHO which encodes rhodopsin; usherin, encoded by USH2A; and Retinitis Pigmentosa GTPase Regulator (RPGR), encoded by the gene of the same name. RHO is also targeted by 2 oligonucleotides and 1 recombinant protein. A further 2 oligonucleotide drug candidates target USH2A, making these targets the most common in the RP pipeline (Figure 3).8
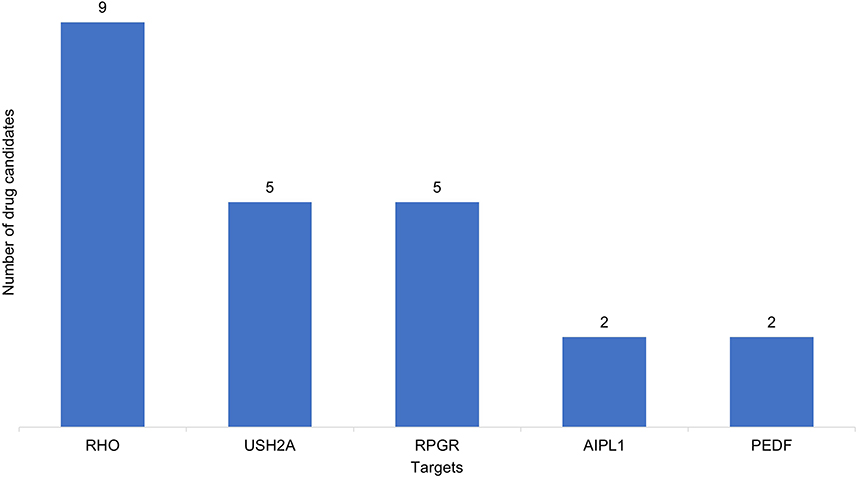 |
Figure 3 The most common targets for RP drug candidates (excludes those targeted by a single product in the pipeline). Note: Data from GlobalData.8 |
Mutations in RHO are predominantly associated with autosomal recessive RP (ARRP) and account for 25% of RP.18 The study of the defects in this gene is particularly relevant to therapeutic approaches to sustain photoreceptor viability without correcting the underlying mutation.18 Mutations in USH2A collectively account for 12–25% of cases of autosomal recessive (AR) non-syndromic RP and result in non-syndromic ARRP or Usher syndrome, where patients also experience hearing impairment. Potential therapeutic strategies either aim to manipulate the levels of USH2A or aim to prevent retinal degeneration by targeting disease pathways such as cell death or oxidative stress. However, the research on USH2A presents several challenges due to a broad spectrum of mutations, a large gene size hindering gene therapy development, and limited knowledge on its pathogenicity.19 In contrast, mutations in RPGR are responsible for over 70% of cases of X-linked RP (XLRP), which itself accounts for 10–15% of all RP cases.20
There are also several cell therapies in development for RP. These cell therapies can be applied in RP with two main therapeutic goals, “rescue” (ie preservation and restoration of function) and “replacement” (ie the direct replacement of dead or dysfunctional cells with healthy ones).21
“Rescue” can be sight-restoring if cells that are required for vision can be returned to normal physiological function. This is generally facilitated by the modulation of metabolic abnormalities eg with the production of trophic factors,21 and there are several cell therapies under development for RP which involve the secretion of neuroprotective factors from transplanted cells. There are also cell therapies under development which are able to engraft and protect the photoreceptor layer of the retina from degeneration, cells which can phagocytose damaging materials, and those which can reduce the activity of immune cells causing inflammation and damage.8
Cell therapies can also mediate the introduction of cells to directly replace damaged and dying cells. As blindness occurs due to photoreceptor death in RP, cell replacement therapies can be sight-restoring even in end-stage disease.21 Cell therapies in the pipeline for RP include those that introduce retinal-pigmented epithelium (RPE) and photoreceptor precursor cells (PRP) or pluripotent stem cells, which undergo in vivo differentiation. These pluripotent stem cells have the ability to integrate with the host retinal tissue and are capable of self-renewal, indefinite propagation, and multipotent and/or retina-specific differentiation.8
As illustrated in Figure 2B, there are currently 3 gene-modified cell therapies in development for RP. These products elicit their therapeutic activity by i) activating glial cell line-derived neurotrophic factor to promote survival and growth of dopamine-producing neurons; ii) replacing the broken copy of the MFRP gene with a functioning gene, encoding the functional membrane frizzled-related protein, to prevent the mutation-induced progressive loss of rod and cone vision; and iii) introducing cells to promote regenerative processes and prevent damage to central nervous neuronal cells.2
Of the small molecules in development, drug candidates target a range of enzymes and receptors in cell signaling pathways involved in sight, or work to restore vision by, for example, reducing inflammation and oxidative stress to combat cell death.8
Leading Candidates in Development for RP
Several leading candidates can be identified in the RP pipeline through secondary research and an extensive analysis of relevant databases.8 As outlined in the methods section, candidates were selected for inclusion based on development stage (phase II and III), drug target, and geography. These products are outlined in Table 2.
