Back to Journals » Journal of Asthma and Allergy » Volume 18
Research Progress on Glycolysis in the Pathogenesis of Asthma
Received 18 March 2025
Accepted for publication 3 July 2025
Published 26 July 2025 Volume 2025:18 Pages 1147—1160
DOI https://doi.org/10.2147/JAA.S528965
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Luis Garcia-Marcos
Jindan Luo,1 Xiaoli Ge1,2
1Department of Pediatrics, The Affiliated Wuxi Second Hospital of Nanjing Medical University, Wuxi, 214000, People’s Republic of China; 2Department of Pediatrics, The Affiliated Wuxi Second People’s Hospital of Jiangnan University, Wuxi, 214000, People’s Republic of China
Correspondence: Xiaoli Ge, Department of Pediatrics, The Affiliated Wuxi Second Hospital of Nanjing Medical University, No. 68 of Zhongshan Road, Wuxi, 214000, People’s Republic of China, Tel +86-15936078575, Email [email protected]
Abstract: Asthma is a chronic, complex, and heterogeneous respiratory condition marked by airway hyperresponsiveness and reversible airflow limitation. Recent evidence highlights the significant role of metabolic changes, particularly glycolytic reprogramming, in the pathogenesis of asthma. Glycolysis, a fundamental pathway for both anaerobic and aerobic oxidation, not only generates adenosine triphosphate (ATP) but also supplies substrates for lipid-based ATP storage. While initially studied in the context of cancer, glycolysis has been associated with asthma through its interplay with programmed cell death, which is crucial in asthma pathophysiology. This study offers a comprehensive overview of current research on glycolytic reprogramming in asthma, emphasizing its potential implications and significance. The findings aim to inform future studies and contribute to the development of asthma-related precision medicine. A deeper understanding of the interplay between glycolysis and the development and progression of asthma may facilitate the development of innovative therapeutic approaches for this complex condition.
Keywords: asthma, glycolysis, immune cells, precision medicine, programmed cell death
Introduction
Asthma is increasingly recognized as a complex and heterogeneous disease. Its incidence varies slightly across countries and can affect up to 20% of a population, placing a significant medical and societal burden.1 The disease exhibits substantial heterogeneity in clinical presentation, severity, pathological biology, natural history, and response to treatment.2 Asthma can manifest at any stage of life, and while there are no current treatments that can alter its long-term effects, precision medicine holds promise for optimizing care and tailoring follow-up plans for patients.3
Children are more frequently affected by asthma than adults, with the condition presenting a broad range of underlying pathophysiology and severity. Poorly managed childhood asthma leads to significant economic consequences and serious physical and psychological effects on both children and caregivers. Recent advancements in asthma classification have led to the identification of distinct asthma endotypes, including type 2-low (non-eosinophilic, occasionally neutrophilic, metabolic) and type 2-high or ultra-high (primarily eosinophilic).4 Notably, epidemiological studies reveal that children with type 2-high asthma tend to experience more severe symptoms and frequent exacerbations compared to those with type 2-low asthma, highlighting the clinical relevance of these classifications.5
A key feature of asthma is airway obstruction, resulting from a reduction in airway diameter. Chronic inflammation of the airway wall, marked by the infiltration and activation of immune cells such as dendritic cells (DCs), eosinophils, neutrophils, lymphocytes, innate lymphoid cells (ILCs), and mast cells, leads to airway stenosis. The clinical manifestations of asthma arise from complex interactions between these immune cells and adjacent structural cells, such as epithelial cells.6
Numerous studies have demonstrated abnormal glucose metabolism in various immune cells, which is associated with both programmed cell death and the pathophysiology of asthma.7 Programmed cell death (PCD), including apoptosis, autophagy, pyroptosis, and ferroptosis, plays a crucial role in maintaining airway homeostasis and regulating inflammatory responses in asthma. Dysregulation of these processes contributes to key pathological features such as airway epithelial damage, excessive mucus production, and airway remodeling. For instance, impaired apoptosis of eosinophils prolongs airway inflammation, while excessive autophagy in airway smooth muscle cells promotes hyperplasia. The growing recognition of PCD’s role in asthma pathogenesis has opened new avenues for therapeutic intervention, particularly in treatment-resistant cases. This review aims to provide a concise overview of the relationship between type 2 inflammatory response-associated cells, glycolysis, programmed cell death, and asthma and it will offer a theoretical foundation for the precise treatment of asthma.
Glycolysis is a highly conserved metabolic pathway that serves as a key hub for cellular energy production and metabolic reprogramming. Recent studies have emphasized glycolysis as a central driver in the pathophysiology of asthma. Both immune cells (eg, mast cells, macrophages, and Th2 cells) and structural cells (eg, airway epithelial cells and smooth muscle cells in asthmatic airways) exhibit enhanced glycolytic activity, which promotes inflammation and remodeling.8 This metabolic shift not only meets the energy demands of overactive immune cells but also sustains chronic inflammation and remodeling, making glycolysis a potential therapeutic target.
Asthma
Epidemiology and Pathophysiologic Mechanisms of Asthma
Asthma affects approximately 10% of children worldwide,9 though its prevalence, symptoms, and disease differ significantly across regions. Mortality rates are highest in middle- and low-income countries, while morbidity is more pronounced in high-income nations.10 A nationwide cross-sectional study conducted in China in 2019 reported an overall asthma prevalence of 4.2% among adults (aged 20 years and older). This indicates that at least 45.7 million adults in China are affected by asthma.11 Factors such as living environment, infections, psychological factors, and the accessibility and quality of healthcare may all exacerbate asthma severity and impact patients’ quality of life.12–14
The fundamental pathological and physiological changes in asthma involve dysfunction of bronchial epithelial cells, hypertrophy and excessive contraction of airway smooth muscle cells, proliferation of goblet cells, and airway remodeling, along with the involvement of various cellular components and inflammatory mediators.15 Notably, the excessive deposition of extracellular matrix and contractile proteins in airway smooth muscle cells (ASMCs) has been implicate as a key contributor to airway remodeling, further exacerbating the chronic nature of asthma.16
Asthma Pathogenesis
In most individuals with asthma, eosinophilic inflammation and Th2 immune responses triggered by allergens are the primary causes of inflammation. Alarmins such as interleukin-25, interleukin-33, and thymic stromal lymphopoietin are typically released following the activation of the bronchial epithelium by allergens, microbes, or pollutants, leading to bronchial eosinophilic airway inflammation. This inflammation is sustained by type 2 cytokines produced by cells of the innate immune system (including natural killer cells, eosinophil and basophil progenitors, type 2 innate lymphoid-like 2 cells), such as Interleukin 4(IL-4), Interleukin 5(IL-5), and Interleukin 13 (IL-13), as well as cells of the adaptive immune system (eg, TH2 cells and innate-like natural killer T cells). Furthermore, IL-4 and IL-13 induce the release of pro-inflammatory mediators, B-cell class switching, IgE production, barrier disruption, and tissue remodeling.17
