")
Back to Journals » Degenerative Neurological and Neuromuscular Disease » Volume 6
Respiratory management of patients with neuromuscular disease: current perspectives
Received 14 September 2016
Accepted for publication 19 October 2016
Published 18 November 2016 Volume 2016:6 Pages 111—118
DOI https://doi.org/10.2147/DNND.S87323
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Thomas Müller
Gerald Pfeffer,1,2 Marcus Povitz3
1Department of Clinical Neurosciences, 2Hotchkiss Brain Institute, University of Calgary, Calgary, AB, 3Department of Medicine, Schulich School of Medicine and Dentistry, Western University, London, ON, Canada
Abstract: Neuromuscular ventilatory weakness can be difficult to recognize because the symptoms can be nocturnal, nonspecific, or attributed to other conditions. The presence of respiratory muscle weakness suggests a number of possible heterogeneous conditions, including neurodegenerative, autoimmune, and genetic neuromuscular diseases. In some conditions, disease-modifying management exists, but in the absence of such intervention, supportive respiratory therapy can improve quality of life and survival. In this review, we discuss the differential diagnosis and diagnostic approach to chronic neuromuscular respiratory weakness. We also review the clinical assessment and management of respiratory failure in these conditions.
Keywords: respiratory failure, hereditary myopathy, amyotrophic lateral sclerosis, myopathy, noninvasive ventilation
Introduction
Patients with neuromuscular disease may develop respiratory failure on account of weakness of respiratory muscles (diaphragm and accessory muscles of respiration), hypotonia of bulbar muscles, coexistent anatomical abnormalities (scoliosis or rigid spine), and decreased central respiratory drive.
In broad categories, respiratory failure in neuromuscular disease can occur as an acute phenomenon or a chronic progressive disorder. This review focuses on chronic progressive neuromuscular disorders causing respiratory failure. Typical examples of disorders that will have acute or subacute onset of respiratory muscle weakness include myasthenia gravis (MG) and acute inflammatory demyelinating polyneuropathy. These conditions have been reviewed in detail elsewhere, and will not be discussed in this review. However, it should be noted that chronic respiratory failure can present acutely in the presence of a superimposed acute illness. In this respect, it is important to recognize the presence of chronic neuromuscular respiratory failure, because it can be a poor prognostic indicator by predisposing patients to development of severe respiratory infections, and as a significant source of disability, on account of dyspnea and sleep disruption.
Conditions that present with gradual (chronic) onset of respiratory failure due to degenerative muscle disease require careful follow-up and long-term treatment. For the purposes of this review, we focus on these degenerative conditions, with the objectives of 1) discussing differential diagnosis and investigation of respiratory muscle weakness according to aspects of the clinical presentation, 2) appropriate screening and follow-up of patients who do not yet have respiratory muscle weakness, but have disorders that predispose them to development of respiratory failure, and 3) the long-term management of patients with chronic respiratory muscle weakness.
Differential diagnosis and investigation of respiratory muscle weakness according to clinical presentation
Patients presenting with respiratory muscle weakness will typically have nocturnal symptoms including orthopnea, sleep disruption, and headache upon awakening. These symptoms are very similar to the presentation of obstructive sleep apnea. Daytime symptoms include hypersomnolence, dyspnea (positional or nonspecific), tachypnea, and dysarthria. Symptoms may have insidious onset, and are often difficult to recognize.1
At initial presentation, the patient should be investigated for any coexistent pathology that may explain the symptoms (pulmonary infection, pneumothorax, or other pulmonary or cardiac pathology). Neuromuscular conditions with the possibility of rapid deterioration should be considered first, such as MG or acute inflammatory demyelinating polyneuropathy. If respiratory failure is impending, supportive management should be instituted. When acute conditions have been excluded and the patient is stable with regard to his/her respiratory condition, they should be investigated for the underlying cause. Subsequent investigations should depend upon the clinical scenario (Table 1).
 | Table 1 Approach to diagnostic investigations Abbreviation: EMG, electromyography; ESR, erythrocyte sedimentation rate; MRI, magnetic resonance imaging; CK, creatine kinase; CRP, C-reactive protein; 2C, D, I: designations for subtypes of LGMD caused by SGCG, SGCA, and FKRP mutations, respectively. |
Bulbar symptoms
These patients should have careful neurologic examination, neurophysiology (nerve-conduction studies and electromyography), and acetylcholine-receptor antibody testing, to consider the possibility of MG or amyotrophic lateral sclerosis (ALS).