Other Advances in the Evolving Treatment Landscape for RP
Alongside advances in cell and gene technologies, research advances in the fields of optogenetics and electronic retinal prosthesis also represent encouraging progress for the treatment of RP and other IRDs. Optogenetic tools and photo-switchable compounds have been explored to make cells light-sensitive and bypass the loss of sensory input, and optogenetics may be applied to re-activate remaining cone receptors.28
Retinal prostheses involve the grafting of artificial light sensing devices that provide electrical stimuli to the inner nuclear layer.28 Retinal prostheses have been shown to restore some basic visual function in affected patients28 and both epiretinal and retinal implants have entered into clinical trials and clinical practice.29
Discussion
Treatment Selection for RP Can Be Informed by Several Factors, Including the Stage of Disease
The RP pipeline is diverse with several drug types in development. However, in clinical practice, the application of the most appropriate therapy type will likely depend, in part, on the stage of disease. Overall, therapeutic approaches that aim to rescue photoreceptors (eg gene therapies) are likely to be applied at an earlier stage of disease as gene replacement requires that non-functional target cells are still present. Therefore, this option may not be appropriate for the treatment of advanced retinal degeneration.27 Other therapeutic approaches, such as neuroprotection and antibody therapy, also require the presence of endogenous photoreceptors and are therefore more likely to be applied in early disease before patients experience severe photoreceptor loss (Figure 4).28
 |
Figure 4 Potential treatment for photoreceptor degeneration based on stage of disease and degree of photoreceptor loss. Notes: Reproduced from: Santos-Ferreira TF, Borsch O, Ader M. Rebuilding the Missing Part-A Review on Photoreceptor Transplantation. Front Syst Neurosci. 2017;10:105. doi:10.3389/fnsys.2016.00105.28 Copyright © 2017 Santos-Ferreira, Borsch and Ader. Creative Commons Attribution License (CC BY); https://creativecommons.org/licenses/by/4.0/legalcode. |
In contrast, other therapies are not reliant on the presence of remaining, viable photoreceptors. As such, cell therapies, optogenetics, retinal prostheses and photo-switchable compounds can be applied to patients in later stages of disease when photoreceptor degeneration is advanced (Figure 4).28,29
Clinical Rationale for Cell and Gene Therapies in RP
Hereditary retinal diseases are suitable for treatment with gene therapies due to the accessibility of the retina and its immune privilege and compartmentalization. This means that gene therapies can be applied with limited immune response or systemic side effects. The development of imaging techniques, including optical coherence tomography (OCT) and adaptive optics, also allows changes in the retina to be monitored after therapy.30
The location of photoreceptors in the eye next to the RPE means that diseases characterised by photoreceptor loss are also amenable to cell replacement via cell therapies. The contact between the RPE and outer segments of the photoreceptors is relatively loose and can be separated when donor cells are injected into the eye. This also means that once photoreceptors have been transplanted, they only have to extend their axons a few micrometres to establish synaptic connections to other cells in the eye.28
The Advantages and Limitations Associated with Cell and Gene Therapies
There are several considerations that could influence the choice of treatment strategy with a cell or gene therapy for RP patients. An overview of the advantages and limitations associated with both cell and gene therapies for RP is detailed in Boxes 1 and 2.