Currently, both glucocorticoids and inhaled bronchodilators are commonly prescribed for asthma management. However, excessive reliance on short-acting β2 receptor agonists increases the risk of asthma-related mortality, and the use of inhaled corticosteroids is often limited due to individual sensitivity to hormones and adverse reactions.1,18 Therefore, more effective and practical approaches are still needed for asthma treatment. Focusing on the molecular characteristics of asthma-related allergens, exploring the potential of molecular allergenics methods to better understand the mechanisms of allergic asthma is crucial for accurate diagnosis and patient management.19 With the rise of metabolomics research, a significant correlation between metabolic reprogramming and inflammatory responses has been identified.20 Studies have demonstrated that dexamethasone prevents asthma by regulating the HIF-1α-glycosylation lactate axis and protein lactylation.21,22 Increasing evidence suggests that certain forms of programmed cell death (PCD), such as autophagy, pyroptosis, ferroptosis, and apoptosis, also play a role in the onset and progression of asthma. These findings may ultimately open new avenues for asthma treatment.7
Glycolysis
Glycolysis Process
Glycolysis refers to the breakdown of one molecule of glucose into two molecules of pyruvate in the cytoplasm. Both anaerobic and aerobic oxidation of glucose utilize glycolysis as a common initial pathway. In conditions of insufficient oxygen supply or when oxygen is not effectively used, pyruvate is converted to lactate in the cytoplasm, a process known as anaerobic oxidation of glucose. Conversely, when oxygen supply is adequate, pyruvate primarily enters the mitochondria for complete oxidation to CO2 and H2O, which is termed aerobic oxidation of glucose. Glycolysis consists of ten steps (Figure 1), primarily involving the phosphorylation of hexose, the cleavage of hexose into triose, and the conversion of triose into pyruvate. Three irreversible metabolic steps, mediated by pyruvate kinase (PK), phosphofructokinase-1 (PFK-1), and hexokinase (HK), serve as rate-limiting steps in the glycolysis pathway. These steps are crucial regulatory points in glycolysis, which not only generates adenosine triphosphate (ATP) but also supplies substrates for ATP storage in the form of lipids.23 Although glycolysis produces a limited amount of ATP, glucose metabolism plays a central role in the three major metabolic processes, making this metabolic pathway highly efficient for the synthesis of nucleotides, lipids, and amino acids.24
 |
Figure 1 Glycolysis process. Created with BioGDP.com. |
Glycolysis in the Occurrence and Progression of Diseases
Glycolysis was first identified in the context of cancer, where tumor cells exhibit a high reliance on this pathway. Consequently, glycolysis is considered a key regulator in the initiation and progression of various tumors, including hepatocellular carcinoma, breast cancer, thyroid cancer, and oral cancer.25–28 Glucose metabolism reprogramming leads to several hallmark characteristics of cancer, including a strong association with cell proliferation, angiogenesis, lymphangiogenesis, metastasis, immune escape, and immune sensitization.29,30 In various types of tumors, m6A modifications activate or inhibit glycolysis-related signaling pathways by affecting the stability of key glycolytic enzyme mRNAs, which ultimately influences tumor biology, therapeutic response, and prognosis31 Beyond cancer, glycolysis also plays a critical role in other diseases, such as atherosclerosis, Alzheimer’s disease, rheumatoid arthritis, osteoarthritis, pathological pregnancy, kidney disease, diabetes, idiopathic pulmonary fibrosis, and bronchial asthma.32–40
Research Progress on Glycolysis and Asthma
Abnormalities of Glucose Metabolism in Airway Epithelial Cells of Individuals with Asthma
Studies have demonstrated impaired glucose metabolism and an imbalance in energy metabolism in the airway epithelial cells of children with asthma.41 Several metabolic pathways, particularly the glucose metabolism pathway, influence the function of airway epithelial cells, which act as the primary defense against inhaled allergens. These pathways are closely associated with inflammation, allergic responses, and compromised epithelial cell barrier function.42 Additionally, one of the primary mechanisms for increased airway resistance in asthma is bronchomotor tone, which is controlled by the shortening of airway smooth muscle. Inhibition of glycolysis alleviates bronchial constriction caused by carbohydrates in the small airways of humans, accompanied by a reduction in mitochondrial respiration and ATP production.43 A study using RNA sequencing of bronchial epithelial cells from individuals with asthma found that genes associated with the glycolysis pathway were downregulated after bronchial thermoplasty compared to how they were before treatment.44 Previous studies have demonstrated a strong correlation between Glutathione S-Transferase Pi (GSTP), M2 pyruvate kinase (PKM2), and the glycolysis pathways in asthma through sputum proteomics and transcriptomics (Table 1).
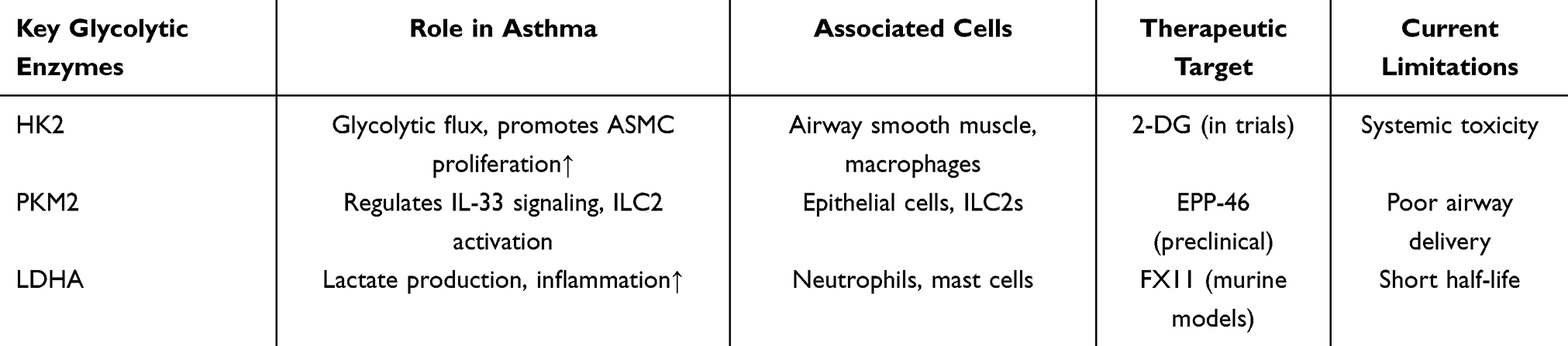 |
Table 1 Key Glycolytic Enzymes in Asthma Pathogenesis and Potential Therapeutic Targets |
Enhanced glycolysis pathways were detected in sputum samples from patients with asthma, and exposure to house dust mites stimulated the expression of PKM2 in mouse lung tissue.45 Moreover, increased expression of glycolysis-related proteins such as Hexokinase 1 (HK1), Hexokinase 2 (HK2), lactate dehydrogenase A (LDHA) was also observed in the lung tissue of allergic asthma model mice induced by house dust mites, as well as in the primary nasal epithelial cells of patients with asthma. Knocking out the expression of the LDHA gene led to a decrease in lactate levels in mouse lung tissue and a reduction in airway inflammation, suggesting that glycolysis may play a key pro-inflammatory role in asthma pathogenesis, contributing to airway remodeling and enhancing airway hyperresponsiveness.46 After exposing asthma model mice to PM2.5, Sun et al observed an increase in the glycolysis rate, upregulation of HK2 and LDHA protein expression, and further exacerbation of airway inflammation, indicating a complex relationship between glycolysis and asthma.47
Abnormalities in Glucose Metabolism in Immune Cells Associated with Asthma
Previous studies have found that during the onset of asthma, various immune cells undergo heightened oxidative stress (Table 2), with disruptions in glycolysis, fatty acid, and amino acid metabolism. These abnormalities can lead to dysregulation of both innate and adaptive immune responses.8
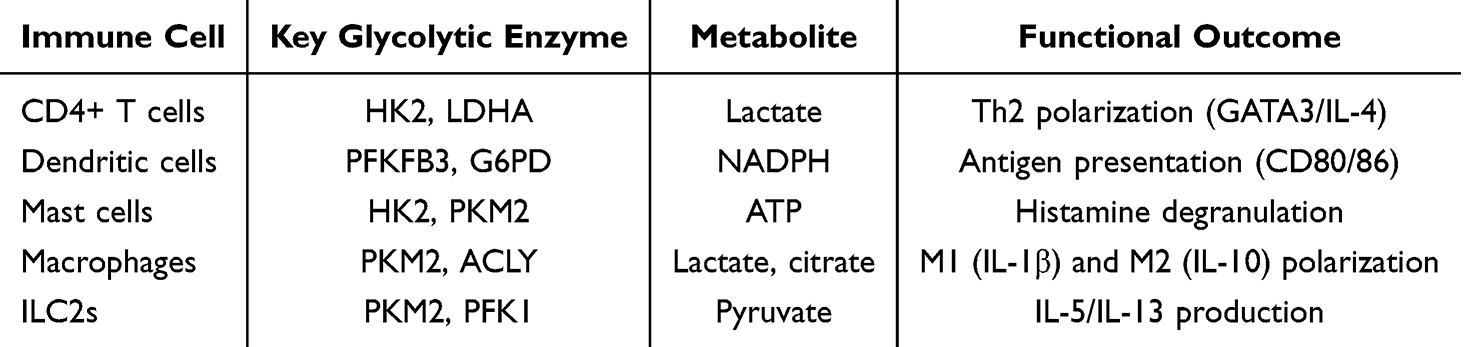 |
Table 2 Glycolysis Regulation of Asthma Related Immune Cells |
Mast Cells
Calcium-binding protein S100A4 plays a crucial role in allergic asthma mouse models by influencing mast cell activation.48 Additionally, Interleukin 33 (IL-33) impacts the energy metabolism of mast cells, leading to an increase in the expression of glycolytic proteins (HK2, M1 pyruvate kinase, and PKM2), as well as elevated lactate levels and oxidative phosphorylation. The glycolysis byproduct, lactate, has the ability to inhibit IL-33-induced mast cell activation.49 Changes in the glycolytic pathway also contribute to mast cell-mediated degranulation and histamine release in allergic reactions.
