Back to Journals » ImmunoTargets and Therapy » Volume 14
Research Progress of Natural Killer Cells in Hashimoto’s Thyroiditis
Authors Wang Y ![]() , Yan Z, Miao J, Que H, Shan W
, Yan Z, Miao J, Que H, Shan W
Received 9 June 2025
Accepted for publication 6 September 2025
Published 12 September 2025 Volume 2025:14 Pages 1029—1040
DOI https://doi.org/10.2147/ITT.S545646
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Flavio Salazar-Onfray
Yanchun Wang,1,* Zhangren Yan,2,* Jianhang Miao,3 Huafa Que,4 Wei Shan4
1College of Clinical Medicine, Jiangxi University of Chinese Medicine, Nanchang, Jiangxi, People’s Republic of China; 2Department of Traditional Chinese Medicine Surgery, The Affiliated Hospital of Jiangxi University of Chinese Medicine, Nanchang, Jiangxi, People’s Republic of China; 3Department of General Surgery,Zhongshan City People’s Hospital, Zhongshan, Guangdong, People’s Republic of China; 4Traditional Chinese Medicine Surgery, Longhua Hospital Affiliated to Shanghai University of Chinese Medicine, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Wei Shan, Traditional Chinese Medicine Surgery, Longhua Hospital Affiliated to Shanghai University of Chinese Medicine, No. 725 Wanping South Road, Shanghai, 200032, People’s Republic of China, Email [email protected]
Abstract: Hashimoto’s thyroiditis is one of the most common autoimmune diseases, characterized by lymphocytic infiltration, thyroid autoantibody production, and progressive thyroid destruction. Natural killer cells, as innate immune effectors, play a dual role in HT pathogenesis through cytotoxicity, death receptor signaling, inflammasome activation, and secretion of proinflammatory cytokines. Recent studies using single-cell RNA sequencing have revealed functional heterogeneity of NK subsets, suggesting stage-specific roles in either amplifying or regulating inflammation. Moreover, peripheral blood from HT patients shows increased expression of activating receptors such as NKG2D and NKp30, positively correlated with thyroid autoantibody titers, while abnormal activation of the NLRP3 inflammasome drives NK cell–mediated IFN-γ release and thyroid follicular cell pyroptosis. These advances highlight NK cells as both contributors to immune imbalance and potential therapeutic targets. A better understanding of NK cell–related mechanisms will provide novel insights into disease monitoring and the development of targeted interventions for HT.
Keywords: natural killer cells, hashimoto’s thyroiditis, receptor, cytotoxicity
Introduction
Hashimoto’s thyroiditis (HT) is a common autoimmune thyroid disease, usually characterized by thyroid enlargement, lymphocyte infiltration and elevated serum autoimmune antibodies. Female HT patients are approximately 8 times more likely than males due to the development of X chromosome inactivation patterns regulated by genetic susceptibility, environmental factors, and microbial composition.1 It is currently believed that its pathogsis is related to the infiltration of T cells and B cells in the thyroid gland,2 ultimately leading to progressive fibrosis and atrophy of thyroid tissue, as well as an increased risk of thyroid malignancy.
Among these immune cells, T cells play a pivotal role in HT through multiple mechanisms involving regulatory T cells (Tregs), cytotoxic T lymphocytes, and helper T cells. Functional impairment of Tregs can result in the breakdown of immune tolerance, thereby triggering uncontrolled autoimmune responses.Cytotoxic T lymphocytes are capable of directly attacking thyroid follicular cells, while helper T cells secrete proinflammatory cytokines, both of which further amplify the inflammatory cascade.3 B cells, on the other hand, represent the principal source of anti-thyroid antibodies in HT. They can function as antigen-presenting cells to present thyroid autoantigens to T cells, thereby enhancing T cell–mediated immune responses. Excessive activation of T cells can, in turn, drive abnormal B-cell differentiation and the production of autoantibodies against thyroid antigens. These antibodies mediate thyroid follicular cell destruction via antibody-dependent cell-mediated cytotoxicity, thereby aggravating thyroid inflammation and perpetuating a vicious cycle.4
Unlike T cells and B cells, NK cells do not express antigen-specific receptors and their cytotoxicity is determined by a delicate balance of activating and inhibitory signals from cell surface receptors.5 With the advancement of NK cell research, single-cell RNA sequencing (scRNA-seq) has been employed to profile their transcriptomes, uncovering the heterogeneity of NK cell subpopulations across different tissues and revealing dynamic functional changes under various physiological and pathological conditions6 Evidence suggests that in the early stages of HT, NK cells may contribute to disease progression by directly killing thyroid cells and promoting inflammatory responses. In contrast, during later stages, NK cells may exert immunomodulatory effects by regulating the activity of other immune cells, thereby alleviating inflammation4. These findings indicate that identifying characteristic alterations in specific NK cell subsets could provide insight into thyroid function and treatment responsiveness, and that targeted interventions tailored to different stages of HT may be developed based on the dual role of NK cells.
In recent years, research on the relationship between HT and NK cells has primarily focused on establishing correlations, confirming their association but leaving the precise mechanisms largely unexplored.7,8 This review will therefore focus on distinct NK cell subsets and their mechanistic roles in HT, with the aim of identifying novel therapeutic targets for clinical management of the disease.
Development and Differentiation of NK Cells
NK cells were first discovered in the spleen of mice in 1975, and are a cytotoxic branch of the innate lymphoid cell family, which are mainly found in bone marrow, lungs, lymph nodes, peripheral blood, spleen and liver.9 Among them, bone marrow is the most important developmental site of NK cells. Its development goes through three stages of early differentiation, maturation and function acquisition, and gradually acquires the expression of surface receptors that define the identity of NK cells and regulates their effector functions.
Mice share a high degree of genetic homology with humans and are easy to breed, making murine models an important tool for studying human NK cells.10 However, there are significant differences between murine and human NK cells in terms of development, subsets, and surface receptor expression. During NK cell maturation, murine NK cells are classified according to the expression levels of CD27 and CD11b, with immature NK cells defined as CD11blowCD27high and mature NK cells as CD27lowCD11b.high11,12 Researchers have analyzed NK cells from different mouse strains and identified unique features. For example, C57BL/6 mice exhibit strong interferon production and complement activity, making them suitable for studying NK cell–mediated immune responses. In contrast, BALB/c mice express high levels of IFN-γ but display impaired NK cell function and are susceptible to MCMV infection, providing a valuable model for investigating the complex relationship between IFN-γ and NK cell function, as well as their role in viral infections. Furthermore, BALB/c nude mice, which lack a thymus and T lymphocytes due to a Foxn1 gene mutation, show age-dependent alterations in NK cell function. This enables the study of NK cell development and function in the absence of T cells, thereby offering important insights into human NK cell developmental mechanisms.13 Collectively, NK cells derived from different murine models exhibit distinct biological characteristics and immune responses, providing valuable references for the study of human NK cells and offering novel strategies and theoretical foundations for the prevention and treatment of human diseases.