 |
Box 1 The Advantages and Potential Limitations Associated with Cell Therapies |
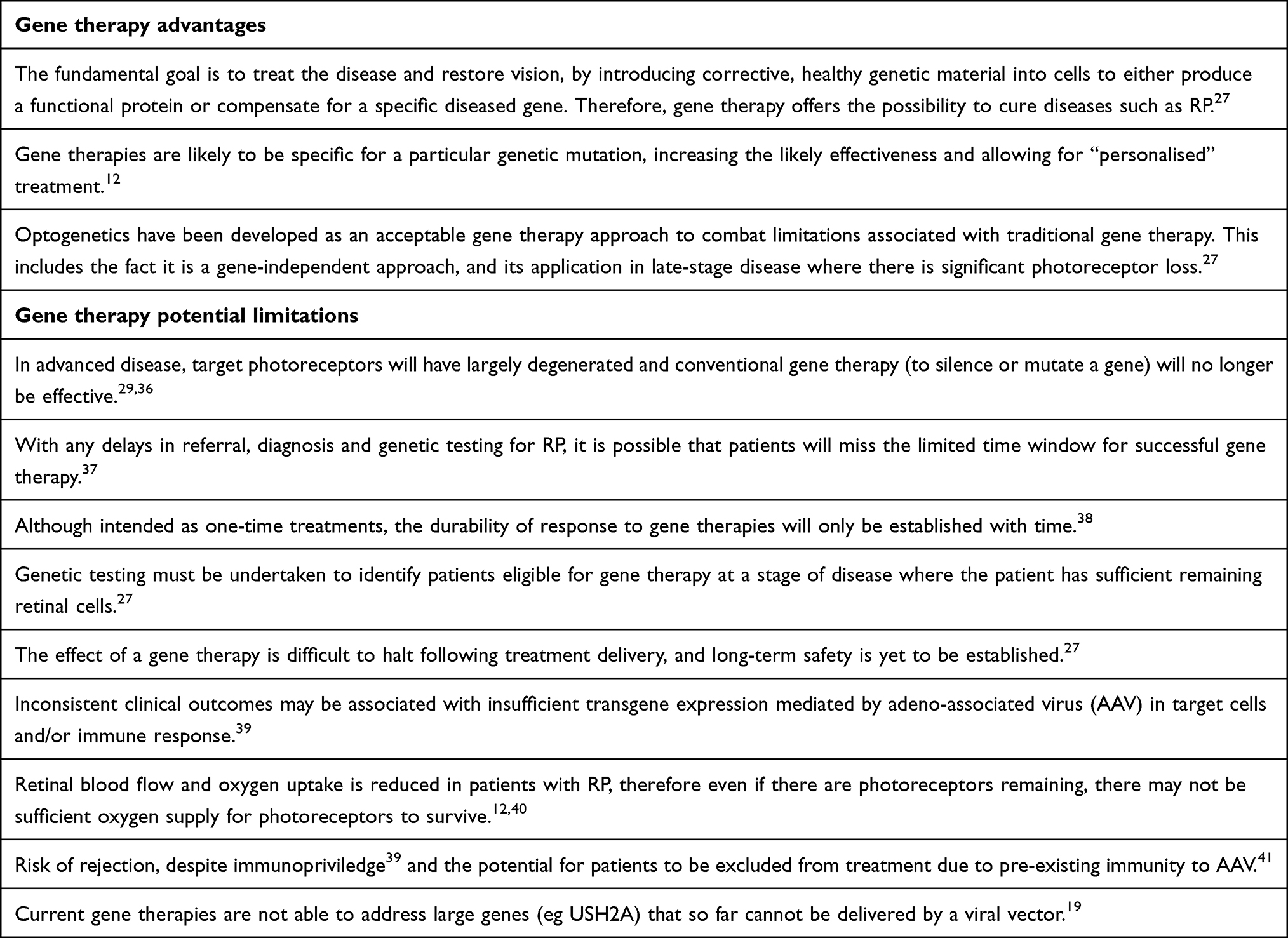 |
Box 2 The Advantages and Potential Limitations Associated with Gene Therapies |
Both gene therapies and cell therapies have important roles to play in the treatment of RP. Gene therapies require the presence of photoreceptors as a non-functional target cell, so are likely to be employed in early to mid-stage disease. Cell therapies do not require the presence of photoreceptors and so could be used throughout the disease course but are likely to be particularly relevant in mid-late stage disease, where severe photoreceptor loss has occurred.
