")
Back to Journals » Cancer Management and Research » Volume 12
Regulation of Autophagy by Glycolysis in Cancer
Authors Chu Y , Chang Y , Lu W, Sheng X, Wang S , Xu H , Ma J
Received 31 August 2020
Accepted for publication 28 October 2020
Published 23 December 2020 Volume 2020:12 Pages 13259—13271
DOI https://doi.org/10.2147/CMAR.S279672
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Eileen O'Reilly
Ying Chu, Yi Chang, Wei Lu, Xiumei Sheng, Shengjun Wang, Huaxi Xu, Jie Ma
Department of Immunology, Jiangsu Key Laboratory of Laboratory Medicine, School of Medicine, Jiangsu University, Zhenjiang 212013, People’s Republic of China
Correspondence: Jie Ma
Department of Immunology, Jiangsu Key Laboratory of Laboratory Medicine, School of Medicine, Jiangsu University, 301 XueFu Road, Zhenjiang, Jiangsu Province 212013, People’s Republic of China
Tel +86-1377-555-5586
Email [email protected]
Abstract: Autophagy is a critical cellular process that generally protects cells and organisms from harsh environment, including limitations in adenosine triphosphate (ATP) availability or a lack of essential nutrients. Metabolic reprogramming, a hallmark of cancer, has recently gained interest in the area of cancer therapy. It is well known that cancer cells prefer to utilize glycolysis rather than oxidative phosphorylation (OXPHOS) as their major energy source to rapidly generate ATP even in aerobic environment called the Warburg effect. Both autophagy and glycolysis play essential roles in pathological processes of cancer. A mechanism of metabolic changes to drive tumor progression is its ability to regulate autophagy. This review will elucidate the role and the mechanism of glycolysis in regulating autophagy during tumor growth. Indeed, understanding how glycolysis can modulate cellular autophagy will enable more effective combinatorial therapeutic strategies.
Keywords: glycolysis, autophagy, lung cancer, lactate, metabolism
Introduction
The processes of autophagy, which induces degradation of proteins and organelles upon cellular stress, are crucial in the pathophysiology of cancers. Biological autophagy includes three types, macroautophagy (hereafter called autophagy), microautophagy and chaperone-mediated autophagy.1 Macroautophagy refers to a process that some of the damaged proteins or organelles are encapsulated by autophagic vesicles and then sent to lysosomes (animals) or vacuoles (yeast and plants) for degradation. The corresponding autophagic cargos are degraded by acidic hydrolases to produce metabolites and biomolecules to maintain the normal biological functions.2,3 In mammalian systems, the levels of basal autophagy are normally low but are profoundly stimulated by starvation and stressful environments.4 Autophagy is mediated by multiple signaling pathways, which establish an interactive network system. The summary is shown in Figure 1.
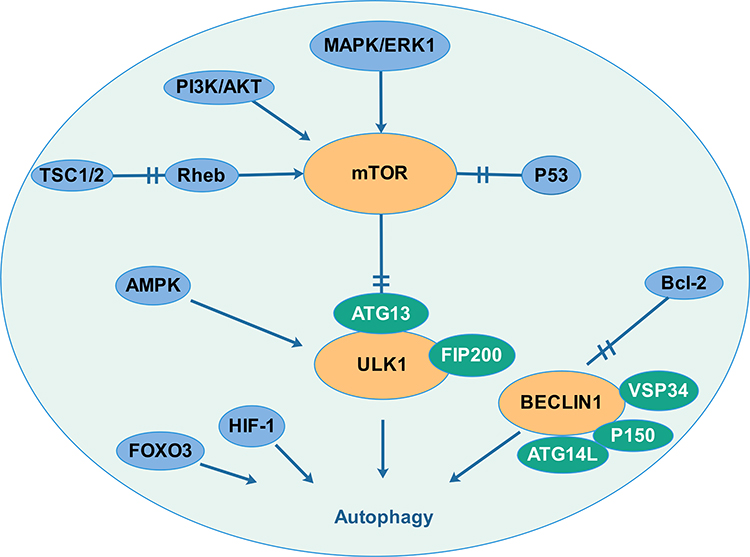 |
Figure 1 Autophagy is mediated by multiple signaling pathways that creating an interaction network system. Several signal molecules (PI3K/AKT pathway, MAPK pathway, TSC1/2 and the p53 tumour suppressor) regulate the mTOR pathway and then regulate autophagy by interacting with the uncoordinated 51-like kinase-1 (ULK1) complex. Autophagy also responds to intracellular energy. The 5′-adenosine monophosphate (AMP)-activated protein kinase (AMPK) which is upregulated by increasing AMP levels inactivates mTORC1 and activates ULK1. Autophagy is also regulated by the Beclin1 complex. Bcl-2, a key regulator of apoptosis, which binds and interacts with beclin1 to inhibits the occurrence of autophagy. Hypoxia-inducible factor (HIF) and FOXO transcription factor also participate in the regulation of autophagy. |
Defects in the autophagy mechanism are associated with many diseases, including neurodegenerative diseases, cancer, cardiovascular diseases, infectious diseases and metabolic diseases.5–7 Cancer cells have higher levels of autophagy, which can provide energy and nutrients to promote survival. Autophagy-defective cancers harbor impaired mitochondrial energy homoeostasis, oxidative stress and inflammatory response.8 Apart from autophagy, it is well known that metabolic abnormality is a hallmark of cancer. During the development of tumors, its metabolic state usually changed, including glucose metabolism, lipid metabolism, biological oxidation and iron metabolism.9 Metabolic changes will cause a series of abnormalities in physiological and biochemical functions.
Among a large number of metabolic pathways, glucose metabolism has been extensively studied. Most cancer cells adopt glycolysis preferentially as their major energy source even in the presence of sufficient oxygen, which is called the “Warburg effect”.10 Glycolysis refers to the process in which glucose is decomposed into pyruvate in the cytoplasm accompanied by a large amount of production of lactate. Glycolysis has a total of ten steps, including three key enzymes (the rate-limiting enzyme): hexokinase (HK), 6-phosphofructokinase (PFK) and pyruvate kinase (PK). Cancer cells undergo glycolysis during cell proliferation11 and migration12 and are immensely involved in radioresistance13 and drug resistance.14 The conversion of metabolism to aerobic glycolysis becomes a feature of cancer, which is related to aggressive clinical behavior.15 Therefore, the inhibition of glycolysis metabolism offers a potential therapeutic target for clinical treatment.
These two biological processes associated with cancer have profound significance of research. Here, we will discuss the regulation of autophagy by glycolysis in cancer cells. Insights from these studies provide the rationale and approach to target the autophagy and glycolysis pathway for cancer therapy.