Macrophages
Macrophages are essential immune cells that play a key role in both innate and adaptive immune responses. By producing pro-inflammatory cytokines, they establish an initial line of defense in collaboration with other phagocytes. Pro-inflammatory cytokines facilitate the activation of the innate immune system, as well as subsequent T- and B-cell responses. M2 macrophages depend on oxidative metabolism, whereas M1 macrophages primarily rely on aerobic glycolysis. Glutathione can enhance inflammatory signaling in macrophages through histone lactylation.50
CD4+ T Cells
CD4+ T cells, a critical subset of lymphocytes, play an essential role in airway inflammation. Research has demonstrated that alterations in glycolysis levels in CD4+ T cells significantly impact cell activation, differentiation, proliferation, and survival, potentially altering their inflammatory phenotype and function.51 CD4+ T cells in asthma exhibit a metabolic shift toward aerobic glycolysis, which fuels their differentiation into pro-inflammatory subsets (eg, Th2, Th17) and suppresses regulatory T cells (Tregs). Key mechanisms include:①HK2/mTOR Activation: Allergen exposure upregulates hexokinase 2 (HK2) via mTOR signaling, increasing glucose uptake and lactate production. This promotes Th2 cytokine secretion (IL-4, IL-5, IL-13) by enhancing the stability of GATA3 mRNA, a master transcription factor for Th2 differentiation.52 ②LDHA-Lactate Axis: Lactate dehydrogenase A (LDHA)-derived lactate induces histone lactylation (eg, H3K18la) at the Il4 gene locus, further amplifying Th2 responses.45 Knockout of Ldha in T cells reduces airway eosinophilia in murine asthma models.
Type 2 Innate Lymphoid Cells (ILC2S)
In an IL-33-induced asthma animal model, prostaglandin E2 exerts a protective effect by inhibiting glycolysis and oxidative phosphorylation, effectively reducing the ILC2 response in mouse lung tissue and regulating the ILC2 response. By activating cAMP to inhibit the energy metabolism of ILC2s and blocking glycolysis, excessive ILC2 responses can be suppressed. Targeting the energy metabolism pathway of ILC2s could help control inflammation in the lungs of patients with type 2 asthma.53 The maturation and activation of ILC2s in allergic reactions are also critically dependent on arginine metabolism and glycolysis. Methylation of its gene ST2 and Hif-1α-driven development of the glycolytic enzyme PKM2 suppresses IL-33-driven ILC2 maturation by reducing the expression of its receptor, Interleukin 1 Receptor-Like 1 (ILR1).54
Dendritic Cells
Research utilizing DCs generated from mouse bone marrow has demonstrated that elevated glycolysis is essential for DC activation. Glycolysis supplies pyruvate for mitochondrial respiration and directs intermediates to the pentose phosphate pathway, which produces NADPH necessary for fatty acid synthesis and maintaining redox balance. This process supports the energy and biosynthesis requirements of activated DCs. DCs rely on glycolytic flux to support their activation and immunostimulatory functions in asthma: ①PFKFB3-Driven Glycolysis: Allergen uptake triggers PFKFB3 (a key glycolytic regulator) via TLR4/MyD88 signaling, increasing ATP production for MHC-II upregulation and costimulatory molecule (CD80/86) expression.55 ②NADPH and Lipid Synthesis: Glycolysis intermediates feed into the pentose phosphate pathway (PPP), generating NADPH to sustain lipid raft formation and antigen presentation. Inhibition of glucose-6-phosphate dehydrogenase (G6PD), the rate-limiting PPP enzyme, impairs DC-mediated Th2 priming.56
Other Related Cells and Cytokines
Yu et al discovered that Thymic Stromal Lymphopoietin (TSLP) enhances aerobic glycolysis in asthmatic airway epithelial cells by stimulating the release of inflammatory cytokines through the miR-223/VHL/HIF-1α pathway.57 In mouse lungs, it has been demonstrated that glycolysis mediates allergen-induced production of IL-33, IL-13, and Chemokine (C-C motif) Ligand 20.46
All of these indicate that glycolysis-related pathways may be involved in the pathogenesis of asthma by affecting the metabolic processes of multiple cellular components.
Glycolytic Reprogramming in Asthma Phenotypes: T2-High vs T2-Low
Emerging evidence suggests that glycolytic changes may drive distinct asthma phenotypes, particularly T2-high (eosinophilic) and T2-low (non-eosinophilic, often neutrophilic or metabolic) endotypes, through divergent metabolic and inflammatory pathways.
T2-High Asthma
In T2-high asthma, glycolysis fuels the activation and proliferation of type 2 immune cells (eg, ILC2s, Th2 cells, eosinophils). For instance, IL-33 and IL-13 signaling upregulates HIF-1α, which enhances glycolytic enzymes (eg, PKM2, HK2) and lactate production. This metabolic shift supports the energetic demands of eosinophil survival and cytokine production (eg, IL-5, IL-13), perpetuating eosinophilic inflammation.58 Knockdown of LDHA in murine models reduces lactate levels and attenuates airway inflammation, directly implicating glycolysis in T2-high pathology. Additionally, HIF-1α-driven glycolysis stabilizes GATA3 expression in Th2 cells, reinforcing type 2 cytokine production.59
T2-Low Asthma
In T2-low asthma, glycolytic reprogramming may promote neutrophilic inflammation via macrophage polarization (M1 phenotype) and NLRP3 inflammasome activation. Elevated glycolysis in airway epithelial cells increases ROS production, which amplifies IL-17/IL-23 pathways and neutrophil recruitment.60 PM2.5 exposure exacerbates T2-low features by upregulating HK2 and LDHA, leading to mitochondrial dysfunction and oxidative stress—key drivers of neutrophilic inflammation.61 Inhibition of glycolysis with 2-deoxyglucose (2-DG) reduces IL-17A secretion in murine models, suggesting a metabolic checkpoint for this phenotype.62
Contradictions in Glycolysis Inhibition
The Impact of Glycolysis Inhibition on Asthma Inflammation
The effects of glycolysis inhibition on asthma inflammation appear to be contradictory across different studies, with some reporting reduced inflammation and others increased inflammation. These discrepancies may stem from differences in experimental models, target cell types, disease stages, and metabolic microenvironments.
Cell-Type Specificity
Anti-inflammatory Effects: In eosinophilic (T2-high) asthma, glycolysis inhibition (eg, via LDHA knockdown or 2-deoxyglucose) reduces lactate production, weakens HIF-1α stability, and dampens Th2/ILC2 activation, thereby alleviating type 2 inflammation.21 Similarly, in airway smooth muscle cells (ASMCs), glycolysis inhibition decreases excitation-contraction coupling and relaxes bronchial contraction.43 Glycolysis promotes the production of pro-inflammatory cytokines (eg, IL-4, IL-13); thus, glycolysis inhibition disrupts this energy-demanding process.
Pro-inflammatory Effects: In neutrophilic (T2-low) asthma, glycolysis inhibition may exacerbate inflammation by impairing anti-inflammatory M2 macrophage polarization50 or increasing ROS due to compensatory oxidative phosphorylation. M2 macrophages rely on oxidative metabolism. Glycolysis inhibition shifts macrophages toward an M1-like pro-inflammatory state, amplifying IL-17/neutrophil responses.
Disease Stage and Metabolic Flexibility
Acute vs Chronic Inflammation: In early asthma, glycolysis inhibition can suppress acute inflammation by limiting immune cell activation (eg, mast cell degranulation49); In chronic remodeling, prolonged glycolysis inhibition may impair epithelial repair mechanisms (eg, autophagy-mediated glucose uptake63), worsening tissue damage. Metabolic Adaptation: Cells can switch to alternative pathways (eg, fatty acid oxidation). For instance, when glycolysis is blocked, ILC2s increase fatty acid oxidation to sustain inflammation.64
Environmental Triggers
PM2.5 exposure upregulates HK2/LDHA, exacerbating glycolysis-driven inflammation.47 However, in models without environmental co-stimulation, glycolysis inhibition may have neutral or beneficial effects. House dust mite (HDM) induces PKM2-dependent glycolysis, promoting inflammation,45 thus inhibition is protective. In contrast, viral infections may require glycolysis for antiviral defense, making inhibition detrimental.