Myasthenia gravis
MG is an autoimmune disease of the neuromuscular junction causing weakness of skeletal muscles. The bulbar and respiratory muscles are commonly involved, and in one series 14% of patients had respiratory failure at initial disease presentation (ie, respiratory failure already present in patients at time of initial diagnosis with MG).2 Respiratory failure in MG often requires intensive monitoring and invasive management, and can be associated with poor outcomes.3–5 MG is diagnosed based upon a combination of clinical features and pharmacologic, neurophysiologic, and serologic tests.6,7
Amyotrophic lateral sclerosis
In ALS, this condition, causing degeneration of motor neurons in the brain and spinal cord, invariably progresses to cause weakness of respiratory muscles.8 Respiratory failure tends to occur earlier in ALS with predominantly bulbar symptoms, and in one series respiratory symptoms were present at onset of ALS in 2.7% of patients.9 When respiratory failure is managed with noninvasive ventilation (NIV), prognosis for these patients appears to be similar to that of other patients with ALS.9 ALS is diagnosed by clinical criteria and neurophysiologic findings.10 Spinobulbar muscular atrophy is another condition associated with respiratory muscle weakness, but this typically occurs late in the disease course.11
Ophthalmoplegia and ptosis
These patients should be investigated as for MG. If no evidence for MG is present, muscle biopsy should be performed to consider the possibility of oculopharyngodistal myopathy or mitochondrial myopathy (MM).
Oculopharyngodistal myopathy
This condition is associated with both autosomal dominant and recessive inheritance, and the genetic etiology is unknown.12 Respiratory failure in ambulant patients is common in this condition, and clinical presentation includes ocular myopathy, bulbar weakness, and distal myopathy.12 Diagnosis is made by exclusion of other similar conditions with genetic testing, and muscle-pathology findings of rimmed and autophagic vacuoles. Recently, muscle magnetic resonance imaging (MRI) abnormalities were described in this condition, showing fatty infiltration of primarily distal leg muscles.13
Mitochondrial myopathy
MM has only rarely been associated with early respiratory muscle weakness in selected cases with mitochondrial transfer-RNA mutations.14,15 However, respiratory muscle weakness in MM may be underdiagnosed, based on a study showing 8.5% of patients with undiagnosed exercise limitation had respiratory muscle weakness and were later confirmed to have MM.16 MM classically has multisystem dysfunction, and is diagnosed with muscle biopsy17 and genetic testing.
Proximal myopathy
Pompe disease
The condition to consider first is adult-onset Pompe disease (glycogen storage disease type II), which may be rapidly screened with dried blood-spot testing for GAA-enzyme activity. If levels are low or intermediate, further testing may include GAA-enzyme activity in other tissues (leukocytes, fibroblasts, or muscle), and muscle-biopsy testing (showing glycogen accumulation of periodic acid–Schiff stains).18 If the diagnosis is indeterminate at this stage, patients should have GAA sequencing to identify recessive mutations. Note that some patients may have low GAA-enzyme activity in the absence of Pompe disease associated with certain polymorphic variants (“pseudodeficiency haplotype”).19 Pompe disease is treatable with alglucosidase-α enzyme-replacement therapy, which has been shown to improve motor function in a randomized controlled trial.20
Inflammatory myopathy
If Pompe disease is not identified, other rare causes for proximal myopathy with respiratory failure should be considered. These include inflammatory myopathies, such as polymyositis, dermatomyositis, and sarcoidosis, which have been associated with respiratory failure due to muscle involvement,21 and in some cases due to coexistent lung disease (antisynthetase syndrome).22 In this situation, testing for creatine-kinase levels, chest X-ray, and autoantibodies is helpful to confirm the diagnosis, although definitive diagnosis is made by muscle biopsy.23
Limb-girdle muscular dystrophy
Limb-girdle muscular dystrophies are a group of hereditary muscle diseases that typically do not have early involvement of respiratory muscles. However in the case of LGMD2I (due to recessive FKRP mutations), respiratory muscle weakness was found in ten of 16 patients24 and may be common. Two of the sarcoglycanopathies (LGMD2C and D) have also been associated with respiratory muscle involvement.25
Other myopathies
Muscle biopsy is required to identify other rare diagnostic possibilities, such as mitochondrial abnormalities (for MM), dystrophic abnormalities (suggesting limb-girdle muscular dystrophy), nemaline pathology (for adult-onset nemaline myopathy),26 or fiber-type disproportion (which can be seen in TPM3 myopathy).27 Although only a single case of colchicine myopathy has been reported with respiratory failure,28 this should be considered in patients taking colchicine, given the potential reversibility after cessation of therapy.
