Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Rescuing apoptotic neurons in Alzheimer’s disease using wheat germ agglutinin-conjugated and cardiolipin-conjugated liposomes with encapsulated nerve growth factor and curcumin
Received 18 December 2014
Accepted for publication 9 February 2015
Published 1 April 2015 Volume 2015:10(1) Pages 2653—2672
DOI https://doi.org/10.2147/IJN.S79528
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Lei Yang
Yung-Chih Kuo, Ching-Chun Lin
Department of Chemical Engineering, National Chung Cheng University, Chia-Yi, Taiwan, Republic of China
Abstract: Liposomes with cardiolipin (CL) and wheat germ agglutinin (WGA) were developed to permeate the blood–brain barrier and treat Alzheimer’s disease. WGA-conjugated and CL-incorporated liposomes (WGA-CL-liposomes) were used to transport nerve growth factor (NGF) and curcumin (CUR) across a monolayer of human brain-microvascular endothelial cells regulated by human astrocytes and to protect SK-N-MC cells against apoptosis induced by ß-amyloid1–42 (Aß1–42) fibrils. An increase in the CL mole percentage in lipids increased the liposomal diameter, absolute zeta potential value, entrapment efficiency of NGF and CUR, release of NGF, biocompatibility, and viability of SK-N-MC cells with Aß1–42, but decreased the atomic ratio of nitrogen to phosphorus and release of CUR. In addition, an increase in the WGA concentration for grafting enhanced the liposomal diameter, atomic ratio of nitrogen to phosphorus, and permeability of NGF and CUR across the blood–brain barrier, but reduced the absolute zeta potential value and biocompatibility. WGA-CL-liposomes carrying NGF and CUR could be promising colloidal delivery carriers for future clinical application in targeting the blood–brain barrier and inhibiting neurotoxicity.
Keywords: Alzheimer’s disease, nerve growth factor, curcumin, wheat germ agglutinin, cardiolipin, liposome
Introduction
Alzheimer’s disease (AD), listed officially as the sixth leading cause of death, is one of the commonly encountered dementias, with an estimated 5.2 million cases in the USA, of which 5 million cases involve people aged 65 years and older.1 With regard to its pathology, the most typical feature of AD is an abnormal accumulation of β-amyloid peptide (Aβ) in the cortex and hippocampus, leading to extensive apoptosis of cholinergic neurons.2,3 Aβ is derived from amyloid precursor protein, a transmembrane glycoprotein for material exchange among neurons and extracellular matrices.4 β-secretase and γ-secretase transform amyloid precursor protein into Aβ1–40 and then into Aβ1–42.5 The latter is a strong neurotoxin with a β-sheet structure and appears as nondecomposable neurofibrillary tangles in senile plaque.6 For AD therapy, nerve growth factor (NGF) can be an efficacious protein on account of its neuroprotective and neurorepair functions.7,8 Discovered about 60 years ago, NGF was demonstrated to improve the survival and differentiation of sensory and sympathetic neurons.9 NGF can regulate the neuronal growth via its two major neurotrophin receptors, neurotrophic tyrosine kinase receptor type 1 and p75.10 However, low permeability across the blood–brain barrier critically diminishes the ability of NGF to treat neurodegenerative disorders in the central nervous system.11 In addition to NGF, curcumin (CUR), a polyphenolic bioflavonoid, can be efficacious in the management of AD.12 CUR is an excellent antioxidant and can prevent cyclo-oxygenase, lipoxygenase, and xanthine oxidase from generating free radicals.13 In regard to its anti-inflammatory function, CUR can obstruct the metabolism of arachidonic acid, preventing production of nitric oxide, tumor necrosis factor-α, and interleukin-12.14 Nevertheless, in a clinical study, daily administration of CUR 1 g for 6 months was found to be unable to recover or improve cognitive ability in AD patients to a normal status.15 This drawback of CUR was due principally to its poor solubility in plasma, high degradability in acidic, basic, and oxidative milieus, rapid metabolism in the intestine and liver, and low bioavailability. Thus, application of apposite vectors to deliver NGF and CUR could be a practical approach to pharmacotherapy for AD.
Liposomes are composed of sealed phospholipids and aqueous cores, and can be an effective system for entrapping hydrophilic and hydrophobic drugs.16 In fact, hydrophilic NGF has been encapsulated in positively charged liposomes to treat focal cerebral ischemia.17 Liposomes comprising cholesterol, soybean phospholipids, and 1,2-dipalmitoyl-sn-glycero-3-phosphoethanol-amine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG[2000]) have been used to carry NGF across the blood–brain barrier.18 To aid in the diagnosis and treatment of AD, incorporation of cardiolipin (CL) was able to stabilize the bilayer structure in liposomes and favored the ligand binding of liposomes to Aβ1–42.19 CL, a unique amphiphilic lipid, comprises four chains of saturated fatty acid and two head groups of charged phosphate. CL was initially found in chloroplasts and eukaryotic mitochondria for membrane protein support and enzyme activation.20 In addition, wheat germ agglutinin (WGA) has distinct carbohydrate specificity and can be efficient in delivery of pharmaceuticals across the blood–brain barrier.21 In a lectin-mediated targeting study, WGA was found to have a strong interaction with human ECV304, a human bladder carcinoma-derived epithelial cell line for mimicking the blood–brain barrier, and to generate only a slight variation in glycosylation pattern.22
The aim of this study was to investigate the ability of WGA-conjugated and CL-incorporated liposomes (WGA-CL-liposomes) to encapsulate nerve-inductive NGF and anti-inflammatory CUR for AD therapy. Hydrophilic NGF and hydrophobic CUR in WGA-CL-liposomes could be able to ameliorate the poor ability of NGF to cross the blood–brain barrier and the low bioavailability of CUR. We examined the physicochemical properties of WGA-CL-liposomes carrying NGF and CUR (WGA-CL-NGF-CUR-liposomes), their cytotoxicity, permeability of NGF and CUR across the blood–brain barrier, and the role of these liposomes in rescuing SK-N-MC cells (a cell line established from a metastatic tumor mass showing cholinergic trait) with Aβ1–42 fibrils.
Materials and methods
Materials
Cholesterol, fluorescein isothiocyanate-conjugated dextran 70,000, Dulbecco’s phosphate buffered saline (DPBS), 2-(N-morpholino)ethanesulfonic acid, a QuantiPro bicinchoninic acid assay kit, phosphotungstic acid, sodium azide, gelatin, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide, human fibronectin, propidium iodide (PI), formalin, trypsin–ethylenediaminetetraacetic acid (EDTA), and 1,1,1,3,3,3-hexafluoro-2-propanol, and 4′,6-diamidino-2-phenylindole were purchased from Sigma-Aldrich (St Louis, MO, USA). 1′,3′-bis[1,2-dimyristoyl-sn-glycero-3-phospho]-sn-glycerol (CL), 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), DSPE-PEG(2000), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[carboxy(polyethylene glycol)-2000] (DSPE-PEG(2000)-CA), and a polycarbonate membrane (pore size 100 nm) were obtained from Avanti Polar Lipids (Alabaster, AL, USA). Chloroform, tetrahydrofuran, sodium bicarbonate, and dimethyl sulfoxide were purchased from JT Baker (Phillipsburg, NJ, USA). Tris hydroxymethyl aminomethane (Tris), and hydrochloric acid were obtained from Riedel-de Haen (Seelze, Germany). Methanol was purchased from Mallinckrodt Baker (Phillipsburg, NJ, USA), a BDS Hypersil C-18 column (with 5 μm microspheres) was sourced from Thermo Hypersil-Keystone (Bellefonte, PA), and ultrapure water from Barnstead (Dubuque, IA, USA), citric acid from Showa (Tokyo, Japan), dialysis tubing (regenerated cellulose membrane, 50 kDa) from Spectrum Laboratories (Rancho Dominguez, CA, USA), N-hydroxysuccinimide from Alfa Aesar (Ward Hill, MA, USA), WGA from Medicago AB (Uppsala, Sweden), human β-NGF from Alomone Labs (Jerusalem, Israel), Sephadex G-100 powders from GE Healthcare (Piscataway, NJ, USA), human brain-microvascular endothelial cells (HBMECs) from Biocompare (South San Francisco, CA, USA), Triton X-100 from Acros (Geel, Belgium), SK-N-MC cells (from human neuroblastoma) from the American Type Tissue Collection (Rockville, MD, USA), penicillin–streptomycin–glutamate solution from Gibco (Carlsbad, CA, USA), serum blocking solution from Zymed (South San Francisco, CA, USA), mounting medium from Bio SB (Santa Barbara, CA, USA), and 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (XTT) from Biological Industries (Beit Haemek, Israel). Human astrocytes (HAs), human brain-vascular pericytes (HBVPs), HBMEC culture medium, HA culture medium, and HBVP culture medium were obtained from Sciencell (Corte Del Cedro Carlsbad, CA, USA). Aβ1–42 powders and SK-N-MC cell culture medium were purchased from Life Technologies (Carlsbad, CA, USA). An anti-O-linked human NGF kit, N-acetylglucosamine antibody, goat polyclonal secondary antibody to mouse immunoglobulin G (H and L), and anti-Aβ monoclonal antibody were obtained from Abcam (Cambridge, MA, USA). The Transwell, polyethylene terephthalate (PET) insert (pore size 0.4 μm, surface area 0.3 cm2), and cryovials (2 mL) were purchased from BD Falcon (Franklin Lakes, NJ, USA). A T75 (75 cm2) tissue culture flask was obtained from Corning Costar (Cambridge, MA, USA), a MicroWell 96-well plate (polystyrene) from Nalge Nunc (Rochester, NY, USA), a 24-well tissue culture microtiter plate (polystyrene, flat bottom) from Midwest Scientific (St Louis, MO, USA), and microscope cover glass from Marienfeld GmbH (Lauda-Koenigshofen, Germany).
Preparation of WGA-CL-NGF-CUR-liposomes
Synthesis of CL-NGF-CUR-liposomes
CL, DPPC, cholesterol, DSPE-PEG(2000), DSPE-PEG(2000)-CA, and CUR were dissolved in 1 mL of chloroform at 25°C. The mole percentage of CL in a lipid mixture of CL, DPPC, and cholesterol was 0%, 5%, 10%, and 20%. An equal mole percentage of DPPC and cholesterol in the lipid mixture was used in every batch. The weight percentage of CUR in the lipid phase was 2%. The mole percentage of DSPE-PEG(2000) and DSPE-PEG(2000)-CA was each controlled at 3% in the lipid phase. For fluorescent liposomes, 0.1% (w/v) fluorescein isothiocyanate-conjugated dextran 70,000 was added to the lipid phase. The organic lipid solution was concentrated using a rotary evaporator (Panchum, Kaohsiung, Taiwan) with a vacuum pump (Panchum) at 75 rpm and 45°C for 15 minutes to deposit a thin lipid layer on a round bottom 50 mL flask. The solid lipid deposition was hydrated with DPBS containing 50 ng/mL human β-NGF and vibrated at 46 kHz and 45°C for 30 minutes. After hydration, the suspension at a lipid concentration of 2 mg/mL was compressed using an extruder set (Avanti Polar Lipids) through a porous polycarbonate membrane 15 times to generate CL-conjugated liposomes carrying NGF and CUR (CL-NGF-CUR-liposomes).
