Back to Journals » Medical Devices: Evidence and Research » Volume 17
Reporting of Demographics & Subgroup Analyses in Premarketing Studies of FDA Approved High-Risk Cardiovascular Devices, 2014–2022
Authors Swanson MJ ![]() , Uyeki CL, Yoder SR, Dhruva SS
, Uyeki CL, Yoder SR, Dhruva SS ![]() , Miller JE, Ross JS
, Miller JE, Ross JS ![]()
Received 29 December 2023
Accepted for publication 21 April 2024
Published 29 April 2024 Volume 2024:17 Pages 165—172
DOI https://doi.org/10.2147/MDER.S457152
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Matthew J Swanson,1,2 Colin L Uyeki,1 Sarah R Yoder,1 Sanket S Dhruva,3 Jennifer E Miller,4 Joseph S Ross4– 6
1Frank H. Netter MD School of Medicine, Quinnipiac University, North Haven, CT, USA; 2Leonard N. Stern School of Business, New York University, New York, NY, USA; 3Department of Medicine, University of California, San Francisco School of Medicine, San Francisco, CA, USA; 4Department of Medicine, Yale School of Medicine, New Haven, CT, USA; 5Department of Health Policy and Management, Yale School of Public Health, New Haven, CT, USA; 6Center for Outcomes Research and Evaluation, Yale-New Haven Hospital, New Haven, CT, USA
Correspondence: Matthew J Swanson, Frank H. Netter MD School of Medicine, Quinnipiac University, 370 Bassett Road, North Haven, CT, 06473-1908, USA, Email [email protected]
Background: Representation of diverse study populations in pivotal clinical trials for medical devices and subgroup analyses for demographic groups to explore differences in safety and effectiveness are essential to understanding the benefits and risks in diverse populations. The US Food and Drug Administration (FDA) has taken many steps to improve transparency and subgroup analyses over the past decade, but there has not been a recent evaluation of demographic reporting and subgroup analyses.
Methods: We reviewed all FDA Premarket Approvals for high-risk cardiovascular devices from 2014 to 2022, focusing on pivotal studies supporting device approval. We abstracted detailed demographic data about the age, sex, race, ethnicity, and socioeconomic position of study participants. We also assessed the presence and results of subgroup analyses to understand the safety and effectiveness of devices across trial populations.
Results: Analysis of 92 pivotal studies revealed that age and sex were reported in 96.7% of the studies, while race and ethnicity were reported in 71.7% and 58.7%, respectively. However, only 7.9% of studies explicitly detailed the participation of older adults (≥ 65 years) and no studies reported patients’ socioeconomic position. Subgroup analyses by sex were conducted in 70.7% of studies, with 12.3% reporting significant differences. In contrast, analyses by race and ethnicity were performed in only 12.0% of the studies, with 9.1% reporting significant differences.
Conclusion: Approximately one-third of pivotal studies for high-risk cardiovascular devices approved by the FDA from 2014 to 2022 did not report the race of study participants, nearly 40% did not report ethnicity, and more than 90% did not report the participation of older adults (≥ 65 years). Subgroup analyses were infrequently conducted by age or race and ethnicity. There is a need for better trial demographic reporting and conduct of subgroup analyses in premarketing studies to ensure the safety and effectiveness of medical devices for all patients.
Keywords: FDA, clinical trial, cardiovascular device, diversity, inclusion, equity
Introduction
Clinical trial diversity has long been an area of concern, with significant underrepresentation of key patient demographic groups such as older adults, women, and racial and ethnic minority patients.1–5 This underrepresentation has great clinical significance, impeding the generalizability of trial results. Policymakers, recognizing the criticality of this issue, have taken substantial steps toward improvement. The Food and Drug Omnibus Reform Act (FDORA) in 2022 and the Food and Drug Administration (FDA) Center for Devices and Radiological Health’s 2022 strategic plan underscore legislative and regulatory commitments to enhancing trial diversity, including mandates for sex and gender analyses in device development.6,7
Efforts to promote inclusive representation in clinical trials have a history of legislative support. The Food and Drug Administration Safety and Innovation Act (FDASIA) in 2012 and the subsequent FDA action plan in 2014 to enhance demographic data collection highlight a commitment to diversity and the accurate assessment of medical interventions across all population segments.8,9 Recognizing the unique challenges and opportunities presented by medical devices and drugs is crucial for designing inclusive trials. Medical devices, due to their specific applications and complexities, and drugs, benefiting from a wider application context, face distinct barriers to achieving diverse recruitment. Yet, the goal remains the same: to ensure trials reflect the broad population for reliable generalizability.