Autophagy in Cancer
The Mechanism of Autophagy
Autophagy is initiated by a small part of the cytoplasm called phagophore, then it undergoes elongation and completion to form a double-membrane structure, which docked and fused with lysosomes to degrade the cargos.16 Many molecules participate in the autophagic process such as autophagy-related genes (ATGs) and the complex unc-51-like kinase 1 (ULK1) in the initiation phase. The class III PtdIns3K (phosphatidylinositol 3-kinase, best known as VPS34) complex initiates nucleation and participates in the formation of a double membrane structure. In the elongation phase, the ATG5-ATG12-ATG16 complex interacts with light chain 3 (LC3)-II to participate in the extension of the membrane, thereby forming a closed double-layer membrane structure. Autophagosomes, driven by dynein, move along the microtubules toward the lysosomes to form autophagolysosomes, which are involved in the subsequent degradation and recycling of the cargo. More details have already been elaborated in other researches.17
The Role of Autophagy in Cancer
As a crucial component of cellular defense mechanisms, autophagy plays major roles in the degradation of damaged organelles, dysfunctional proteins and the recycling of metabolites to maintain the stability of the internal environment.18 The role of autophagy in cancer has been explored extensively. In cancer biology, autophagy plays dual roles in tumor promotion and suppression. Basal autophagy in organisms is low and almost considered as a tumor-suppressive mechanism during the early stage of tumorigenesis by suppressing reactive oxygen species (ROS), DNA damage, tissue damage, inflammation, and genome instability.19 Therefore, lack of key genes in autophagy can lead to carcinogenesis. For instance, deletion of ATG5 and ATG7 could develop benign liver adenomas from autophagy-deficient hepatocytes due to mitochondrial swelling and oxidative stress.20 Autophagy-related gene BECN1 (encoded for Beclin 1) is important in the formation of the phagophore. Loss of autophagy-related gene Beclin 1 could develop spontaneous tumors and cause an increase in cell proliferation in different types of cancer.21,22 Recent study showed that autophagy as a tumor suppressor was also associated with replicative crisis. Replicative crisis is a final barrier before tumorigenesis, it culminates in mitotic delay, amplified telomere deprotection and extensive cell death. Emerging studies have shown that autophagy was an integral component of the tumor suppressive crisis mechanism and that loss of autophagy could promote continued proliferation and accumulation of genome instability, which was required for the initiation of cancer.23 Autophagy functions as a tumor suppressor also due to the removal of oncogenic proteins involved in oncogenesis. For example, BCR-ABL1 is a critical oncogenic protein responsible for the progression of leukemia, which can be degraded by autophagy.24
Paradoxically, even though autophagy decreases tumor initiation, there is mounting evidence showing that autophagy inhibition could be an effective approach in advanced cancer. Genetic ablation of essential autophagy genes in genetically engineered mouse models (GEMMs) for cancer has revealed an important role of autophagy in promoting tumor to a malignant stage.25,26 This is not hard to understand as autophagy supplies nutrients and energy to cancer cells and helps tumors overcome stress conditions such as hypoxia and nutritional deprivation.22 Autophagy also promotes angiogenesis, metastasis and invasion during tumorigenesis. For instance, autophagy up-regulated vascular endothelial growth factor-A (VEGFA) through JAK2/STAT3 pathway and played a crucial role in angiogenesis in the pathological process of non-small cell lung cancer (NSCLC).27 Recent studies established the autophagy-yes-associated protein (YAP)-metastasis axis in TNBC. When autophagy was triggered, the effector of the Hippo signaling pathway YAP translocated into the nucleus to promote cell invasion and migration of triple-negative breast cancer (TNBC).28 Accumulating evidence supported that autophagy confers drug resistance in established tumors in several cases. Recent studies have demonstrated that autophagy inhibition with chloroquine reverses paclitaxel resistance in human non-small lung adenocarcinoma A549 cells. Of note, autophagy inhibition followed by paclitaxel treatment caused a time-dependent increase in intracellular ROS generation and caspase-dependent apoptosis, further increasing paclitaxel sensitivity.29 As mentioned above, autophagy has a context-dependent and stage-dependence role associated with cancers.22
Glycolysis in Cancer
Glycolysis refers to the process of decomposing glucose or glycogen into pyruvate to produce ATP. This process is carried out in the cytoplasm and consists of a total of 10 consecutive steps, all catalyzed by the corresponding enzymes. The differences in energy supply between normal cells and cancer cells were described by Otto Warburg in the 1920s. Dr Warburg reported that cancer cells have high glycolysis rate and lactate production, even under conditions of sufficient oxygen called the Warburg effect.10 In normal cells, glucose is converted to pyruvate via the glycolytic pathway, accompanied with two ATPs producing. After that, pyruvate enters the tricarboxylic acid cycle with 36 ATPs producing. However, the difference is that pyruvate is catalyzed by lactate dehydrogenase to produce lactate with no ATP production in cancer cells. Cancer cells require more energy than normal cells, so they consume more glucose and then produce more excessive lactate.
Glycolysis is a high-rate but inefficient process in cancer and glycolysis-related genes were overexpressed in more than 70% of cancers.30,31 This faster rate of ATP production is thought to provide an advantage for proliferation and survival in cancer cells. In addition to providing ATP, glycolysis also provides intermediates for biosynthesis of other important molecules such as lipids, nucleotides, and amino acids.32 More and more research has focused on the role of glycolytic enzymes and the metabolic intermediates such as lactate in cancer since the 1920s. Additionally, a higher glycolytic rate in malignant cells gains growth advantages over normal cells in invasion, tumorigenesis and resistance to chemotherapeutics.33 As a result, abnormally high aerobic glycolysis has been recognized as one of the “hallmarks of cancer”.
Glycolysis Regulates Autophagy During Tumor Growth
Cancer cells are apt to use metabolic reprogramming to maintain their growth, survival, proliferation and metastasis. In order to meet the demands of metabolic changes in cancer cells, autophagy is usually changed. The relationship between autophagy and metabolism has been extensively studied in recent years. Glucose is the main nutrient source for energy production in cells. ATP production requires a constant glucose supply. As a key biological process of glucose metabolism, glycolysis is inseparable from autophagy. A mechanism of metabolic changes to drive tumor progression is its ability to regulate autophagy.
Glycolysis regulates autophagy and participates in the survival of cancer cells. Research showed that when glycolysis was suppressed, autophagy was enhanced and became a driving force of oxidative phosphorylation for ATP production to support leukemia cell survival.34 RSL3 inhibited the viabilities of glioma cells with induced glycolysis dysfunction and autophagic cell death. Moreover, supplement of pyruvate prevented RSL3-induced cell death, which indicated that glycolysis dysfunction could induce autophagic cell death in glioma cells.35 Ji Hye Kim et al demonstrated that EGFR mutation-mediated enhancement of glycolysis sustains EGFR stability and is critical to EGFR-mutant NSCLC survival. Glucose metabolism inhibition is able to overcome T790M-mediated resistance because inhibition of glucose-derived ATP production could result in ROS-mediated c-Jun N-terminal kinase (JNK) activation, leading to autophagy-mediated degradation of EGFR.36
Glycolysis regulates autophagy and modulates anti-tumor immunity. Myeloid-derived suppressor cells (MDSCs), a collection of myeloid‐derived heterogenetic cells, endowed with immune suppressive properties via various effector molecules such as arginase1 (Arg1), reactive oxygen species (ROS) and inducible carbon monoxide synthase (INOS).37 High glycolytic rate in TNBC cells prevented the AMP-activated protein kinase (AMPK)-unc-51-like kinase 1 (ULK1) signaling activation and autophagy formation, which reduced autophagy-mediated partial LAP reduction and, in turn, LAP enhanced granulocyte-colony stimulating factor (G-CSF) and granulocyte-macrophage colony stimulating factor (GM-CSF) expression to support MDSC development and maintain tumor immunosuppression.38
Glycolysis regulates autophagy and plays a key role in chemo-resistance in cancer. For instance, hexokinase 2 (HK2) conferred resistance to cisplatin in ovarian cancer cells by increasing ERK1/2 phosphorylation as well as autophagic activity. Blocking autophagy with the autophagy inhibitor 3-MA sensitized resistant ovarian cancer cells to cisplatin.39 As an inhibitor of glycolysis, 2-Deoxy-D-Glucose (2DG) is an anti-tumor drug with limited efficacy in single drug therapy, mainly due to its ability to induce pro-survival autophagy. Propranolol, a widely used non-selective epinephrine receptor blocker, has recently shown anticancer properties. Propranolol +2DG can effectively inhibit the proliferation of prostate cancer cells in vitro, induce cell apoptosis, change mitochondrial morphology, inhibit mitochondrial bioenergy, aggravate endoplasmic reticulum stress, and inhibit tumor growth in vivo.40 Human NSCLC with activating mutations in EGFR was resistant to EGFR-targeted tyrosine kinase inhibitors (TKIs). Increased glucose uptake and high glycolysis rates can be found in resistant cell lines. And drug-resistant cell lines showed activated autophagy during glucose starvation. Combining glucose deprivation and autophagy inhibitor could overcome the resistance against EGFR-targeted therapy for NSCLCs.41 Thus, it can be seen that the combined therapy of targeted glycolysis and autophagy is effective in cancer treatment. Additionally, the molecules involved in regulating glycolysis and autophagy have also been extensively studied in recent years, which are listed in Table 1. Targeting these molecules may provide novel ideas for cancer treatment.