Entrapment efficiency of NGF and CUR in CL-NGF-CUR-liposomes
CL-NGF-CUR-liposomes were isolated from the suspension containing free NGF and CUR using an upright, home-made 1.5 cm ×50 cm glass column packed with DPBS-swollen gel of Sephadex G-100 powders at 25°C. The suspension containing 3 mL of CL-NGF-CUR-liposomes and 150 mL of DPBS was added to the column. Every effluent of 2 mL was collected. To evaluate the quantity of NGF, the effluents of CL-NGF-CUR-liposomes were immersed in 1% (v/v) Triton X-100 at 4°C for one hour and analyzed with the human NGF kit using an enzyme-linked immunosorbent assay (ELISA) spectrophotometer (Bio-Tek, Winooski, VT, USA) at 450 nm. To evaluate the quantity of CUR, the effluents of CL-NGF-CUR-liposomes were treated with methanol at 37°C for 30 minutes, separated using high-performance liquid chromatography (HPLC, Jasco, Tokyo, Japan) with a reverse phase BDS Hypersil C-18 column, and detected using an ultraviolet (UV) detector (UV-2075 Plus, Jasco) at 430 nm. The C-18 column was warmed using a column heater (Alltech, Deerfield, IL, USA) at 37°C. The mobile phase was ultrapure water adjusted to pH 3 with citric acid, contained tetrahydrofuran gradient from 5% to 40% (v/v), and was impelled using two high pressure pumps (PU-2080 Plus, Jasco) in series at a flow rate of 1 mL per minute for 20 minutes. The entrapment efficiency of NGF, ENGF, and that of CUR, ECUR, was defined, respectively, as ENGF (%) = (We,NGF/Wt,NGF) ×100% and ECUR (%) = (We,CUR/Wt,CUR) ×100%. In these expressions, We,NGF, Wt,NGF, We,CUR, and Wt,CUR are the weight of NGF entrapped in CL-NGF-CUR-liposomes, the weight of total NGF, the weight of CUR entrapped in CL-NGF-CUR-liposomes, and the weight of total CUR, respectively.
Grafting efficiency of WGA on WGA-CL-NGF-CUR-liposomes
CL-NGF-CUR-liposomes were suspended in DPBS containing 20% (w/v) 2-(N-morpholino)ethanesulfonic acid at 80 rpm and mixed with 1 mM 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide and 1.5 mM N-hydroxysuccinimide at 80 rpm and 25°C for 1.5 hours. The suspension containing CL-NGF-CUR-liposomes with functionalized carboxyl groups was mixed with WGA 2.5 or 5 mg/mL at 80 rpm and 25°C for 12 hours. Next, the suspension containing 3 mL of WGA-conjugated CL-NGF-CUR-liposomes and 150 mL of DPBS was added to the upright Sephadex G-100 column at 25°C to separate the free WGA from the WGA-CL-NGF-CUR-liposomes in every 2 mL effluent. The quantity of free WGA was assayed using a QuantiPro bicinchoninic acid assay kit and analyzed using the ELISA spectrophotometer at 562 nm. The efficiency of WGA grafting on WGA-CL-NGF-CUR-liposomes, GEWGA, was defined as GEWGA (%) = [(Wt,WGA − Wf,WGA)/Wt,WGA] ×100%, where Wt,WGA and Wf,WGA are the weight of total WGA and the weight of free WGA after gel chromatography, respectively. Figure 1 shows a schematic illustration of a WGA-CL-NGF-CUR-liposome.
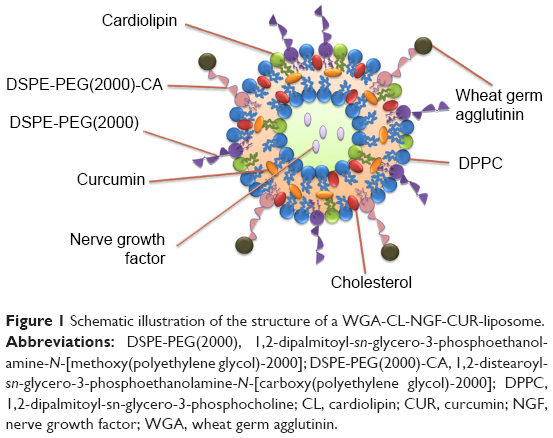 | Figure 1 Schematic illustration of the structure of a WGA-CL-NGF-CUR-liposome. |
Characterization of WGA-CL-NGF-CUR-liposomes
Particle size and zeta potential
The cumulant Z-average diameter and zeta potential of the WGA-CL-NGF-CUR-liposomes were analyzed using a Zetasizer 3000 HSA with a photon correlation spectroscope and a laser Doppler velocimeter (Malvern Instruments, Malvern, UK) at 25°C. The suspension containing WGA-CL-NGF-CUR-liposomes was added to a buffer containing Tris 0.1 M at pH 7.4 and became a liposomal concentration of 1 mg/mL. In any independent detection, 3 mL of the fresh preparation was used.
Morphology
The structure of WGA-CL-NGF-CUR-liposomes was identified using a transmission electron microscope (JEM-1400, JEOL, Tokyo, Japan) and a field emission scanning electron microscope (JSM-6330 TF, JEOL). The suspension containing WGA-CL-NGF-CUR-liposomes was controlled at a liposomal concentration of 2 mg/mL in Tris buffer. For analysis by transmission electron microscopy, 10 μL of the suspension was uniformly loaded on a carbon-coated 200-mesh copper grid for one minute and interacted with 10 μL of 2% (w/v) phosphotungstic acid solution for 2 minutes. The excess fluid was removed by filter paper. For analysis by scanning electron microscopy, 10 μL of the suspension was ultrasonically vibrated for one minute, dripped down on a cover slide, dehydrated at 25°C for 24 hours, vacuum-dried, glued with carbon paint, sputter-coated with platinum at an accelerating voltage of 2 kV for 90 seconds, and scanned with a voltage of 3 kV.
Atomic distribution in the surface layer
The atomic distribution in the surface layer of WGA-CL-NGF-CUR-liposomes to a depth of 10 nm was evaluated by X-ray photoelectron spectroscopy (XPS, Kratos, Kanagawa, Japan) and a light beam area of 300 μm ×700 μm. The concentration of suspended WGA-CL-NGF-CUR-liposomes was fixed at 2 mg/mL. Next, 10 μL of the suspension was dropped onto a 5 mm ×5 mm cover slide, dehydrated at 25°C, and dried with a vacuum grade of 2×10−7 Pa at 300 W for 15 minutes.
Dissolution of NGF and CUR from WGA-CL-NGF-CUR-liposomes
WGA-CL-NGF-CUR-liposomes were suspended in DPBS containing 0.05% (w/v) sodium azide at a liposomal concentration of 1 mg/mL and a pH of 7.4. A 3.4 cm ×30 cm dialysis tube was pretreated with 2% (w/v) sodium bicarbonate for 20 minutes, immersed in 1 mM EDTA at pH 8 for 20 minutes, and washed with ultrapure water at 4°C for 20 minutes. The refined tube with the suspension containing 2 mL of WGA-CL-NGF-CUR-liposomes was added, placed in a 100 mL flask containing 20 mL of DPBS at pH 7.4, and shaken in a bath-reciprocal shaker at 50 rpm and 37°C for 48 hours. Next, 100 μL of liquid was sampled from the dissolution medium at specific time points. At the point of sampling, 100 μL of fresh DPBS was added immediately to the flask to replenish the volume of medium. To estimate the level of NGF released, the sample was analyzed with a human NGF kit using the ELISA spectrophotometer at 450 nm. To estimate the level of CUR released, the sample was analyzed using the HPLC-UV system (at 430 nm) described in the “Entrapment efficiency of NGF and CUR in CL-NGF-CUR-liposomes” section. The cumulative percentage of NGF released, PNGF, and that of CUR, PCUR, were defined, respectively, as PNGF (%) = Wd,NGF/We,NGF ×100% and PCUR (%) = Wd,CUR/We,CUR ×100%. In these expressions, Wd,NGF and Wd,CUR are the cumulative released quantity of NGF and that of CUR, respectively.
Toxicity of WGA-CL-NGF-CUR-liposomes to HBMECs, HAs, HBVPs, and SK-N-MC cells
Cell culture
The methods for propagating HBMECs, HAs, and SK-N-MC cells have been described previously.23 The scheme for expanding HBVPs was as follows. Briefly, HBVPs were seeded in a T75 flask pretreated with hydrofluoric acid at a density of 7.5×104 cells/cm2 and cultivated in a humidified CO2 incubator (NuAire, Plymouth, MN, USA) at 37°C for 7 days. The culture medium used was pericyte medium supplemented with pericyte growth supplement and 1% (v/v) penicillin–streptomycin–glutamate solution, and was replaced every day. The multiplied HBVPs were washed with 5 mL of DPBS, detached with 2 mL of trypsin-EDTA solution, rinsed with 4 mL of pericyte medium, added to a 15 mL sterile conical tube, and centrifuged at 150× g for 5 minutes. The HBVP pellet at the bottom of the centrifuge tube was resuspended in fresh culture medium, and equally divided and distributed in three hydrofluoric acid-pretreated T75 flasks. Excess HBVPs were suspended in the culture medium containing 10% (v/v) dimethyl sulfoxide, added to cryovials, placed in a freezer (Sanyo, Osaka, Japan) at −80°C for 24 hours, and preserved in liquid nitrogen.
Viability
Aβ1–42 powders were dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol to form a protein solution at a concentration of 1 mM. To eliminate the original Aβ1–42 structures, the solution was agitated using a bath-reciprocal shaker at 50 rpm for 10 minutes, vacuum-dried at 4°C, and stored in a low temperature freezer (Frigidaire, Augusta, GA, USA) at −20°C. The dried Aβ1–42 deposit was used to produce Aβ1–42 fibrils by dissolving in 10 mM hydrochloric acid to form a protein solution with a concentration of 100 μM and placing in the humidified CO2 incubator at 37°C for 24 hours. The acidic protein solution was vacuum-dried at 4°C and dissolved in the culture medium used for SK-N-MC cells.