Understanding the distinctions between medical device and drug trials is key to addressing population diversity. Drug trials, organized into four phases, assess safety and efficacy, whereas device trials, typically conducted in feasibility and pivotal stages, focus on safety and performance, as well as effectiveness. The scale and global reach of drug trials often exceed those of device trials, affecting the diversity and applicability of results. This contrast highlights the necessity of targeted recruitment and inclusion strategies to cultivate representative and diverse study populations.
Effective representation requires consistent demographic reporting, a point underscored by the FDA in 2020 with an effort to support greater consideration of a wide range of demographic factors in clinical trial design.10 Adding to this, the inclusion of socioeconomic status, although not currently mandated, could provide valuable insights into inclusivity and identify socio-demographic barriers to participation. While these recommendations aimed at expanding clinical trial diversity through more inclusive eligibility criteria and recruitment practices, translating these policies into practice, particularly for high-risk cardiovascular devices that are most common among FDA Premarket Approvals (PMA),11 remains inadequately documented. This gap is particularly concerning given the high stakes of these devices, where demographic-specific responses can significantly impact patient outcomes.12–15 For medical devices, the FDA also has specific guidance about the evaluation and reporting of sex7 as well as age-, race-, and ethnicity-specific data in clinical studies.16
One notable example highlighting the importance of representative study populations is seen with left atrial appendage occlusion (LAAO) devices, such as the WatchmanTM device, approved by the FDA in 2015. These devices are used to occlude the part of the left atrium where clots commonly form in patients with atrial fibrillation with the goal of reducing stroke risk. However, the study populations in the two key comparative trials that supported FDA approval were comprised of approximately 70% men,17,18 leaving a gap in understanding the device’s safety and effectiveness in women. An independent study later revealed that women had two-fold higher odds of major adverse events (MAE) and a two-fold higher risk of death in-hospital post-procedure.19 About six weeks after publication of this study, in September 2021, the FDA sent a letter alerting healthcare providers about these risks.20 This example underscores the consequences of under-enrolling women in clinical trials and highlights the risks that patients may face due to incomplete data on device safety and effectiveness in diverse populations.
This study endeavors to fill this critical knowledge gap. The recent legislative and policy efforts have aimed to address the disparities in clinical trial participation, but challenges persist in implementing these policies, especially in the context of high-risk medical devices. By analyzing demographic data from pivotal premarketing studies that supported FDA approvals of original high-risk therapeutic cardiovascular devices between 2014 and 2022, we aim to shed light on FDA transparency and the real-world implications of its diversity initiatives. We specifically focused on pivotal clinical studies, as they are the foundational clinical evidence for the FDA’s approval decisions. Although the FDA’s “Clinical Trial Snapshots” program provides clinical trial data for pharmaceuticals and biologics, focusing on the demographic composition of trial participants and highlighting how well the drug worked in various demographic groups, such reporting is unavailable for medical device approvals.21 Therefore, this study is poised to offer a comprehensive overview of the progress made and challenges that persist in ensuring diversity in clinical trials for medical devices. Doing so will provide valuable insights into the effectiveness of policy interventions and identify areas where further efforts are needed to achieve truly inclusive and representative clinical research.
Materials and Methods
We comprehensively reviewed all original high-risk therapeutic cardiovascular devices approved from 2014 to 2022, utilizing the FDA’s publicly accessible PMA database.22 One author (MJS) selected devices using the following filters: “Cardiovascular” under advisory committee and “Originals Only” under supplement type. We excluded feasibility studies, studies missing data, summaries using a meta-analysis for the pivotal study, and automated external defibrillators (AEDs). We excluded AEDs because the FDA approves these devices leveraging existing clinical data. Two authors (CLU and SRY) abstracted data from pivotal studies listed in the FDA’s Summary of Safety and Effectiveness Data about patients historically underrepresented in clinical trials: older age (age ≥65 years), female sex, from racial and ethnic minority groups, and of low socioeconomic position. We exclusively extracted this data because these groups are often underrepresented in clinical trials, aiming to ensure medical equity and understand device impacts across diverse populations. The inclusion of socioeconomic status addresses an important yet underexplored characteristic of patients historically underrepresented in clinical trials. To complement data from the FDA’s PMA database when pivotal study details were insufficient, we sought additional information from ClinicalTrials.gov and PubMed’s listing of MEDLINE-indexed journals. For the studies that reported race information, we noted the specific races included. Additionally, we determined whether these studies conducted subgroup analyses to identify significant demographic differences. We documented the presence of subgroup analyses focusing on age, sex, race, and ethnicity within each pivotal study. Furthermore, we identified any instances where these subgroup analyses yielded significant findings, highlighting differences in device safety or effectiveness across diverse patient groups. Where significant subgroup findings were reported, we carefully extracted direct quotes from the FDA’s Summary of Safety and Effectiveness Data, capturing the essence of the significant results and the precise language used, thereby shedding light on the implications of these findings. We used descriptive statistics to analyze the data. We prepared this study in accordance with the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guideline for observational studies.