Distal myopathy
Inclusion-body myopathy
Inclusion body myopathy (IBM) is an adult-onset myopathy that has recently been associated with respiratory failure.29 Typically, IBM presents with early finger flexor and quadricep weakness, and is diagnosed by clinical findings and muscle biopsy, with the assistance of formal diagnostic criteria.30 Sleep-disordered breathing with subclinical respiratory dysfunction is common in IBM,31,32 and does not appear to correspond with the degree of limb weakness.31
Myotonic dystrophy type I
Myotonic dystrophy type I has a highly variable phenotype, and may come to medical attention at any age. The classical clinical presentation in adulthood includes distal weakness, frontal balding, facial muscle weakness, cataracts, endocrinopathy, and cardiac dysfunction.33 Myotonic dystrophy type I is an autosomal-dominant disorder, and diagnosis is established by identification of an expanded CTG trinucleotide repeat in DMPK. The common manifestation of respiratory failure is hypersomnolence associated with severe sleep apnea in nearly 70% of patients,34 although respiratory muscle weakness is also present in a substantial number of patients, including patients with only distal weakness.35
Titinopathy and myofibrillar myopathy
Hereditary myopathy with early respiratory failure (HMERF) is an adult-onset autosomal-dominant myopathy. HMERF typically presents with respiratory muscle weakness in patients who are still ambulant. Limb weakness is most prominent in the tibialis anterior. The condition is caused by mutations in the 119th fibronectin-3 domain of titin.36 Diagnosis is established by genetic testing, but can be aided by muscle-biopsy findings or muscle MRI of the lower legs.37 HMERF has been demonstrated to meet pathologic criteria for myofibrillar myopathy (MFM).36 In other subtypes of MFM, early respiratory failure is also a possible presentation, particularly MFM due to mutations in desmin,38 αβ-crystallin,39 and BAG3.40 Diagnosis is confirmed by genetic testing or muscle biopsy, and again muscle MRI can be quite useful to guide the differential diagnosis.41
Other conditions
Multifocal motor neuropathy is an autoimmune motor neuropathy preferentially involving the distal upper limbs.42 There exists a single case report of presentation with respiratory failure.43 In the appropriate clinical context, this diagnosis should be considered, given its response to therapy with intravenous immunoglobulins.42
It should be noted that patients with these disorders may not always have respiratory failure at their initial presentation. Even in this situation, these patients remain at high risk of developing respiratory failure, and should undergo regular screening for respiratory muscle weakness, as described in the following sections.
Screening and follow-up of patients at risk of developing respiratory muscle weakness
Early identification of respiratory muscle weakness is desirable, given the availability of therapies that have been shown to improve survival and quality of life.44 Since the progression of these diseases may be subacute, asymmetric screening testing is recommended to supplement the clinical interview.45–47 The approach to the history, physical examination, and diagnostic testing in this situation is summarized in Table 2.
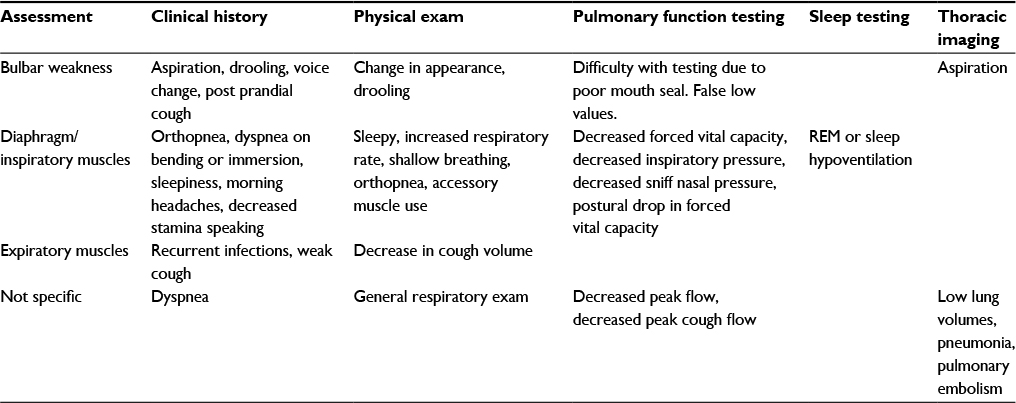 | Table 2 Assessment for respiratory muscle weakness Abbreviation: REM, rapid eye movement. |
The clinical interview
During the clinical interview, bulbar symptoms should be assessed. This is done by questioning difficulty in swallowing, postprandial coughing, sputtering, or choking, and this should be assessed for different food consistencies, as well as solids and liquids. Other symptoms of bulbar weakness will include drooling and changes in voice volume, pitch, or clarity. Diaphragm weakness is assessed by symptoms of orthopnea, dyspnea on bending, or dyspnea on immersion to water. These symptoms may be preceded by symptoms of sleep-disordered breathing, such as nonrestorative sleep, nocturnal dyspnea, excessive daytime sleepiness, and morning headaches. With a reduction in vital capacity volume of the voice singing particularly may be impacted. A history of recurrent chest infection may suggest either bulbar weakness-related aspiration or inadequate cough due to failure of the respiratory muscles, particularly for expiration.