The ontogeny of human NK cells has not yet been fully elucidated, mainstream evidence indicates that the maturation of NK cells is most commonly classified by the expression density of CD56 molecules.14 Based on the characterization of NK cells in peripheral blood, human NK cells are generally subdivided into two major subpopulations: CD56brightCD16dim/−, which is generally considered less mature and a potent cytokine producer, and CD56dimCD16+, which is more mature and the most cytotoxic.15
Studies have shown that the The CD56bright subset is mainly located in secondary lymphoid tissues, playing a role in immune homeostasis under both normal and pathological conditions, and is capable of regulating immune responses through cytokine secretion. In autoimmune diseases, excessive proliferation of CD4+ T cells may exacerbate autoimmune responses, whereas CD56bright NK cells can mitigate inflammation and tissue damage by eliminating these over-proliferating CD4+ T cells, thereby exerting a protective effect.16 On the other hand, under conditions of immune dysregulation, CD56^bright NK cells may sustain inflammation by secreting GM-CSF and CD154 to drive monocyte differentiation into dendritic cells. Prolonged chronic inflammation may subsequently lead CD56bright cells to acquire features of immune exhaustion, a state in which the immune system fails to efficiently clear viral infections, thereby maintaining chronic infection and perpetuating a vicious cycle.17
CD56dim NK cells possess stronger cytotoxic activity, enabling them to play a critical role in antitumor immunity and antiviral defense.18 Under conditions of immune dysregulation, however, CD56dim NK cells can exhibit hyperactivation. Studies have shown that in patients with systemic lupus erythematosus (SLE), CD56dim NK cells display enhanced IFN-γ secretion, and their phenotypic characteristics are negatively correlated with disease activity indices.19 Other studies suggest that the ability of CD56dim NK cells to produce IFN-γ depends on the presence of T cells.20 Furthermore, single-cell multi-omics analyses have revealed a more complex relationship among these immune components: in SLE patients with high IFN expression, CD56^dim NK cells exhibit heightened cytotoxicity, which may be attributable to IFN-driven activation.21 These findings indicate that in T cell-dominated autoimmune diseases, CD56dim NK cells may be hyperactivated by overactive T cells or T cell-derived cytokines, thereby augmenting the production of proinflammatory mediators such as IFN-γ, which exacerbates tissue inflammation and damage.
In addition, significant ethnic differences have been observed in the distribution of NK cell subsets. Regarding NK cell absolute counts, Han Chinese individuals show higher average counts (442 cells/μL) compared with Caucasians and individuals of African descent. Among people younger than 40 years, NK cell counts in individuals of African descent are comparable to those of Han Chinese but higher than those of Caucasians; however, in individuals over 40 years, NK cell counts are higher in individuals of African descent compared with Han Chinese. With respect to subsets, both Caucasians and Han Chinese show higher proportions of CD56dim cells, with slightly higher frequencies in Caucasians, whereas Caucasians have the highest proportion of CD56bright cells and individuals of African descent display the highest proportion of NK cells with undefined function.22 Even within the same ethnic group, however, there are marked differences in killer-cell immunoglobulin-like receptor (KIR) gene expression. For example, significant variation has been identified between Southern and Northern Han Chinese populations, particularly in the frequency of activating KIR genes. The activating gene KIR2DS4, among others, is more frequent in Southern Han Chinese, while Northern Han Chinese show relatively lower frequencies.23 Suchgenetic differences may reflect population-specific adaptive responses to locally prevalent pathogens. Collectively, these findings highlight the adaptive potential of NK cells and underscore the importance of considering regional and ethnic factors in future NK cell research.
The Main Functions of NK Cells
NK cells play a significant role in innate immunity, and achieve immune protection by mediating the killing of target cells and secreting various cytokines. Its functional activation benefits from activating and inhibiting receptors. Under physiological conditions, inhibitory receptors play a dominant role in preventing NK cells from killing normal cells. When the inhibitory signal weakens and the activation signal strengthens, NK cells will exhibit killing effects.24 Therefore, the balance between the two determines the outcome of NK cell activation.
Activated Receptor
Activated receptors mainly include KIR (2DS, 3DS), NCR, NKG2D, 2B4, CD226, etc.
KIR is a transmembrane glycoprotein expressed by natural killer cells and T cell subsets, which typically has two or three extracellular Ig-like domains (2D or 3D) and long (L) or short (S) cytoplasmic tails.25 Hasan26 confirmed through animal experiments that KIR containing a short cytoplasmic tail can bind to related adaptor molecule such as FcRγ to transduce activation signals. Based on the current KIR gene content, two KIR haplotype groups A and B have been identified, with most of the KIR genes constituting haplotype B having activating function.27 The research by Ashouri E28 showed that six activated KIR genes appear to play a role in HT susceptibility, and suggested that the functional significance of the KIR-HLA combination in the pathogenesis of HT requires further study. A study on the KIR gene of HT in the Chinese population by Li29 showed that the frequency of activated KIR2D/HLA-C1 increased significantly.
This pro-inflammatory role of activating KIRs, however, presents a fascinating paradox when considered in the context of thyroid cancer, for which HT is a known risk factor.30 Interestingly, studies have shown that the frequencies of activating KIR2DS2 and KIR2DS4 are significantly lower in thyroid cancer patients compared to healthy controls,31 implying a protective role against oncogenesis.
This apparent contradiction underscores a critical principle in NK cell biology: the functional outcome of KIR signaling is not intrinsic but is entirely context-dependent, determined by the ligand landscape on target cells and the overall immune milieu. In the autoimmune setting of HT, activating KIRs may recognize stress-induced or inappropriately presented self-ligands on thyroid follicular cells, driving aberrant cytotoxicity. In contrast, in the immunosuppressive tumor microenvironment of thyroid cancer, the same activating KIRs may be crucial for recognizing downregulated HLA classes or stress ligands on malignant cells, thereby restoring immune surveillance. This dichotomy highlights the dual nature of NK cell activation as both a driver of autoimmunity and a defender against malignancy.
Natural cytotoxicity receptor (NCR) was discovered about 20 years ago by Pende D32 in a series of redirected lysis experiments using human NK cells. It consists of three type I transmembrane receptors, including NKp46, NKp30, and NKp44. The three are only expressed on the surface of NK cells. After binding to their specific ligands, they can induce strong activation of NK cell-mediated cytotoxicity. Studies have shown that NKp30 and NKp46 are highly expressed in 73.6% and 82.7% in NK cells respectively.33 And this cytotoxicity seems to advance only towards the target cells, which proves that NK cells have some form of resistance to the cytotoxicity mediated by them and avoid self-damage.34 Park A35 demonstrated that the soluble factors secreted by thyroid cancer cells slowed the killing function of NK cells by inhibiting the expression of NCR receptors, which also indicated that NCR can protect the thyroid gland by killing target cells in diseases. Intriguingly, the role of NCRs is diametrically opposed in Hashimoto’s thyroiditis. In patients with HT, the frequency of NKp46+ NK cells is significantly increased, and their cytotoxic capacity is enhanced, correlating positively with serum anti-thyroid antibody levels.36 These findings suggest that aberrant activation of NCRs may be a key driver of thyroid tissue destruction, transforming a primary defense mechanism into an autoimmune liability.