HBMECs, HAs, HBVPs, and SK-N-MC cells were seeded, respectively, on a 96-well plate pretreated with gelatin at a density of 7.5×103 cells/well. The cells were cultivated with 150 μL of HBMEC culture medium, HA culture medium, HBVP culture medium, and SK-N-MC cell culture medium per well in a humidified CO2 incubator at 37°C for 8 hours and then interacted with 0.025% (w/v) WGA-CL-NGF-CUR-liposomes in the humidified CO2 incubator at 37°C for 8 hours. Cell viability was assayed with XTT by reacting with 50 μL of XTT per well containing 2% (v/v) activation solution, culturing in the humidified CO2 incubator at 37°C for 4 hours, and analyzed using the ELISA spectrophotometer at 450 nm. The viability of HBMECs, HAs, HBVPs, and SK-N-MC cells, PCV, was defined as PCV (%) = [(ODcell,t − ODXTT)/(ODcell − ODXTT)] ×100%, where ODcell,t, ODcell, and ODXTT are, respectively, the optical density of HBMECs, HAs, HBVPs, and SK-N-MC cells after treatment with WGA-CL-NGF-CUR-liposomes, the optical density of HBMECs, HAs, HBVPs, and SK-N-MC cells before treatment with WGA-CL-NGF-CUR-liposomes, and the optical density of XTT. In addition, analogous procedures were adopted for SK-N-MC cells incubated with fibrillar Aβ1–42 at 5 or 10 μM. The incubation period for rescuing SK-N-MC cells after an insult with Aβ1–42 fibrils was 6, 12, 24, and 36 hours. The viability of SK-N-MC cells after an insult with Aβ1–42 fibrils and treatment with WGA-CL-NGF-CUR-liposomes, PCV,SK-N-MC, was defined as PCV,SK-N-MC (%) = [(ODSK,t − ODXTT)/(ODcell − ODXTT)] ×100%, where ODSK,t is the optical density of SK-N-MC cells with Aβ1–42 fibrils and WGA-CL-NGF-CUR-liposomes.
Transport of NGF and CUR across the blood–brain barrier using WGA-CL-NGF-CUR-liposomes
Transendothelial electrical resistance of HBMEC/HA with impact of WGA-CL-NGF-CUR-liposomes
The protocol for developing a monolayer of HBMECs regulated by HAs (HBMEC/HA) was described in a previous study.24 0.025% (w/v) WGA-CL-NGF-CUR-liposomes in culture medium for HBMECs were added into the upper donor chamber, which contacted HBMECs in the Transwell. The receiver chamber (the lower chamber facing HAs) was filled with fresh HA culture medium. HBMEC/HA was cultivated in the humidified CO2 incubator at 37°C for 4 hours. A Millicell electrical resistance system (Millipore, Billerica, MA, USA) was used to evaluate the electrical resistance of a gelatin-pretreated PET insert with and without HBMEC/HA. The transendothelial electrical resistance (TEER) of HBMEC/HA was defined as TEER (Ω × cm2) = (Re,cell − Re,blank) × Ae, where Re,cell, Re,blank, and Ae, are the electrical resistance of the PET insert with HBMEC/HA, the cell-free PET insert, and the surface area of the PET insert, respectively. In addition, the medium in the donor chamber was replaced with HBMEC culture medium containing PI 0.25 mg/mL and cultivated in the CO2 incubator at 37°C for 5 hours. The quantity of PI transported to the receiver chamber was evaluated using a microplate fluorescent reader (Synergy HT, Bio-Tek) at an excitation of 485 nm and an emission of 590 nm. The permeability coefficient for PI across HBMEC/HA, PPI, was calculated by 1/PPI = 1/Pe,PI − 1/Pm,PI, where Pe,PI and Pm,PI are, respectively, the permeability coefficient for PI across the PET insert with HBMEC/HA and the permeability coefficient for PI across the cell-free PET insert.
Permeability of NGF and CUR across HBMEC/HA
0.025% (w/v) WGA-CL-NGF-CUR-liposomes were added into the donor chamber for delivering NGF and CUR across the PET insert in the humidified CO2 incubator at 37°C for 5 hours. Next, 20 μL of fluid in the receiver chamber was drawn out at 2.5 and 5 hours. To evaluate the quantity of NGF transported to the receiver chamber, the sample was treated with 1% (v/v) Triton X-100 at 4°C for one hour and analyzed with a human NGF kit using the ELISA spectrophotometer at 450 nm. To evaluate the quantity of CUR transported to the receiver chamber, the sample was treated with methanol at 37°C for 30 minutes and analyzed using the HPLC-UV system (at 430 nm), described in the Entrapment efficiency of NGF and CUR in CL-NGF-CUR-liposomes section. The volume in the receiver chamber was replenished immediately with 20 μL of fresh HA culture medium at 2.5 hours. The permeability coefficient of NGF across HBMEC/HA, PNGF, and that of CUR, PCUR, was defined, respectively, as 1/PNGF = 1/Pe,NGF − 1/Pm,NGF and 1/PCUR = 1/Pe,CUR − 1/Pm,CUR. In these expressions, Pe,NGF, Pm,NGF, Pe,CUR, and Pm,CUR are the permeability coefficient of NGF across the PET insert with HBMEC/HA, the permeability coefficient of NGF across the blank PET insert, the permeability coefficient of CUR across the PET insert with HBMEC/HA, and the permeability coefficient of CUR across the blank PET insert, respectively.
Immunochemical staining of WGA-CL-NGF-CUR-liposomes in AD model
Internalization of WGA-CL-NGF-CUR-liposomes by HBMECs
HBMECs were seeded on gelatin-pretreated microscope cover glass at a density of 1×105 cells/cm2, placed in a 24-well tissue culture microtiter plate, and cultured in the humidified CO2 incubator for 8 hours. After seeding, HBMECs were treated with 0.025% (w/v) fluorescent WGA-CL-NGF-CUR-liposomes, cultivated in the humidified CO2 incubator at 37°C for 3 hours, and washed with DPBS three times. Subsequently, the HBMECs were fixed with 10% (v/v) formalin at 25°C for 10 minutes, treated with 0.5% (v/v) Triton X-100 at 25°C for 10 minutes, and immersed in 200 μL of serum blocking solution at 25°C for 30 minutes. The pretreated sample was reacted with 10 μg/mL anti-O-linked N-acetylglucosamine antibody at 4°C for 12 hours, incubated with goat polyclonal secondary antibody to mouse immunoglobulin G (H and L, 1:100) in darkness at 25°C for one hour, counterstained with 0.5% (w/v) 4′,6-diamidino-2-phenylindole in 0.1% (v/v) Triton X-100 in darkness at 25°C for 3 minutes, and immersed in aqueous mounting medium. Images of HBMEC nuclei, WGA-CL-NGF-CUR-liposomes, and O-linked N-acetylglucosamine (for WGA binding) were obtained using a confocal laser scanning microscope (LSM 510, Zeiss, Oberkochen, Germany) with an argon laser light source. The blue, green, and red excitation wavelengths were 350 nm, 490 nm, and 555 nm, respectively, and the corresponding emission wavelengths were 475 nm, 520 nm, and 565 nm, respectively. The green intensity in the red–green–blue color model was assessed using Image-Pro Plus (version 4.5, Media Cybernetics, Bethesda, MD, USA) between 120 and 180.
Recognition of WGA-CL-NGF-CUR-liposomes by fibrillar Aβ1-42 near SK-N-MC cells
The experimental procedures for culturing SK-N-MC cells, treating with WGA-CL-NGF-CUR-liposomes, preparing immunochemical staining samples, staining Aβ1–42 receptors, and visualizing fluorescent images were analogous to those for HBMECs described in the “Internalization of WGA-CL-NGF-CUR-liposomes by HBMECs” section. Two differences between the two staining methods were the addition of 5 μM and 10 μM fibrillar Aβ1–42 before treatment with WGA-CL-NGF-CUR-liposomes and replacement for the primary antibody of anti-O-linked N-acetylglucosamine antibody by anti-Aβ monoclonal antibody (1:400) for SK-N-MC cells.
Statistical analysis
The data are presented as the mean ± standard deviation. One-way analysis of variance followed by Tukey’s honest significant difference test was used to evaluate the statistical significance between groups.