Physical examination
The physical exam of the respiratory system should begin with a general impression of patient posture, appearance, and affect. Patients with poor sleep may appear fatigued and have reduced affect and difficulty concentrating. The patients may adopt a rapid shallow-breathing pattern to conserve energy.48 Accessory muscles may be used, and patients may adopt a tripod position to improve the mechanical advantage of their respiratory muscles. Pulse oximetry should be performed when the vitals are assessed, and hypoventilation or atelectasis are associated with a reduction in resting pulse oximetry.49 The patient should be assessed in the supine position for further use of accessory muscles or the presence of diaphragmatic paradox. The patient should be asked to cough forcefully. Absence of an audible cough is abnormal. Other elements of the respiratory exam may be helpful in completing the assessment, but are not specific to neuromuscular weakness.
Pulmonary function testing
Baseline testing should include assessment of spirometry, lung volume, diffusion capacity, and arterial, arterialized venous, or capillary blood-gas testing. These will establish a baseline and exclude other relevant pulmonary diseases. Patients with primarily bulbar weakness may be unable to form a mouth seal or have falsely low measures due to leak. Patients with respiratory muscle weakness will classically present with a restrictive pattern (reductions in both forced expiratory volume in 1 second and forced vital capacity [FVC] with a preserved ratio).50 Subtle abnormalities may be present, as well such a disproportionate reduction in peak expiratory flow or a sudden drop-off in expiratory flow.49 Total lung capacity will be reduced, while residual volume will be increased. Functional residual capacity is affected variably. Diffusion capacity may be normal or low if vital capacity is reduced. An arterial or capillary carbon dioxide level of ≥45 mmHg is consistent with hypoventilation.
Additional testing may assist with the diagnosis of respiratory muscle weakness. This would include measurement of FVC in the supine position.51 A decrease of greater than 20%–30% is indicative of diaphragm weakness.50 Sniff nasal pressure can also be measured, and may have increased prognostic value in comparison to FVC. A sniff pressure of less than 40 was prognostic in ALS patients for mortality and nocturnal hypoxemia.52 Measurement of inspiratory and expiratory mouth pressures may reveal weakness if they are less than –30 cmH2O and 40 cmH2O, respectively. These tests may often have indeterminate results below the lower limit of normal, but not clearly demonstrating respiratory muscle weakness. Peak cough flow should be monitored: a measurement of less than 270 L/min is an indication of significant respiratory muscle weakness, while a level less than 160 L/min suggests cough is severely compromised.53
Sleep testing
Either overnight oximetry or polysomnography has been described in this population. This should be considered as part of the assessment in any patient with nocturnal symptoms described earlier or progressive decline in FVC. A 5-minute period with an oxygen saturation less than 90% has been considered an indication of respiratory muscle weakness.45–47 Certain conditions may also predispose to obstructive sleep apnea, such as myotonic dystrophy due to weight gain, or others due to upper-airway flaccidity. Addition of monitoring for carbon dioxide levels may reveal sleep hypoventilation due to muscle weakness. Several modalities of capnography exist, namely end-tidal capnography or transcutaneous capnography. Transcutaneous capnography is currently recommended by the American Academy of Sleep Medicine.54 These measures can also be used in wakefulness, when arterial or arterialized-venous blood sampling is unavailable for assessment for hypercapnia.