Route of Administration
The different routes of administration for ATMPs are important and can play a role in the selection of the most appropriate treatments for patients.12
Intravitreal injection is a minimally invasive route of administration with less chance of damaging the retina than other methods, such as subretinal injection. It can be performed in a clinic setting, therefore increasing access to cell and gene therapies to a wider pool of patients.39 It has been reported that due to the physical barrier between the vitreous cavity and the inner limiting membrane, there have been some challenges in the use of intravitreal injections for gene delivery for the treatment of IRDs.27 However, it has been demonstrated that intravitreal injection of human retinal progenital cells (hRPCs) is effective and safe in protecting photoreceptor cells, with grafted cells in the vitreous cavity being well tolerated, with no adverse effects.42
In contrast, subretinal injection brings the injected vector into direct contact with target cells, increasing vectorisation efficiency and decreasing the chance of eliciting an immune response, when compared to systematically delivered adeno-associated virus (AAV).39,43 Physicians also acknowledge that for a cell therapy, a subretinal injection of an implant may suggest a longer duration of effect with sustained delivery, subject to supporting data.12 However, subretinal injection is invasive and safety concerns have been reported for this route of administration.12,43 Furthermore, the subretinal space is not immune-privileged and is prone to immune rejection.42 Even when performed by an experienced vitreoretinal surgeon, associated risks include uveitis, cataract, retinal tear, retinal detachment, endophthalmitis, macular fold, and macular hole.43 With the subretinal injection of cell and gene therapies, visual acuity may also be diminished for several months, which will impact the functionality and quality of life of the patient.12 Depending on the treatment centre and healthcare system, capacity constraints for vitreoretinal surgeons have also been reported, which may limit the potential for delivering new treatments with this route of administration.12
Drivers for Investment in the Development of New Cell and Gene Therapies for RP
Demand for effective medicines is one of the drivers of pharmaceutical investment, and investment in areas where there is a high unmet need is important for realising the social value of pharmaceutical innovations.44 Since voretigene neparvovec is only available for a small proportion of RP patients with the specific RPE65 mutation, there is no effective curative treatment for most individuals with RP, with high remaining levels of unmet need.2,14
The approval of voretigene neparvovec has led to a paradigm shift in the therapeutic approach to monogenic retinal diseases, paving the way for pharmaceutical innovation in the ATMP space.45 However, clinical development for ATPMs is highly complex, being subject to rapid technical and scientific advancements and often personalized for individual patients. As such, clinical development is resource-intensive and requires substantial investment. Furthermore, manufacturing ATMPs requires skilled personnel and highly controlled development facilities, both of which are associated with high costs.46
Several factors may contribute to the decision for a manufacturer to invest in either a cell or gene therapy for the treatment of RP. With a broader application, the patient population for cell therapies is larger than for most gene therapies, which target a specific gene or a particular mutation.27,29,43 However, costs for the development of cell therapies can be driven higher by the requirements for highly specialized materials such as tissue culture, and material wastage can be high due to short expiration dates.46 Gene therapies are also associated with additional complexities, including high development costs, requirements for genetic testing and the need for highly skilled and specially trained healthcare providers to perform the procedure, which can be associated with potential surgical complications.27
The majority (80%) of ATMPs in development are gene therapies, therefore it is clear that gene therapies are the current focus of pharmaceutical investment.8 In order to ensure effective therapeutic options are available for a broad range of patient types and disease stages, the continued advancement of a diversified pipeline with a range of therapeutic approaches, including cell therapies, is highly important.