CD226 is an activated receptor expressed mainly in CD8 + and NK cells and functions as an adhesion molecule, which has become an attractive biomarker and immunotherapeutic target through its involvement in the regulation of multiple anti-tumor immune cell activities.37 However, at present, the detailed function and role of CD226 in pan-cancer are still unclear, and further investigation is need.38 2B4 is an Ig Superfamily Signaling Lymphocyte Activation Molecule family receptor whose cytoplasmic domain contains four immunoreceptor tyrosine-based switch motifs that interact with a variety of signaling adaptor molecules and transmit activating signals.39
Natural killer group 2 member D (NKG2D) is a C-type lectin-like receptor, which was discovered as a characteristic and novel receptor in the 1990s.40 It mainly binds to its corresponding ligand to make NK cells transmit activation signals. Notably, the reduction of NKG2D ligand expression can impair the cytotoxicity of NK cells against malignant cells, leading to immune evasion and disruption of the tumor immune surveillance system.41 Guo H42 found that the frequency of activated receptor expression of NKG2D and NKp30 was significantly higher in HT patients than in healthy people, and the concentration of NKG2D was positively correlated with TPOAb. In summary, NK cells drive immune dysregulation in HT through activating receptors, among which activating KIRs and NCRs confer high susceptibility to HT while exerting protective effects in thyroid carcinoma. Both the expression frequency and concentration of peripheral NKG2D are significantly increased in HT patients. Collectively, NCR represents a key therapeutic target for HT intervention, while receptor effects are highly dependent on the immune context and disease type. NKG2D may serve as a potential biomarker for assessing disease activity.
Inhibitory Receptor
NK cells protect normal tissues from autoimmunity by expressing inhibitory receptors, mainly including KIR (2DL, 3DL), CD94/NKG2A, and TIGIT. These signals are primarily transmitted by recognizing the binding of Major Histocompatibility Complex 1 (MHC I) molecules and immune checkpoints. Human MHC is usually called human leukocyte antigen (HLA) genes or HLA complexes. The interaction between these receptors and discrete groups of their own HLA-class I alleles prevents NK cells from attacking normal cells by recruiting tyrosine phosphatase.43
As an inhibitory receptor, the role of KIR2DL in autoimmune diseases is primarily mediated through its effects on CD8 regulatory T cells (CD8 Tregs). Studies have shown that in autoimmune disorders, the inhibitory function of KIR2DL limits the capacity of CD8 Tregs to effectively eliminate autoreactive CD4 T cells. This impairment leads to the expansion of autoreactive CD4 T cells, which subsequently attack host tissues.44 On the other hand, KIR regulates the function of NK cells by interacting with various ligands on the cell surface. Inhibitory KIR has an immunoreceptor tyrosine-based inhibitory motif (ITIM) within its cytoplasmic domain. ITIM exhibits immunosuppressive activity by recruiting tyrosine phosphatases containing SH2 domains, such as SHP-1 or SHP-2, thereby reducing the ability of NK cells to degranulate and produce cytokines and chemokines.45 Among them, SHP-1 down-regulates multiple activation signaling molecules through dephosphorylation, and reduced SHP-1 capacity can lead to mutations in leukocyte immunoglobulin-like receptor B1, which in turn generates autoimmune diseases including HT.46 It indicates SHP-1 plays a crucial role in initiating inhibitory signals and blocking activation signals.
CD94/NKG2A is a type I transmembrane protein that binds to HLA-E, leading to phosphorylation of the tyrosine residues within the ITIM motif in its cytoplasmic tail. The phosphorylated ITIM motif recruits SHP-1 and/or SHP-2 phosphatases, which dephosphorylate Vav and ERM proteins, thereby disrupting activating receptor signaling at the NK cell contact site,47 This mechanism helps maintain NK cell homeostasis and prevents autoreactive immune responses. However, in tumors, high NKG2A expression impairs NK cell infiltration, reducing their numbers within tumor tissue and weakening antitumor immunity. Tumor cells often overexpress HLA-E, which binds to CD94/NKG2A on NK cells, allowing tumors to evade NK cell recognition and cytotoxicity. In contrast, studies in HT patients have shown reduced NKG2A expression and increased frequencies of NK cells expressing activating receptors.36 These findings highlight the differential roles of NKG2A under distinct pathological conditions and underscore the complexity of the immune system in maintaining homeostasis.
T cell immunoglobulin and ITIM domain (TIGIT) is an inhibitory receptor expressed on T cells and NK cells. It engages multiple ligands and triggers inhibitory signaling, competitively binding CD155 to suppress DNAM-1–mediated co-stimulatory signals. This process impedes dendritic cell–mediated T cell activation, prevents NK cells and cytotoxic T lymphocytes from killing malignant cells, and enhances the immunosuppressive activity of regulatory T cells48 Štefanić reported that TIGIT can directly suppress NK cell function, and its high expression in hypothyroid patients may reflect impaired NK cell activity.49 This inhibitory role of TIGIT has also been confirmed in HT. Compared with the healthy group, TIGIT expression in lymphoid follicles of HT is about 2–3 times greater than in the equivalent regions.50
In conclusion, inhibitory receptors primarily transduce inhibitory signals by binding to HLA molecules and inducing phosphorylation of their ITIM. SHP-1, a key cytoplasmic tyrosine phosphatase recruited to phosphorylated ITIMs, plays a critical role in initiating these inhibitory signals and blocking activation pathways by dephosphorylating downstream targets. In the autoimmune milieu of HT, reduced expression of inhibitory receptors like NKG2A and impaired SHP-1 signaling may unleash autoreactive responses. Given its broad regulatory functions, SHP-1 represents a promising potential therapeutic target for HT. Furthermore, the elevated expression of TIGIT observed in HT suggests its potential association with disease severity (Table 1).
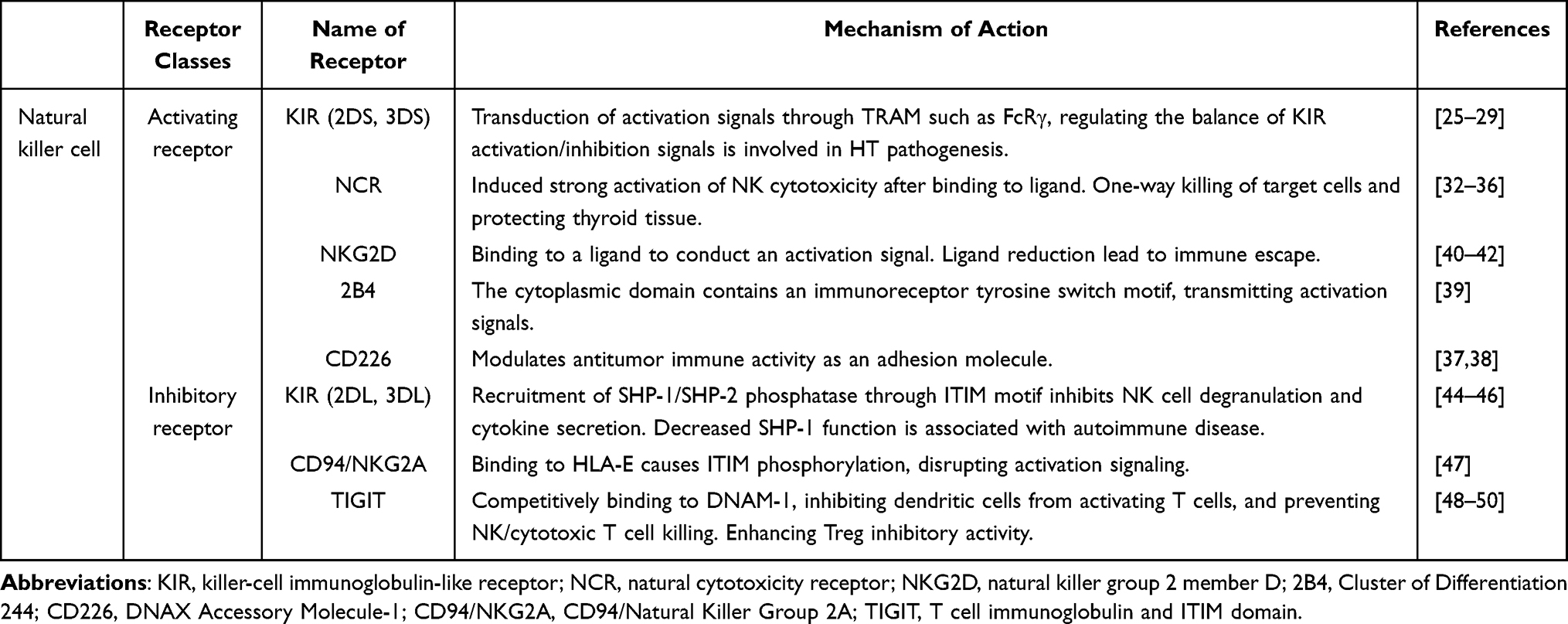 |
Table 1 The Main Functions of NK Cells |
Pathogenesis of NK Cells in HT
Cytotoxicity
Cytotoxicity has the ability to directly kill other cells, which is essential for infected or transformed cells. Therefore, cytotoxicity is the core tool of the immune system in combating inflammation. NK cells are one of the most effective mediators for exerting cytotoxicity, and they are involved in antibody-dependent cell-mediated cytotoxicity, which acts through the expression level of perforin.4
Perforin, as an effective pore-forming protein, undergoes direct cleaved when pores are formed on the cell membrane of cancer cells, allowing lysozymes (such as granzyme A and B) to be released into the cytoplasm, which in turn mediates target cell death.51 Studies have shown that during the progression of HT, cytotoxic T cells and NK cells exhibit increased activity, producing perforin, granzymes, and proteoglycans, which induce cytotoxic death of thyroid cells and contribute to thyroid tissue damage and chronic inflammation.4 However, Interestingly, Popko8 observed a significant decrease in perforin expression in HT patients, despite the fact that NK cells from these patients displayed significantly higher spontaneous cytotoxicity compared with healthy controls. This suggests that cytotoxic mechanisms beyond perforin may also contribute to thyroid tissue damage in HT.