Results
From 2014 to 2022, 92 pivotal studies supported FDA approval of 92 original high-risk therapeutic cardiovascular devices. Age was reported in 89 studies (96.7%; Table 1), but only 7 (7.9%) explicitly reported the proportion of older age patients ≥65 years. Sex was reported in 89 studies (96.7%). Race was reported in 66 studies (71.7%), although 2 of these studies reported only the percentage of white patients. Additionally, of studies that reported specific racial minority groups, 3 (5.0%) reported zero Asian patients, 19 (41.3%) reported zero American Indian or Alaska Native patients, 18 (46.2%) reported zero Native Hawaiian or Other Pacific Islander patients, and 7 (13.5%) reported zero patients from other races. Ethnicity was reported in 54 studies (58.7%) as Hispanic or Latino and non-Hispanic or Latino, although race and ethnicity were often combined (eg, listing “Hispanic” as race or “First Nations/White”), and the categories were at times not inclusive or all-encompassing. No studies reported patients’ socioeconomic position.
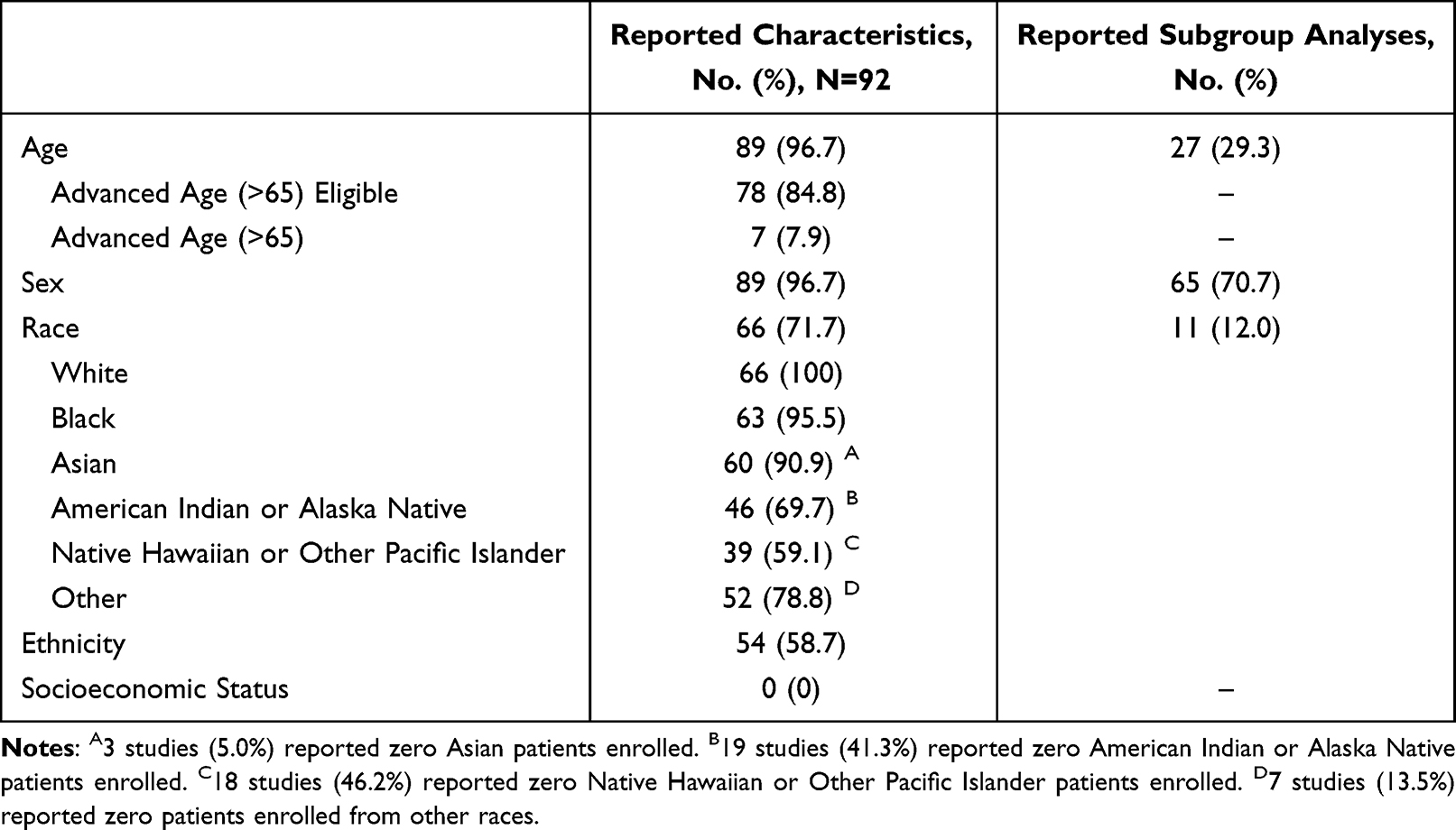 |
Table 1 Pivotal Studies Supporting the FDA Approval of 92 High-Risk Therapeutic Cardiovascular Devices that Reported Characteristics of Patients Historically Underrepresented in Clinical Trials and Subgroup Analyses, 2014–2022 |
Subgroup analyses by age were performed for 27 studies (29.3%), among which 2 (7.4%) reported any statistically significant difference in device safety or effectiveness. Subgroup analyses by sex were performed for 65 studies (70.7%), among which 8 (12.3%) reported statistically significant differences. Subgroup analyses by race and ethnicity were performed for 11 studies (12.0%), among which 1 (9.1%) reported a statistically significant difference. In total, 10 studies (10.9%) had statistically significant subgroup findings, including 2 for age, 8 for sex, and 1 for race and ethnicity (Table 2). For example, for BIOTRONIK Inc’s