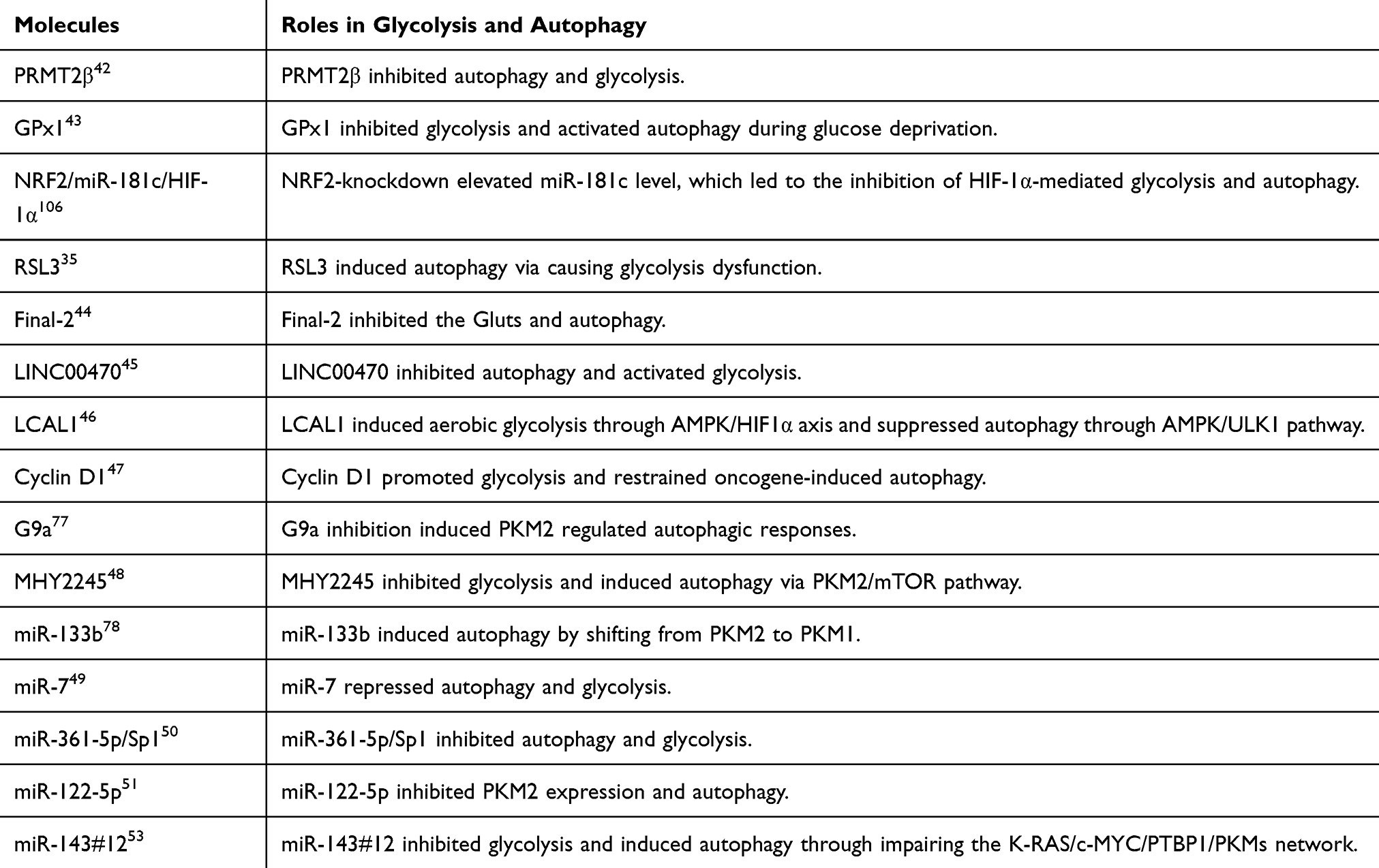 |
Table 1 Molecules Involved in Regulating Glycolysis and Autophagy in Recent Years |
Regulation of Autophagy by Glycolysis
Previous research has shown that in the most glycolytic-dependent cells, autophagy is usually triggered due to adenosine monophosphate (AMP) accumulation (increased AMP/ATP ratio) and activation of AMPK.52 Metabolic shift from glycolysis to OXPHOS induced autophagy through increasing the intracellular level of ROS.53 However, the mechanism by which glycolysis regulates autophagy in cancer cells is not clear enough, and further research is required. We summarized the previous research and found that glycolysis-related enzymes and glycolysis metabolic intermediates are involved in the regulation of autophagy. The mechanism network of glycolysis in regulating autophagy in cancer cells is shown in Figure 2.
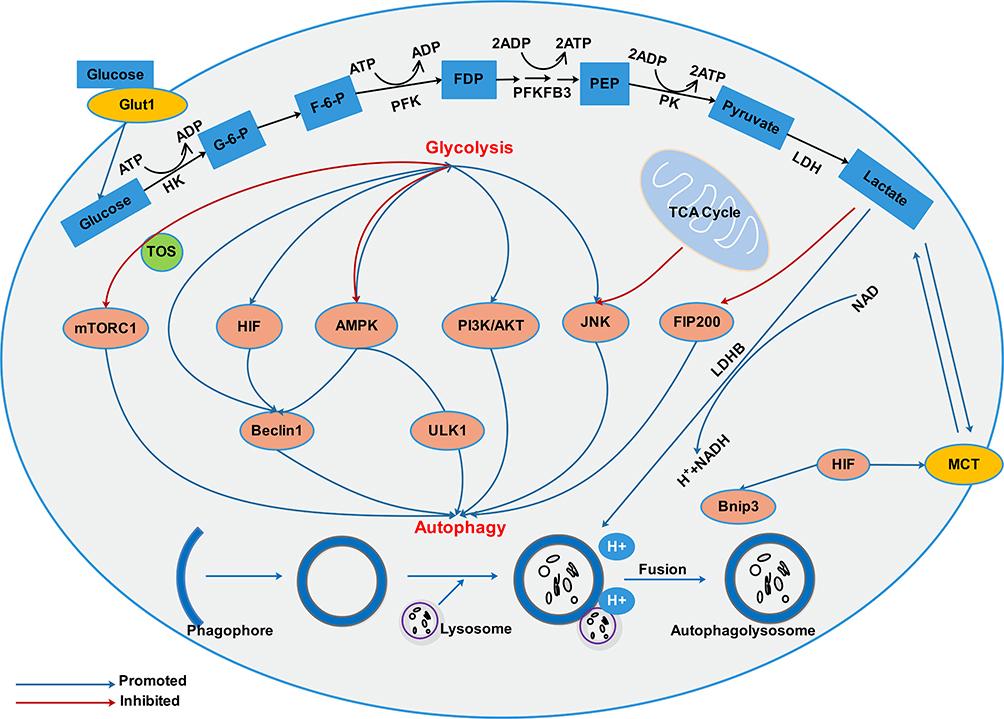 |
Figure 2 There exists an interacting network between autophagy and glycolysis in cancer cells. Glycolysis and autophagy are connected through multiple mechanisms including mTOR, HIF, AMPK, PI3K/AKT, JNK signaling pathways. In addition, protons in acidic environment promotes lysosomal acidification, which is a key step in vesicle maturation and protease activation during autophagy. |
Hexokinase
Hexokinase catalyzes the conversion of glucose to glucose 6-phosphate, which is the first step in the glycolytic pathway. HK has 4 isoenzymes (HK1-4), HK2 is a major regulated subtype, which is overexpressed in various cancers. It not only participates in glycolysis to meet the metabolic energy requirements of cancer cells, but also participates in chemoradiotherapy and apoptosis resistance.54 HK2 selection knockout mice revealed that HK2 was critical for tumorigenesis and maintenance in the KRAS-driven mouse lung cancer model.55
HK2 participation in autophagy regulation. In the lung cancer cell lines KP2 and H23, inhibition of HK2 enzyme activity with 2-DG could inhibit tumor growth, induce cell cycle arrest, and regulate autophagy and apoptotic pathways.56 HK2 regulation of autophagy is also found in tongue squamous cell carcinoma (TSCC) under hypoxia. Deregulation of HK2 resulted in an obvious decrease in autophagic activity and blocked the metastatic potential and epithelial mesenchymal transition (EMT) ability of TSCC.57 HK2 directly promoted autophagy by physically interacting and hence inhibiting mammalian target of rapamycin complex-1 (mTORC1) through the TOS motif during glucose deprivation, while the binding is inhibited by glucose 6-phosphate (G6P) generated by HK2 catalytic activity in the presence of glucose.58,59
Phosphofructo-Kinase/Fructose Biphosphatases (PFKFBs)
The family of PFKFBs control the conversion between fructose-6-phosphate and fructose-2,6-bisphosphate, a key regulator of the glycolytic enzyme phosphofructokinase-1.