Death Receptor
Death receptors are a class of type I transmembrane receptors expressed on the cell surface. They are members of the Tumor Necrosis Factor (TNF) receptor superfamily and are characterized by the presence of death domains in the cytoplasmic region. Death receptors play a central role in directed apoptosis by initiating apoptotic signals after receiving specific death ligands. These receptors can be activated within seconds after ligand binding, resulting in cell apoptosis within hours.52 Among them, TNF receptor 1, Fas and TNF-related apoptosis-inducing ligand receptors receptors (TRAILR) are typical death receptors that play pleiotropic functions in cell death, inflammation and immune surveillance.53
Fas was first identified in 1989 by Trauth et al as an antigen expressed on activated or malignant lymphocytes. Engagement of Fas with the monoclonal antibody anti-APO-1 was found to induce apoptosis in these cells via a cell surface–mediated signaling pathway that is independent of complement or antibody-dependent cellular cytotoxicity. Trauth et al further suggested that Fas induces cell death through the formation of a death-inducing signaling complex (DISC), although the precise mechanisms were not fully elucidated in their study.54 Subsequent research by Frazzette et al demonstrated that binding of Fas to its ligand, FasL, recruits Fas-associated death domain (FADD) proteins to form the DISC, which in turn activates caspase-8 and induces apoptosis in target cells, thereby clarifying the mechanistic role of Fas as a death receptor.55 Wen J56 observed an increase in Caspase protein content in HT patients. Furthermore, some scholars have further verified through experiments that the higher cell mortality rate of the glandular species in HT patients is closely related to the overexpression of Fas57 This further confirms that the Fas pathway is conducive to the cell death of thyroid cells and thyroid destruction.
However, TRAIL has highly similar homology with Fas ligand, which also induces apoptotic signal transduction through binding to its receptor TRAIL-R to activate caspase-8 and downstream caspase cascade, and ultimately leads to cell death. Notably, TRAIL induces apoptosis in many tumor cell lines, but not in most primary cells. Among them, CD4 + cell infiltration is closely related to HT, which can be involved in stimulating cytotoxicity of effector cells and stimulating humoral responses. If TRAIL fails to trigger effective apoptotic signals in CD4T cells, alternative signaling pathways need to be activated.58 SHP-1 has been demonstrated as an inhibitory receptor signal above. Chyuan IT59 confirmed through animal experiments that the TRAIL-R/SHP-1/Lck complex can inhibit proximal TCR signaling and subsequent T cell activation through co-inhibitory signal transduction. It suggests that the binding of TRAIL-R to SHP-1 is a novel pathway different from the traditional apoptotic signaling pathway and can be used as a potential therapeutic target for immune mediation in HT.
In conclusion, NK cells can intervene in the development of HT by regulating the Fas and TRAIL signaling pathways in terms of death receptors, and the TRAIL-R and SHP-1 axes may become new apoptotic pathways of NK cells in HT.
Inflammasome
Inflammasome is a large protein complex composed of three parts. The first is the pattern recognition receptor (PRR), which acts as an intracellular receptor. The second is the cytokine activation system, that is, apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC). One end of ASC is linked to the PRR, and pro-caspase-1 is recruited through its CARD domain at the other end. Once activated, pro-caspase-1 becomes caspase-1, which is the third important component of the inflammasome.60 Caspase-1 can mediate the inflammatory response, promote the cleavage of IL-1β, IL-18 and Gasdermin-D, and then lead to local inflammation and inflammatory pyroptosis. It has been confirmed that nucleotide-binding domain and leucine-rich repeat receptors (NLR) family members NLRP1, NLRP3, NLRC4 and AIM2 are involved in the formation of inflammasomes.60,61
HT is an autoimmune disease. Some scholars have found that the expression of inflammasome components such as NLRP3, AIM2, NLRC4 and NLRP in thyroid follicular cells of HT patients is higher than that in normal tissues. Among them, the most significant mediators of pyroptosis are NLRP3 and AIM2.62 It suggests a potential role for inflammasome-associated pyroptosis mediators in the pathogenesis of HT. NLRP3 is the most definite inflammasome in structure and function so far. In HT mouse models, inhibition of NLRP3 activation or pyroptosis significantly attenuates disease progression, including reductions in thyroid volume, lymphocyte infiltration, and serum levels of thyroid peroxidase antibodies and thyroglobulin antibodies, indicating that targeting NLRP3 activation or pyroptosis may represent a potential therapeutic strategy.63 Further analysis of the relationship between NLRP3 and NK cells has shown that PRR recognizes PAMP and DAMP through Toll-like receptors in the membrane or cytoplasm, thereby activating NLRP3 in macrophages, NLRP3 inflammasome activation stimulates NK cells and INF-γ secretion by releasing IL-1β and IL-18.64 These findings indicate that inflammasomes act through NK cells in HT, amplifying autoimmune cascades in an IFN-γ-dependent manner, ultimately leading to thyroid follicular cell destruction and autoantibody production.
Proinflammatory Factor
In addition to cytotoxicity, NK cell activation is accompanied by the secretion of proinflammatory cytokines such as GM-CSF, IFN-γ, TNF-α, etc.65 Proinflammatory factors in HT not only attack the thyroid gland, but also cause thyroid follicular cell damage.
