Back to Journals » Patient Related Outcome Measures » Volume 10
Reporter’s occupation and source of adverse device event reports contained in the FDA’s MAUDE database
Authors Kavanagh KT ![]() , Brown RE Jr
, Brown RE Jr ![]() , Kraman SS, Calderon LE, Kavanagh SP
, Kraman SS, Calderon LE, Kavanagh SP
Received 21 April 2019
Accepted for publication 10 June 2019
Published 2 July 2019 Volume 2019:10 Pages 205—208
DOI https://doi.org/10.2147/PROM.S212991
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lynne Nemeth
Kevin T Kavanagh,1 Raeford E Brown Jr,2 Steve S Kraman,3 Lindsay E Calderon,4 Sean P Kavanagh5
1Health Watch USA, Somerset, KY, USA; 2Department of Anesthesiology, University of Kentucky Medical Center/Kentucky Children’s Hospital, Lexington, KY, USA; 3Department of Internal Medicine, University of Kentucky College of Medicine, Health Watch USA, Lexington, KY, USA; 4Department of Biological Sciences, Eastern Kentucky University, Health Watch USA, Lexingtion, KY, USA; 5Health Watch USA, Solon, OH, USA
Introduction: A review of the medical device adverse events submitted to the United States Food & Drug Administration (FDA) Manufacturer and User Facility Device Experience (MAUDE) database was undertaken to determine the major sources of the information.
Methods: The reporter’s occupation and source of the medical device report were determined for acquisition dates Jan 1, 1997 to Dec 31, 2018. A total of 7,766,737 adverse event records were analyzed.
Results: 96.6% of reports originated with the manufacturer. Patients (patients/family/friend) were the most frequent submitter of reports directly to the FDA, almost five times as often as physicians. Nurses submitted reports directly to the FDA 2.77 times as often as physicians. Only 0.49% of physician reports were submitted directly to the FDA, representing 0.09% of total MAUDE reports.
Conclusion: Increasing physician reporting directly to the FDA and MAUDE through the MedWatch reporting system is an imperative. Incorporating information from the perspective of the physician has the potential of increasing the quality of the data and improving the reliability of post-market surveillance.
Keywords: FDA, MAUDE, reporter, occupation, medical device, adverse events
Introduction
One element in the United States Food & Drug Administration’s (FDA’s) post-market surveillance of devices is the Manufacturer and User Facility Device Experience (MAUDE) database. The database represents reports of adverse events relating to biomedical devices and is used to identify harms that were not common or not observed in premarket testing. Because of the requirement for accuracy, it is imperative to understand who reports events. A previous study observed a preponderance of reports from manufacturers, with few physicians reporting directly to the FDA.1 However, a relatively small subset of data, comprised of two highly litigated companies and one product code, was studied; and the requirement to include the company’s name could result in a potential bias depending on the reporter’s occupation. Because of the importance of these observations, a repeat analysis of the entire MAUDE database was undertaken to determine the reporting source and reporter occupation.
Methods
The entire MAUDE database from Jan 1, 1997 to Dec 31, 2018, was downloaded from the FDA’s website2 and uploaded to a PostgreSQL server. Adverse event records, which were contained in the Master Event File, were analyzed for Reporting Source and Reporter Occupation.
Results
A total of 7,766,737 adverse event records were analyzed. The manufacturer submitted 96.62% of the reports. Table 1 shows a breakdown of the top 16 reporter occupation codes which comprise 97.9% of the total records. Only 3.38% of reports to this dataset were independent of the manufacturer; physician reporting directly to the FDA (voluntary reporting) represented a very small proportion (0.09%) of total MAUDE reports.