Overall, whether glycolysis inhibition exacerbates or alleviates asthma inflammation depends on multiple factors. Precise targeting (eg, cell-specific inhibitors or stage-specific interventions) could reconcile these contradictions.
Glycolysis-Related Genes, Proteins, and Metabolites in Asthma
Key Genes and Proteins
HK1/2: Upregulated in asthmatic airway epithelial cells and smooth muscle cells, driving glucose phosphorylation and glycolytic flux46; PKM2: Promotes lactate production and IL-33 release in epithelial cells; Its oxidation by GSTP enhances glycolysis in asthma; LDHA: Elevated in asthma models; knockout reduces lactate levels and airway inflammation; PFKFB3 (6-Phosphofructo-2-Kinase/Fructose-2,6-Biphosphatase 3): Links glycolysis to HIF-1α stabilization, exacerbating type 2 inflammation.55
Metabolites
Lactate: A Key Immunomodulatory Metabolite Accumulates in asthmatic airways. ① Pro-inflammatory Role: Elevated lactate levels in asthmatic airways promote histone lactylation (eg, Vps34, HIF-1α), sustaining autophagy and cytokine production (IL-33, IL-13) in epithelial cells.65 ② Macrophage Polarization: Lactate induces M1 macrophage polarization via histone lactylation (eg, H3K18la), amplifying oxidative stress and NLRP3 inflammasome activation.50 ③ Mast Cell Regulation: Lactate inhibits IL-33-induced mast cell degranulation but enhances glycolysis-driven histamine release, exacerbating allergic responses.49
ATP: Energy Supply and Signaling. ① Airway Smooth Muscle (ASM) Contraction: Glycolysis-derived ATP fuels ASM hyperresponsiveness by maintaining Ca2+ oscillations.43 ② Immune Cell Activation: ATP acts as a danger signal (via P2X7 receptors), promoting Th2 cytokine release (IL-4, IL-5) and eosinophil recruitment.66
NADPH: Redox Balance and Lipid Synthesis. ① Oxidative Stress: NADPH from the pentose phosphate pathway (PPP) supports ROS production in epithelial cells, exacerbating barrier dysfunction.67 ② Lipid Mediators: NADPH fuels synthesis of pro-inflammatory lipids (eg, PGE2) in dendritic cells, driving type 2 inflammation.56
Pyruvate: A Metabolic Switch.Mitochondrial Shuttling: Under hypoxia, pyruvate is diverted to lactate (via LDHA), while normoxia favors mitochondrial oxidation. In asthma, HIF-1α stabilizes LDHA, skewing metabolism toward lactate production21 (Table 3).
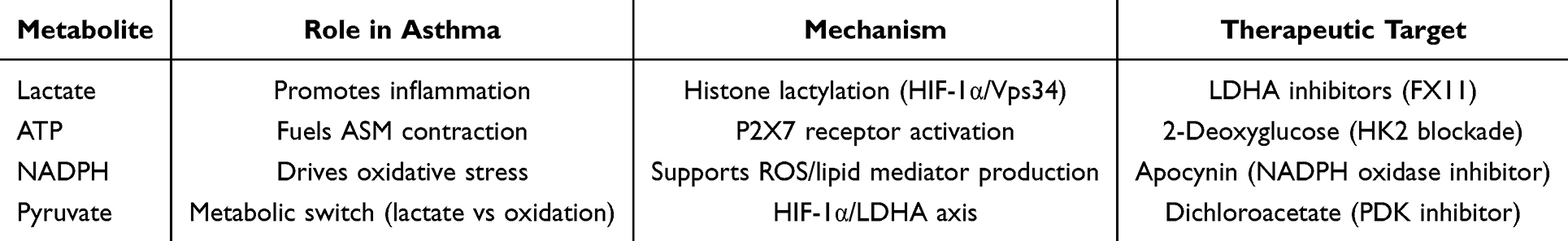 |
Table 3 Glycolytic Metabolites in Asthma: Roles and Therapeutic Targets |
Upstream Regulators of Glycolysis in Asthma
HIF-1α: Activated by hypoxia/allergens, upregulates HK2, LDHA, and GLUT1 to enhance glycolysis;21 MTOR/c-Myc: Drives HK2 expression via p62-mediated autophagy, promoting smooth muscle proliferation;68 IL-33/TSLP: Alarmins stimulate glycolysis through miR-223/VHL/HIF-1α and PI3K/Akt pathways.57
Downstream Metabolic Pathways Linked to Glycolysis
Lactate-Shuttling: Lactate induces M2 macrophage polarization via histone lactylation, amplifying inflammation;50 Pentose Phosphate Pathway (PPP): Supplies NADPH for lipid synthesis in dendritic cells, sustaining their activation56; Mitochondrial Dysfunction: Glycolysis-driven ROS (eg, via PKM2) exacerbates oxidative stress and airway remodeling.
Glycolysis, Programmed Cell Death, and Asthma
A growing body of research has demonstrated that certain types of PCD, including pyroptosis, autophagy, apoptosis, and ferroptosis, are related to the pathogenesis of asthma.7 Numerous studies on autophagy have gradually revealed its role in the physiology of asthma.
Patients with asthma can reduce the generation of NLRP3 (nucleotide-binding oligomeric domain-like receptor protein 3) -mediated pulmonary epithelial inflammation by blocking the activation of the ULK1/Atg9a/Rab9 signaling pathways, thereby inhibiting Golgi apparatus fragmentation and mitochondrial oxidative stress.69 Mitochondria are the primary source of reactive oxygen species (ROS); however, excessive ROS production can result in airway inflammation and remodeling in patients with asthma. The Th2-like cytokine IL-25 can trigger M2 macrophage polarization in monocytes by inducing ROS generation, increasing the activity of the mitochondrial respiratory chain complex, activating AMPK, and inducing mitochondrial respiration.70 Transcription factor EB (TFEB) activation restores damaged autophagy, alleviates NLRP3-driven lung inflammation, and improves the severe asthma (SA) phenotype.71 Studies have demonstrated that particulate matter (PM) may cause immune imbalance through MAPK and autophagy pathways, induce and exacerbate lung diseases, lead to increased smooth muscle thickness and mucus production, and result in decreased lung function.72 By stabilizing High Mobility Group Box 1 (HMGB1) increased Ring finger protein 125 (RNF125) methylation encourages autophagy-induced oxidative stress in asthma.73 In airway epithelial cells, apigenin (API) can reverse the decline in mitochondrial membrane potential (MMP) and apoptosis caused by House Dust Mite (HDM), while boosting cell viability and reducing ROS production.74
Numerous studies have demonstrated a link between autophagy and glucose, with low glucose levels promoting cellular autophagy. However, little is known about how these two highly conserved biological processes—autophagy and glycolysis—interact within both healthy and pathological cellular programs. In cases of malnutrition, UNC-51 like kinase 1 (ULK1) activation triggers the glycolytic enzyme LDHA, according to a study by Jia et al. Specifically, ULK1 stimulates lactate synthesis, phosphorylates serine-196 during nutritional shortages, and directly interacts with LDHA. Through Vps34 lactylation (at lysine 356 and lysine 781) mediated by acyltransferase Lysine Acetyltransferase 5 (KAT5) / Tat-interactive protein 60 (TIP60), lactic acid is linked to autophagy and glycolysis. Lactylation of Vps34 results in increased Vps34 lipid kinase activity and enhanced binding of Vps34 to Beclin1, Atg14L, and UVRAG. This process stimulates lysosomal transport and autophagic flux.65
Autophagy is typically activated under stress conditions, such as nutrient deprivation, which includes amino acids and glucose. It has been demonstrated that the glucose starvation response involves autophagy triggered by AMP-activated protein kinase (AMPK).75 On the other hand, autophagy also stimulates glucose absorption via the glucose transporter 1 (Glut1).76 Knocking down the autophagy gene Atg5 leads to a reduction in glucose uptake by Club cells. Additionally, autophagy promotes the proliferation of Club cells while inhibiting their differentiation into ciliated and goblet cells, indicating that autophagy and glucose metabolism play important regulatory roles in the differentiation and proliferation of airway progenitor cells.63
In addition, asthma and bleomycin-induced pulmonary fibrosis are two conditions in which autophagy drives metabolic reprogramming of the lung epithelium, controlling the energy balance of metabolic pathways.77 ILC2, in the absence of autophagy, is impaired in its ability to utilize fatty acid oxidation, which significantly promotes glycolysis, as demonstrated by Treger et al through transcriptomics and metabolite analysis.64 In the bronchial smooth muscle cells of patients with asthma, Yu et al found that the autophagy protein p62 regulates glycolysis through the mTOR/c-Myc/HK2 pathway, promoting smooth muscle cell proliferation and migration, thereby exacerbating chronic airway remodeling.68
Precision Medicine for Asthma Treatment
Asthma management involves controlling asthma symptoms, assessing risk factors through periodic cycles, and adjusting medication accordingly. The development of biotherapy has introduced the era of precision medicine in the management of severe asthma, driving clinical advancements toward disease remission.12 Immune metabolism mechanisms encompass multiple signaling pathways, offering new therapeutic targets for asthma.