Previous studies demonstrated that inhibition of PFKFB3 suppressed autophagy, which showed an important role of PFKFBs in regulating autophagy.60 Subcellular localization of PFKFB3, especially in the nucleus, was essential for its regulatory role in H2O2-induced autophagy through the AMPK signaling pathway.61 And, Inhibition of PFKFB3 in HCT-116 colon adenocarcinoma cells by either siRNA transfection or derivatives of a PFKFB3 inhibitor treatment could trigger autophagy as a pro-survival mechanism.62 PFKFB4 can also positively or negatively regulate autophagy. Strohecker et al demonstrated PFKFB4 as a novel autophagy regulator for that PFKFB4 could suppress autophagy and p62 accumulation by mitigating oxidative stress.63 While another research demonstrated that PFKFB4 could promote autophagy flux. Epithelial and endothelial tyrosine kinase (Etk) was found to be directly interacted with PFKFB4 and was found to be critical in modulating the chemoresistance of small-cell lung cancer (SCLC). Targeting either Etk or PFKFB4 significantly reduced autophagic flux, which may be a potential strategy for drug-resistant SCLC.64
Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH)
GAPDH, a glycolytic enzyme that catalyzes the reversible conversion of glyceraldehyde-3-phosphate to 1.3-bisphosphoglycerate in glycolysis. GAPDH plays a vital role in maintaining aerobic glycolysis and is up-regulated in several cancer types.65
Besides playing the glycolytic role in the cytosol, GAPDH participates in several non-glycolytic functions including autophagy based mainly on its subcellular localization. Under glucose deprivation, cytoplasmic GAPDH phosphorylated at serine 122 by AMPK and was redistributed into the nucleus. In the nucleus, GAPDH interacts directly with sirt1 to stimulate Sirt1 activation and autophagy, mechanistically increasing SIRT1 deacetylase activity and disassociating its inhibitor DBC1.66 Mitochondrial uncoupling protein 2 (UCP2) inhibition triggered ROS/Akt/mTOR axis in pancreatic adenocarcinoma cells. ROS production stimulated the nuclear translocation of GAPDH to promote autophagy.67
Phosphoglycerate Kinase (PGK)
PGK is a rate-limiting enzyme that coordinates energy production during aerobic glycolysis in cancer cells. PGK has two isoforms: PGK1 and PGK2. PGK1 catalyzes the transfer of phosphate from 1, 3-bisphosphoglycerate to ADP, which leads to producing 3-phosphoglycerate and ATP.68
PGK1 plays important dual roles in cell metabolism and autophagy to maintain cell homeostasis. Glutamine deprivation or hypoxia condition resulted in inhibition of mTOR and mTOR-mediated the acetyltransferase NAA10 S228 phosphorylation, then PGK1 was acetylated at K388 by NAA10. This acetylation led to the interaction between PGK1 and BECN1 and the subsequent phosphorylation of BECN1 at S30 by PGK1, which enhanced ATG14-associated PIK3C3/VPS34-BECN1-PIK3R4/VPS15 complex activity to promote autophagy initiation.69 Long non-coding RNA GBCDRlnc1 activated autophagy mainly by binding with PGK1 and inhibiting its ubiquitination, so the overexpression of PGK1 increased expression of ATG5-ATG12 conjugate.70
Pyruvate Kinase
During the glycolysis process, pyruvate kinase is a rate-limiting enzyme catalyzing the conversion of phosphoenolpyruvate (PEP) to pyruvate. PK has two isoforms, PKM and PKL. PKM has two subtypes: PKM1 and PKM2. M1 is mainly distributed in myocardium, skeletal muscle and brain tissue and M2 is mainly distributed in tissues such as the brain and liver. L-form isoenzymes are mainly found in liver, kidney and red blood cells. PK was highly expressed in various malignancies and was widely used in the clinical diagnosis of acute myocardial infarction, myogenic diseases and hematological diseases.30 PKM2 exists in two ways, one is an active tetramer with high affinity for PEP, the other is an inactive dimer with low affinity for PEP. Cancer cells preferred the dimeric form, resulting in inefficient glycolysis and large accumulation of intermediates, synthesized a large number of nucleic acids, amino acids and lipids to promote tumor survival and growth.71 For this reason, as a promising anti-tumor drug, 0089–0022 could be directly used as an activator of PKM2 to effectively induce apoptosis by inhibiting AKT phosphorylation, thereby treating NSCLC.72
Mount of studies provided evidence that PKM was implicated in the autophagic process. Studies demonstrated that PKM2 knockdown could enhance the radiosensitivity of lung cancer cell lines and xenografts via enhancing ionizing radiation (IR)-induced apoptosis and autophagy both in vitro and in vivo.73 The PKM1/2 knockdown in H1299 cells activated AMPK signaling to promote mitochondrial biogenesis and autophagy to maintain energy homeostasis.74 It was found that overexpression of PKM2 activated mTORC1 signaling through phosphorylating mTORC1 inhibitor AKT1 substrate 1 (AKT1S1). Phosphorylation of S202/203 of AKT1S1 by PKM2 dissociated AKT1S1 from raptor and facilitated its binding to 14-3-3, which resulted in activation of mTORC1 signaling independent of growth factors and amino acids signals and led to autophagy inhibition in cancer cells.75 Further experiments demonstrated that PKM2 functioned as a tumor supporter in leukemia to promote cell survival for the reason that PKM2 could trigger autophagy via the phosphorylation of key autophagy protein Beclin-1.76 Histone methyltransferase G9a inhibition induced elevated PKM2 levels regulated LC-3II and YAP-1 expression, which indicated the key role of PKM2-YAP1 crosstalk in autophagic responses in glioma cells.77 Knockdown of polypyrimidine tract-binding protein 1 (PTBP1) in gastric cancer cell lines induced the switching of PKM isoform expression from PKM2 to PKM1. This switching triggered oxidative phosphorylation, resulting in ROS production and leading to autophagic cell death.78
Lactic Dehydrogenase (LDH)
The reduction of pyruvate to lactate by LDH recycles the NAD+ (Nicotinamide adenine dinucleotide) to NADH during glycolysis, allowing glycolysis to persist. LDHA preferentially converts pyruvate to lactate, while LDHB supports conversion of lactate to pyruvate in cells.79 Compared to normal tissues, LDHA expression elevated in many types of spontaneous cancers such as pancreatic cancer, prostate cancer, glioma, and cutaneous melanoma metastases, which was thought to be their metabolic adaptation to anaerobic glycolysis.80 While LDHB level differs among different tumor cells types.81
LDH participation in autophagy regulation. LDHA could modulate autophagy and tamoxifen resistance by interaction with Beclin-1 in MCF-7/TAM-R cells.82 When treating A549 cells with oxamate to inhibit LDHA, the appearance of acidic vesicles was observed in the cytoplasm accompanied by the changed expression of LC-3. These observations indicated that LDHA inhibition could trigger protective autophagy against apoptosis in NSCLC.83 Recent experiments demonstrated that LDHB, but not LDHA, controlled lysosomal activity and basal autophagy of cancer cells. LDHB catalyzed the conversion of lactic acid to pyruvate and protons in acidic environment promoted lysosomal acidification, which was a key step in vesicle maturation and protease activation during autophagy.84 Research showed that SIRT5 induced deacetylated LDHB triggered hyperactivation of autophagy in tumor cells, facilitated lysosomal acidification and autolysosomal maturation, which was a key event in tumorigenesis.85
Lactate