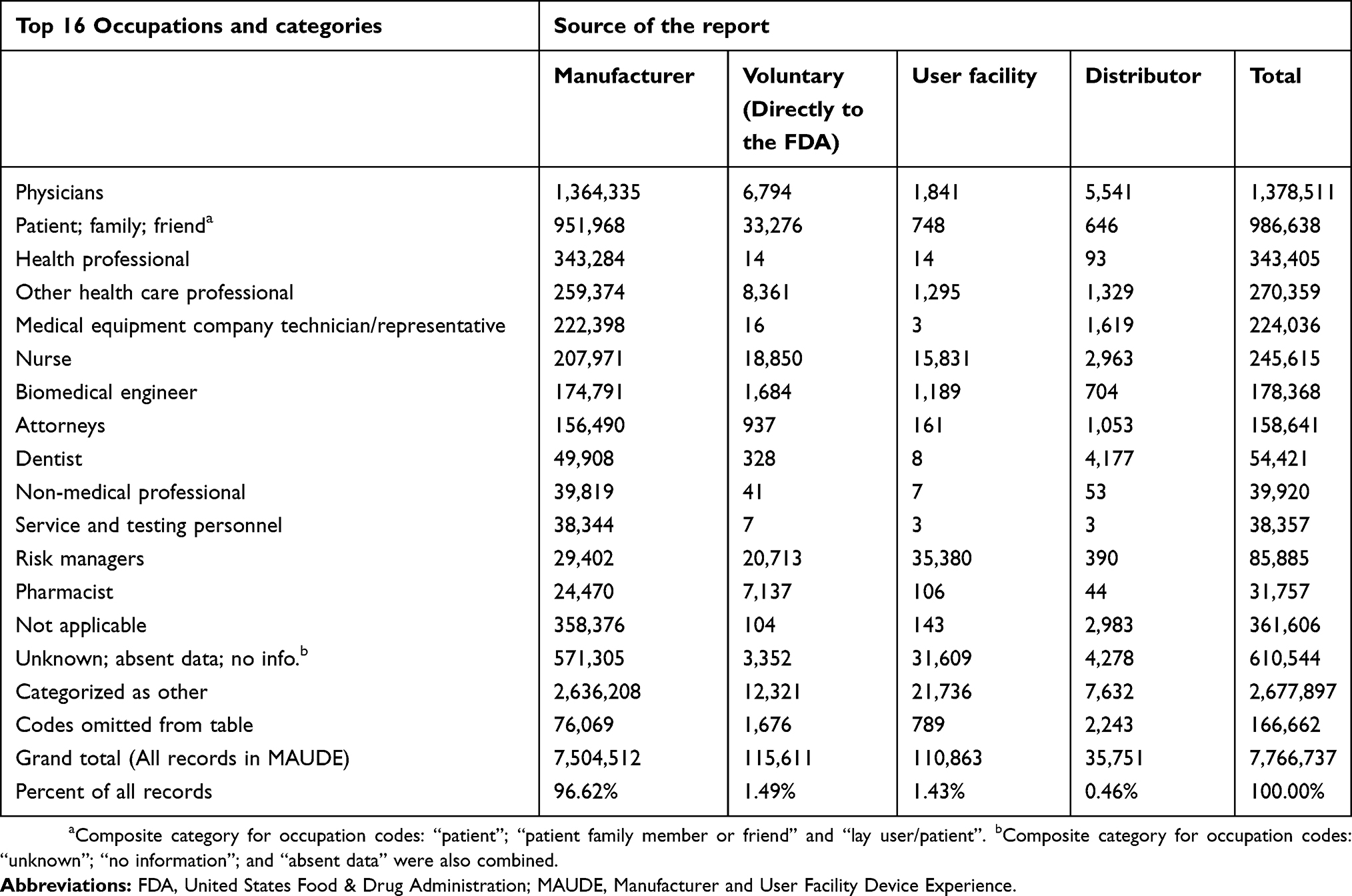 |
Table 1 Reporting source and occupation of reporter – Jan 1, 1997 to Dec 31, 2018 |
Physician
A total 17.75% adverse event reports list the reporter’s occupation as a physician. Only 0.49% of these reports were submitted directly to the FDA. The manufacturer submitted 98.97% of physician reports.
Nurse
In this analysis, “Nurse” was listed as the occupation in 3.16% of total reports. Nurses made reports directly to the FDA over twice (2.77 times) as often as physicians.
Patient/family/friend
The “patient/family/friend” category was listed as the occupation in 12.7% of the total reports; 96.5% of these reports were submitted by the manufacturer. Patients most frequently submitted reports directly to the FDA, almost five times as often as physicians.
Attorney
Attorney was listed as the occupation in 2.04% of the total reports. The manufacturer submitted 98.6% of attorney reports, possibly prompted by litigation.
Risk managers
Risk managers were the second most frequent submitter of reports directly to the FDA, three times as often than physicians. 24.1% of the total risk manager reports were submitted directly to the FDA.
In 2018, there were 88,090 reports submitted the MAUDE. Physicians made 262 or 30% of 861 reports submitted directly to the FDA (0.30% of total 2018 MAUDE reports).