Results and discussion
Average diameter of WGA-CL-NGF-CUR-liposomes
Figure 2 shows the average particle diameter of the WGA-CL-NGF-CUR-liposomes. As indicated in this figure, an increase in the CL mole percentage enlarged the liposomes from 105 nm to 165 nm, indicating a significant increase in diameter. In addition, incorporation of CUR and WGA increased the liposomal diameter. This was because the ingredients of CL and CUR might extend the bilayer of WGA-CL-NGF-CUR-liposomes. However, entrapment of NGF did not apparently alter the liposomal diameter. This was mainly because the hydrophilic NGF was entrapped in the aqueous core of the CL-NGF-liposomes. Thus, NGF could not affect the surface tension or the bilayer or the liposome size. In a study on electrokinetic capillary chromatography of liposomes, an increase in the CL mole percentage to 10% in lipids primarily comprising 1-palmitoyl-2-oleyl-sn-glycerophosphatidylcholine increased the particle size.25 It has also been observed that inclusion of CUR in the lipid bilayer of sphingomyelin and cholesterol increased the particle size.26 Moreover, modification with transferrin on the particle surface was found to increase the surface layer thickness of liposomes comprising L-α-soy phosphatidyl choline, stearylamine, and cholesterol.27
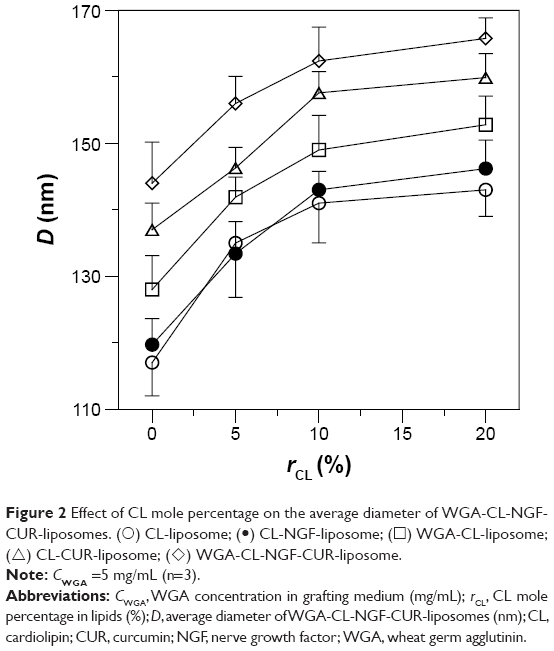 | Figure 2 Effect of CL mole percentage on the average diameter of WGA-CL-NGF-CUR-liposomes. (○) CL-liposome; (•) CL-NGF-liposome; (◻) WGA-CL-liposome; (△) CL-CUR-liposome; (◊) WGA-CL-NGF-CUR-liposome. |
Zeta potential of WGA-CL-NGF-CUR-liposomes
Table 1 shows the zeta potential of the WGA-CL-NGF-CUR-liposomes. As indicated in this table, WGA-CL-NGF-CUR-liposomes were negatively charged, with a zeta potential between −5 mV and −15 mV. This was mainly because the negatively charged DSPE-PGE(2000) and DSPE-PGE(2000)-CA were coated on the external liposomal layer. In addition, an increase in the CL mole percentage enhanced the absolute zeta potential value. This was because one CL molecule contains two negative charges, which is higher than on the other phospholipids used.28 Moreover, incorporation of CUR and NGF did not significantly alter the zeta potential. This is consistent with the literature because CUR and NGF are embedded in the bilayer and aqueous core, respectively.26 Further, conjugation of WGA neutralized the negative surface charge. This was because WGA carries a positive charge in the grafting medium.29
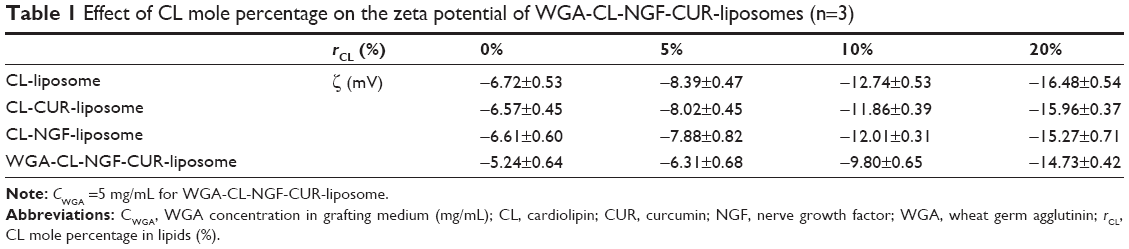 | Table 1 Effect of CL mole percentage on the zeta potential of WGA-CL-NGF-CUR-liposomes (n=3) |
Scanning and transmission electron micrographs of WGA-CL-NGF-CUR-liposomes
Figure 3 shows the structure of the WGA-CL-NGF-CUR-liposomes. As indicated in this figure, the particle diameter was about 110–170 nm with appropriate polydispersity, which is consistent with the data shown in Figure 2. As seen in Figure 3A–D, typical bilayer vesicles were obtained. This morphology is comparable with the geometry of liposomes comprising 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-[poly(ethylene glycol)-2000], DPPC, cholesterol, and palmitic acid.30 As indicated in Figure 3E–H, a higher WGA concentration yielded a thicker and rougher dark external layer of WGA-CL-NGF-CUR-liposomes. This was because conjugated WGA could extend the exterior of the liposomes. As shown in Figure 3I–L, spherical CL-NGF-CUR-liposomes with a smooth surface were observed, which was quite similar to liposomes comprising DPPC and 1,2-distearoyl-sn-glycero-3-phosphocholine.31
 | Figure 3 Micrographs of WGA-CL-NGF-CUR-liposomes. |
XPS spectra of WGA-CL-NGF-CUR-liposomes
Figure 4 shows the XPS spectra for the WGA-CL-NGF-CUR-liposomes. The peaks at 132 eV, 282 eV, 401 eV, and 532 eV belonged to the signals of phosphorus, carbon, nitrogen, and oxygen, respectively, on the surface of WGA-CL-NGF-CUR-liposomes. As indicated in Figure 4, the phosphorus intensity was roughly a constant, when the CL mole percentage increased from 10% to 20% and the WGA concentration increased from 2.5 mg/mL to 5 mg/mL. However, the nitrogen intensity fluctuated, when the CL mole percentage and WGA concentration varied. Table 2 shows the effect of the CL mole percentage and WGA concentration on the atomic ratio of nitrogen to phosphorus. As seen in this table, an increase in the CL mole percentage reduced the atomic percentage of nitrogen. This was because CL contains phosphorus but does not bear nitrogen. In addition, an increase in the WGA concentration increased the atomic ratio of nitrogen to phosphorus. This was because WGA carries ample nitrogen atoms. Table 2 also shows the grafting efficiency of WGA on WGA-CL-NGF-CUR-liposomes. This table further shows that an increase in the CL mole percentage enhanced the grafting efficiency. This was mainly because the negatively charged CL in the liposomal bilayer could attract positively charged WGA to the vicinity of WGA-CL-NGF-CUR-liposomes via electrical interaction. Based on the WGA grafting efficiency, an increase in the CL mole percentage could increase the quantity of nitrogen in the WGA-CL-NGF-CUR-liposomes. However, based on the atomic ratio of nitrogen to phosphorus, an increase in the CL mole percentage reduced the overall nitrogen quantity on the WGA-CL-NGF-CUR-liposomes. Moreover, an increase in the WGA concentration decreased the grafting efficiency. This was because a high level of WGA increased the competition among WGA molecules for surface carboxyl groups. In fact, the self-competition to occupy the nanoparticle surface was intensified by the more active molecules involved, such as WGA and transferrin.32 It has been observed that the hydrophilic Beclin 1 evolutionarily conserved domain could be preferably conjugated on a lipid membrane enriched with CL.33 This was due to small head groups and large hydrophobic tails in the molecular structure of CL.
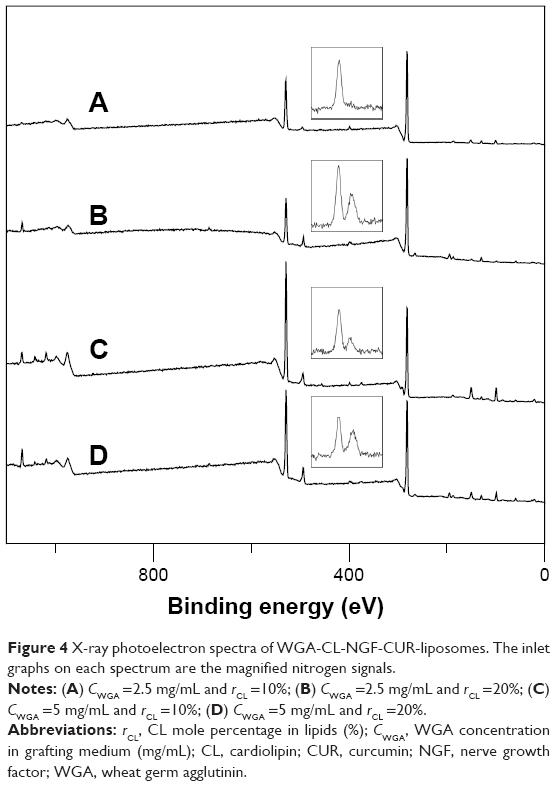 | Figure 4 X-ray photoelectron spectra of WGA-CL-NGF-CUR-liposomes. The inlet graphs on each spectrum are the magnified nitrogen signals. |
 | Table 2 Atomic ratio of nitrogen to phosphorus on WGA-CL-NGF-CUR-liposomes and WGA grafting efficiency (n=3) |
NGF and CUR entrapment efficiency in CL-NGF-CUR-liposomes
Figure 5 shows the entrapment efficiency of NGF and CUR in the CL-NGF-CUR-liposomes. As indicated in Figure 5A, an increase in the CL mole percentage enhanced the entrapment efficiency of NGF. This was because negatively charged CL could attract cationic NGF to the aqueous interior. In addition, incorporation of CUR reduced the entrapment efficiency of NGF. This was because the interaction between CUR and the membrane lipids might extend the bilayer region and shrink the core space in the CL-NGF-CUR-liposomes. As shown in Figure 5B, an increase in the CL mole percentage enhanced the entrapment efficiency of CUR. This could be explained as follows. First, a CL molecule contains four hydrophobic tails and has a strong affinity for hydrophobic CUR. Second, an equal mole percentage of DPPC and cholesterol was used in every batch of lipid mixture. An increase in the CL mole percentage decreased the cholesterol fraction in the liposome formulation, leading to a decrease in the rigidity of the lipid bilayer of the liposomes and less entrapment efficiency of the CUR bilayer inside. Moreover, incorporation of NGF enhanced the entrapment efficiency of CUR. This was because positively charged NGF emerged uniformly near the inner bilayer, leading to a uniform distribution of negatively charged phospholipids in the CL-NGF-CUR-liposomes and accelerating the CUR encapsulation rate. It has been observed that addition of phosphatidylcholine to lipids increases the entrapment of CUR in the phospholipid bilayer.34 In fact, soybean phospholipids, egg yolk phospholipids, and hydrogenated soybean phospholipids could be effective in encapsulating CUR.35 It has also been found that CUR could be accommodated in liposomal cores via its high solubility in 2-hydroxypropyl-γ-cyclodextrin.36 Further, CL in anionic liposomes could benefit an encapsulation of cationic drug.37
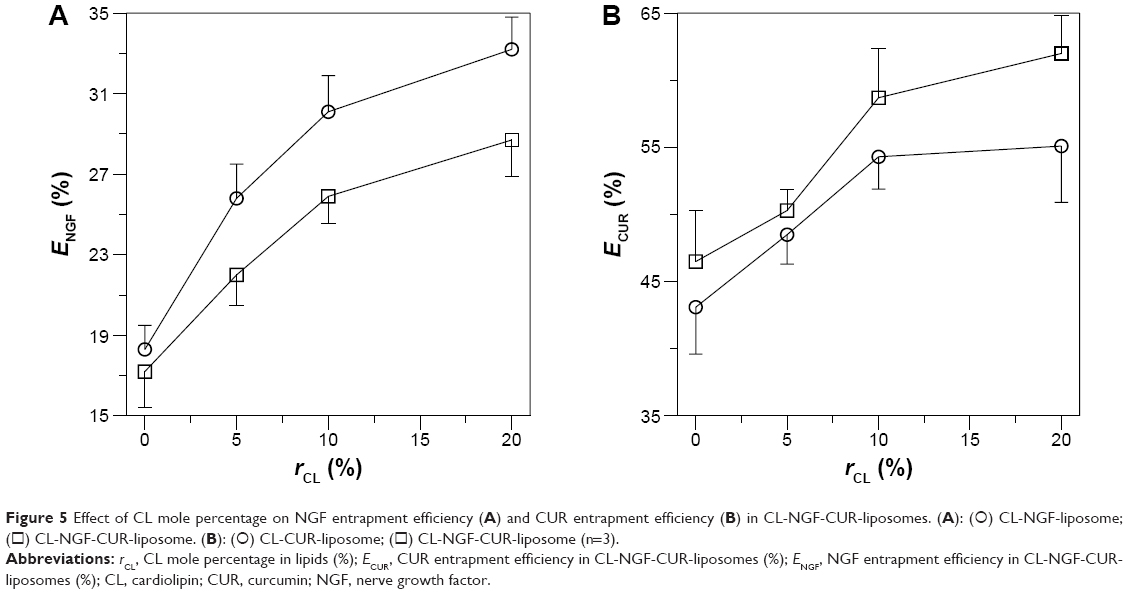 | Figure 5 Effect of CL mole percentage on NGF entrapment efficiency (A) and CUR entrapment efficiency (B) in CL-NGF-CUR-liposomes. (A): (○) CL-NGF-liposome; (◻) CL-NGF-CUR-liposome. (B): (○) CL-CUR-liposome; (◻) CL-NGF-CUR-liposome (n=3). |
Profiles of NGF and CUR release from WGA-CL-NGF-CUR-liposomes
Figure 6 shows the dissolution kinetics of NGF and CUR from WGA-CL-NGF-CUR-liposomes. As indicated in this figure, no initial burst of NGF and CUR was obtained. The molecular weight of human β-NGF is about 27 kDa.38 The NGF liberated from WGA-CL-NGF-CUR-liposomes could permeate a cellulose membrane of 50 kDa. The accumulated percentage of NGF released from WGA-CL-NGF-CUR-liposomes over 6 hours was about 20%–32%, which was lower than the corresponding CUR release percentage of 35%–55%. In addition, the release rate decelerated over 48 hours. The accumulated percentage of NGF released from WGA-CL-NGF-CUR-liposomes over 48 hours was about 50%–72%, which is comparable with the 55%–75% for CUR. In this stage, the dissolution behavior resulted from collision between WGA-CL-NGF-CUR-liposomes and migration of NGF through the membrane or detachment of CUR from the liposomal bilayer. As indicated in Figure 6A, an increase in the CL mole percentage accelerated the release of NGF from the WGA-CL-NGF-CUR-liposomes. This acceleration was because the high level of CL reduced the cholesterol content available for lipid assembly, decreased the tightness and closure of the sealed configuration in the WGA-CL-NGF-CUR-liposomes, and enhanced the fluidity of the liposomal bilayer. It has been suggested that cholesterol with its particular planar ring structure is an important component of the cell membrane and can generate a compact configuration in the bilayer via hydrogen bonding between its hydroxyl group and the head groups of phospholipids.39 As indicated in Figure 6B, an increase in the CL mole percentage slowed the release of CUR from the WGA-CL-NGF-CUR-liposomes. Two reasons could explain this deceleration. First, the affinity of CUR for CL was stronger than that for other phospholipids. Second, an increase in the CL mole percentage increased the absolute zeta potential (data shown in Table 1), decreased the possibility of liposomal collision, and reduced the deterioration of the bilayer structure where CUR was entrapped.