Lactate is mainly produced in tumor microenvironment (TME) and serves as an energy-rich substrate, signaling molecule and important immunosuppressive factor for tumors.86 Glycolysis is much less efficient than oxidative phosphorylation to produce ATP. For this reason, cancer cells prefer to increase their glucose uptake and glycolytic rate, and then more lactate is released. Excreting lactate maintains pH homeostasis inside the cell and acidifies the extracellular space. Evidence indicates that lactate and/or protons leading to pH 6–6.5 in tumor microenvironment, a characteristic phenotype of more aggressive tumors, modulate cell proliferation and promote angiogenesis, invasion,87,88 metastasis89,90 and poor prognosis.91–93
Emerging studies showed the evidence that an acidic tumor microenvironment was common in cancers, partly due to the production of lactate by the Warburg effect. It has been found that acute low pH-stimulated autophagy functioned as a protective role in lung cancer cells, with increasing LC3 II and reducing p62 protein expression levels.94 Lactic acid-induced extracellular acidification suppressed glucose deprivation-induced autophagy in B16 melanoma cells.95 Gefitinib-induced autophagy is related to cancer cell survival and chemoresistance. GPR119 agonists enhanced glycolysis accompanied by reduced mitochondrial oxidative phosphorylation, which inhibited gefitinib-induced autophagosome formation due to increased intracellular and extracellular lactate in MCF-7 and MDA-MB-231 cells.96 FIP200 is required for autophagosome formation.97 Tumor-derived lactate suppressed FIP200 and LC-3II expression and increased apoptosis in human naïve T cells. Mechanisms involved are that tumor-derived lactate translationally inhibited FIP200 expression by downregulating nicotinamide adenine dinucleotide (NAD) levels and offering more dehydrogenases binding within the adenylate-uridylate–rich elements (AREs) of Fip200 mRNA.98 And it is worth mentioning that lactate acidosis promotes the survival and proliferation of Lewis lung carcinoma cells and inhibits apoptosis and autophagy through energy system reprogramming.99
MCT
Lactate itself cannot pass through the plasma membrane by free diffusion. Hence, it requires a specific transport mechanism provided by monocarboxylate transporters (eg MCT1, 4). MCT1 and MCT4 play important roles in metabolic reprogramming and have been demonstrated to be overexpressed in the majority of malignancies.100
Studies demonstrated that osimertinib (OSI) with potent anti-CRC effects upregulated the expression of MCT1 and induced autophagy flux via LKB1/AMPK signaling in CRC cells.101 MCT4 depletion enhanced the cytotoxicity of NK cells by blocking lactate flux and reversing the acidified tumor microenvironment, of note, the MCT4 inhibition with a pronounced impact on autophagy induction.101,102
Hypoxia-Inducible Factor-1 (HIF-1)
HIF-1 accumulates during hypoxia and directly up-regulates the expression of many molecules involved in glycolysis, including glucose transporter-1 (GLUT1), HK2, LDHA and so forth. HIF-1 promotes autophagy by enhancing transcription of adenovirus (E1B)-19KD-interacting protein 3 (BNIP3)/BNIP3L. BNIP3 can compete with lymphoma 2 (Bcl-2) to bind to beclin1, which leads to dissociation of the beclin1-BCL complex and triggers autophagy.103 Furthermore, hypoxia-induced upregulation of HIF-1 could trigger autophagy by phosphorylation of downstream C-JUN to upregulate Beclin 1 mRNA and protein expression.104 The mechanisms by which HIF-1 regulates glycolysis and autophagy have been extensively studied, respectively. However, whether HIF-1 acts as a “bridge” or “trigger” role in these two biological processes requires further study.
It is worth noting that glucose deprivation could trigger HIF-1 α -independent autophagy by the induction of AMPK and inhibition of mTOR, which indicated the relationship between glucose metabolism and autophagy.105 The latest research showed that the nuclear factor erythroid 2-like-2 (NFE2L2; NRF2)-silencing elevated the level of miR-181c, which blocked HIF-1α accumulation in hypoxic breast cancer cells. HIF-1α dysregulation blocked hypoxia-induced autophagy, glycolysis and pentose phosphate pathway (PPP) activation.106 HIF-1α knockdown in A549 cells (human alveolar type II epithelial cell line) reduced glycolysis by regulating transcription of glycolysis-related enzymes, which subsequently activated the AMPKα-ULK1 signalling pathway to promote autophagy.107
Discussion
Autophagy and metabolic reprogramming are essential for maintaining cellular and organismal homeostasis. Recently, the role of energy metabolism and autophagy in cancer has become increasingly prominent. This review attempts to elaborate the role and the mechanism of glycolysis in regulating autophagy during tumor growth.
However, it is worth mentioning that autophagy also plays an important role in regulating glycolysis. Autophagy could be triggered by nutrient limitation and the accumulation of specific metabolites or metabolic byproducts.108 Given the research that autophagy could act on a variety of substrate molecules to fuel nearly all aspects of central carbon metabolism,109 autophagy can degrade carbohydrates into glucose, DNA into nucleosides to fuel glycolysis.9 The regulation of glycolysis by autophagy has also been extensively studied in cancer cells. Emerging studies have shown that autophagy induced MCT1 expression and promoted glycolysis via activating Wnt/β-catenin signaling in hepatocellular carcinoma (HCC) cells.110 During metabolic stress, autophagy-dependent shuttling of TBC1D5 controlled plasma membrane translocation of GLUT1 and facilitates glucose uptake and glycolytic flux.111 In addition, the autophagic degradation of HK2 through the TRAF6- and SQSTM1-mediated ubiquitination system demonstrated the regulation of glycolysis by autophagy.112
To sum up, it is clear that autophagy and glycolysis can be mutually regulated. We attempted to elucidate the role of metabolism, especially glycolysis, in the regulation of autophagy. We found that enzymes and molecules involved in glycolysis and metabolic intermediates such as lactate can regulate autophagy. Moreover, glycolysis regulates autophagy and plays an important role in tumor survival, anti-tumor immunity and drug resistance. This suggests that autophagy may act as a bridge between metabolism and cancer, which provides a new mechanistic viewpoint for future therapeutic approaches. Although the regulation of autophagy by glycolysis in cancer has been widely reported, the exact molecular mechanism is still highly controversial and needs further exploration. We had a series of questions when we collated existing research results. For example, whether the molecules in the glycolysis process could be directly combined with the molecules in the autophagy process? As a metabolite of glycolysis, lactate has received extensive attention in recent years. We know that the acidic environment caused by lactate contributed to the formation of autophagy, and lactate could also inhibit autophagy-related molecules to regulate autophagy. But so far, there are few studies on the regulation of autophagy by lactate, which deserves further investigation. In fact, when we discussed the role of regulation of autophagy by glycolysis during cancer development, we questioned whether the physiological and pathological significance of glycolysis regulating autophagy is consistent in different types of cancer, and even whether the molecular mechanism of glycolysis regulating autophagy is different due to different cancer types. All the questions need to be answered by further research.