Conclusion
RP is a retinal degenerative disease; progressive photoreceptor degeneration means that patients experience worsening symptoms as the disease advances and RP is associated with a high burden of disease for patients. As the age of onset of symptoms varies widely and symptoms often go unnoticed in the early stages of disease, diagnosis can be challenging. Due to common delays between initial presentation of symptoms and referral to a specialist ophthalmologist, patients may be in the advanced stage of disease before they obtain a formal diagnosis. Consequently, many RP patients experience a significant impact on their visual function and quality of life.7,12
There are currently 131 drugs in clinical development for RP, with around 50% considered to be ATMPs. These therapies represent a highly important advance for the treatment of RP, with the potential for a curative effect and a significant reduction in the burden of disease.
The clinical development pipeline is diverse with several drug types in development which are suitable to treat RP patients across each stage of the disease. Gene therapies are likely to be applied in earlier stages of disease when viable target photoreceptors still remain. In contrast, cell therapies are not reliant on the presence of remaining, viable photoreceptors, can work independently of gene mutation, and could be used throughout the course of the disease.
Ensuring that novel and effective therapeutic options are available for a range of RP patients across disease stages will require the continued diversification and advancement of the clinical development pipeline, and sustained efforts to promote early patient identification and timely referral and diagnosis of RP.
Abbreviations
AAV, Adeno-Associated Virus; AR, Autosomal Recessive; ARRP, Autosomal Recessive RP; ATMP, Advanced Therapy Medicinal Product; ETDRS, Early Treatment Diabetic Retinopathy Study; EU, European Union; hRPC, human Retinal Progenital Cell; IRD, Inherited Retinal Dystrophy; OCT, Optical Coherence Tomography; RNAi, Ribonucleic acid interference; RP, Retinitis Pigmentosa; RPE, Retinal-Pigmented Epithelium; RPGR, Retinitis Pigmentosa GTPase Regulator; PRP, Photoreceptor Precursor Cell; UK, United Kingdom; US, United States; XLRP, X-linked RP.
Disclosure
Conflict of interest: Luigi Angelillo and Cécile van Steen are employees of Santen GmBH.
The views expressed in this article are the authors’ own and do not necessarily reflect the views of the company. Nancy Cross, Yasmina Zegaoui, and Andrew Satherley are employees of Lightning Health. Financial support: this research was funded by Santen GmbH, München, Germany.
References
1. Hamel C. Retinitis pigmentosa. Orphanet J Rare Dis. 2006;1:40. doi:10.1186/1750-1172-1-40
2. O’Neal TB, Luther EE. Retinitis Pigmentosa. StatPearls Publishing; 2021.
3. Narayan DS, Wood JPM, Chidlow G, Casson RJ. A review of the mechanisms of cone degeneration in retinitis pigmentosa. Acta Ophthalmol. 2016;94(8):748–754. doi:10.1111/aos.13141
4. Orphanet. Prevalence and incidence of rare diseases: bibliographic data. Available from: https://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_decreasing_prevalence_or_cases.pdf.
5. Verbakel SK, van Huet RAC, Boon CJF, et al. Non-syndromic retinitis pigmentosa. Prog Retin Eye Res. 2018;66:157–186.
6. Jangra D, Ganesh A, Thackray R, et al. Psychosocial adjustment to visual loss in patients with retinitis pigmentosa. Ophthalmic Genet. 2007;28(1):25–30.