Granulocyte-macrophage colony stimulating factor (GM-CSF) is a multifunctional cytokine originally described as a growth factor that induces the differentiation and proliferation of myeloid progenitor cells in the bone marrow. As an important participant in the innate immunity of NK cells, GM-CSF can stimulate macrophages to produce antibacterial and antitumor effects in the context of local inflammatory responses.66 Thyroglobulin-immunized mice treated with GM-CSF showed inhibition of thyroglobulin effector T cell responses and no thyroid changes occurred, suggesting that GM-CSF has the ability to prevent and inhibit the formation of a mouse model of HT.67 Moreover, GM-CSF–treated mice exhibited higher frequencies of CD4+CD25+ T cells in the thyroid, while the expression of death receptors in the thyroid remained unchanged. This indicates that GM-CSF primarily modulates immune cell function rather than directly affecting thyroid cells.68 These findings further elucidate the mechanism of GM-CSF in HT and provide theoretical support for its potential application in the treatment of Hashimoto’s thyroiditis.
IFN-γ is a pleiotropic molecule secreted by lymphocytes including activated T cells and NK cells, which has bidirectional properties in immunosuppression and activation. NK cells recognize non-self targets and produce IFN-γ-mediated cytotoxic effects to intervene and regulate diseases.69 Some scholars have demonstrated that increased levels of IFN-γ can lead to HT production through evidence of cytokine secretion in the thyroid gland and experimental evidence in animal models and cultured cells, which may help prevent disease progression by controlling IFN-γ expression.70,71 Other scholars have stated that there is no significant difference in IFN-γ levels between HT patients and healthy groups, which indicates that whether there is a rel39 hip between IFN-γ and HT still requires further research.72 Through a Mendelian randomization analysis between inflammatory cytokines and autoimmune thyroid disease, it was found that high levels of IFN-γ may lead to a higher risk of HT, but HT doe40 lead to further changes in IFN-γ.63 It indicates that early intervention of IFN-γ level in future treatment has positive significance in reducing the incidence of HT. TNF-α can be responsible for various signaling events within cells, leading to necrosis or apoptosis. This protein is also important for resisting infections and cancer. NK cells, as innate immune cells, can activate cytotoxic granules of granzymes including TNF-α and cytokines during acute inflammation.73 It is worth noting that two cytokines, IFN-γ and TNF-α, are closely related to inflammasomes in HT. Studies have shown that both can lead to upregulation of mRNA levels of NLRP3 and AIM2, and suggest that abnormal cytoplasmic DAMP triggered by TNF-α or IFN-γ may trigger pyroptosis of thyroid follicular cells during HT progression and be directly or indirectly sensed by NLRP3 or AIM2.62 Figure 1 shows the pathogenesis of NK cells in HT.
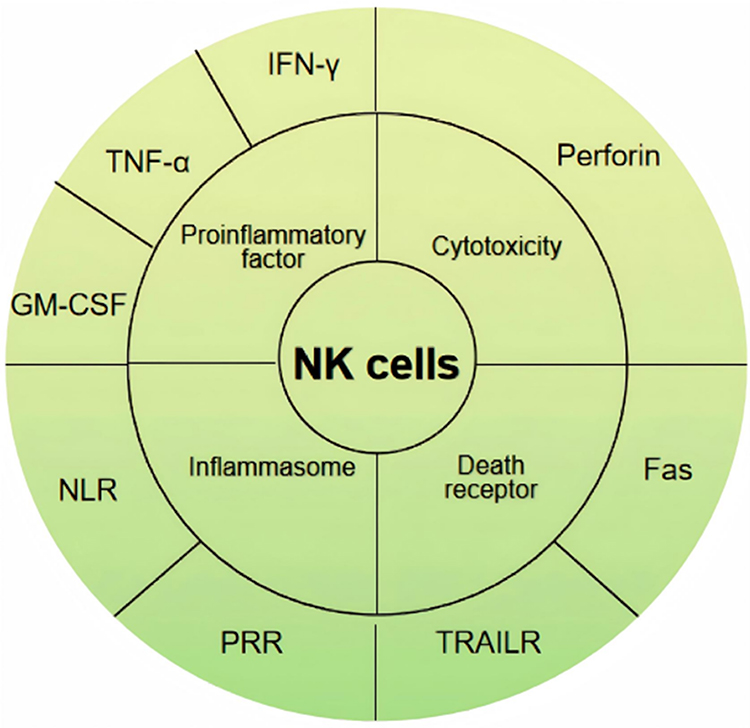 |
Figure 1 Mechanisms of NK Cell-Mediated Thyroid Damage in Hashimoto’s Thyroiditis. Abbreviations: TNF-α, Tumor Necrosis Factor-α; GM-CSF, Granulocyte-Macrophage Colony-Stimulating Factor; IFN-γ, Interferon-γ; NLR, nucleotide-binding domain and leucine-rich repeat receptors; PRR, pattern recognition receptor; TRAILR, Tumor Necrosis Factor-related apoptosis-inducing ligand receptors, TRAILR; Fas, Factor-related Apoptosis. |
In addition, inhibition of proinflammatory factors is an important way to intervene in HT. The immunomodulatory effects of metformin as an antiglycemic drug are now increasingly well known, and it can ameliorate disease in HT by inhibiting the number and function of Th17 cell and macrophage polarization.74 Data suggest that metformin not only effectively inhibits TNF-α, but also blocks caspase-1 and GSDMD-N associated with NLRP3 inflammasome activation.75,76 It suggests that metformin has great potential in the treatment of HT. Inositol is the first reported subtype of inositol, accounting for more than 99% of intracellular inositol. It has various effects on hormones that affect brain and metabolism, mainly from legumes, citrus fruits, nuts and grains with high bran content. Inositol depletion may lead to the development of thyroid diseases.77,78 In recent years, studies have shown that inositol reduces the secretion of CXCL10 chemokine induced by IFN-γ and TNF-α, which has positive implications for45 cting thyroid cells.79 Vitamin D is a fat-soluble vitamin that plays a crucial role in bone health and calcium regulation in the body. After weekly vitamin D supplementation to female patients with HT, the serum levels of IFN-γ and TNF-α decreased significantly, indicating that vitamin D has the potential to control the inflammatory axis of IFN-γ and TNF-α in HT.80
In conclusion, NK cells play an important role in promoting inflammation in HT, and play a role through inflammasomes and proinflammatory factors such as IFN-γ, TNF-α and GM-CSF. These abnormal activation pathways are not only harmful factors that cause inflammasomes and pyroptosis of thyroid follicular cells during HT development, but also provide potential therapeutic targets for NK cells to intervene in HT from different inflammatory angles, and corresponding drug therapy and dietary guidance can be selected through the above mechanisms to reduce the severity of the disease.
Conclusion and Prospect
Previous studies have suggested that T cells recognize thyroid autoantigens through interactions with MHC molecules on thyroid epithelial cells, thereby triggering autoimmune responses. In HT, thyroid cells may act as antigen-presenting cells, further amplifying autoreactive T-cell activation and accelerating disease progression. A hallmark of HT is the development of hypothyroidism, which can be delayed through early intervention. Unlike T cells, NK cells do not require antigen-specific priming and thus exert effector functions at early disease stages, highlighting their potential importance in HT pathogenesis and intervention.