Glycolytic Inhibitors in Preclinical Asthma Models
Recent studies have explored glycolysis inhibitors as potential therapeutic agents for asthma:
2-Deoxy-D-Glucose (2-DG): A competitive inhibitor of hexokinase (HK), 2-DG has been shown to reduce airway hyperresponsiveness and inflammation in mouse models of asthma by inhibiting glycolysis in immune cells and airway smooth muscle cells.21 However, its clinical application is limited by systemic toxicity and off-target effects on non-glycolytic pathways.
Dichloroacetate (DCA): By inhibiting pyruvate dehydrogenase kinase (PDK). Preclinical studies demonstrate that DCA attenuates airway remodeling and inflammation in allergic asthma models78. However, its efficacy is variable, and long-term use may lead to peripheral neuropathy.
3-Bromopyruvate (3-BP): This alkylating agent targets HK2 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH), effectively reducing lactate production and inflammation in asthma models. However, 3-BP’s poor selectivity and potential hepatotoxicity pose significant challenges for clinical translation.79
Although these inhibitors show promise, they also have certain limitations: ① The lack of cell specificity may lead to unintended metabolic disruptions in non-target tissues. ② An incomplete understanding of metabolic redundancy, where compensatory pathways may diminish therapeutic efficacy. ③ Toxicity profiles, which necessitate further optimization for safe human use.
Future research should focus on developing targeted delivery systems (eg, nanoparticle-based carriers) and combination approaches to enhance specificity and reduce side effects. Targeting glycolysis in asthma offers transformative potential but requires careful consideration of endotype specificity, delivery methods, and biomarker development. Collaborative efforts between metabolomics and clinical research are essential to advance this paradigm.
“Two-Phase Glycolytic Modulation” Approach for Asthma
Phase 1 (Acute Exacerbations): Short-term inhibition of glycolysis in immune cells (eg, using HK2 inhibitors) to suppress inflammation. Phase 2 (Chronic Management): Long-term modulation of glycolysis in airway structural cells (eg, ASMCs) to prevent remodeling. This strategy addresses the dual role of glycolysis in inflammation (acute) and tissue remodeling (chronic), offering a more nuanced approach than current therapies.
Glycolytic Profiling of Patients Could Guide Personalized Therapy
Type 2-high asthma: Target PKM2 to reduce eosinophilic inflammation. Obesity-associated asthma: Combine glycolysis inhibitors (eg, 2-DG) with metabolic modulators (eg, AMPK activators). Additionally, single-cell metabolomics could be employed in the future to map glycolytic heterogeneity in asthma airways and organ-on-a-chip models to test metabolic therapies. Future therapies should not only focus on inhibiting glycolysis but also aim to rewire metabolic pathways to restore the crosstalk between immune and structural cells. Through mTOR/c-Myc/HK2-mediated glycolysis, autophagy protein p62 promotes the growth and migration of bronchial smooth muscle cells (BSMCs), providing a novel target for airway remodeling therapy.68 Human airway epithelial cells release siderophores as fatality signals under type 2 (T2) inflammatory conditions, activating the combination of 15-lipoxygenase-1 (15LO1) and PE binding protein-1 (PEBP1). PEBP1 drives the dynamic interaction between siderophores and 15LO1, while microtubule-associated light chain-3 (LC3), an autophagy protein, also interacts with this complex. In T2 asthma, low mitochondrial DNA levels in bronchoalveolar lavage fluid and disease progression correlate with high expression of 15LO1-PEBP1 and LC3-II in airway epithelial cells. The concurrent activation of iron-induced apoptosis and autophagy by the 15LO1-PEBP1 complex presents a potential therapeutic target and a novel pathogenic molecular mechanism related to allergic asthma.80
Inhibition of HMGB1, by blocking the ROS/AMPK/autophagy pathway, may alleviate asthma caused by toluene diisocyanate (TDI), providing important insights into the etiology and potential treatment targets for TDI-induced asthma.81 DEK deficiency alleviates airway inflammation in asthma by downregulating PINK1/Parkin mitosis, NLRP3 inflammasome activation, and apoptosis, potentially through the DEK/ATAD3A/DRP1 signaling axis. This discovery may offer a new therapeutic target for asthma management.82
Research indicates that exogenous Ang-(1-7) can inhibit autophagy through the HIF-1α/THB1/BECN1 axis, alleviating airway remodeling caused by chronic intermittent hypoxia (CIH) and suggesting its potential therapeutic effect on obstructive sleep apnea-associated asthma.83 Hu et al demonstrated that high methylation of RNF125 reduces its expression, increasing the stability of HMGB1 and promoting autophagy-induced oxidative stress in asthma.73 These findings provide a theoretical and experimental basis for understanding the etiology of asthma.
Celery extract interferes with the ROS-ASK1-MAPK pathway, enhancing cell viability, mitochondrial function, inhibiting cell apoptosis, and clarifying its potential therapeutic targets in chronic obesity-related asthma.74 Sesamin decreases PINK1/Parkin expression, mitochondrial translocation, and cell apoptosis, increases mitochondrial membrane potential, and blocks TNF-α/IL-4-induced mitochondrial ROS. By blocking mitochondrial autophagy and apoptosis, sesamin may reduce airway inflammation in asthma, making it a potential asthma therapy.84
Wnt5a is associated with several lung disorders, and the epithelial-mesenchymal transition (EMT) of human bronchial epithelial cells (HBECs) plays a crucial role in airway remodeling in asthma. Recent research has revealed a novel link between the Wnt5a-Ca2+/CaMKII autophagy axis and the initiation of airway remodeling.85 FOXK2, a key regulator of glycolysis, inhibits TGF-β1-induced EMT and glycolysis in BEAS-2B cells when absent, offering new approaches to treating diseases associated with EMT.86
Conclusion
Overall, glycolysis-related pathways play a pivotal role in the onset and progression of asthma; however, current research remains in its early stages, with limited literature available. The feasibility of targeting glycolysis pathways for asthma treatment requires further research to validate its effectiveness. While targeted glycolysis has yielded promising results in the treatment of tumor diseases, it may eventually serve as a therapeutic target in asthma research, offering a theoretical foundation for personalized asthma treatment. Despite increasing evidence linking glycolytic reprogramming to asthma pathogenesis, several critical questions remain unanswered. First, we still do not fully understand how different airway cells, including immune and structural cells, specifically utilize glycolysis during the disease. Second, most studies only capture metabolic changes in asthma patients at a single time point, failing to reflect the long-term evolution of the metabolic state. Third, while animal experiments offer hope, clinical trials are needed to confirm whether targeting glycolysis can safely and effectively treat human asthma patients. To address these gaps, future research should: (1) develop targeted therapies that precisely alter glycolysis in specific cell types; (2) identify metabolic biomarkers to better classify asthma subtypes and monitor treatment responses; (3) test combination therapies pairing glycolytic inhibitors with existing drugs; (4) use advanced multi-omics approaches to map the complete metabolic network of asthma; and (5) explore personalized treatment strategies based on patients’ unique metabolic profiles. These studies will not only deepen our understanding of the metabolic basis of asthma but also potentially lead to breakthrough therapies. By focusing on these key areas, we can hopefully translate laboratory findings into real clinical benefits for asthma patients.