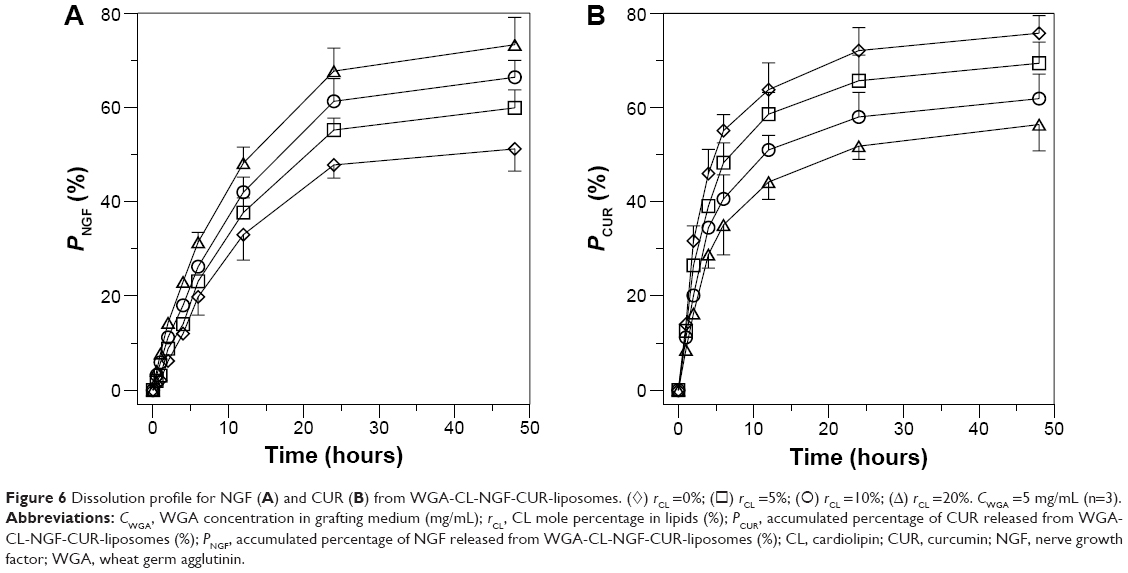 | Figure 6 Dissolution profile for NGF (A) and CUR (B) from WGA-CL-NGF-CUR-liposomes. (◊) rCL =0%; (◻) rCL =5%; (○) rCL =10%; (Δ) rCL =20%. CWGA =5 mg/mL (n=3). |
Viability of HBMECs, HAs, HBVPs, and SK-N-MC cells after treatment with WGA-CL-NGF-CUR-liposomes
Figure 7 shows the viability of HBMECs, HAs, HBVPs, and SK-N-MC cells. As indicated in this figure, the mean of four independent experiments using the control was defined as 100%. Thus, the presentation for the standard deviation of the control could be higher than 100%. Treatment with WGA-CL-NGF-CUR-liposomes slightly reduced the cell viability in general. This was mainly because WGA-CL-NGF-CUR-liposomes stimulated the secretion of inflammatory cytokines such as tumor necrosis factor-α.40 For bare liposomes and CL-liposomes, the viability of HBMECs, HAs, HBVPs, and SK-N-MC cells was at a comparable level. This suggests that inclusion of CL in liposomes did not jeopardize the drug delivery system. In addition, no significant difference in viability of HBMECs, HAs, HBVPs, and SK-N-MC cells was found between bare liposomes and CUR-liposomes. This suggests that the current liposomes could avoid a strong cytotoxicity induced by CUR. However, WGA on the surface slightly reduced the viability of HBMECs, HAs, HBVPs, and SK-N-MC cells. This was because the positive charge of WGA could trigger electrical intervention in the negatively charged cell systems. It has been observed that the viability of L1210 murine leukemia cells treated with liposomes containing CL for 24 hours was at the same level as the untreated control group.37 In a rodent toxicity study of liposomes containing a cationic CL analog, no rat deaths occurred after daily injection of 100 mg/kg for 3 days.41 Moreover, it has been found that modification with WGA slightly increased the toxicity of PEG-poly(lactic acid) nanoparticles in rats.29 It has been well known for decades that lipids can induce apoptosis of endothelial cells via oxidation products such as 7-oxysterols.42,43 A lipid accumulation in endothelial cells could induce apoptosis. However, the low toxicity of WGA-CL-NGF-CUR-liposomes to HBMECs, HAs, and HBVPs demonstrated a reasonable biocompatibility to normal blood–brain barrier cells. It has also been observed that anti-inflammatory capsaicin can protect against endothelial injury.44 In this study, anti-inflammatory CUR was embedded in the liposomal bilayer and improved the viability of HBMECs, HAs, and HBVPs, showing a difference between this work and a recent publication.23
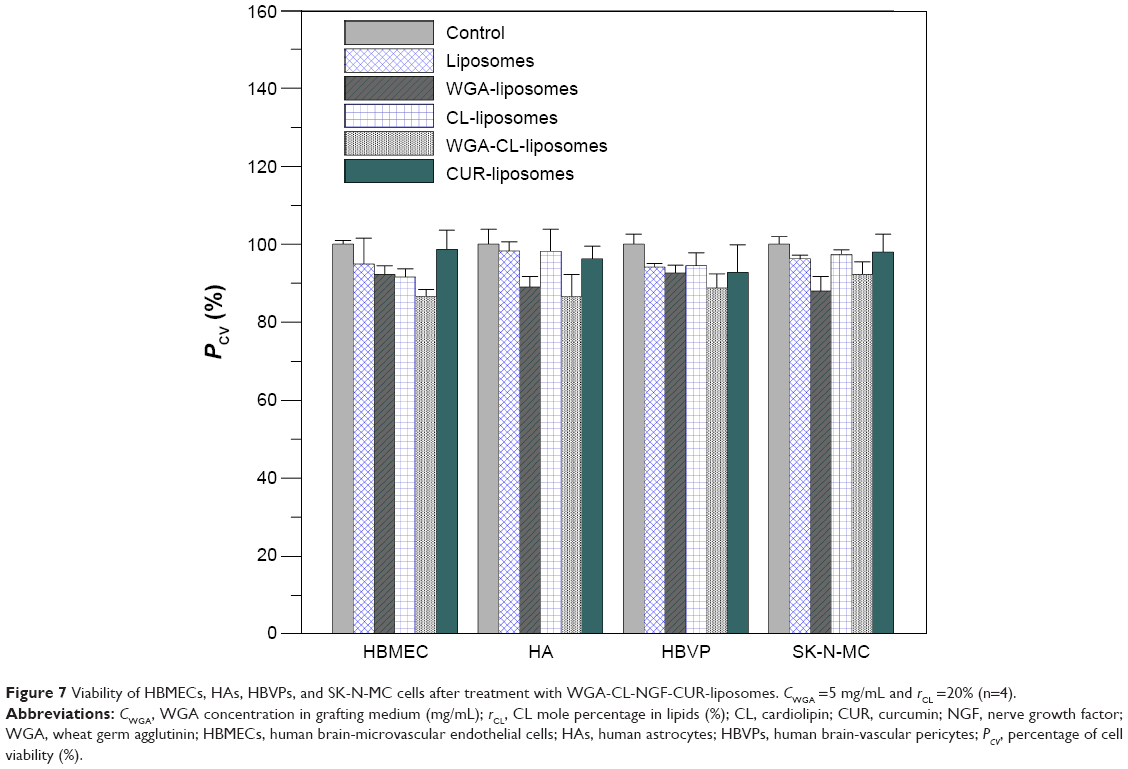 | Figure 7 Viability of HBMECs, HAs, HBVPs, and SK-N-MC cells after treatment with WGA-CL-NGF-CUR-liposomes. CWGA =5 mg/mL and rCL =20% (n=4). |
Permeability of NGF and CUR across HBMEC/HA using WGA-CL-NGF-CUR-liposomes
Figure 8 shows the permeability of NGF and CUR across HBMEC/HA. In this study, the TEER of HBMEC/HA treated with liposomal carriers varied from 191 Ω × cm2 to 233 Ω × cm2, demonstrating satisfactory tightness for investigating delivery across the blood–brain barrier. The control TEER value (no treatment with nanocarriers) was 243±4 Ω × cm2 (n=4). The control PI permeability coefficient across HBMEC/HA was (3.8±0.3) ×10−6 cm/sec (n=4), defining a compact monolayer with a high level of tight junctions that does not allow small molecules to pass by paracellular transport. As indicated in Figure 8A, CL-NGF-liposomes (indicated by black bars in Figure 8Ab–e) enhanced the permeability for NGF when compared with free NGF (Figure 8Aa). Modification with WGA markedly increased the permeability for NGF. As shown in Figure 8B, CL-CUR-liposomes reduced the permeability for CUR when compared with free CUR. Conjugation of WGA on the surface of WGA-CL-CUR-liposomes enhanced the permeability of CUR. However, an increase in the CL mole percentage did not significantly change the permeability of NGF and CUR. There are five possible reasons for these results. First, free hydrophilic NGF with a molecular weight of about 27 kDa was not able to infiltrate HBMEC/HA. The slight penetration of NGF across HBMEC/HA resulted from its affinity for neurotrophic tyrosine kinase receptor type 1 expressed on HBMECs.45 Second, free hydrophobic CUR with a molecular weight of 368 Da infiltrates HBMEC/HA easily. It has been shown that hydrophobic drugs with a molecular weight smaller than 400 Da are likely to permeate