Discussion
The presence of a large number of manufacturer reports in the MAUDE is to be expected, since manufacturers and distributors are required to submit a report to the FDA, if their medical device caused or contributed to a severe injury or fatality. However, the exact definition of a severe event is not well defined. These two entities are responsible for 96.62% and 0.46% of the reports submitted to the MAUDE, respectively. Facilities are required to submit adverse event reports for medical devices to both the manufacturer and the FDA.3,4 However, user facility reports accounted for only 1.43% of the total MAUDE reports, which raises questions of the current regulation’s effectiveness.1 Physicians and physician offices are not required by federal law to submit reports to the FDA of events involving biomedical hardware.
Reporting directly to the FDA
Patients (patients/family/friend) were the most frequent submitter of reports directly to the FDA, almost five times as often as physicians. Nurses made reports directly to the FDA 2.77 times as often as physicians. Attorney was listed as the occupation in 2.0% of total reports in the MAUDE but infrequently submitted directly to the FDA—see Table 1. The vast majority of attorney reports were manufacturer reports. In a highly litigated subset studied by Kavanagh, et al.,1 attorney was the most frequent occupation, comprising 42.2% of total MAUDE reports and 99.3% of these were submitted by the manufacturer.
Kavanagh, et al, also observed that MAUDE adverse event reports were unstructured with a paucity of objective data.1 For example: They observed that in records which indicated the elevation of blood cobalt, less than 4% had units for reporting ion concentrations. Our analysis found that the vast majority of physician reports were submitted by the manufacturer where there is no guarantee that significant redaction will not take place. Physicians reporting directly to the FDA through their MedWatch reporting system will bring an important perspective and has the potential of increasing the quality of the data and improving the reliability of post-market surveillance and improving patient safety.
Conclusion
The FDA needs objective, unbiased, complete data relating to any adverse device-related incident. Physicians bring a unique perspective and can provide vital information which is critical to post-market surveillance of approved devices. Unfortunately, physicians rarely submit a report directly to the FDA. This may be due to a relatively unstructured and time-consuming reporting process. Building reporting functions into electronic medical records, including ready access to a device’s Unique Device Identification (UDI) code, could encourage reporting and improve the quality of MAUDE adverse event reports. In addition, educational institutions and professional associations should educate students and physicians on the importance of submitting reports to the FDA and how to access and input data into MedWatch.
Acknowledgments
The conclusions in this review do not represent the policy of the FDA.
Disclosure
Dr Kevin Kavanagh has received partial conference attendance and meeting support from the US Department of Health and Human Services, National Quality Forum, National Patient Safety Foundation (NPSF), The Leapfrog Group, Consumer Union and the Anesthesia Patient Safety Foundation. He has served on the Centers for Medicare and Medicaid Services’ Technical Expert Panel for Hospital Acquired Conditions, and most recently on the Strategic Working Group for Agency for Healthcare Research and Quality (AHRQ) for quality indicators, and AHRQ Health Care Effectiveness and Outcomes Research (HEOR) Study Section. He is an Associate Editor for the Journal of Patient Safety for which he receives an honorarium. He has a first degree relative, who is employed by a state university, and is involved with the development of cancer chemotherapeutic and diagnostic agents. Dr Raeford E Brown Jr is past Chair of the FDA Anesthetic and Analgesic Drug Products Advisory Committee, and Chair of the American Academy of Pediatrics Section on Anesthesiology and Pain Medicine. The authors report no other conflicts of interest in this work.
References
1. Kavanagh KT, Kraman SS, Kavanagh SP. An analysis of the FDA MAUDE database and the search for cobalt toxicity in metal on metal hip implants. J Patient Saf. 2018;14(4):e89–e96. doi:10.1097/PTS.0000000000000534
2. Manufacturer and User Facility Device Experience Database - (MAUDE). U.S. food & drug administration; January 14, 2019. Available from: https://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/PostmarketRequirements/ReportingAdverseEvents/ucm127891.htm.
3. Center for Devices and Radiological Health, MDR Policy Branch. Mandatory reporting requirements: manufacturers, importers, and device user facilities. food and drug administration; March 27, 2018. Available from: https://www.fda.gov/medicaldevices/deviceregulationandguidance/postmarketrequirements/reportingadverseevents/default.htm.
4. Title 21, Food and Drugs Chapter I. Food and Drug Administration Department of Health and Human Services Subchapter H. Medical devices; April 1, 2018. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=803&showFR=1. Accessed June 17, 2019.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.