Imaging
Thoracic imaging may be helpful in excluding underlying cardiopulmonary diseases. It should also be performed in assessment of conditions known to impact the lung parenchyma, such as drug reactions or poly/dermatomyositis. In patients with newly diagnosed hypoxemia, chest imaging can help to exclude aspiration, intercurrent pneumonia, or atelectasis. Patients with significant immobility are also at increased risk of pulmonary embolism, and this may be more common in certain conditions.55
Long-term management of established neuromuscular weakness
Once a diagnosis of respiratory muscle weakness has been made, supportive respiratory therapy should be initiated, even if the underlying neurological diagnosis is pending. The approach to management is similar in these conditions, although the level of evidence supporting the various interventions, such as NIV, cough assist, lung-volume recruitment, and respiratory muscle training, varies.45–47 Follow-up of patients with early respiratory muscle weakness not requiring therapy should be determined from the established rate of progression. Patients requiring therapy should be seen at least every 3 months or more frequently if new therapies are being instituted. During the course of therapy, discussions should be had to establish the patient’s wishes in regard to feeding tubes, invasive ventilation, and cardiopulmonary resuscitation, given the high probability of disease progression. Patients with bulbar involvement or where bulbar symptoms are anticipated should be considered for early placement of a feeding tube if this is in keeping with the patient’s wishes. Placement of a percutaneous feeding tube is associated with a risk of respiratory complications. The risk is thought to be greatest when the FVC has fallen below 50%, particularly in patients not yet stabilized on supportive respiratory therapy. For patients for whom supportive respiratory therapies are not appropriate or not desired, palliative management of dyspnea and secretions should be considered.56,57
Noninvasive ventilation
Many criteria have been proposed for the timing of initiation of NIV. These include the presence of symptoms of sleep-disordered breathing, daytime hypercapnia ≥45 mmHg, hypoxemia during sleep, FVC of less than 40%–60% (depending on the diagnosis) or rapid decline in FVC, and low sniff nasal pressure. Significant bulbar symptoms have been proposed as a contraindication or a reason to delay initiation until respiratory failure is more advanced.58 Initiation of NIV at earlier stages has been attempted, but has not proven beneficial and may even be harmful.59 NIV can be initiated through polysomnography or clinic- or hospital-based titration, depending on the resources available.60–62 Each has its advantages, and a combination may prove to be ideal, such as clinic-based initiation followed by polysomnography to ensure adequacy and for fine tuning. NIV settings should be set to target adequate minute ventilation and provide rest to the respiratory system by minimizing NIV triggering. Adherence to NIV should be encouraged with increasing use as the disease progresses. Patients using NIV during daytime hours for relief of dyspnea should be provided with a second machine, preferably with a battery backup. Alternatively, such patients may benefit from mouthpiece ventilation via ventilator or nasal mask ventilation, which allows for more mobility with a motorized wheelchair if necessary.45–47
Role of invasive ventilation
Patients with copious secretions or unable to tolerate NIV but wishing to prolong life may be considered for invasive ventilation via tracheostomy. Alternatively, mouthpiece or sip ventilators may delay the need for tracheostomy and provide an alternative to NIV during the day. Patients in a recent survey reported this mode of ventilation helped with dyspnea and fatigue, as well as speech and eating. This also provides a break from the mask interface required for NIV.63
Cough assist
Once FVC has begun to fall, patients are at risk of developing atelectasis with loss of lung compliance. Breath stacking or lung-volume recruitment may improve lung function by increasing VC with the use of an Ambu bag and oronasal interface with a one-way valve. This has been proposed as a method to decrease pulmonary infections by augmenting cough, as well as improving oxygenation. This is particularly useful for patients with respiratory tract infections.45–47
If patients are unable to coordinate breath stacking or lack the hand dexterity required, an alternative is use of mechanical insufflation–exsufflation. This device provides a positive-pressure breath followed by a negative-pressure exsufflation, and has been shown to be the most potent method of cough augmentation.64 This can be instituted preemptively to reduce the frequency of infection or to assist with secretion clearance. There is a possible role for this device in the management of intubated patients to assist with extubation.65 This device has superior properties to catheter suction, as it is not preferentially directed into the right bronchus. Caution should be exercised with this device in individuals with bullous lung disease, as there is a potential risk of pneumothorax.
Respiratory muscle training
Respiratory muscle training is an area of interest, but is not supported by many studies, and is still considered experimental by expert guidelines.46 This is performed with inspiratory and expiratory resistors and repeated forced maneuvers. Studies have considered the potential benefits for slowly66,67 and rapidly progressive conditions.68 These devices can augment mouth pressures and improve cough. These exercises also improve bulbar function. To date, these exercises have not been shown to improve spirometric measures.
Conclusion
Early recognition of the clinical symptoms and signs of neuromuscular respiratory failure aids in the diagnosis and management of patients with the aforementioned rare chronic muscle diseases. Some of these conditions have disease-modifying treatment available, but even in the absence of such therapy, the management of respiratory failure with supportive care such as NIV and cough assist is recommended to improve quality of life and prevent secondary complications.
Disclosure
The authors report no conflicts of interest in this work.