the blood–brain barrier.46 Third, the bilayer structure and phospholipid composition of CL-NGF-liposomes is similar to that of cell membranes. Therefore, the membrane fusion of CL-NGF-liposomes with HBMECs enabled permeation of NGF across HBMEC/HA. However, CL-CUR-liposomes physically hindered the ability of CUR to traverse the blood–brain barrier. Although CL-CUR-liposomes reduced the permeability of CUR across the blood–brain barrier, they could reduce the toxicity of CUR and enhance its bioavailability. Fourth, positively charged WGA could preferentially interact with the negatively charged HBMECs via electrostatic attraction and activate adsorptive-mediated transcytosis.47 In addition, HBMECs carry N-acetylglucosamine, which is recognized by surface WGA.22 It has also been demonstrated that WGA has a high affinity for porcine capillary endothelial cells and has low toxicity.48 Fifth, HBMECs did not carry ligands for conjugating CL. However, it has been observed that CL is widely distributed in the inner membranes of liver mitochondria for specific molecular regulation.49 In a study of the permeation characteristics of calcium, CL-enriched liposomes were shown to decrease calcium uptake, as estimated by cytochrome oxidase vesicles.50
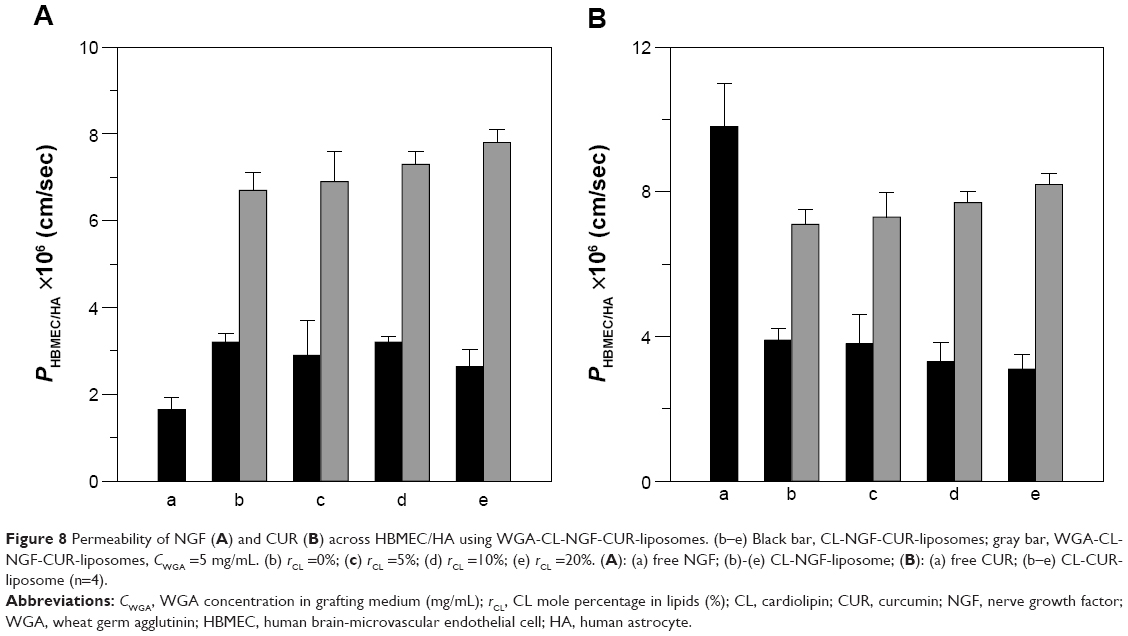 | Figure 8 Permeability of NGF (A) and CUR (B) across HBMEC/HA using WGA-CL-NGF-CUR-liposomes. (b–e) black bar, CL-NGF-CUR-liposomes; gray bar, WGA-CL-NGF-CUR-liposomes, CWGA =5 mg/mL. (b) rCL =0%; (c) rCL =5%; (d) rCL =10%; (e) rCL =20%. (A): (a) free NGF; (b)-(e) CL-NGF-liposome; (B): (a) free CUR; (b–e) CL-CUR-liposome (n=4). |
Rescue of degenerating SK-N-MC cells using WGA-CL-NGF-CUR-liposomes
Figure 9 shows the effect of WGA-CL-NGF-CUR-liposomes on the viability of SK-N-MC cells with Aβ1–42-induced cytotoxicity. As indicated in this figure, the viability of SK-N-MC cells decreased with time. In addition, treatment with WGA-CUR-liposomes for 24 and 36 hours significantly enhanced the viability of SK-N-MC cells. However, no significant difference was found between viability of SK-N-MC cells without treatment and that with free CUR. The order of cell viability was WGA-CL-NGF-CUR-liposomes >WGA-CL-CUR-liposomes > WGA-CUR-liposomes > free CUR ≈ no treatment. This manifested a neuroprotection of the current liposomal formulations against the degeneration induced by Aβ1–42. Comparing Figure 9A and B, the viability of SK-N-MC cells with Aβ1–42 5 μg/mL was higher than that with Aβ1–42 10 μg/mL. An explanation for these results is as follows. First, CUR could inhibit aggregation of Aβ1–42 and destroy Aβ1–42 fibrils.51 It has also been found that application of CUR for 7 days could reduce senile plaques in APPswe/PS1dE9 mice.52 Moreover, neuronal apoptosis associated with Aβ1–42 toxicity is commonly found in neuronal degeneration.53 In fact, the frequency of apoptosis in AD was found to be high because AD neurons were immunopositive for caspase 3.54 Also, c-Jun, a protein activated during the apoptotic cascade, is expressed in AD.55 Thus, encapsulation of CUR in WGA-CUR-liposomes can efficiently rescue SK-N-MC cells from apoptosis. Second, NGF plays an important role in neuronal survival and phenotypic maintenance. In an organotypic brain slice model, NGF was found to protect cholinergic neurons from degeneration.56 Third, CL has a high affinity for Aβ1–42 fibrils and its oligomers. It has been observed that CL enhances the viability of neuroblastoma cells due to its strong affinity for Aβ1–42.57 In this study, CL was included in the liposomal bilayer for Aβ1–42 binding, which was different from the targeting scheme reported in a recent publication.23 Fourth, SK-N-MC cells expressed several neurochemical markers, possessed typical cholinergic characteristics, and were recognized as a neuronal phenotype. Aβ1–42 fibrils are neurotoxic to SK-N-MC cells and have been used to induce neuronal apoptosis in an AD model.58 Although SK-N-MC cells were reported to be unresponsive to NGF, it was found later that SK-N-MC cells express neurotrophic tyrosine kinase receptor type 1, which is the major receptor for NGF.59–61 Thus, SK-N-MC cells could be responsive to NGF.45,62 In addition, an AD model could be established using an in vitro neuroblastoma cell line with a synthetic compound such as Aβ1–42.63 Since SK-N-MC cells are derived from neuroepithelioma, a culture of SK-N-MC cells with Aβ1–42 could represent AD.