Abbreviations
DCs, dendritic cells; ILCs, innate lymphoid cells; ASMCs, airway smooth muscle cells; PCD, programmed cell death; PK, pyruvate kinases; PFK-1, phosphofructokinases-1; HK, hexokinases; EC, excitation-contraction; ILC2S, Type 2 Innate Lymphoid Cells; SA, severe asthma; ROS, reactive oxygen species; IL-13, Interleukin 13; IL-33, Interleukin 13; PKM2, Pyruvate kinase isozyme type M2; HK1, Hexokinase 1; HK2, Hexokinase 1; IL1RL1, Recombinant interleukin 1 receptor like protein 1; NADPH, Nicotinamide adenine dinucleotide phosphate; CCL20, C-C chemokine ligand 20; NLRP3, NOD-like receptor protein 3; TFEB, Transcription factor EB; AMPK, AMP-activated protein kinase; RNF125, Ring finger protein 125; API, apigenin; MMP, Mitochondrial membrane potential; HDM, House dust mite; ULK1, UNC-51 like kinase 1; LDHA, lactate dehydrogenase A; Glut1, glucose transporter 1; 15LO1, 15 lipoxygenase-1; PEBP1, PE binding protein-1; LC3, light chain-3; TDI, toluene diisocyanate; CIH, chronic intermittent hypoxia; HBECs, human bronchial epithelial cells; EMT, epithelial mesenchymal transition.
Data Sharing Statement
All data generated or analysed during this study are included in this article. Further enquiries can be directed to the corresponding author.
Ethics Approval and Consent to Participate
This study is a literature review and does not require the informed consent of patients, and does not require ethical approval.
Acknowledgments
We would like to acknowledge the hard and dedicated work of all the staff that implemented the intervention and evaluation components of the study.
Funding
Scientific Research Program of Wuxi health Commission (grant number:M202223).
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Reddel HK, Bacharier LB, Bateman ED, et al. Global initiative for asthma strategy 2021: executive summary and rationale for key changes. Eur Respir J. 2022;59(1):2102730. doi:10.1183/13993003.02730-2021
2. Lommatzsch M, Brusselle GG, Levy ML, et al. A2BCD: a concise guide for asthma management. Lancet Respir Med. 2023;11(6):573–576. doi:10.1016/S2213-2600(22)00490-8
3. Melén E, Zar HJ, Siroux V, et al. Asthma inception: epidemiologic risk factors and natural history across the life course. Am J Respir Crit Care Med. 2024;210(6):737–754. doi:10.1164/rccm.202312-2249SO
4. Peters MC, Ringel L, Dyjack N, et al. A transcriptomic method to determine airway immune dysfunction in t2-high and t2-low asthma. Am J Respir Crit Care Med. 2019;199(4):465–477. doi:10.1164/rccm.201807-1291OC
5. Levy ML, Bacharier LB, Bateman E, et al. Key recommendations for primary care from the 2022 Global Initiative for Asthma (GINA) update. NPJ Prim Care Respir Med. 2023;33(1):7. doi:10.1038/s41533-023-00330-1
6. Hammad H, Lambrecht BN. The basic immunology of asthma. Cell. 2021;184(9):2521–2522. doi:10.1016/j.cell.2021.04.019
7. Liu L, Zhou L, Wang LL, et al. Programmed cell death in asthma: apoptosis, autophagy, pyroptosis, ferroptosis, and necroptosis. J Inflamm Res. 2023;16:2727–2754. doi:10.2147/JIR.S417801
8. Qin Z, Chen Y, Wang Y, et al. Immunometabolism in the pathogenesis of asthma. Immunology. 2024;171(1):1–17. doi:10.1111/imm.13688
9. Global Asthma Network. The global asthma report 2022. Int J Tuberc Lung Dis. 2022;26:S1–104.
10. Mortimer K, Reddel HK, Pitrez PM, Bateman ED. Asthma management in low and middle income countries: case for change. Eur Respir J. 2022;60(3):2103179. doi:10.1183/13993003.03179-2021
11. China Pulmonary Health (CPH) Study Group. Huang K, Yang T, Xu J, et al. Prevalence, risk factors, and management of asthma in China: a national cross-sectional study. Lancet. 2019;394(10196):407–418. doi:10.1016/S0140-6736(19)31147-X.
12. Cheng PP, Yu F, Chen SJ, et al. PM2.5 exposure-induced senescence-associated secretory phenotype in airway smooth muscle cells contributes to airway remodeling. Environ Pollut. 2024;347:123674. doi:10.1016/j.envpol.2024.123674
13. Rosas-Salazar C, Chirkova T, Gebretsadik T, et al. Respiratory syncytial virus infection during infancy and asthma during childhood in the USA (INSPIRE): a population-based, prospective birth cohort study. Lancet. 2023;401(10389):1669–1680. doi:10.1016/S0140-6736(23)00811-5
14. Shipp CL, Gergen PJ, Gern JE, Matsui EC, Guilbert TW. Asthma Management in Children. J Allergy Clin Immunol Pract. 2023;11(1):9–18. doi:10.1016/j.jaip.2022.10.031
15. Porsbjerg C, Melén E, Lehtimäki L, Shaw DA. Asthma. Lancet. 2023;401(10379):858–873. doi:10.1016/S0140-6736(22)02125-0
16. Cheng F, Lu T, Wang Y, et al. Expression of airway smooth muscle contractile proteins in children with acute interstitial pneumonia. Int J Exp Pathol. 2022;103(5):190–197. doi:10.1111/iep.12443
17. Maspero J, Adir Y, Al-Ahmad M, et al. Type 2 inflammation in asthma and other airway diseases. ERJ Open Res. 2022;8(3):00576–2021. doi:10.1183/23120541.00576-2021
18. García-Marcos L, Chiang CY, Asher MI, et al. et al.Global Asthma Network Phase I Study Group. Asthma management and control in children, adolescents, and adults in 25 countries: a global asthma network phase I cross-sectional study. Lancet Glob Health. 2023;11(2):e218–e228. doi:10.1016/S2214-109X(22)00506-X
19. Agache I, Palmer E, Sanver D, Kirtland M, Shamji MH. Molecular allergology approach to allergic asthma. Mol Aspects Med. 2022;85:101027. doi:10.1016/j.mam.2021.101027
20. Pålsson-McDermott EM, O’Neill LAJ. Targeting immunometabolism as an anti-inflammatory strategy. Cell Res. 2020;30(4):300–314. doi:10.1038/s41422-020-0291-z
21. Chen N, Xie QM, Song SM, et al. Dexamethasone protects against asthma via regulating Hif-1α-glycolysis-lactate axis and protein lactylation. Int Immunopharmacol. 2024;131:111791. doi:10.1016/j.intimp.2024.111791
22. Gan PXL, Liao W, Lim HF, Wong WSF. Dexamethasone protects against Aspergillus fumigatus-induced severe asthma via modulating pulmonary immunometabolism. Pharmacol Res. 2023;196:106929. doi:10.1016/j.phrs.2023.106929
23. Chandel NS. Glycolysis. Cold Spring Harb Perspect Biol. 2021;13(5):a040535. doi:10.1101/cshperspect.a040535
24. Reinfeld BI, Rathmell WK, Kim TK, Rathmell JC. The therapeutic implications of immunosuppressive tumor aerobic glycolysis. Cell Mol Immunol. 2022;19(1):46–58. doi:10.1038/s41423-021-00727-3
25. Xu M, Zhou C, Weng J, et al. Tumor associated macrophages-derived exosomes facilitate hepatocellular carcinoma malignance by transferring lncMMPA to tumor cells and activating glycolysis pathway. J Exp Clin Cancer Res. 2022;41(1):253. doi:10.1186/s13046-022-02458-3
26. Lin J, Fang W, Xiang Z, et al. Glycolytic enzyme HK2 promotes PD-L1 expression and breast cancer cell immune evasion. Front Immunol. 2023;14:1189953. doi:10.3389/fimmu.2023.1189953