References
Aboussouan LS. Sleep-disordered breathing in neuromuscular disease. Am J Respir Crit Care Med. 2015;191(9):979–989. | ||
Qureshi AI, Choundry MA, Mohammad Y, et al. Respiratory failure as a first presentation of myasthenia gravis. Med Sci Monit. 2004;10(12):CR684–CR689. | ||
Rabinstein AA, Mueller-Kronast N. Risk of extubation failure in patients with myasthenic crisis. Neurocrit Care. 2005;3(3):213–215. | ||
O’Riordan JI, Miller DH, Mottershead JP, Hirsch NP, Howard RS. The management and outcome of patients with myasthenia gravis treated acutely in a neurological intensive care unit. Eur J Neurol. 1998;5(2):137–142. | ||
Berrouschot J, Baumann I, Kalischewski P, Sterker M, Schneider D. Therapy of myasthenic crisis. Crit Care Med. 1997;25(7):1228–1235. | ||
Jaretzki A 3rd, Barohn RJ, Ernstoff RM, et al. Myasthenia gravis: recommendations for clinical research standards. Neurology. 2000;55(1):16–23. | ||
Juel VC, Massey JM. Myasthenia gravis. Orphanet J Rare Dis. 2007;2:44. | ||
Salameh JS, Brown RH Jr, Berry JD. Amyotrophic lateral sclerosis: review. Semin Neurol. 2015;35(4):469–476. | ||
Shoesmith CL, Findlater K, Rowe A, Strong MJ. Prognosis of amyotrophic lateral sclerosis with respiratory onset. J Neurol Neurosurg Psychiatry. 2007;78(6):629–631. | ||
Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(5):293–299. | ||
Atsuta N, Watanabe H, Ito M, et al. Natural history of spinal and bulbar muscular atrophy (SBMA): a study of 223 Japanese patients. Brain. 2006;129(Pt 6):1446–1455. | ||
Durmus H, Laval SH, Deymeer F, et al. Oculopharyngodistal myopathy is a distinct entity: clinical and genetic features of 47 patients. Neurology. 2011;76(3):227–235. | ||
Zhao J, Liu J, Xiao J, et al. Clinical and muscle imaging findings in 14 mainland Chinese patients with oculopharyngodistal myopathy. PLoS One. 2015;10(6):e0128629. | ||
Bindoff LA, Howell N, Poulton J, et al. Abnormal RNA processing associated with a novel tRNA mutation in mitochondrial DNA: a potential disease mechanism. J Biol Chem. 1993;268(26):19559–19564. | ||
Hadjigeorgiou GM, Kim SH, Fischbeck KH, et al. A new mitochondrial DNA mutation (A3288G) in the tRNALeu(UUR) gene associated with familial myopathy. J Neurol Sci. 1999;164(2):153–157. | ||
Flaherty KR, Wald J, Weisman IM, et al. Unexplained exertional limitation: characterization of patients with a mitochondrial myopathy. Am J Respir Crit Care Med. 2001;164(3):425–432. | ||
Bernier FP, Boneh A, Dennett X, Chow CW, Cleary MA, Thorburn DR. Diagnostic criteria for respiratory chain disorders in adults and children. Neurology. 2002;59(9):1406–1411. | ||
Dasouki M, Jawdat O, Almadhoun O, et al. Pompe disease: literature review and case series. Neurol Clin. 2014;32(3):751–776, ix. | ||
Labrousse P, Chien YH, Pomponio RJ, et al. Genetic heterozygosity and pseudodeficiency in the Pompe disease newborn screening pilot program. Mol Genet Metab. 2010;99(4):379–383. | ||
van der Ploeg AT, Clemens PR, Corzo D, et al. A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N Engl J Med. 2010;362(15):1396–1406. | ||
Kalluri M, Oddis CV. Pulmonary manifestations of the idiopathic inflammatory myopathies. Clin Chest Med. 2010;31(3):501–512. | ||
Dejardin Botelho A, Perez T, Pouwels S, Wallaert B, Hachulla E, Tillie-Leblond I. [Inspiratory muscle myopathy and antisynthetase syndrome]. Rev Med Interne. 2008;29(4):325–327. | ||
Dalakas MC. Inflammatory muscle diseases. N Engl J Med. 2015;373(4):393–394. | ||
Poppe M, Bourke J, Eagle M, et al. Cardiac and respiratory failure in limb-girdle muscular dystrophy 2I. Ann Neurol. 2004;56(5):738–741. | ||
Politano L, Nigro V, Passamano L, et al. Evaluation of cardiac and respiratory involvement in sarcoglycanopathies. Neuromuscul Disord. 2001;11(2):178–185. | ||
Chahin N, Selcen D, Engel AG. Sporadic late onset nemaline myopathy. Neurology. 2005;65(8):1158–1164. | ||