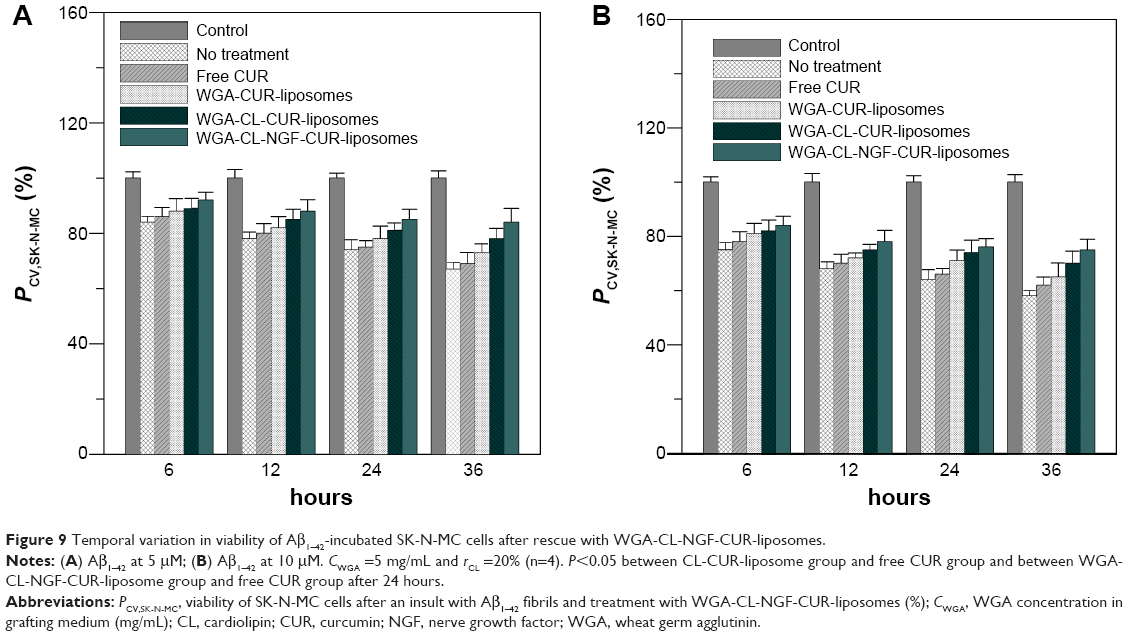 | Figure 9 Temporal variation in viability of Aβ1–42-incubated SK-N-MC cells after rescue with WGA-CL-NGF-CUR-liposomes. |
Fluorescent images of WGA-CL-NGF-CUR-liposomes internalized by HBMECs
Figure 10A shows immunochemical staining images of binding and uptake of WGA-CL-NGF-CUR-liposomes by HBMECs via N-acetylglucosamine expressed on the cell membrane. As seen in Figure 10A(a–d), HBMECs expressed abundant N-acetylglucosamine moieties. In addition, a few CL-NGF-CUR-liposomes emerged around the HBMECs. Although the interaction between CL-NGF-CUR-liposomes and HBMECs was weak, there was some permeability of NGF across HBMEC/HA. The green intensity ratios in Figure 10Aa to d, Figure 10Ab to d, and Figure 10Ac to d were 1.18, 0.87, and 0.92, respectively. Thus, the green intensity in Figure 10Aa–d was at a similar level. As shown in Figure 10Ae–h, internalization of WGA-CL-NGF-CUR-liposomes by HBMECs was prevalent owing to the recognition of N-acetylglucosamine. Moreover, the green intensity in Figure 10Ae was 0.95 times that in Figure 10Af. Based on Figure 10Ah, the green intensity in Figure 10Ag is 0.92 times that in Figure 10Ah. Further, an increase in WGA concentration enhanced the uptake quantity, suggesting that WGA on the surface of WGA-CL-NGF-CUR-liposomes could target HBMECs and deliver NGF and CUR across the blood–brain barrier. These images are consistent with the data shown in Figure 8. It has been demonstrated that modification of liposomes with anti-transferrin receptor antibody RI7217 increased the cellular uptake and showed no intracellular colocalization with early/late endosomes or early lysosomes during delivery across the blood–brain barrier.64
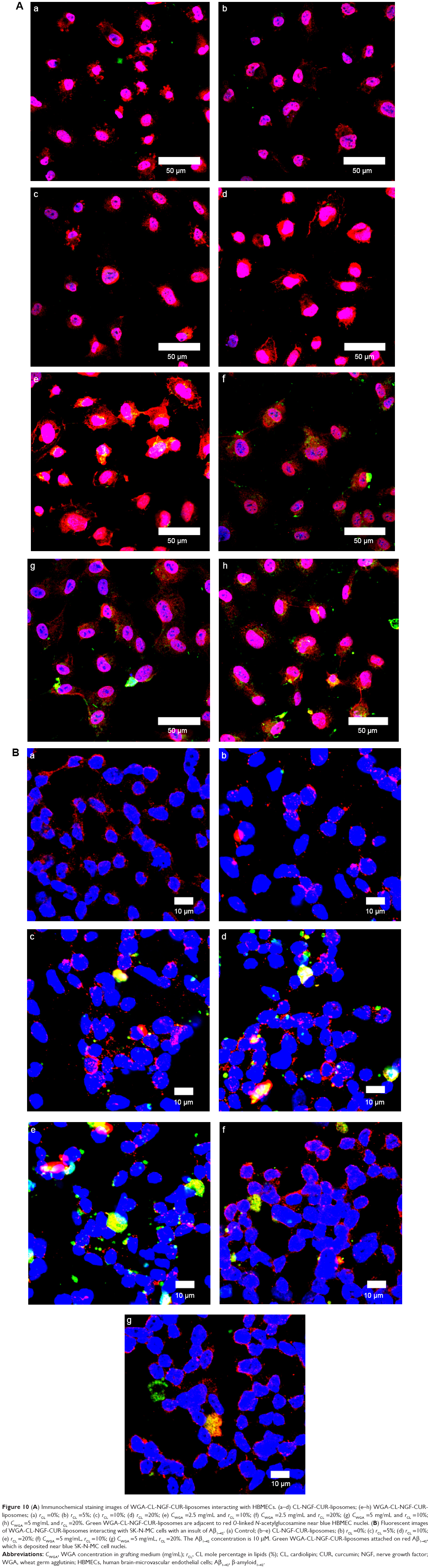 | Figure 10 (A) Immunochemical staining images of WGA-CL-NGF-CUR-liposomes interacting with HBMECs. (a–d) CL-NGF-CUR-liposomes; (e–h) WGA-CL-NGF-CUR-liposomes; (a) rCL =0%; (b) rCL =5%; (c) rCL =10%; (d) rCL =20%; (e) CWGA =2.5 mg/mL and rCL =10%; (f) CWGA =2.5 mg/mL and rCL =20%; (g) CWGA =5 mg/mL and rCL =10%; (h) CWGA =5 mg/mL and rCL =20%. Green WGA-CL-NGF-CUR-liposomes are adjacent to red O-linked N-acetylglucosamine near blue HBMEC nuclei. (B) Fluorescent images of WGA-CL-NGF-CUR-liposomes interacting with SK-N-MC cells with an insult of Aβ1–42. (a) Control; (b–e) CL-NGF-CUR-liposomes; (b) rCL =0%; (c) rCL =5%; (d) rCL =10%; (e) rCL =20%; (f) CWGA =5 mg/mL, rCL =10%; (g) CWGA =5 mg/mL, rCL =20%. The Aβ1–42 concentration is 10 μM. Green WGA-CL-NGF-CUR-liposomes attached on red Aβ1–42, which is deposited near blue SK-N-MC cell nuclei. |
Fluorescent images of WGA-CL-NGF-CUR-liposomes adjacent to SK-N-MC cells
Figure 10B shows immunochemical staining images of WGA-CL-NGF-CUR-liposomes adhering to Aβ1–42 and interacting with SK-N-MC cells. As seen in Figure 10Ba, ample Aβ1–42 fibrils deposited in the vicinity of SK-N-MC cells. As shown in Figure 10Bb–e, fibrillar Aβ1–42 could conjugate with CL-NGF-CUR-liposomes. In addition, SK-N-MC cells could internalize a portion of CL-NGF-CUR-liposomes, suggesting that CL-NGF-CUR-liposomes could carry NGF and CUR to rescue cholinergic neurons from apoptosis. Moreover, an increase in the CL mole percentage enhanced the quantity of CL-NGF-CUR-liposomes in and near SK-N-MC cells. This demonstrates that CL in the phospholipid bilayer could promote the delivery of NGF and CUR from CL-NGF-CUR-liposomes to SK-N-MC cells. Since CL-NGF-CUR-liposomes could only bind to fibrillary Aβ1–42, the delivery of CL-NGF-CUR-liposomes might via passive uptake by SK-N-MC cells. In addition to the targeting effect, it has been observed that incorporation of CL in liposomes could reduce the dose-limiting toxicity of mitoxantrone.37 It has also been found that Aβ1–42 can be endocytosed by SK-N-MC cells via association with the α7 nicotinic acetylcholine receptor.65 As seen in Figure 10Bf–g, the green intensity of the WGA-CL-NGF-CUR-liposomes was a little weaker than that of the corresponding CL-NGF-CUR-liposomes in Figure 10Bd–e. This was because surface WGA could slightly hinder the attachment of WGA-CL-NGF-CUR-liposomes to Aβ1–42 fibrils and SK-N-MC cells.
Conclusion
WGA-CL-NGF-CUR-liposomes were prepared and evaluated as a treatment for AD. The toxicity of WGA-CL-NGF-CUR-liposomes to HBMECs, HAs, HBVPs, and SK-N-MC cells is acceptable. Surface WGA contributes targeting ability to enhance the permeability of NGF and CUR across the blood–brain barrier. In addition, CL in the liposomal bilayer carries WGA-CL-NGF-CUR-liposomes to Aβ1–42 fibrils and protects degenerating SK-N-MC cells against neurotoxicity. As a result, WGA-CL-NGF-CUR-liposomes slow neuronal apoptosis and are potential pharmacotherapy vehicles for AD therapy.
Acknowledgment
This work is supported by a grant from the Ministry of Science and Technology of the Republic of China (MOST 103–2221–E–194–043–MY3).
Disclosure
The authors report no conflicts of interest in this work.
References
Gaugler J, James B, Johnson T, Scholz K, Weuve J. 2014 Alzheimer’s disease facts and figures. Alzheimers Dement. 2014;10:1–75. | ||
Citron M. Alzheimer’s disease: strategies for disease modification. Nat Rev Drug Discov. 2010;9:387–398. | ||
Duyckaerts C, Delatour B, Potier MC. Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 2009;118:5–36. | ||
Turner PR, O’Connor K, Tate WP, Abraham WC. Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog Neurobiol. 2003;70:1–32. | ||
van Oijen M, Hofman A, Soares HD, Koudstaal PJ, Breteler MM. Plasma Aβ1–40 and Aβ1–42 and the risk of dementia: a prospective case-cohort study. Lancet Neurol. 2006;5:655–660. | ||
Gandy S. The role of cerebral amyloid β accumulation in common forms of Alzheimer disease. J Clin Invest. 2005;115:1121–1129. | ||
Kuo YC, Chou PR. Neuroprotection against degeneration of SK-N-MC cells using neuron growth factor-encapsulated liposomes with surface cereport and transferrin. J Pharm Sci. 2014;103:2484–2497. | ||
Sofroniew MV, Howe CL, Mobley WC. Nerve growth factor signaling, neuroprotection, and neural repair. Annu Rev Neurosci. 2001;24:1217–1281. | ||
Frielingsdorf H, Simpson DR, Thal LJ, Pizzo DP. Nerve growth factor promotes survival of new neurons in the adult hippocampus. Neurobiol Dis. 2007;26:47–55. | ||
Niewiadomska G, Mietelska-Porowska A, Mazurkiewicz M. The cholinergic system, nerve growth factor and the cytoskeleton. Behav Brain Res. 2011;221:515–526. | ||
Friden PM, Walus LR, Watson P, et al. Blood–brain barrier penetration and in vivo activity of an NGF conjugate. Science. 1993;259:373–377. | ||
Ray B, Bisht S, Maitra A, Lahiri DK. Neuroprotective and neurorescue effects of a novel polymeric nanoparticle formulation of curcumin (NanoCurc) in the neuronal cell culture and animal model: implications for Alzheimer’s disease. J Alzheimers Dis. 2011;23:61–77. | ||
Chen YF, Jobanputra P, Barton P, et al. Cyclooxygenase-2 selective non-steroidal anti-inflammatory drugs (etodolac, meloxicam, celecoxib, rofecoxib, etoricoxib, valdecoxib and lumiracoxib) for osteoarthritis and rheumatoid arthritis: a systematic review and economic evaluation. Health Technol Assess. 2008;12:1–278. | ||
Aggarwal BB, Harikumar KB. Potential therapeutic effects of curcumin, the anti-inflammatory agent, against neurodegenerative, cardiovascular, pulmonary, metabolic, autoimmune and neoplastic diseases. Int J Biochem Cell Biol. 2009;41:40–59. | ||
Baum L, Lam CW, Cheung SK, et al. Six-month randomized, placebo-controlled, double-blind, pilot clinical trial of curcumin in patients with Alzheimer disease. Clin Psychopharmacol. 2008;28:110–113. | ||
Bangham AD, Standish MM, Watkins JC. Diffusion of univalent ions across the lamellae of swollen phospholipids. J Mol Biol. 1965;13:238-252. | ||
Luk YO, Chen WY, Wong WJ, et al. Treatment of focal cerebral ischemia with liposomal nerve growth factor. Drug Deliv. 2004;11:319–324. | ||
Xie Y, Ye L, Zhang X, Cui W, Lou J. Transport of nerve growth factor encapsulated into liposomes across the blood–brain barrier: in vitro and in vivo studies. J Control Release. 2005;105:106–119. | ||
Gobbi M, Re F, Canovi M, et al. Lipid-based nanoparticles with high binding affinity for amyloid-β1–42 peptide. Biomaterials. 2010;31:6519–6529. | ||
Lewis RN, McElhaney RN. The physicochemical properties of cardiolipin bilayers and cardiolipin-containing lipid membranes. Biochim Biophys Acta. 2009;1788:2069–2079. | ||
Gao X, Wu B, Zhang Q, et al. Brain delivery of vasoactive intestinal peptide enhanced with the nanoparticles conjugated with wheat germ agglutinin following intranasal administration. J Control Release. 2007;121:156–167. | ||