Astron Pulsar Stent System, a notable instance was observed where the reported data showed a statistically significant increase in MAE between genders after 12 months: 9.6% of male subjects experienced an MAE compared to 19.8% of female subjects. However, a common issue with these subgroup analyses was the lack of specificity, often resulting in broad conclusions, such as reports of “lower efficacy for women” without clarity whether further investigation was pursued to understand these sex-based differences in outcomes. No approvals were restricted or specified to a particular patient population based on demographic factors.
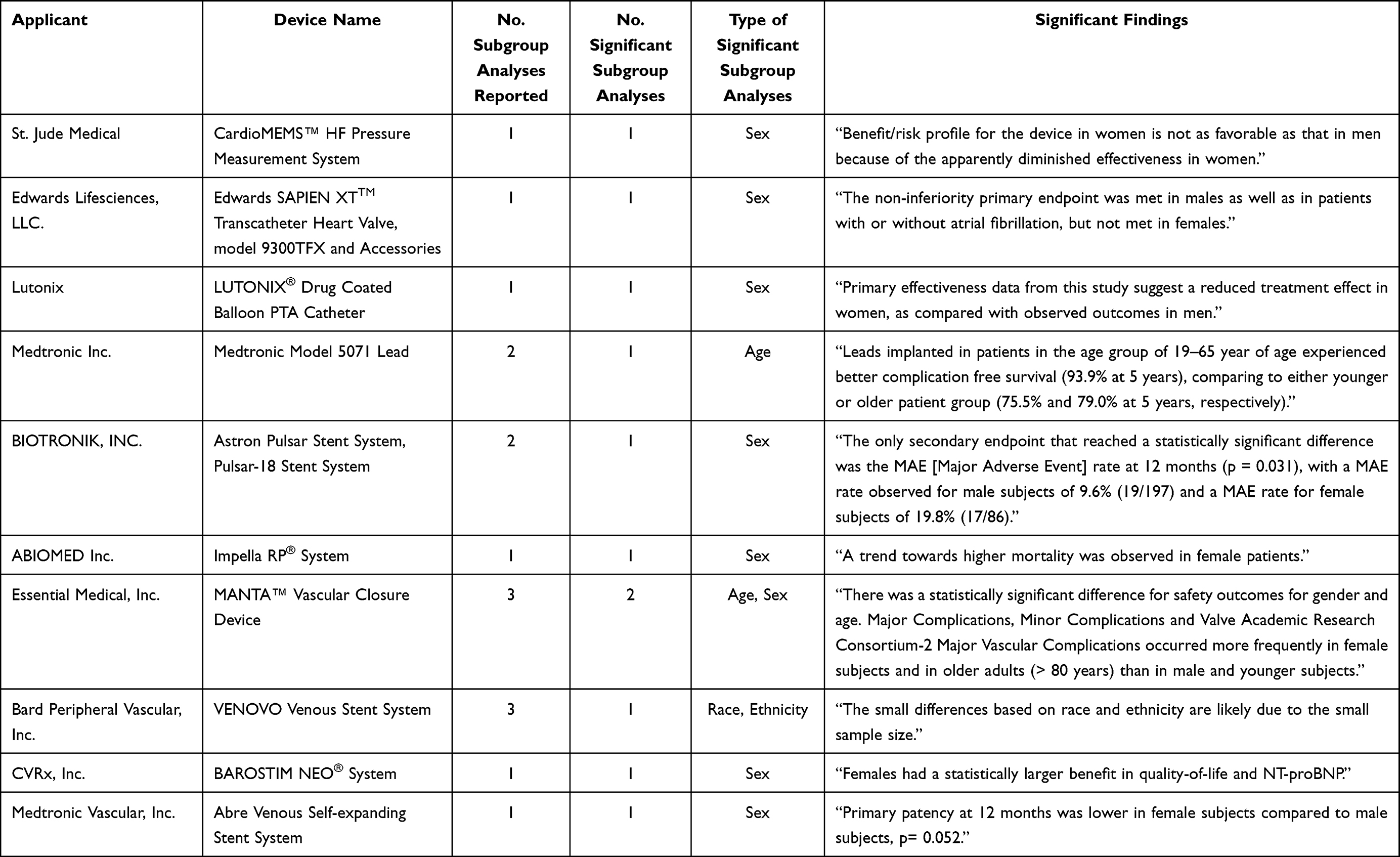 |
Table 2 Descriptions of Statistically Significant Subgroup Findings in Pivotal Studies for FDA Approved High-Risk Cardiovascular Devices, 2014–2022 |
Discussion
Among the 92 pivotal studies supporting FDA approval of 92 original high-risk therapeutic cardiovascular devices between 2014 and 2022, patient age and sex were nearly always reported, although few studies reported the explicit participation of older adults (≥65 years). In contrast, only three-quarters and three-fifths of studies reported patient race and ethnicity, respectively. And no studies reported patients’ socioeconomic position. The inconsistency in reporting is further underscored by substantial disparities in the enrollment of racial minority groups, where the degree of underrepresentation varied widely. For example, while 3 studies (5.0%) reported no Asian patients, as many as 18 studies (46.2%) lacked any Native Hawaiian or Other Pacific Islander patients, revealing a striking range of underrepresentation from 5% to nearly 50% in the studies. Similar findings have been shown in orthopedic23 and neurosurgical24 medical devices, highlighting the need for stronger federal mandates and enforcement of study inclusivity.