27. Shi L, Duan R, Sun Z, et al. LncRNA GLTC targets LDHA for succinylation and enzymatic activity to promote progression and radioiodine resistance in papillary thyroid cancer. Cell Death Differ. 2023;30(6):1517–1532. doi:10.1038/s41418-023-01157-6
28. Hou Z, Wu C, Tang J, Liu S, Li L. CLSPN actives Wnt/β-catenin signaling to facilitate glycolysis and cell proliferation in oral squamous cell carcinoma. Exp Cell Res. 2024;435(2):113935. doi:10.1016/j.yexcr.2024.113935
29. Paul S, Ghosh S, Kumar S. Tumor glycolysis, an essential sweet tooth of tumor cells. Semin Cancer Biol. 2022;86(Pt 3):1216–1230. doi:10.1016/j.semcancer.2022.09.007
30. Wu L, Jin Y, Zhao X, et al. Tumor aerobic glycolysis confers immune evasion through modulating sensitivity to T cell-mediated bystander killing via TNF-α. Cell Metab. 2023;35(9):1580–1596.e9. doi:10.1016/j.cmet.2023.07.001
31. Yue SW, Liu HL, Su HF, et al. m6A-regulated tumor glycolysis: new advances in epigenetics and metabolism. Mol Cancer. 2023;22(1):137. doi:10.1186/s12943-023-01841-8
32. Li L, Wang M, Ma Q, Ye J, Sun G. Role of glycolysis in the development of atherosclerosis. Am J Physiol Cell Physiol. 2022;323(2):C617–C629. doi:10.1152/ajpcell.00218.2022
33. Yang F, Zhao D, Cheng M, et al. mTOR-mediated immunometabolic reprogramming nanomodulators enable sensitive switching of energy deprivation-induced microglial polarization for alzheimer’s disease management. ACS Nano. 2023;17(16):15724–15741. doi:10.1021/acsnano.3c03232
34. Jia N, Gao Y, Li M, et al. He Q, et al.Metabolic reprogramming of proinflammatory macrophages by target delivered roburic acid effectively ameliorates rheumatoid arthritis symptoms. Signal Transduct Target Ther. 2023;8(1):280. doi:10.1038/s41392-023-01499-0
35. Jiang D, Guo J, Liu Y, Li W, Lu D. Glycolysis: an emerging regulator of osteoarthritis. Front Immunol. 2024;14:1327852. doi:10.3389/fimmu.2023.1327852
36. Gou R, Zhang X. Glycolysis: a fork in the path of normal and pathological pregnancy. FASEB J. 2023;37(12):e23263. doi:10.1096/fj.202301230R
37. Gu M, Tan M, Zhou L, et al. Protein phosphatase 2Acα modulates fatty acid oxidation and glycolysis to determine tubular cell fate and kidney injury. Kidney Int. 2022;102(2):321–336. doi:10.1016/j.kint.2022.03.024
38. Atawia RT, Batori RK, Jordan CR, et al. Fukai T, et al.type 1 diabetes impairs endothelium-dependent relaxation via increasing endothelial cell glycolysis through advanced glycation end products, PFKFB3, and nox1-mediated mechanisms. Hypertension. 2023;80(10):2059–2071. doi:10.1161/HYPERTENSIONAHA.123.21341
39. Yan P, Liu J, Li Z, et al. Glycolysis reprogramming in idiopathic pulmonary fibrosis: unveiling the mystery of lactate in the lung. Int J Mol Sci. 2023;25(1):315. doi:10.3390/ijms25010315
40. Xuan L, Ren L, Zhang W, Du P, Li B, An Z. Formaldehyde aggravates airway inflammation through induction of glycolysis in an experimental model of asthma exacerbated by lipopolysaccharide. Sci Total Environ. 2024;912:168947. doi:10.1016/j.scitotenv.2023.168947
41. Berdnikovs S, Newcomb DC, Gebretsadik T, et al. Cellular and systemic energy metabolic dysregulation in asthma development-a hypothesis-generating approach. J Allergy Clin Immunol. 2022;149(5):1802–1806.e2. doi:10.1016/j.jaci.2021.10.024
42. Queener AM, Chiarella SE, Cuervo-Pardo L, Coden ME, Abdala-Valencia H, Berdnikovs S. Metabolism of epithelial cells in health and allergic disease: collegium internationale allergologicum update 2021. Int Arch Allergy Immunol. 2021;182(8):663–678. doi:10.1159/000516809
43. Xu S, Karmacharya N, Woo J, et al. Starving a cell promotes airway smooth muscle relaxation: inhibition of glycolysis attenuates excitation-contraction coupling. Am J Respir Cell Mol Biol. 2023;68(1):39–48. doi:10.1165/rcmb.2021-0495OC
44. Ravi A, Goorsenberg AWM, Dijkhuis A, et al. Metabolic differences between bronchial epithelium from healthy individuals and patients with asthma and the effect of bronchial thermoplasty. J Allergy Clin Immunol. 2021;148(5):1236–1248. doi:10.1016/j.jaci.2020.12.653
45. van de Wetering C, Manuel AM, Sharafi M, et al. Glutathione-S-transferase P promotes glycolysis in asthma in association with oxidation of pyruvate kinase M2. Redox Biol. 2021;47:102160. doi:10.1016/j.redox.2021.102160
46. Qian X, Aboushousha R, van de Wetering C, et al. Casey DT, et al.IL-1/inhibitory κB kinase ε-induced glycolysis augment epithelial effector function and promote allergic airways disease. J Allergy Clin Immunol. 2018;142(2):435–450.e10. doi:10.1016/j.jaci.2017.08.043
47. Sun L, Fu J, Lin SH, et al. Particulate matter of 2.5 μm or less in diameter disturbs the balance of TH17/regulatory T cells by targeting glutamate oxaloacetate transaminase 1 and hypoxia-inducible factor 1α in an asthma model. J Allergy Clin Immunol. 2020;145(1):402–414. doi:10.1016/j.jaci.2019.10.008
48. Wu T, Ma L, Jin X, et al. S100A4 is critical for a mouse model of allergic asthma by impacting mast cell activation. Front Immunol. 2021;12:692733. doi:10.3389/fimmu.2021.692733
49. Caslin HL, Taruselli MT, Haque T, et al. Inhibiting glycolysis and ATP production attenuates il-33-mediated mast cell function and peritonitis. Front Immunol. 2018;9:3026. doi:10.3389/fimmu.2018.03026
50. Rujillo MN, Jennings EQ, Hoffman EA, et al. Lactoylglutathione promotes inflammatory signaling in macrophages through histone lactoylation. Mol Metab. 2024;81:101888. doi:10.1016/j.molmet.2024.101888
51. Liu S, Liao S, Liang L, Deng J, Zhou Y. The relationship between CD4+ T cell glycolysis and their functions. Trends Endocrinol Metab. 2023;34(6):345–360. doi:10.1016/j.tem.2023.03.006
52. Borghi SM, Domiciano TP, Rasquel-Oliveira FS, et al. Sphagneticola trilobata (L.) Pruski-derived kaurenoic acid prevents ovalbumin-induced asthma in mice: effect on Th2 cytokines, STAT6/GATA-3 signaling, NFκB/Nrf2 redox sensitive pathways, and regulatory T cell phenotype markers. J Ethnopharmacol. 2022;283:114708. doi:10.1016/j.jep.2021.114708
53. Robb CT, Zhou Y, Felton JM, et al. Metabolic regulation by prostaglandin E2 impairs lung group 2 innate lymphoid cell responses. Allergy. 2023;78(3):714–730. doi:10.1111/all.15541
54. Li Q, Li D, Zhang X, et al. E3 Ligase VHL Promotes Group 2 innate lymphoid cell maturation and function via glycolysis inhibition and induction of interleukin-33 receptor. Immunity. 2018;48(2):258–270.e5. doi:10.1016/j.immuni.2017.12.013
55. Shen Z, Lu P, Jin W, et al. MOTS-c Promotes Glycolysis via AMPK-HIF-1α-PFKFB3 pathway to ameliorate cpb-induced lung injury. Am J Respir Cell Mol Biol. doi:10.1165/rcmb.2024-0533OC
56. Wang Y, Ding Z, Tu Y, et al. Poldip2/Nox4 mediates lipopolysaccharide-induced oxidative stress and inflammation in human lung epithelial cells. Mediators Inflamm. 2022;2022:6666022. doi:10.1155/2022/6666022