Clarke NF, Kolski H, Dye DE, et al. Mutations in TPM3 are a common cause of congenital fiber type disproportion. Ann Neurol. 2008;63(3):329–337. | ||
Tanios MA, El Gamal H, Epstein SK, Hassoun PM. Severe respiratory muscle weakness related to long-term colchicine therapy. Respir Care. 2004;49(2):189–191. | ||
Needham M, Mastaglia FL. Sporadic inclusion body myositis: a review of recent clinical advances and current approaches to diagnosis and treatment. Clin Neurophysiol. 2016;127(3):1764–1773. | ||
Rose MR. 188th ENMC International Workshop: Inclusion Body Myositis, 2-4 December 2011, Naarden, the Netherlands. Neuromuscul Disord. 2013;23(12):1044–1055. | ||
Rodriguez Cruz PM, Needham M, Hollingsworth P, Mastaglia FL, Hillman DR. Sleep disordered breathing and subclinical impairment of respiratory function are common in sporadic inclusion body myositis. Neuromuscul Disord. 2014;24(12):1036–1041. | ||
Della Marca G, Sancricca C, Losurdo A, et al. Sleep disordered breathing in a cohort of patients with sporadic inclusion body myositis. Clin Neurophysiol. 2013;124(8):1615–1621. | ||
Turner C, Hilton-Jones D. The myotonic dystrophies: diagnosis and management. J Neurol Neurosurg Psychiatry. 2010;81(4):358–367. | ||
Laberge L, Begin P, Dauvilliers Y, et al. A polysomnographic study of daytime sleepiness in myotonic dystrophy type 1. J Neurol Neurosurg Psychiatry. 2009;80(6):642–646. | ||
Kaminsky P, Poussel M, Pruna L, Deibener J, Chenuel B, Brembilla-Perrot B. Organ dysfunction and muscular disability in myotonic dystrophy type 1. Medicine (Baltimore). 2011;90(4):262–268. | ||
Pfeffer G, Barresi R, Wilson IJ, et al. Titin founder mutation is a common cause of myofibrillar myopathy with early respiratory failure. J Neurol Neurosurg Psychiatry. 2014;85(3):331–338. | ||
Birchall D, von der Hagen M, Bates D, Bushby KM, Chinnery PF. Subclinical semitendinosus and obturator externus involvement defines an autosomal dominant myopathy with early respiratory failure. Neuromuscul Disord. 2005;15(9–10):595–600. | ||
Walter MC, Reilich P, Huebner A, et al. Scapuloperoneal syndrome type Kaeser and a wide phenotypic spectrum of adult-onset, dominant myopathies are associated with the desmin mutation R350P. Brain. 2007;130(Pt 6):1485–1496. | ||
Selcen D, Engel AG. Myofibrillar myopathy caused by novel dominant negative αB-crystallin mutations. Ann Neurol. 2003;54(6):804–810. | ||
Selcen D, Muntoni F, Burton BK, et al. Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Ann Neurol. 2009;65(1):83–89. | ||
Wattjes MP, Kley RA, Fischer D. Neuromuscular imaging in inherited muscle diseases. Eur Radiol. 2010;20(10):2447–2460. | ||
Lawson VH, Arnold WD. Multifocal motor neuropathy: a review of pathogenesis, diagnosis, and treatment. Neuropsychiatr Dis Treat. 2014;10:567–576. | ||
Kyriakides T, Papacostas S, Papanicolaou E, Bagdades E, Papathanasiou ES. Sleep hypoventilation syndrome and respiratory failure due to multifocal motor neuropathy with conduction block. Muscle Nerve. 2011;43(4):610–614. | ||
Annane D, Orlikowski D, Chevret S. Nocturnal mechanical ventilation for chronic hypoventilation in patients with neuromuscular and chest wall disorders. Cochrane Database Syst Rev. 2014;(12):CD001941. | ||
Windisch W, Walterspacher S, Siemon K, Geiseler J, Sitter H. Guidelines for non-invasive and invasive mechanical ventilation for treatment of chronic respiratory failure. Pneumologie. 2010;64(10):640–652. | ||
McKim DA, Road J, Avendano M, et al. Home mechanical ventilation: a Canadian Thoracic Society clinical practice guideline. Can Respir J. 2011;18(4):197–215. | ||
Agency for Clinical Innovation. Non-invasive ventilation guidelines for adult patients with acute respiratory failure. Available from: https://www.aci.health.nsw.gov.au/networks/intensive-care/intensive-care-manual/statewide-guidelines/non-invasive-ventilation-guidelines. Accessed October 21, 2016. | ||
Ambrosino N, Carpene N, Gherardi M. Chronic respiratory care for neuromuscular diseases in adults. Eur Respir J. 2009;34(2):444–451. | ||