Moreover, demographic subgroup analyses, which are crucial for understanding the safety and effectiveness of devices across trial populations and a key component of the FDA’s action plan to enhance the collection and availability of demographic subgroup data, was conducted in fewer than 30% of all studies by age and just 12% by race and ethnicity, consistent with prior research.25 This contrasts with analysis by sex, which was done by more than 70% of studies. These findings highlight that while reporting of and subgroup analyses by sex have improved,26 there are still gaps in multiple other key demographic categories. Additionally, the ambiguity in subgroup differences reported raises concerns about the depth and power of analysis conducted.27 Furthermore, the lack of targeted approvals raises questions about if and how subgroup analyses are used in regulatory decision-making.
Our findings illustrate a lack of uniformity in demographic reporting across studies, suggesting that the FDA’s Summary of Safety and Effectiveness Data template could include comprehensive race and ethnicity categories. This change would ensure structured and complete demographic reporting, with explicit documentation of zero participants in specific categories, thereby enhancing transparency and the utility of these data for regulatory and public health analyses.
The FDORA represents a meaningful stride towards diverse inclusivity by requiring sponsors to submit diversity action plans (DAPs) alongside their study protocols or Investigational Device Exemption (IDE) applications for pivotal drug studies and device trials.1 These DAPs are designed to set clear target goals for participant diversity, reflecting the population’s demographics most likely to use the medical device or drug being tested.28 Moreover, the FDORA mandates include detailed strategies for achieving these goals, such as outreach efforts to underrepresented communities, collaborations with community organizations, and using culturally tailored recruitment materials.