Abbreviations
ATP, adenosine triphosphate; ADP, adenosine diphosphate; AKT1S1, AKT1 substrate 1; AMP, adenosine monophosphate; AMPK, AMP-activated proteinkinase; AREs, adenylate-uridylate–rich elements; Arg1, arginase1; ROS, reactive oxygen species; INOS, inducible carbon monoxide synthase; ATG, autophagy related gene; BCL-2, B-cell lymphoma 2; BECN1, beclin1; EGFR, epidermal growth factor receptor; EMT, epithelial mesenchymal transition; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; G-CSF, granulocyte-colony stimulating factor; GEMMs, genetically engineered mouse models; GLUT, glucose transporter; GM-CSF, granulocyte-macrophage colony stimulating factor; GPx1, glutathione peroxidase-1; G6P, glucose 6-phosphate; H +, hydrogen ion; HCC, hepatocellular carcinoma; HIF-1, hypoxia inducible factor-1; HK, hexokinase; INOS, inducible carbon monoxide synthase; IR, ionizing radiation; JAK2, Janus activating kinase 2; LCAL1, lung cancer-associated lncRNA 1; LC3, microtubule-associated proteins light chain 3; LDH, lactic dehydrogenase; LINC00470, long intergenic non-protein coding RNA 470; LUAD, lung adenocarcinoma; MAPK, mitogen-activated protein kinase; MCT, monocarboxylate transporter; MDSCs, myeloid-derived suppressor cells; mTOR, mammalian target of rapamycin; NAD+, nicotinamide adenine dinucleotide (oxidized form); NADH, nicotinamide adenine dinucleotide (reduced form); NFE2L2/NRF2, nuclear factor erythroid 2-like-2; NSCLC, non-small cell lung cancer; OSI, osimertinib; OXPHOS, oxidative phosphorylation; PEP, phosphoenolpyruvate; PFKFBs, 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatases; PGK, phosphoglycerate kinase; PI3K, phosphoinositide-3-kinase; PK, pyruvate kinase; PPP, pentose phosphate pathway; PRMT2, protein arginine N-methyltransferase 2; PTBP1, polypyrimidine tract-binding protein 1; SCLC, small-cell lung cancer; STAT3, signal transducer and activator of transcription 3; TCA, tricarboxylic acid cycle; TIGAR, Tp53 induced glycolysis and apoptosis regulator; TKIs, tyrosine kinase inhibitors; TME, tumor microenvironment; TSCC, tongue squamous cell carcinoma; UCP2, uncoupling protein 2; ULK1, unc-51-like kinase 1; VEGFA, vascular endothelial growth factor-A.
Funding
Zhenjiang Key R&D Program – Social Development, number: SH2020026. Natural Science Foundation of Jiangsu Province, number: BK20191429.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Schneider JL, Cuervo AM. Autophagy and human disease: emerging themes. Curr Opin Genet Dev. 2014;26(26):16–23. doi:10.1016/j.gde.2014.04.003
2. Beth L, Guido K. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42. doi:10.1016/j.cell.2007.12.018
3. Chiara M M, Einat Z, Adi K, Guido K. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8(9):741–752. doi:10.1038/nrm2239
4. Galluzzi L. Autophagy in malignant transformation and cancer progression. EMBO J. 2015;34(7):856–880. doi:10.15252/embj.201490784
5. Onorati AV, Dyczynski M, Ojha R, Amaravadi RK. Targeting autophagy in cancer. Cancer. 2018;124(16):3307–3318. doi:10.1002/cncr.31335
6. Leidal AM, Levine B, Debnath J. Autophagy and the cell biology of age-related disease. Nat Cell Biol. 2018;20(12):1338–1348. doi:10.1038/s41556-018-0235-8
7. Guo F, Liu X, Cai H, Le W. Autophagy in neurodegenerative diseases: pathogenesis and therapy. Brain Pathol. 2018;28(1):3–13. doi:10.1111/bpa.12545
8. Ryter SW, Choi AMK. Autophagy in lung disease pathogenesis and therapeutics. Redox Biol. 2015;4:215–225. doi:10.1016/j.redox.2014.12.010
9. Kimmelman AC, White E. Autophagy and tumor metabolism. Cell Metab. 2017;25(5):1037–1043. doi:10.1016/j.cmet.2017.04.004
10. Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–314. doi:10.1126/science.123.3191.309
11. Fu H, Gao H, Qi X, et al. Aldolase A promotes proliferation and G1/S transition via the EGFR/MAPK pathway in non-small cell lung cancer. Cancer Communications. 2018;38(1):18. doi:10.1186/s40880-018-0290-3
12. Liu T, Yin H. PDK1 promotes tumor cell proliferation and migration by enhancing the Warburg effect in non-small cell lung cancer. Oncol Rep. 2017;37(1):193–200. doi:10.3892/or.2016.5253
13. Li L, Liu H, Du L, et al. miR-449a suppresses LDHA-mediated glycolysis to enhance the sensitivity of non-small cell lung cancer cells to ionizing radiation. Oncol Res. 2018;26(4):547–556. doi:10.3727/096504017X15016337254605
14. Gong T, Cui L, Wang H, Wang H, Han N. Knockdown of KLF5 suppresses hypoxia-induced resistance to cisplatin in NSCLC cells by regulating HIF-1alpha-dependent glycolysis through inactivation of the PI3K/Akt/mTOR pathway. J Transl Med. 2018;16(1):164. doi:10.1186/s12967-018-1543-2
15. Giatromanolaki A, Sivridis E, Arelaki S, Koukourakis MI. Expression of enzymes related to glucose metabolism in non-small cell lung cancer and prognosis. Exp Lung Res. 2017;43(4–5):167–174. doi:10.1080/01902148.2017.1328714
16. Maho H, Nobumichi F, Atsushi M, et al. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013;495(7441):389–393. doi:10.1038/nature11910
17. Galluzzi L, Baehrecke EH, Ballabio A, et al. Molecular definitions of autophagy and related processes. EMBO J. 2017;36(13):1811–1836. doi:10.15252/embj.201796697
18. Eileen W. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12(6):401–410. doi:10.1038/nrc3262
19. Poillet-Perez L, White E. Role of tumor and host autophagy in cancer metabolism. Genes Dev. 2019;33(11–12):610–619. doi:10.1101/gad.325514.119
20. Takamura A, Komatsu M, Hara T, et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25(8):795–800. doi:10.1101/gad.2016211
21. Qu X, Yu J, Bhagat G, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112(12):1809–1820.
22. Kocaturk NM, Akkoc Y, Kig C, Bayraktar O, Gozuacik D, Kutlu O. Autophagy as a molecular target for cancer treatment. Eur J Pharm Sci. 2019;134:116–137. doi:10.1016/j.ejps.2019.04.011
23. Joe N, Robert R, Adriana C, et al. Autophagic cell death restricts chromosomal instability during replicative crisis. Nature. 2019;565:7741.
24. Goussetis DJ, Gounaris E, Wu EJ, et al. Autophagic degradation of the BCR-ABL oncoprotein and generation of antileukemic responses by arsenic trioxide. Blood. 2012;120(17):3555–3562.
25. Guo JY, Teng X, Laddha SV, et al. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 2016;30(15):1704–1717. doi:10.1101/gad.283416.116
26. Yang A, Kimmelman AC. Inhibition of autophagy attenuates pancreatic cancer growth independent of TP53/TRP53 status. Autophagy. 2014;10(9):1683–1684. doi:10.4161/auto.29961
27. Liang L, Hui K, Hu C, et al. Autophagy inhibition potentiates the anti-angiogenic property of multikinase inhibitor anlotinib through JAK2/STAT3/VEGFA signaling in non-small cell lung cancer cells. J Exp Clin Cancer Res. 2019;38(1):71. doi:10.1186/s13046-019-1093-3
28. Chen W, Bai Y, Patel C, Geng F. Autophagy promotes triple negative breast cancer metastasis via YAP nuclear localization. Biochem Biophys Res Commun. 2019;520(2):263–268. doi:10.1016/j.bbrc.2019.09.133
29. Datta S, Choudhury D, Das A, et al. Autophagy inhibition with chloroquine reverts paclitaxel resistance and attenuates metastatic potential in human nonsmall lung adenocarcinoma A549 cells via ROS mediated modulation of β-catenin pathway. Apoptosis. 2019;24(5–6):414–433. doi:10.1007/s10495-019-01526-y
30. Altenberg B, Greulich KO. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics. 2004;84(6):1014–1020. doi:10.1016/j.ygeno.2004.08.010
31. Martins SF, Amorim R, Viana-Pereira M, et al. Significance of glycolytic metabolism-related protein expression in colorectal cancer, lymph node and hepatic metastasis. BMC Cancer. 2016;16:535.
32. Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–464. doi:10.1146/annurev-cellbio-092910-154237
33. Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4(11):891–899. doi:10.1038/nrc1478
34. Kawaguchi M, Aoki S, Hirao T, Morita M, Ito K. Autophagy is an important metabolic pathway to determine leukemia cell survival following suppression of the glycolytic pathway. Biochem Biophys Res Commun. 2016;474(1):188–192. doi:10.1016/j.bbrc.2016.04.098
35. Wang X, Lu S, He C, et al. RSL3 induced autophagic death in glioma cells via causing glycolysis dysfunction. Biochem Biophys Res Commun. 2019;518(3):590–597. doi:10.1016/j.bbrc.2019.08.096
36. Kim JH, Nam B, Choi YJ, et al. Enhanced glycolysis supports cell survival in EGFR-mutant lung adenocarcinoma by inhibiting autophagy-mediated EGFR degradation. Cancer Res. 2018;78(16):4482–4496. doi:10.1158/0008-5472.CAN-18-0117
37. Mantovani A. The growing diversity and spectrum of action of myeloid-derived suppressor cells. Eur J Immunol. 2010;40(12):3317–3320. doi:10.1002/eji.201041170
38. Li W, Tanikawa T, Kryczek I, et al. Aerobic glycolysis controls myeloid-derived suppressor cells and tumor immunity via a specific cebpb isoform in triple-negative breast cancer. Cell Metab. 2018;28(1):87–103.e106. doi:10.1016/j.cmet.2018.04.022
39. Zhang XY, Zhang M, Cong Q, et al. Hexokinase 2 confers resistance to cisplatin in ovarian cancer cells by enhancing cisplatin-induced autophagy. Int J Biochem Cell Biol. 2018;95:9–16. doi:10.1016/j.biocel.2017.12.010
40. Brohée L, Peulen O, Nusgens B, et al. Propranolol sensitizes prostate cancer cells to glucose metabolism inhibition and prevents cancer progression. Sci Rep. 2018;8(1):7050. doi:10.1038/s41598-018-25340-9
41. Ye M, Wang S, Wan T, et al. Combined inhibitions of glycolysis and akt/autophagy can overcome resistance to EGFR-targeted therapy of lung cancer. J Cancer. 2017;8(18):3774–3784. doi:10.7150/jca.21035
42. Chen Y, Dai X, Yao Y, et al. PRMT2β suppresses autophagy and glycolysis pathway in human breast cancer MCF-7 cell lines. Acta Biochim Biophys Sin. 2019;51(3):335–337. doi:10.1093/abbs/gmz006
43. Meng Q, Xu J, Liang C, et al. GPx1 is involved in the induction of protective autophagy in pancreatic cancer cells in response to glucose deprivation. Cell Death Dis. 2018;9(12):1187. doi:10.1038/s41419-018-1244-z
44. Chen Z, Yang D, Jiang X, et al. Final-2 targeted glycolysis mediated apoptosis and autophagy in human lung adenocarcinoma cells but failed to inhibit xenograft in nude mice. Food Chemical Toxicol. 2019;130:1–11. doi:10.1016/j.fct.2019.04.054
45. Liu C, Zhang Y, She X, et al. A cytoplasmic long noncoding RNA LINC00470 as a new AKT activator to mediate glioblastoma cell autophagy. J Hematol Oncol. 2018;11(1):77. doi:10.1186/s13045-018-0619-z
46. Li JY, Luo ZQ. LCAL1 enhances lung cancer survival via inhibiting AMPK-related antitumor functions. Mol Cell Biochem. 2019;457(1–2):11–20. doi:10.1007/s11010-019-03507-w
47. Casimiro MC, Di Sante G, Di Rocco A, et al. Cyclin D1 restrains oncogene-induced autophagy by regulating the AMPK-LKB1 signaling axis. Cancer Res. 2017;77(13):3391–3405. doi:10.1158/0008-5472.CAN-16-0425
48. Tae IH, Son JY, Lee SH, et al. A new SIRT1 inhibitor, MHY2245, induces autophagy and inhibits energy metabolism via PKM2/mTOR pathway in human ovarian cancer cells. Int J Biol Sci. 2020;16(11):1901–1916. doi:10.7150/ijbs.44343
49. Ye ZQ, Zou CL, Chen HB, Jiang MJ, Mei Z, Gu DN. MicroRNA-7 as a potential biomarker for prognosis in pancreatic cancer. Dis Markers. 2020;2020:2782101. doi:10.1155/2020/2782101
50. Ling Z, Liu D, Zhang G, et al. miR-361-5p modulates metabolism and autophagy via the Sp1-mediated regulation of PKM2 in prostate cancer. Oncol Rep. 2017;38(3):1621–1628. doi:10.3892/or.2017.5852
51. Wang S, Zheng W, Ji A, Zhang D, Zhou M. Overexpressed miR-122-5p promotes cell viability, proliferation, migration and glycolysis of renal cancer by negatively regulating PKM2. Cancer Manag Res. 2019;11:9701–9713. doi:10.2147/CMAR.S225742
52. Grahame H D, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13(4):251–262. doi:10.1038/nrm3311
53. Takai T, Tsujino T, Yoshikawa Y, et al. Synthetic miR-143 exhibited an anti-cancer effect via the downregulation of K-RAS networks of renal cell cancer cells in vitro and in vivo. Mol Therapy. 2019;27(5):1017–1027. doi:10.1016/j.ymthe.2019.03.004
54. Mathupala SP, Ko YH, Pedersen PL. Hexokinase II: cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene. 2006;25(34):4777–4786. doi:10.1038/sj.onc.1209603
55. Patra K, Wang Q, Bhaskar P, et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell. 2013;24(2):213–228. doi:10.1016/j.ccr.2013.06.014
56. Wang H, Wang L, Zhang Y, Wang J, Deng Y, Lin D. Inhibition of glycolytic enzyme hexokinase II (HK2) suppresses lung tumor growth. Cancer Cell Int. 2016;16:9. doi:10.1186/s12935-016-0280-y
57. Chen G, Zhang Y, Liang J, et al. Deregulation of hexokinase ii is associated with glycolysis, autophagy, and the epithelial-mesenchymal transition in tongue squamous cell carcinoma under hypoxia. Biomed Res Int. 2018;2018:8480762.
58. Roberts D, Tan-Sah V, Ding E, Smith J, Miyamoto S. Hexokinase-II positively regulates glucose starvation-induced autophagy through TORC1 inhibition. Mol Cell. 2014;53(4):521–533. doi:10.1016/j.molcel.2013.12.019
59. Tan VP, Miyamoto S. HK2/hexokinase-II integrates glycolysis and autophagy to confer cellular protection. Autophagy. 2015;11(6):963–964. doi:10.1080/15548627.2015.1042195
60. Lu Q, Yan S, Sun H, et al. Akt inhibition attenuates rasfonin-induced autophagy and apoptosis through the glycolytic pathway in renal cancer cells. Cell Death Dis. 2015;6(12):e2005. doi:10.1038/cddis.2015.344
61. Yan S, Wei X, Xu S, et al. 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase isoform 3 spatially mediates autophagy through the AMPK signaling pathway. Oncotarget. 2017;8(46):80909–80922. doi:10.18632/oncotarget.20757
62. Klarer AC, O’Neal J, Imbert-Fernandez Y, et al. Inhibition of 6-phosphofructo-2-kinase (PFKFB3) induces autophagy as a survival mechanism. Cancer Metab. 2014;2(1):2. doi:10.1186/2049-3002-2-2
63. Strohecker AM, Joshi S, Possemato R, Abraham RT, Sabatini DM, White E. Identification of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase as a novel autophagy regulator by high content shRNA screening. Oncogene. 2015;34(45):5662–5676. doi:10.1038/onc.2015.23
64. Wang Q, Zeng F, Sun Y, et al. Etk Interaction with PFKFB4 modulates chemoresistance of small-cell lung cancer by regulating autophagy. Clin Cancer Res. 2018;24(4):950–962. doi:10.1158/1078-0432.CCR-17-1475
65. Ripple MO, Wilding G. Alteration of glyceraldehyde-3-phosphate dehydrogenase activity and messenger RNA content by androgen in human prostate carcinoma cells. Cancer Res. 1995;55(19):4234–4236.