7. Chaumet-Riffaud AE, Chaumet Riffaud P, Cariou A, et al. Impact of retinitis pigmentosa on quality of life, mental health, and employment among young adults. Am J Ophthalmol. 2017;177:169–174.
8. GlobalData. A leading data and analytics company, “Retinitis Pigmentosa Drug Search”; 2022.
9. AGTC. X-linked retinitis pigmentosa. Available from: https://agtc.com/programs/x-linked-retinitis-pigmentosa/.
10. Hurse P. Retinitis pigmentosa: visual function and multidisciplinary management. Clin Exp Optometry. 2005;88:335–350. doi:10.1111/j.1444-0938.2005.tb06717.x
11. RNIB. Understanding retinitis pigmentosa and other inherited retinal dystrophies. Available from: https://www.rnib.org.uk.
12. European Physician Research. Lightning health European ophthalmologist research; 2021.
13. Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368(9549):1795–1809. doi:10.1016/S0140-6736(06)69740-7
14. Ferrari S, Iorio ED, Barbaro V, et al. Retinitis pigmentosa: genes and disease mechanisms. Curr Genomics. 2011;12(4):238–249. doi:10.2174/138920211795860107
15. Zhao Y, Feng K, Liu R, Pan J, Zhang L, Xuejing L. Vitamins and mineral supplements for retinitis pigmentosa. J Ophthalmol. 2019;2019:1–11. doi:10.1155/2019/8524607
16. Globaldata. Retinitis pigmentosa: competitive landscape to 2026. Available from: https://store.globaldata.com/report/retinitis-pigmentosa-competitive-landscape-to-2026/.
17. Wang H, Thompson TC. Gene-modified bone marrow cell therapy for prostate cancer. Gene Ther. 2008;15:787–796. doi:10.1038/gt.2008.37
18. Massengill MT, Lewin AS. Gene therapy for rhodopsin-associated autosomal dominant retinitis pigmentosa. Int Ophthalmol Clin. 2021;61(4):79–96. doi:10.1097/IIO.0000000000000383
19. Toualbi L, Toms M, Moosajee M. USH2A-retinopathy: from genetics to therapeutics. Exp Eye Res. 2020;201:108330. doi:10.1016/j.exer.2020.108330
20. De Silva S, Arno G, Robson AG, et al. The X-linked retinopathies: physiological insights, pathogenic mechanisms, phenotypic features and novel therapies. Prog Retin Eye Res. 2021;82. doi:10.1016/j.preteyeres.2020.100898