As critical components of immune surveillance, NK cells contribute to thyroid follicular cell injury not only via cytotoxicity, death receptor pathways, and inflammasome activation, but also by secreting pro-inflammatory cytokines such as IFN-γ, TNF-α, and GM-CSF, thereby amplifying inflammatory cascades and tissue destruction. Notably, alterations in NK cell phenotype, function, and abundance have been consistently observed in HT patients, underscoring their role in immune dysregulation and supporting their potential as biomarkers of disease activity. Emerging evidence suggests that NK cells may exert dual roles: promoting inflammatory responses during early disease, while displaying immunoregulatory properties at later stages. Characterizing the dynamic shifts of NK cell subsets may therefore provide valuable insights for predicting disease progression and monitoring therapeutic response.
To date, most studies have focused on correlations between NK cell phenotypes or receptor expression and HT, without establishing direct causality. Given their strong interferon-producing capacity and complement activity, C57BL/6 mice represent an ideal model to investigate NK cell functions in HT. Future studies leveraging this model may help determine whether NK cell alterations precede disease onset and thereby clarify their causal contribution to HT. Furthermore, population-based differences in NK cell subset distribution, combined with the genetic susceptibility of HT, pose challenges for developing universally applicable therapies. Advances in scRNA-seq now enable high-resolution profiling of NK cell transcriptomes across disease stages and populations, providing an opportunity to define NK cell subset signatures in diverse ethnic groups. Such efforts may ultimately inform precision medicine strategies for HT.
Funding
This work was supported by the Science and Technology Program Project (No. 2024A0051) of Jiangxi Province Administration of Traditional Chinese Medicine.
Disclosure
All authors declare no conflicts of interest in this work.
References
1. Klubo-Gwiezdzinska J, Wartofsky L. Hashimoto thyroiditis: an evidence-based guide to etiology, diagnosis and treatment. Pol Arch Intern Med. 2022;132(3). doi:10.20452/pamw.16222
2. Ralli M, Angeletti D, Fiore M, et al. Hashimoto’s thyroiditis: an update on pathogenic mechanisms, diagnostic protocols, therapeutic strategies, and potential malignant transformation. Autoimmun Rev. 2020;19(10):102649. doi:10.1016/j.autrev.2020.102649
3. Weetman AP. An update on the pathogenesis of Hashimoto’s thyroiditis. J Endocrinol Invest. 2021;44(5):883–890. doi:10.1007/s40618-020-01477-1
4. Wronska K, Halasa M, Szczuko M. The role of the immune system in the course of hashimoto’s thyroiditis: the current state of knowledge. Int J Mol Sci. 2024;25(13). doi:10.3390/ijms25136883
5. Kucuksezer UC, Aktas CE, Esen F, et al. The role of natural killer cells in autoimmune diseases. Front Immunol. 2021;12:622306. doi:10.3389/fimmu.2021.622306
6. Liu H, Zhao R, Qin R, et al. Panoramic comparison between NK cells in healthy and cancerous liver through single-cell RNA sequencing. Cancer Biol Med. 2022;19(9):1334–1351. doi:10.20892/j.issn.2095-3941.2022.0050
7. Liu Y, You R, Yu N, et al. Increased proportions of Tc17 cells and NK cells may be risk factors for disease progression in Hashimoto’s thyroiditis. Int Immunopharmacol. 2016;40:332–338. doi:10.1016/j.intimp.2016.09.016
8. Popko K, Osinska I, Kucharska A, Demkow U. Cytometric analysis of perforin expression in NK cells, CD8+, and CD4+ lymphocytes in children with autoimmune Hashimoto’s thyroiditis--a preliminary study. J Pediatr Endocr Met. 2015;28(7–8):789–792. doi:10.1515/jpem-2014-0520
9. Kiessling R, Klein E, Wigzell H. “Natural” killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur J Immunol. 1975;5(2):112–117. doi:10.1002/eji.1830050208
10. Waterston RH, Lindblad-Toh K, Birney E, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420(6915):520–562. doi:10.1038/nature01262
11. Quatrini L, Della CM, Sivori S, Mingari MC, Pende D, Moretta L. Human NK cells, their receptors and function. Eur J Immunol. 2021;51(7):1566–1579. doi:10.1002/eji.202049028
12. Del ZG, Antonini F, Pesce S, Moretta L, Marcenaro E, Marcenaro E. Comprehensive phenotyping of human PB NK cells by flow cytometry. Cytom Part A. 2020;97(9):891–899. doi:10.1002/cyto.a.24001
13. Qin J, Zhang Z, Cui H, Yang J, Liu A. Biological characteristics and immune responses of NK Cells in commonly used experimental mouse models. Front Immunol. 2024;15:1478323. doi:10.3389/fimmu.2024.1478323
14. Crinier A, Narni-Mancinelli E, Ugolini S, Vivier E. SnapShot: natural killer cells. Cell. 2020;180(6):1280–1280.e1. doi:10.1016/j.cell.2020.02.029
15. Liu S, Galat V, Galat Y, Lee Y, Wainwright D, Wu J. NK cell-based cancer immunotherapy: from basic biology to clinical development. J Hematol Oncol. 2021;14(1):7. doi:10.1186/s13045-020-01014-w
16. Lee M, Bell C, Rubio GA, et al. CD56(bright) natural killer cells preferentially kill proliferating CD4(+) T cells. Discov Immunol. 2023;2(1):kyad012. doi:10.1093/discim/kyad012
17. Chen S, Zhu H, Jounaidi Y. Comprehensive snapshots of natural killer cells functions, signaling, molecular mechanisms and clinical utilization. Signal Transduction Tar. 2024;9(1):302. doi:10.1038/s41392-024-02005-w
18. Ding Y, Lavaert M, Grassmann S, et al. Distinct developmental pathways generate functionally distinct populations of natural killer cells. Nat Immunol. 2024;25(7):1183–1192. doi:10.1038/s41590-024-01865-2
19. Liu M, Liu J, Zhang X, Xiao Y, Jiang G, Huang X. Activation status of CD56(dim) natural killer cells is associated with disease activity of patients with systemic lupus erythematosus. Clin Rheumatol. 2021;40(3):1103–1112. doi:10.1007/s10067-020-05306-x
20. He XS, Draghi M, Mahmood K, et al. T cell-dependent production of IFN-gamma by NK cells in response to influenza A virus. J Clin Invest. 2004;114(12):1812–1819. doi:10.1172/JCI22797
21. Trzupek D, Lee M, Hamey F, Wicker LS, Todd JA, Ferreira RC. Single-cell multi-omics analysis reveals IFN-driven alterations in T lymphocytes and natural killer cells in systemic lupus erythematosus. Wellcome Open Res. 2021;6:149. doi:10.12688/wellcomeopenres.16883.2
22. Feng YM, Zhang RJ, Zhu H, et al. Comparison of the quantities and subset distributions of natural killer cells among different races. Chinese Med J-Peking. 2010;123(22):3272–3276.