Plattner VE, Germann B, Neuhaus W, Noe CR, Gabor F, Wirth M. Characterization of two blood–brain barrier mimicking cell lines: distribution of lectin-binding sites and perspectives for drug delivery. Int J Pharm. 2010;387:34–41. | ||
Kuo YC, Wang CT. Protection of SK-N-MC cells against β-amyloid peptide-induced degeneration using neuron growth factor-loaded liposomes with surface lactoferrin. Biomaterials. 2014;35:5954–5964. | ||
Kuo YC, Lu CH. Effect of human astrocytes on the characteristics of human brain-microvascular endothelial cells in the blood–brain barrier. Colloids Surf B Biointerfaces. 2011;86:225–231. | ||
Muhonen J, Holopainen JM, Wiedmer SK. Interactions between local anesthetics and lipid dispersions studied with liposome electrokinetic capillary chromatography. J Chromatogr A. 2009;1216:3392–3397. | ||
Taylor M, Moore S, Mourtas S, et al. Effect of curcumin-associated and lipid ligand-functionalized nanoliposomes on aggregation of the Alzheimer’s Aβ peptide. Nanomed Nanotechnol Biol Med. 2011;7:541–550. | ||
Soni V, Kohli DV, Jain SK. Transferrin coupled liposomes as drug delivery carriers for brain targeting of 5-florouracil. J Drug Target. 2005;13:245–250. | ||
Lutter M, Fang M, Luo X, Nishijima M, Xie X, Wang X. Cardiolipin provides specificity for targeting of tBid to mitochondria. Nat Cell Biol. 2000;2:754–756. | ||
Liu Q, Shao X, Chen J, et al. In vivo toxicity and immunogenicity of wheat germ agglutinin conjugated poly(ethylene glycol)-poly(lactic acid) nanoparticles for intranasal delivery to the brain. Toxicol Appl Pharmacol. 2011;251:79–84. | ||
Centisa V, Vermette P. Physico-chemical properties and cytotoxicity assessment of PEG-modified liposomes containing human hemoglobin. Colloids Surf B Biointerfaces. 2008;65:239–246. | ||
Begum MY, Abbulu K, Sudhakar M. Flurbiprofen-loaded stealth liposomes: studies on the development, characterization, pharmacokinetics, and biodistribution. J Young Pharm. 2012;4:209–219. | ||
Kuo YC, Wang LJ. Transferrin-grafted catanionic solid lipid nanoparticles for targeting delivery of saquinavir to the brain. J Taiwan Inst Chem Eng. 2014;45:755–763. | ||
Huang W, Choi W, Hu W, et al. Crystal structure and biochemical analyses reveal beclin 1 as a novel membrane binding protein. Cell Res. 2012;22:473–489. | ||
Takahashi M, Uechi S, Takara K, Asikin Y, Wada K. Evaluation of an oral carrier system in rats: bioavailability and antioxidant properties of liposome-encapsulated curcumin. J Agric Food Chem. 2009;57:9141–9146. | ||
Chen Y, Wu Q, Zhang Z, Yuan L, Liu X, Zhou L. Preparation of curcumin-loaded liposomes and evaluation of their skin permeation and pharmacodynamics. Molecules. 2012;17:5972–5987. | ||
Dhule SS, Penfornis P, Frazier T, et al. Curcumin-loaded γ-cyclodextrin liposomal nanoparticles as delivery vehicles for osteosarcoma. Nanomedicine. 2012;8:440–451. | ||
Chang RS, Kim J, Lee HY, et al. Reduced dose-limiting toxicity of intraperitoneal mitoxantrone chemotherapy using cardiolipin-based anionic liposomes. Nanomedicine. 2010;6:769–776. | ||
Hefti F. Is Alzheimer disease caused by lack of nerve growth factor? Ann Neurol. 1983;13:109–110. | ||
Ivankin A, Kuzmenko I, Gidalevitz D. Cholesterol-phospholipid interactions: new insights from surface X-ray scattering data. Phys Rev Lett. 2010;104:108101. | ||
Kuo YC, Lin PI, Wang CC. Targeting nevirapine delivery across human brain microvascular endothelial cells using transferrin-grafted poly(lactide-co-glycolide) nanoparticles. Nanomedicine. 2011;6:1011–1026. | ||
Chien PY, Wang J, Carbonaro D, et al. Novel cationic cardiolipin analogue-based liposome for efficient DNA and small interfering RNA delivery in vitro and in vivo. Cancer Gene Ther. 2005;12:321–328. | ||
Sata M, Walsh K. Oxidized LDL activates Fas-mediated endothelial cell apoptosis. J Clin Invest. 1998;102:1682–1689. | ||
Li W, Ghosh M, Eftekhari S, Yuan XM. Lipid accumulation and lysosomal pathways contribute to dysfunction and apoptosis of human endothelial cells caused by 7-oxysterols. Biochem Biophys Res Commun. 2011;409:711–716. | ||
Chen KS, Chen PN, Hsieh YS, Lin CY, Lee YH, Chu SC. Capsaicin protects endothelial cells and macrophage against oxidized low-density lipoprotein-induced injury by direct antioxidant action. Chem Biol Interact. 2015;228:35–45. | ||
Tsuruda A, Suzuki S, Maekawa T, Oka S. Constitutively active Src facilitates NGF-induced phosphorylation of TrkA and causes enhancement of the MAPK signaling in SK-N-MC cells. FEBS Lett. 2004;560:215–220. | ||
Zhang EY, Knipp GT, Ekins S, Swaan PW. Structural biology and function of solute transporters: implications for identifying and designing substrates. Drug Metab Rev. 2002;34:709–750. | ||
Kuo YC, Su FL. Transport of stavudine, delavirdine, and saquinavir across the blood–brain barrier by polybutylcyanoacrylate, methylmethacrylate–sulfopropylmethacrylate, and solid lipid nanoparticles. Int J Pharm. 2007;340:143–152. | ||
Fischer D, Kissel T. Histochemical characterization of primary capillary endothelial cells from porcine brains using monoclonal antibodies and fluorescein isothiocyanate-labelled lectins: implications for drug delivery. Eur J Pharm Biopharm. 2001;52:1–11. | ||
Nicolay K, Timmers RJ, Spoelstra E, et al. The interaction of adriamycin with cardiolipin in model and rat liver mitochondrial membranes. Biochim Biophys Acta. 1984;778:359–371. | ||
Zazueta C, Ramirez J, Garcia N, Baeza I. Cardiolipin regulates the activity of the reconstituted mitochondrial calcium uniporter by modifying the structure of the liposome bilayer. J Membr Biol. 2003;191:113–122. | ||
Ono K, Hasegawa H, Naiki H, Yamada M. Curcumin has potent anti-amyloidogenic effects for Alzheimer’s β-amyloid fibrils in vitro. J Neurosci Res. 2004;75:742–750. | ||
Garcia-Alloza M, Borrelli LA, Rozkalne A, Hyman BT, Bacskai BJ. Curcumin labels amyloid pathology in vivo, disrupts existing plaques, and partially restores distorted neurites in an Alzheimer mouse model. J Neurochem. 2007;102:1095–1104. | ||
Stadelmann C, Deckwerth TL, Srinivasan A, et al. Activation of caspase-3 in single neurons and autophagic granules of granulovacuolar degeneration in Alzheimer’s disease: evidence for apoptotic cell death. Am J Pathol. 1999;155:1459–1466. | ||
Broe M, Shepherd CE, Milward EA, Halliday GM. Relationship between DNA fragmentation, morphological changes and neuronal loss in Alzheimer’s disease and dementia with Lewy bodies. Acta Neuropathol. 2001;101:616–624. | ||
MacGibbon GA, Lawlor PA, Walton M, et al. Expression of Fos, Jun, and Krox family proteins in Alzheimer’s disease. Exp Neurol. 1997;147:316–332. | ||
Bottger D, Ullrich C, Humpel C. Monocytes deliver bioactive nerve growth factor through a brain capillary endothelial cell-monolayer in vitro and counteract degeneration of cholinergic neurons. Brain Res. 2010;1312:108–119. | ||
Bereczki E, Re F, Masserini ME, Winblad B, Pei JJ. Liposomes functionalized with acidic lipids rescue Aβ-induced toxicity in murine neuroblastoma cells. Nanomedicine. 2011;7:560–571. | ||
Shaykhalishahi H, Taghizadeh M, Yazdanparast R, Chang YT. Anti-amyloidogenic effect of AA3E2 attenuates β-amyloid induced toxicity in SK-N-MC cells. Chem Biol Interact. 2010;186:16–23. | ||
Sonnenfeld KH, Ishii DN. Nerve growth factor effects and receptors in cultured human neuroblastoma cell lines. J Neurosci Res. 1982;8:375–391. | ||
Snider WD. Function of the neurotrophins during nervous system development: what the knockouts are teaching us. Cell. 1994;77:627–638. | ||
Kuo YC, Lin CY. Targeting delivery of liposomes with conjugated p-aminophenyl-α-D-manno-pyranoside and apolipoprotein E for inhibiting neuronal degeneration insulted with β-amyloid peptide. J Drug Target. 2015;23:147–158. | ||
Kuo YC, Liu YC. Cardiolipin-incorporated liposomes with surface CRM197 for enhancing neuronal survival against neurotoxicity. Int J Pharm. 2014;473:334–344. | ||
Carolindah MN, Rosli R, Adam A, Nordin N. An overview of in vitro research models for Alzheimer’s disease (AD). Regenerative Research. 2013;2:8–13. | ||
Salvati E, Re F, Sesana S, et al. Liposomes functionalized to overcome the blood–brain barrier and to target amyloid-β peptide: the chemical design affects the permeability across an in vitro model. Int J Nanomedicine. 2013;8:1749–1758. | ||
Nagele RG, D’Andrea MR, Anderson WJ, Wang HY. Intracellular accumulation of β-amyloid1–42 in neurons is facilitated by the α7 nicotinic acetylcholine receptor in Alzheimer’s disease. Neuroscience. 2002;110:199–211. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.