Laghi F, Tobin MJ. Disorders of the respiratory muscles. Am J Respir Crit Care Med. 2003;168(1):10–48. | ||
American Thoracic Society/European Respiratory Society. ATS/ERS Statement on respiratory muscle testing. Am J Respir Crit Care Med. 2002;166(4):518–624. | ||
Poussel M, Kaminsky P, Renaud P, Laroppe J, Pruna L, Chenuel B. Supine changes in lung function correlate with chronic respiratory failure in myotonic dystrophy patients. Respir Physiol Neurobiol. 2014;193:43–51. | ||
Morgan RK, McNally S, Alexander M, Conroy R, Hardiman O, Costello RW. Use of sniff nasal-inspiratory force to predict survival in amyotrophic lateral sclerosis. Am J Respir Crit Care Med. 2005;171(3):269–274. | ||
Morrow B, Zampoli M, van Aswegen H, Argent A. Mechanical insufflation-exsufflation for people with neuromuscular disorders. Cochrane Database Syst Rev. 2013;(12):CD010044. | ||
Berry RB, Budhiraja R, Gottlieb DJ, et al. Rules for scoring respiratory events in sleep: update of the 2007 AASM Manual for the Scoring of Sleep and Associated Events. J Clin Sleep Med. 2012;8(5):597–619. | ||
Qureshi MM, Cudkowicz ME, Zhang H, Raynor E. Increased incidence of deep venous thrombosis in ALS. Neurology. 2007;68(1):76–77. | ||
Baxter SK, Baird WO, Thompson S, et al. The use of non-invasive ventilation at end of life in patients with motor neurone disease: a qualitative exploration of family carer and health professional experiences. Palliat Med. 2013;27(6):516–523. | ||
Bede P, Oliver D, Stodart J, et al. Palliative care in amyotrophic lateral sclerosis: a review of current international guidelines and initiatives. J Neurol Neurosurg Psychiatry. 2011;82(4):413–418. | ||
Bourke SC, Tomlinson M, Williams TL, Bullock RE, Shaw PJ, Gibson GJ. Effects of non-invasive ventilation on survival and quality of life in patients with amyotrophic lateral sclerosis: a randomised controlled trial. Lancet Neurol. 2006;5(2):140–147. | ||
Simonds AK. Recent advances in respiratory care for neuromuscular disease. Chest. 2006;130(6):1879–1886. | ||
Berry RB, Chediak A, Brown LK, et al. Best clinical practices for the sleep center adjustment of noninvasive positive pressure ventilation (NPPV) in stable chronic alveolar hypoventilation syndromes. J Clin Sleep Med. 2010;6(5):491–509. | ||
Chatwin M, Nickol AH, Morrell MJ, Polkey MI, Simonds AK. Randomised trial of inpatient versus outpatient initiation of home mechanical ventilation in patients with nocturnal hypoventilation. Respir Med. 2008;102(11):1528–1535. | ||
Jaye J, Chatwin M, Dayer M, Morrell MJ, Simonds AK. Autotitrating versus standard noninvasive ventilation: a randomised crossover trial. Eur Respir J. 2009;33(3):566–571. | ||
Khirani S, Ramirez A, Delord V, et al. Evaluation of ventilators for mouthpiece ventilation in neuromuscular disease. Respir Care. 2014;59(9):1329–1337. | ||
Chatwin M, Ross E, Hart N, Nickol AH, Polkey MI, Simonds AK. Cough augmentation with mechanical insufflation/exsufflation in patients with neuromuscular weakness. Eur Respir J. 2003;21(3):502–508. | ||
Bach JR, Sinquee DM, Saporito LR, Botticello AL. Efficacy of mechanical insufflation-exsufflation in extubating unweanable subjects with restrictive pulmonary disorders. Respir Care. 2015;60(4):477–483. | ||
Jones HN, Crisp KD, Robey RR, Case LE, Kravitz RM, Kishnani PS. Respiratory muscle training (RMT) in late-onset Pompe disease (LOPD): effects of training and detraining. Mol Genet Metab. 2016;117(2):120–128. | ||
Aslan GK, Gurses HN, Issever H, Kiyan E. Effects of respiratory muscle training on pulmonary functions in patients with slowly progressive neuromuscular disease: a randomized controlled trial. Clin Rehabil. 2014;28(6):573–581. | ||
Plowman EK, Watts SA, Tabor L, et al. Impact of expiratory strength training in amyotrophic lateral sclerosis. Muscle Nerve. 2016;54(1):48–53. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.