23. Shi L, Zhang H, Shen Y, et al. Distribution of KIR genes in Han population in Yunnan Province: comparison with other Han populations in China. Int J Immunogenet. 2013;40(5):361–368. doi:10.1111/iji.12046
24. Chen Y, Lu D, Churov A, Fu R. Research progress on NK cell receptors and their signaling pathways. Mediators Inflammation. 2020;2020:6437057. doi:10.1155/2020/6437057
25. Castrillon M, Marin ND, Karduss-Urueta AJ, Velasquez SY, Alvarez CM. Killer-cell immunoglobulin-like receptor diversity in an admixed South American population. Cells-Basel. 2022;11(18). doi:10.3390/cells11182776
26. Hasan MZ, Walter L. Rhesus macaque activating killer immunoglobulin-like receptors associate with fc receptor gamma (FCER1G) and not with DAP12 adaptor proteins resulting in stabilized expression and enabling signal transduction. Front Immunol. 2021;12:678964. doi:10.3389/fimmu.2021.678964
27. Gomez-Luque JM, Urrutia-Maldonado E, Munoz DRP, Abril-Molina A, Ocete-Hita E. Killer immunoglobulin-like receptor and cancer. An Pediatr. 2021. doi:10.1016/j.anpedi.2021.02.001
28. Ashouri E, Dabbaghmanesh MH, Ranjbar OG. Presence of more activating KIR genes is associated with Hashimoto’s thyroiditis. Endocrine. 2014;46(3):519–525. doi:10.1007/s12020-013-0080-2
29. Li JT, Guo C, Li ML, et al. Killer cell immunoglobulin-like receptor genes and their hla-c ligands in hashimoto thyroiditis in A Chinese Population. Endocr Pract. 2016;22(8):935–940. doi:10.4158/EP151175.OR
30. Hu X, Wang X, Liang Y, et al. Cancer risk in hashimoto’s thyroiditis: a systematic review and meta-analysis. Front Endocrinol. 2022;13:937871. doi:10.3389/fendo.2022.937871
31. Altalhi RA, Aljuaimlani A, Alswayyed M, et al. Association of the genetic diversity of killer cell immunoglobulin-like receptor genes and HLA-C ligand in saudi women with thyroid cancer. Cancer Control. 2024;31:10732748241274495. doi:10.1177/10732748241274495
32. Pende D, Parolini S, Pessino A, et al. Identification and molecular characterization of NKp30, a novel triggering receptor involved in natural cytotoxicity mediated by human natural killer cells. J Exp Med. 1999;190(10):1505–1516. doi:10.1084/jem.190.10.1505
33. Wang F, Zhang G, Xu T, et al. High and selective cytotoxicity of ex vivo expanded allogeneic human natural killer cells from peripheral blood against bladder cancer: implications for natural killer cell instillation after transurethral resection of bladder tumor. J Exp Clin Canc Res. 2024;43(1):24. doi:10.1186/s13046-024-02955-7
34. Mace EM. Human natural killer cells: form, function, and development. J Allergy Clin Immun. 2023;151(2):371–385. doi:10.1016/j.jaci.2022.09.022
35. Park A, Lee Y, Kim MS, et al. Prostaglandin E2 secreted by thyroid cancer cells contributes to immune escape through the suppression of natural killer (NK) cell cytotoxicity and NK cell differentiation. Front Immunol. 2018;9:1859. doi:10.3389/fimmu.2018.01859
36. Ortega-Rodriguez AC, Martinez-Hernandez R, Monsivais-Urenda A, et al. Quantitative and functional analysis of PD-1+ NK cells in patients with autoimmune thyroid disease. J Clin Endocr Metab. 2020;105(11):e4001–e4011. doi:10.1210/clinem/dgaa569
37. Chiang EY, Mellman I. TIGIT-CD226-PVR axis: advancing immune checkpoint blockade for cancer immunotherapy. J Immunother Cancer. 2022;10(4):e004711. doi:10.1136/jitc-2022-004711
38. Ma P, Sun W. Integrated single-cell and bulk sequencing analyses with experimental validation identify the prognostic and immunological implications of CD226 in pan-cancer. J Cancer Res Clin. 2023;149(16):14597–14617. doi:10.1007/s00432-023-05268-y
39. Huang Y, Zeng J, Liu T, Xu Q, Song X, Zeng J. DNAM1 and 2B4 costimulatory domains enhance the cytotoxicity of Anti-GPC3 chimeric antigen receptor-modified natural killer cells against hepatocellular cancer cells in vitro. Cancer Manag Res. 2020;12:3247–3255. doi:10.2147/CMAR.S253565
40. Houchins JP, Yabe T, Mcsherry C, Bach FH. DNA sequence analysis of NKG2, a family of related cDNA clones encoding type II integral membrane proteins on human natural killer cells. J Exp Med. 1991;173(4):1017–1020. doi:10.1084/jem.173.4.1017
41. Kim HJ, Jeon S, Lee HJ, et al. Effects of sevoflurane on metalloproteinase and natural killer group 2, member D (NKG2D) ligand expression and natural killer cell-mediated cytotoxicity in breast cancer: an in vitro study. Korean J Anesthesiol. 2023;76(6):627–639. doi:10.4097/kja.23323
42. Guo H, Xu B, Yang X, et al. A high frequency of peripheral blood NKG2D+NK and NKT cells in euthyroid patients with new onset hashimoto’s thyroiditis--a pilot study. Immunol Invest. 2014;43(4):312–323. doi:10.3109/08820139.2013.854377
43. Poggi A, Zocchi MR. Natural killer cells and immune-checkpoint inhibitor therapy: current knowledge and new challenges. Mol Ther Oncolytics. 2022;24:26–42. doi:10.1016/j.omto.2021.11.016
44. Gardell JL, Maurer ME, Childs MM, et al. Preclinical characterization of MTX-101: a novel bispecific CD8 Treg modulator that restores CD8 Treg functions to suppress pathogenic T cells in autoimmune diseases. Front Immunol. 2024;15:1452537. doi:10.3389/fimmu.2024.1452537
45. Wei Y, Ren X, Galbo PJ, et al. KIR3DL3-HHLA2 is a human immunosuppressive pathway and a therapeutic target. Sci Immunol. 2021;6(61). doi:10.1126/sciimmunol.abf9792
46. Sinthuwiwat T, Buranapraditkun S, Kamolvisit W, et al. A LILRB1 variant with a decreased ability to phosphorylate SHP-1 leads to autoimmune diseases. Sci Rep-Uk. 2022;12(1):15420. doi:10.1038/s41598-022-19334-x
47. Peruzzi G, Masilamani M, Borrego F, Coligan JE. Endocytosis as a mechanism of regulating natural killer cell function: unique endocytic and trafficking pathway for CD94/NKG2A. Immunol Res. 2009;43(1–3):210–222. doi:10.1007/s12026-008-8072-7
48. Harjunpaa H, Guillerey C. TIGIT as an emerging immune checkpoint. Clin Exp Immunol. 2020;200(2):108–119. doi:10.1111/cei.13407
49. Stefanic M, Tokic S, Suver-Stevic M, Glavas-Obrovac L. Expression of TIGIT and FCRL3 is altered in t cells from patients with distinct patterns of chronic autoimmune thyroiditis. Exp Clin Endocr Diab. 2019;127(5):281–288. doi:10.1055/a-0597-8948
50. Blessin NC, Simon R, Kluth M, et al. Patterns of TIGIT expression in lymphatic tissue, inflammation, and cancer. Dis Markers. 2019;2019:5160565. doi:10.1155/2019/5160565