In response to these challenges, legislative measures like FDORA represent a crucial step toward rectifying these disparities. However, the journey from policy to practice in clinical trial diversity, particularly in medical device research, requires a concerted effort from all stakeholders in the medical device ecosystem. The effectiveness of these regulatory efforts hinge on rigorous enforcement and careful monitoring. As such, continued vigilance and assessment of these policies in clinical trials and post-marketing studies are essential to gauge their impact and identify areas for further improvement.
Our findings also highlight the critical role of post-marketing real-world studies. Essential for broadening our understanding of device performance across diverse populations, these studies complement pre-marketing efforts and align with FDA initiatives to increase clinical trial inclusivity. These post-marketing evaluations are crucial for capturing longitudinal data on safety and efficacy among underrepresented groups, potentially uncovering adverse reactions or efficacy differences not apparent in the controlled environments of pre-marketing trials. Emphasizing rigorous methodologies and multidisciplinary collaboration is key to generating reliable evidence and adapting regulatory practices to better protect and serve all population segments.29
It is important to note that our study’s focus on high-risk therapeutic cardiovascular devices may limit the generalizability of our findings. We relied on pivotal trial identification and demographic data from FDA Summaries, acknowledging that additional details might be accessible via ClinicalTrials.gov or related publications. Furthermore, the retrospective nature of our analysis, while informative, does not allow for real-time assessment of ongoing trials and may not fully capture the dynamic changes in trial demographics and policies that occur after our review period.
Conclusions
The lack of sufficient representation in clinical trial populations severely hampers the possibility of conducting robust subgroup analyses crucial for assessing population-specific risks and benefits.30 This shortfall is especially critical in medical devices, where demographic-specific responses are inadequately documented and can have significant implications for safety and effectiveness. Our findings reveal a large gap in the reporting of race and ethnicity in pivotal studies, with approximately 30% not reporting race and 40% not reporting ethnicity, as well as more than 90% not reporting participation of older adults (≥65 years). Additionally, subgroup analyses were infrequently conducted, especially by age, race, and ethnicity. To overcome these challenges, it is critical to adopt practices that involve consistent reporting of patient demographics and comprehensive subgroup analyses. Implementing such practices, aligned with the directives of FDORA, is not only a regulatory requirement but also a step towards meaningful change in medical research. By prioritizing the collection and analysis of detailed demographic data and performing rigorous subgroup analyses, we can enhance the integrity and fairness of healthcare, ensuring that medical devices are effectively and safely applicable to all segments of the US population.31 Ensuring that clinical trials accurately reflect the populations they intend to serve is essential for developing medical interventions that are both effective and equitable.
Funding
This project was not supported by any external grants or funds.
Disclosure
Drs. Dhruva, Miller, and Ross currently receive research support through the University of California-San Francisco (Dhruva) and Yale University (Miller, Ross) from Arnold Ventures. Dr. Dhruva receives research funding from the Department of Veterans Affairs. Dr. Dhruva also reports having received, in the past 36 months, funding from the Food and Drug Administration, Greenwall Foundation, National Evaluation System for Health Technology Coordinating Center, and National Institute for Health Care Management. Dr. Dhruva further reports serving on the Medicare Evidence Development and Coverage Advisory Committee and Institute for Clinical and Economic Review California Technology Assessment Forum. Dr. Miller receives research support from the Food and Drug Administration for the Yale-Mayo Clinic Center for Excellence in Regulatory Science and Innovation (CERSI) program and Scientific American through Yale University. Dr. Miller is a Board Member for Bioethics International and Galatea Bio. Dr. Ross currently receives research support through Yale University from Johnson and Johnson to develop methods of clinical trial data sharing, from the Medical Device Innovation Consortium as part of the National Evaluation System for Health Technology (NEST), from the Food and Drug Administration for the Yale-Mayo Clinic Center for Excellence in Regulatory Science and Innovation (CERSI) program (U01FD005938), from the Agency for Healthcare Research and Quality (R01HS022882), and from the National Heart, Lung and Blood Institute of the National Institutes of Health (NIH) (R01HS025164, R01HL144644); in addition, Dr. Ross is an expert witness at the request of Relator’s attorneys, the Greene Law Firm, in a qui tam suit alleging violations of the False Claims Act and Anti-Kickback Statute against Biogen Inc that was settled in September 2022. The authors report no other conflicts of interest in this work.