57. Yu C, Huang W, Zhou Z, et al. Short isoform thymic stromal lymphopoietin reduces inflammation and aerobic glycolysis of asthmatic airway epithelium by antagonizing long isoform thymic stromal lymphopoietin. Respir Res. 2022;23(1):75. doi:10.1186/s12931-022-01979-x
58. Park JW. Asthma phenotype with metabolic dysfunction. Yonsei Med J. 2022;63(1):1–7. doi:10.3349/ymj.2022.63.1.1
59. Islam R, Dash D, Singh R. An antioxidant ameliorates allergic airway inflammation by inhibiting HDAC 1 via HIF-1α/VEGF axis suppression in mice. Sci Rep. 2023;13(1):9637. doi:10.1038/s41598-023-36678-0
60. Bao C, Liu C, Liu Q, et al. Liproxstatin-1 alleviates LPS/IL-13-induced bronchial epithelial cell injury and neutrophilic asthma in mice by inhibiting ferroptosis. Int Immunopharmacol. 2022;109:108770. doi:10.1016/j.intimp.2022.108770
61. Wang H, Wang G, Meng Y, Liu Y, Yao X, Feng C. Modified Guo-Min decoction ameliorates PM2.5-induced lung injury by inhibition of PI3K-AKT and MAPK signaling pathways. Phytomedicine. 2024;123:155211. doi:10.1016/j.phymed.2023.155211
62. Hsieh B, Biancur DE, Larsen SB, et al. Interleukin-17 governs hypoxic adaptation of injured epithelium. Science. 2022;377(6602):eabg9302. doi:10.1126/science.abg9302
63. Li K, Li M, Li W, et al. Airway epithelial regeneration requires autophagy and glucose metabolism. Cell Death Dis. 2019;10(12):875. doi:10.1038/s41419-019-2111-2
64. Galle-Treger L, Hurrell BP, Lewis G, et al. Autophagy is critical for group 2 innate lymphoid cell metabolic homeostasis and effector function. J Allergy Clin Immunol. 2020;145(2):502–517.e5. doi:10.1016/j.jaci.2019.10.035
65. Jia M, Yue X, Sun W, et al. ULK1-mediated metabolic reprogramming regulates Vps34 lipid kinase activity by its lactylation. Sci Adv. 2023;9(22):eadg4993. doi:10.1126/sciadv.adg4993
66. Srisomboon Y, Ohkura N, Iijima K, et al. Airway exposure to polyethyleneimine nanoparticles induces type 2 immunity by a mechanism involving oxidative stress and ATP release. Int J Mol Sci. 2021;22(16):9071. doi:10.3390/ijms22169071
67. Meganathan V, Hamilton CE, Natarajan K, Keshava S, Boggaram V. NADPH and xanthine oxidases control induction of inflammatory mediator expression by organic dust in the lung. FASEB J. 2022;36(7):e22381. doi:10.1096/fj.202100732
68. Yu H, Cheng Y, Zhang G, Wang X, Gu W, Guo X. p62-dependent autophagy in airway smooth muscle cells regulates metabolic reprogramming and promotes airway remodeling. Life Sci. 2021;266:118884. doi:10.1016/j.lfs.2020.118884
69. Xu C, Song Y, Liu W, et al. IL-4 activates ULK1/Atg9a/Rab9 in asthma, NLRP3 inflammasomes, and Golgi fragmentation by increasing autophagy flux and mitochondrial oxidative stress. Redox Biol. 2024;71:103090. doi:10.1016/j.redox.2024.103090
70. Tsai ML, Tsai YG, Lin YC, et al. IL-25 Induced ROS-Mediated M2 Macrophage Polarization via AMPK-Associated Mitophagy. Int J Mol Sci. 2021;23(1):3. doi:10.3390/ijms23010003
71. Theofani E, Semitekolou M, Samitas K, et al. TFEB signaling attenuates NLRP3-driven inflammatory responses in severe asthma. Allergy. 2022;77(7):2131–2146. doi:10.1111/all.15221
72. Kim EY, Ji Kim E, Park H, et al. A study on specific factors related to inflammation and autophagy in BEAS-2B cells induced by urban particulate matter (PM, 1648a) and histological evaluation of PM-induced bronchial asthma model in mice. Int Immunopharmacol. 2023;123:110730. doi:10.1016/j.intimp.2023.110730
73. Hu J, Ding R, Liu S, Wang J, Li J, Shang Y. Hypermethylation of RNF125 promotes autophagy-induced oxidative stress in asthma by increasing HMGB1 stability. iScience. 2023;26(8):107503. doi:10.1016/j.isci.2023.107503
74. Yu H, Huang X, Zhu HH, et al. Apigenin ameliorates non-eosinophilic inflammation, dysregulated immune homeostasis and mitochondria-mediated airway epithelial cell apoptosis in chronic obese asthma via the ROS-ASK1-MAPK pathway. Phytomedicine. 2023;111:154646. doi:10.1016/j.phymed.2023.154646
75. Zhang D, Wang W, Sun X, et al. AMPK regulates autophagy by phosphorylating BECN1 at threonine 388. Autophagy. 2016;12(9):1447–1459. doi:10.1080/15548627.2016.1185576
76. Chen H, Matsumoto K, Brockway BL, et al. Airway epithelial progenitors are region specific and show differential responses to bleomycin-induced lung injury. Stem Cells. 2012;30(9):1948–1960. doi:10.1002/stem.1150
77. Li X, Zhao F, Wang A, Cheng P, Chen H. Role and mechanisms of autophagy in lung metabolism and repair. Cell Mol Life Sci. 2021;78(12):5051–5068. doi:10.1007/s00018-021-03841-7
78. Nolan KE, Baer LA, Karekar P, et al. Metabolic shifts modulate lung injury caused by infection with H1N1 influenza A virus. Virology. 2021;559:111–119. doi:10.1016/j.virol.2021.03.008
79. Ji X, Nie C, Yao Y, Ma Y, Huang H, Hao C. S100A8/9 modulates perturbation and glycolysis of macrophages in allergic asthma mice. PeerJ. 2024;12:e17106. doi:10.7717/peerj.17106
80. Zhao J, Dar HH, Deng Y, et al. PEBP1 acts as a rheostat between prosurvival autophagy and ferroptotic death in asthmatic epithelial cells. Proc Natl Acad Sci U S A. 2020;117(25):14376–14385. doi:10.1073/pnas.1921618117
81. Meng X, Guo S, Zhang X, et al. HMGB1 inhibition reduces TDI-induced occupational asthma through ROS/AMPK/autophagy pathway. Ecotoxicol Environ Saf. 2023;266:115575. doi:10.1016/j.ecoenv.2023.115575
82. Bai Q, Liu R, Quan C, et al. DEK deficiency suppresses mitophagy to protect against house dust mite-induced asthma. Front Immunol. 2024;14:1289774. doi:10.3389/fimmu.2023.1289774
83. Zhou JP, Wang Y, Li SQ, et al. Exogenous Ang-(1-7) inhibits autophagy via HIF-1α/THBS1/BECN1 axis to alleviate chronic intermittent hypoxia-enhanced airway remodelling of asthma. Cell Death Discov. 2023;9(1):366. doi:10.1038/s41420-023-01662-0
84. Bai Q, Wang Z, Piao Y, et al. Sesamin alleviates asthma airway inflammation by regulating mitophagy and mitochondrial apoptosis. J Agric Food Chem. 2022;70(16):4921–4933. doi:10.1021/acs.jafc.1c07877
85. Liu YB, Tan XH, Yang HH, et al. Wnt5a-mediated autophagy contributes to the epithelial-mesenchymal transition of human bronchial epithelial cells during asthma. Mol Med. 2024;30(1):93. doi:10.1186/s10020-024-00862-3
86. Shan L, Chen L, Shen W, et al. FOXK2 facilitates the airway remodeling during chronic asthma by promoting glycolysis in a SIRT2 -dependent manner. FASEB J. 2024;38(13):e23756. doi:10.1096/fj.202302284R
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Awareness and Predictors of the Use of Bioinformatics in Genome Research in Saudi Arabia
Alomair L, Abolfotouh MA
International Journal of General Medicine 2023, 16:3413-3425
Published Date: 11 August 2023
Precision Medicine in Asthma: The Role of Biomarkers
Quek E, Horn N, Siddiqui S
ImmunoTargets and Therapy 2025, 14:1479-1513
Published Date: 28 December 2025