51. Fantini M, Arlen PM, Tsang KY. Potentiation of natural killer cells to overcome cancer resistance to NK cell-based therapy and to enhance antibody-based immunotherapy. Front Immunol. 2023;14:1275904. doi:10.3389/fimmu.2023.1275904
52. Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281(5381):1305–1308. doi:10.1126/science.281.5381.1305
53. Moriwaki K, Chan F, Miyoshi E. Sweet modification and regulation of death receptor signalling pathway. J Biochem. 2021;169(6):643–652. doi:10.1093/jb/mvab034
54. Trauth BC, Klas C, Peters AM, et al. Monoclonal antibody-mediated tumor regression by induction of apoptosis. Science. 1989;245(4915):301–305. doi:10.1126/science.2787530
55. Frazzette N, Cruz AC, Wu X, et al. Super-Resolution Imaging of Fas/CD95 reorganization induced by membrane-bound fas ligand reveals nanoscale clustering upstream of FADD recruitment. Cells-Basel. 2022;11(12). doi:10.3390/cells11121908
56. Wen J, Zhang W, Shi L, et al. Amiodarone-drove XBP1s aggravates endoplasmic reticulum stress and apoptosis in Hashimoto’s thyroiditis through regulating LINC00842/miR-214/FASL axis. Int Immunopharmacol. 2022;113(Pt A):109298. doi:10.1016/j.intimp.2022.109298
57. Vasconcelos JC, Siqueira IB, Maia F, Parisi M, Zantut-Wittmann DE. Influence of thyroid hormone in the expression of the marker pro-apoptosis Bid, in spite of the predominance of anti-apoptosis activation in intratiroidal lymphocytic infiltration in Hashimoto’s thyroiditis. Mol Cell Endocrinol. 2021;537:111421. doi:10.1016/j.mce.2021.111421
58. Janyga S, Marek B, Kajdaniuk D, Ogrodowczyk-Bobik M, Urbanek A, Buldak L. CD4+ cells in autoimmune thyroid disease. Endokrynol Pol. 2021;72(5):572–583. doi:10.5603/EP.a2021.0076
59. Chyuan IT, Liao HJ, Tan TH, et al. Association of TRAIL receptor with phosphatase SHP-1 enables repressing T cell receptor signaling and T cell activation through inactivating Lck. J Biomed Sci. 2024;31(1):33. doi:10.1186/s12929-024-01023-8
60. Deng Z, Lu L, Li B, Shi X, Jin H, Hu W. The roles of inflammasomes in cancer. Front Immunol. 2023;14:1195572. doi:10.3389/fimmu.2023.1195572
61. Jiao Z, Zhang J. Interplay between inflammasomes and PD-1/PD-L1 and their implications in cancer immunotherapy. Carcinogenesis. 2023;44(12):795–808. doi:10.1093/carcin/bgad072
62. Zhao X, Ni W, Zheng W, et al. Multi-regulatory potency of USP1 on inflammasome components promotes pyroptosis in thyroid follicular cells and contributes to the progression of Hashimoto’s thyroiditis. Mol Med. 2024;30(1):121. doi:10.1186/s10020-024-00885-w
63. Yao Z, Guo F, Tan Y, et al. Causal relationship between inflammatory cytokines and autoimmune thyroid disease: a bidirectional two-sample Mendelian randomization analysis. Front Immunol. 2024;15:1334772. doi:10.3389/fimmu.2024.1334772
64. Batiha GE, Al-Gareeb AI, Rotimi D, Adeyemi OS, Al-Kuraishy HM. Common NLRP3 inflammasome inhibitors and Covid-19: divide and conquer. Sci Afr. 2022;18:e01407. doi:10.1016/j.sciaf.2022.e01407
65. Karvouni M, Vidal-Manrique M, Lundqvist A, Alici E. Engineered NK cells against cancer and their potential applications beyond. Front Immunol. 2022;13:825979. doi:10.3389/fimmu.2022.825979
66. Souza-Fonseca-Guimaraes F, Parlato M, de Oliveira RB, et al. Interferon-gamma and granulocyte/monocyte colony-stimulating factor production by natural killer cells involves different signaling pathways and the adaptor stimulator of interferon genes (STING). J Biol Chem. 2013;288(15):10715–10721. doi:10.1074/jbc.M112.435602
67. Lotfi N, Thome R, Rezaei N, et al. Roles of GM-CSF in the pathogenesis of autoimmune diseases: an update. Front Immunol. 2019;10:1265. doi:10.3389/fimmu.2019.01265
68. Gangi E, Vasu C, Cheatem D, Prabhakar BS. IL-10-producing CD4+CD25+ regulatory T cells play a critical role in granulocyte-macrophage colony-stimulating factor-induced suppression of experimental autoimmune thyroiditis. J Immunol. 2005;174(11):7006–7013. doi:10.4049/jimmunol.174.11.7006
69. Gocher AM, Workman CJ, Vignali D. Interferon-gamma: teammate or opponent in the tumour microenvironment? Nat Rev Immunol. 2022;22(3):158–172. doi:10.1038/s41577-021-00566-3
70. Liu B, Li L, Wang X. Petunidin suppresses Hashimoto’s thyroiditis by regulating Th1/Th17 homeostasis and oxidative stress. Cell Immunol. 2024;403-404:104858. doi:10.1016/j.cellimm.2024.104858
71. Nodehi M, Ajami A, Izad M, et al. The Frequency of CD4(+) T cells in women with hashimoto’s thyroiditis. Int J Endocrinol Met. 2021;19(4):e110013. doi:10.5812/ijem.110013
72. Ozisik H, Cekin A, Suner A, et al. Evaluation of IL-10, MCP-1, IFN gamma, and protectin D1 levels in patients with Hashimoto’s thyroiditis. Irish J Med Sci. 2023;192(1):177–184. doi:10.1007/s11845-022-03231-3
73. Fan Z, Han D, Fan X, Zhao L. Ovarian cancer treatment and natural killer cell-based immunotherapy. Front Immunol. 2023;14:1308143. doi:10.3389/fimmu.2023.1308143
74. Jia X, Zhai T, Zhang JA. Metformin reduces autoimmune antibody levels in patients with Hashimoto’s thyroiditis: a systematic review and meta-analysis. Autoimmunity. 2020;53(6):353–361. doi:10.1080/08916934.2020.1789969
75. Yang F, Qin Y, Wang Y, et al. Metformin Inhibits the NLRP3 Inflammasome via AMPK/mTOR-dependent Effects in Diabetic Cardiomyopathy. Int J Biol Sci. 2019;15(5):1010–1019. doi:10.7150/ijbs.29680
76. Song H, Zhang X, Zhai R, et al. Metformin attenuated sepsis-associated liver injury and inflammatory response in aged mice. Bioengineered. 2022;13(2):4598–4609. doi:10.1080/21655979.2022.2036305
77. Paparo SR, Ferrari SM, Patrizio A, et al. Myoinositol in Autoimmune Thyroiditis. Front Endocrinol. 2022;13:930756. doi:10.3389/fendo.2022.930756
78. Ferrari SM, Fallahi P, Di Bari F, Vita R, Benvenga S, Antonelli A. Myo-inositol and selenium reduce the risk of developing overt hypothyroidism in patients with autoimmune thyroiditis. Eur Rev Med Pharmaco. 2017;21(2 Suppl):36–42.
79. Ferrari SM, Elia G, Ragusa F, et al. The protective effect of myo-inositol on human thyrocytes. Rev Endocr Metab Dis. 2018;19(4):355–362. doi:10.1007/s11154-018-9476-x
80. Lebiedzinski F, Lisowska KA. Impact of vitamin D on Immunopathology of hashimoto’s thyroiditis: from theory to practice. Nutrients. 2023;15(14):3174. doi:10.3390/nu15143174
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.