References
1. National Academies of Sciences, Engineering, and Medicine. Improving Representation in Clinical Trials and Research: Building Research Equity for Women and Underrepresented Groups. Washington, DC: The National Academies Press; 2022. doi:10.17226/26479
2. Varma T, Wallach JD, Miller JE, et al. Reporting of study participant demographic characteristics and demographic representation in premarketing and postmarketing studies of novel cancer therapeutics [published correction appears in JAMA Netw Open. 2021 May 3;4(5):e2114399]. JAMA Network Open. 2021;4(4):e217063. doi:10.1001/jamanetworkopen.2021.7063
3. Varma T, Jones CP, Oladele C, Miller J. Diversity in clinical research: public health and social justice imperatives. J Med Ethics. 2023;49(3):200–203. doi:10.1136/medethics-2021-108068
4. Varma T, Mello M, Ross JS, Gross C, Miller J. Metrics, baseline scores, and a tool to improve sponsor performance on clinical trial diversity: retrospective cross sectional study. BMJ Med. 2023;2(1):e000395. doi:10.1136/bmjmed-2022-000395
5. Downing NS, Shah ND, Neiman JH, Aminawung JA, Krumholz HM, Ross JS. Participation of the elderly, women, and minorities in pivotal trials supporting 2011–2013 U.S. Food and Drug Administration approvals. Trials. 2016;17(1):199. doi:10.1186/s13063-016-1322-4
6. US Food & Drug Administration. Diversity plans to improve enrollment of participants from underrepresented racial and ethnic populations in clinical trials guidance for industry; 2022. Available from: https://www.fda.gov/media/157635/download.
7. US Food & Drug Administration. Health of women strategic plan; 2022. Available from: https://www.fda.gov/media/155461/download.
8. US Food & Drug Administration. Food and Drug Administration Safety and Innovation Act (FDASIA); 2012. Available from: https://www.fda.gov/regulatory-information/selected-amendments-fdc-act/food-and-drug-administration-safety-and-innovation-act-fdasia.
9. US Food & Drug Administration. FDA action plan to enhance the collection and availability of demographic subgroup data; 2014. Available from: https://www.fda.gov/media/89307/download.
10. US Food & Drug Administration. Enhancing the diversity of clinical trial populations — eligibility criteria, enrollment practices, and trial designs guidance for industry; 2020. Available from: https://www.fda.gov/media/127712/download.
11. Swanson MJ, Johnston JL, Ross JS. Registration, publication, and outcome reporting among pivotal clinical trials that supported FDA approval of high-risk cardiovascular devices before and after FDAAA. Trials. 2021;22(1):817–819. doi:10.1186/s13063-021-05790-9
12. Moneer O, Rathi VK, Johnston JL, Ross JS, Dhruva SS. Aligning US agency policies for cardiovascular devices through the breakthrough devices program. JAMA Cardiol. 2023;8(12):1174–1181. doi:10.1001/jamacardio.2023.3819
13. Dhruva SS, Bero LA, Redberg RF. Strength of study evidence examined by the FDA in premarket approval of cardiovascular devices. JAMA. 2009;302(24):2679–2685. doi:10.1001/jama.2009.1899
14. Jones LC, Dhruva SS, Redberg RF. Assessment of clinical trial evidence for high-risk cardiovascular devices approved under the food and drug administration priority review program. JAMA Intern Med. 2018;178(10):1418–1420. doi:10.1001/jamainternmed.2018.3649
15. Rathi VK, Krumholz HM, Masoudi FA, Ross JS. Characteristics of clinical studies conducted over the total product life cycle of high-risk therapeutic medical devices receiving FDA premarket approval in 2010 and 2011. JAMA. 2015;314(6):604–612. doi:10.1001/jama.2015.8761
16. US Food & Drug Administration. Evaluation and reporting of age-, race-, and ethnicity-specific data in medical device clinical studies; 2017. Available from: https://www.fda.gov/media/98686/download.