21. Zarbin M. Cell-based therapy for retinal disease: the new frontier. Methods Mol Biol. 2019;1834:367–381.
22. GlobalData. AGN-151597 drug overview; 2021.
23. GlobalData. GS-030 drug overview; 2021.
24. GlobalData. Jcell drug overview; 2021.
25. GlobalData. ReN-003 drug overview; 2021.
26. GlobalData. VMCO-1 drug overview; 2021.
27. Prado DA, Acosta-Acero M, Maldonado RS. Gene therapy beyond luxturna: a new horizon of the treatment for inherited retinal disease. Curr Opin Ophthalmol. 2020;31(3):147–154. doi:10.1097/ICU.0000000000000660
28. Santos-Ferreira TF, Borsch O, Ader M. Rebuilding the missing Part-A review on photoreceptor transplantation. Front Syst Neurosci. 2017;10:105. doi:10.3389/fnsys.2016.00105
29. Gagliardi G, M’Barek KB, Goureau O. Photoreceptor cell replacement in macular degeneration and retinitis pigmentosa: a pluripotent stem cell-based approach. Prog Retin Eye Res. 2019;71:1–25. doi:10.1016/j.preteyeres.2019.03.001
30. Dias MF, Joo K, Kemp J, et al. Molecular genetics and emerging therapies for retinitis pigmentosa: basic research and clinical perspectives. Prog Retin Eye Res. 2018;63:107–131. doi:10.1016/j.preteyeres.2017.10.004
31. Rafi M. Gene and stem cell therapy: alone or in combination? Bioimpacts. 2011;1(4):213–218. doi:10.5681/bi.2011.030
32. Barnstable CJ. Pluripotential stem cells as replacement therapy in degenerative diseases of the eye. Ann Transl Med. 2019;7(S3):S156–S156. doi:10.21037/atm.2019.06.50
33. Garg A, Yang J, Lee W, Tsang SH. Stem cell therapies in retinal disorders. Cells. 2017;6(1):4. doi:10.3390/cells6010004
34. Gasparini SJ, Llonch S, Borsch O, Ader M. Transplantation of photoreceptors into the degenerative retina: current state and future perspectives. Prog Retin Eye Res. 2019;69:1–37. doi:10.1016/j.preteyeres.2018.11.001
35. Sharma A, Jaganathan BG. Stem cell therapy for retinal degeneration: the evidence to date. Biologics. 2021;15:299–306. doi:10.2147/BTT.S290331
36. Reh TA. Photoreceptor transplantation in late stage retinal degeneration. Invest Ophthalmol Vis Sci. 2016;57(5):ORSFg1. doi:10.1167/iovs.15-17659
37. Datta P, Ruffcorn A, Seo S. Limited time window for retinal gene therapy in a preclinical model of ciliopathy. Hum Mol Genet. 2020;29(14):2337–2352. doi:10.1093/hmg/ddaa124
38. Salzman R, Cook F, Hunt T. Addressing the value of gene therapy and enhancing patient access to transformative treatments. Mol Ther. 2018;26(12):2717–2726. doi:10.1016/j.ymthe.2018.10.017
39. Mendell JR, Al-Zaidy SA, Rodino-Klapac LR, et al. Current clinical applications of in vivo gene therapy with AAVs. Mol Ther. 2021;29(2):464–488. doi:10.1016/j.ymthe.2020.12.007
40. Limoli PG, Vingolo EM, Limoli C, Nebbiosi M. Stem cell surgery and growth factors in retinitis pigmentosa patients: pilot study after literature review. Biomedicines. 2019;7(4):94. doi:10.3390/biomedicines7040094
41. Verdera HC, Kuranda K, Mingozzi F. AAV vector immunogenicity in humans: a long journey to successful gene transfer. Mol Ther. 2020;28(3):723–746.
42. Wang Z, Gao F, Zhang M. Intravitreal injection of human retinal progenitor cells for treatment of retinal degeneration. Med Sci Monit. 2020;26:e921184–1.
43. Ducloyer J-B, Meur G, Cronin T, Adjali O, Weber M. Gene therapy for retinitis pigmentosa. Med Sci (Paris). 2020;36(6–7):607–615.
44. Morgan SG, Bathula HS, Moon S. Pricing of pharmaceuticals is becoming a major challenge for health systems. BMJ. 2020;368:4627.
45. Dulla K, Slijkerman R, van Diepen HC, et al. Antisense oligonucleotide-based treatment of retinitis pigmentosa caused by USH2A exon 13 mutations. Mol Ther. 2021;29(8):2441–2455.
46. Ten Ham RMT, Nievaart JC, Hoekman J. Estimation of manufacturing development cost of cell-based therapies: a feasibility study. Cytotherapy. 2021;23(8):730–739.
47. Page MJ, McKenzie JE, Bossuyt PM, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372:n71. doi:10.1136/bmj.n71
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Retinitis Pigmentosa: Burden of Disease and Current Unmet Needs
Cross N, van Steen C, Zegaoui Y, Satherley A, Angelillo L
Clinical Ophthalmology 2022, 16:1993-2010
Published Date: 20 June 2022
Assessment of the Lifetime Costs of Severe Visual Impairment Due to Retinitis Pigmentosa
Milentijevic D, Ferro C, Rosenblum S, Dieguez G
ClinicoEconomics and Outcomes Research 2026, 18:571305
Published Date: 24 April 2026