17. Reddy VY, Sievert H, Halperin J, et al. Percutaneous left atrial appendage closure vs warfarin for atrial fibrillation: a randomized clinical trial. JAMA. 2014;312(19):1988–1998. doi:10.1001/jama.2014.15192
18. Holmes DR, Kar S, Price MJ, et al. Prospective randomized evaluation of the watchman left atrial appendage closure device in patients with atrial fibrillation versus long-term warfarin therapy: the PREVAIL trial [published correction appears in J Am Coll Cardiol. 2014 Sep 16;64(11):1186]. J Am Coll Cardiol. 2014;64(1):1–12. doi:10.1016/j.jacc.2014.04.029
19. Darden D, Duong T, Du C, et al. Sex differences in procedural outcomes among patients undergoing left atrial appendage occlusion: insights from the NCDR LAAO Registry. JAMA Cardiol. 2021;6(11):1275–1284. doi:10.1001/jamacardio.2021.3021
20. US Food & Drug Administration. Left Atrial Appendage Occlusion (LAAO) Devices potentially associated with procedural outcome differences between women and men Letter to Health Care Providers; 2021. Available from: https://www.fda.gov/medical-devices/letters-health-care-providers/left-atrial-appendage-occlusion-laao-devices-potentially-associated-procedural-outcome-differences.
21. Dhruva SS, Redberg RF. FDA drug trials snapshots-a clearer picture. JAMA Intern Med. 2017;177(5):727. doi:10.1001/jamainternmed.2017.0037
22. U.S. Food and Drug Administration. Premarket Approval (PMA) Database. Available from: http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpma/pma.cfm.
23. Issa TZ, Lambrechts MJ, Lin JS, et al. Diversity in orthopaedic surgery medical device clinical trials: an analysis of the food and drug administration safety and innovation act. J Am Acad Orthop Surg. 2023;31(3):155–165. doi:10.5435/JAAOS-D-22-00704
24. Siddiqui N, Chiu RG, Nunna RS, Glastris G, Mehta AI. Effect of the FDA Safety and Innovation Act on racial and gender diversity in neurosurgical device trials. J Neurosurg. 2021;136(1):274–281. doi:10.3171/2020.10.JNS202155
25. Dhruva SS, Mazure CM, Ross JS, Redberg RF. Inclusion of demographic-specific information in studies supporting US food & drug administration approval of high-risk medical devices. JAMA Intern Med. 2017;177(9):1390–1391. doi:10.1001/jamainternmed.2017.3148
26. Dhruva SS, Bero LA, Redberg RF. Gender bias in studies for Food and Drug Administration premarket approval of cardiovascular devices. Circ Cardiovasc Qual Outcomes. 2011;4(2):165–171. doi:10.1161/CIRCOUTCOMES.110.958215
27. Sleight P. Debate: subgroup analyses in clinical trials: fun to look at - but don’t believe them! Curr Control Trials Cardiovasc Med. 2000;1(1):25–27. doi:10.1186/cvm-1-1-025
28. Kozlov M. FDA to require diversity plan for clinical trials. Nature. 2023. doi:10.1038/d41586-023-00469-4
29. Crisafulli S, Khan Z, Karatas Y, Tuccori M, Trifirò G. An overview of methodological flaws of real-world studies investigating drug safety in the post-marketing setting. Expert Opin Drug Saf. 2023;22(5):373–380. doi:10.1080/14740338.2023.2219892
30. Varma T, Gross CP, Miller JE. Clinical trial diversity-will we know it when we see it? JAMA Oncol. 2023;9(6):765–767. doi:10.1001/jamaoncol.2023.0143
31. Schwartz AL, Alsan M, Morris AA, Halpern SD. Why diverse clinical trial participation matters. N Engl J Med. 2023;388(14):1252–1254. doi:10.1056/NEJMp2215609
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Medical Professionalism in the Provision of Clinical Care in Healthcare Organizations
Bhardwaj A
Journal of Healthcare Leadership 2022, 14:183-189
Published Date: 26 October 2022
Gender Equality Stock-Take into the WHO Regional Office for Africa’s Transformation Agenda: Progress and Opportunities
Chingarande SD, Rwamatwara E, Kithinji D, Nsarhaza K
Journal of Healthcare Leadership 2025, 17:705-712
Published Date: 11 November 2025
Medical Students’ Perspectives on Inclusivity Within Clinical Environments: A Pilot Study
Haque E, Lavin JM, Farrington R
Advances in Medical Education and Practice 2026, 17:543053
Published Date: 23 January 2026
Reporting of Race and Ethnicity in SLE Studies in High-Impact Rheumatology Journals
Eroglu I, Suter LG, Baker H
Open Access Rheumatology: Research and Reviews 2026, 18:526618
Published Date: 25 April 2026