66. Chang C, Su H, Zhang D, et al. AMPK-dependent phosphorylation of GAPDH triggers sirt1 activation and is necessary for autophagy upon glucose starvation. Mol Cell. 2015;60(6):930–940. doi:10.1016/j.molcel.2015.10.037
67. Dando I, Pacchiana R, Pozza ED, et al. UCP2 inhibition induces ROS/Akt/mTOR axis: role of GAPDH nuclear translocation in genipin/everolimus anticancer synergism. Free Radic Biol Med. 2017;113:176–189. doi:10.1016/j.freeradbiomed.2017.09.022
68. Wang S, Jiang B, Zhang T, et al. Insulin and mTOR pathway regulate HDAC3-mediated deacetylation and activation of PGK1. PLoS Biol. 2015;13(9):e1002243. doi:10.1371/journal.pbio.1002243
69. Qian X, Li X, Lu Z. Protein kinase activity of the glycolytic enzyme PGK1 regulates autophagy to promote tumorigenesis. Autophagy. 2017;13(7):1246–1247. doi:10.1080/15548627.2017.1313945
70. Cai Q, Wang S, Jin L, et al. Long non-coding RNA GBCDRlnc1 induces chemoresistance of gallbladder cancer cells by activating autophagy. Mol Cancer. 2019;18(1):82. doi:10.1186/s12943-019-1016-0
71. Christofk HR, Heiden MGV, Harris MH, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452(7184):230–233. doi:10.1038/nature06734
72. Li R-Z, Fan -X-X, Shi D-F, et al. Identification of a new pyruvate kinase M2 isoform (PKM2) activator for the treatment of non-small-cell lung cancer (NSCLC). Chem Biol Drug Des. 2018;92(5):1851–1858. doi:10.1111/cbdd.13354
73. Meng M-B, Wang -H-H, Guo W-H, et al. Targeting pyruvate kinase M2 contributes to radiosensitivity of non-small cell lung cancer cells in vitro and in vivo. Cancer Lett. 2015;356(2Pt B):985–993. doi:10.1016/j.canlet.2014.11.016
74. Prakasam G, Singh RK, Iqbal MA, Saini SK, Tiku AB, Bamezai RNK. Pyruvate kinase M knockdown-induced signaling via AMP-activated protein kinase promotes mitochondrial biogenesis, autophagy, and cancer cell survival. J Biol Chem. 2017;292(37):15561–15576.
75. He CL, Bian YY, Xue Y, et al. Pyruvate Kinase M2 activates mTORC1 by phosphorylating AKT1S1. Sci Rep. 2016;6:21524.
76. Wang L, Yang L, Yang Z, et al. Glycolytic Enzyme PKM2 mediates autophagic activation to promote cell survival in NPM1-mutated leukemia. Int J Biol Sci. 2019;15(4):882–894. doi:10.7150/ijbs.30290
77. Ahmad F, Dixit D, Joshi SD, Sen E. G9a inhibition induced PKM2 regulates autophagic responses. Int J Biochem Cell Biol. 2016;78:87–95. doi:10.1016/j.biocel.2016.07.009
78. Sugiyama T, Taniguchi K, Matsuhashi N, et al. MiR-133b inhibits growth of human gastric cancer cells by silencing pyruvate kinase muscle-splicer polypyrimidine tract-binding protein 1. Cancer Sci. 2016;107(12):1767–1775. doi:10.1111/cas.13091
79. Dawson DM, Goodfriend TL, Kaplan NO. Lactic dehydrogenases: functions of the two types rates of synthesis of the two major forms can be correlated with metabolic differentiation. Science. 1964;143(3609):929–933. doi:10.1126/science.143.3609.929
80. Urbańska K, Orzechowski A. Unappreciated role of LDHA and LDHB to control apoptosis and autophagy in tumor cells. Int J Mol Sci. 2019;20:9.
81. Sun W, Zhang X, Ding X, et al. Lactate dehydrogenase B is associated with the response to neoadjuvant chemotherapy in oral squamous cell carcinoma. PLoS One. 2015;10(5):e0125976. doi:10.1371/journal.pone.0125976
82. Das CK, Parekh A, Parida PK, Bhutia SK, Mandal M. Lactate dehydrogenase A regulates autophagy and tamoxifen resistance in breast cancer. Biochimica Et Biophysica Acta Mol Cell Res. 2019;1866(6):1004–1018.
83. Yang Y, Su D, Zhao L, et al. Different effects of LDH-A inhibition by oxamate in non-small cell lung cancer cells. Oncotarget. 2014;5(23):11886–11896. doi:10.18632/oncotarget.2620
84. Brisson L, Bański P, Sboarina M, et al. Lactate dehydrogenase b controls lysosome activity and autophagy in cancer. Cancer Cell. 2016;30(3):418–431. doi:10.1016/j.ccell.2016.08.005
85. Shi L, Yan H, An S, et al. SIRT5-mediated deacetylation of LDHB promotes autophagy and tumorigenesis in colorectal cancer. Mol Oncol. 2019;13(2):358–375. doi:10.1002/1878-0261.12408
86. Ippolito L, Morandi A, Giannoni E, Chiarugi P. Lactate: A metabolic driver in the tumour landscape. Trends Biochem Sci. 2019;44(2):153–166. doi:10.1016/j.tibs.2018.10.011
87. An J, Zhang Y, He J, et al. Lactate dehydrogenase A promotes the invasion and proliferation of pituitary adenoma. Sci Rep. 2017;7(1):4734. doi:10.1038/s41598-017-04366-5
88. Martﺃﺝnez-Zaguilﺃ-n R, Seftor EA, Seftor RE, Chu YW, Gillies RJ, Hendrix MJ. Acidic pH enhances the invasive behavior of human melanoma cells. Clin Exp Metastasis. 1996;14(2):176–186. doi:10.1007/BF00121214
89. Rizwan A, Serganova I, Khanin R, et al. Relationships between LDH-A, lactate, and metastases in 4T1 breast tumors. Clin Cancer Res. 2013;19(18):5158–5169. doi:10.1158/1078-0432.CCR-12-3300
90. Chen P, Zuo H, Xiong H, et al. Gpr132 sensing of lactate mediates tumor-macrophage interplay to promote breast cancer metastasis. Proc Natl Acad Sci U S A. 2017;114(3):580–585. doi:10.1073/pnas.1614035114
91. Liu R, Cao J, Gao X, et al. Overall survival of cancer patients with serum lactate dehydrogenase greater than 1000 IU/L. Tumour Biol. 2016;37(10):14083–14088. doi:10.1007/s13277-016-5228-2
92. Li G, Wang Z, Xu J, Wu H, Cai S, He Y. The prognostic value of lactate dehydrogenase levels in colorectal cancer: a meta-analysis. BMC Cancer. 2016;16:249. doi:10.1186/s12885-016-2276-3
93. García-Cañaveras JC, Chen L, Rabinowitz JD. The tumor metabolic microenvironment: lessons from lactate. Cancer Res. 2019;79(13):3155–3162. doi:10.1158/0008-5472.CAN-18-3726
94. Xie WY, Zhou XD, Li Q, Chen LX, Ran DH. Acid-induced autophagy protects human lung cancer cells from apoptosis by activating ER stress. Exp Cell Res. 2015;339(2):270–279. doi:10.1016/j.yexcr.2015.11.005
95. Matsuo T, Sadzuka Y. Extracellular acidification by lactic acid suppresses glucose deprivation-induced cell death and autophagy in B16 melanoma cells. Biochem Biophys Res Commun. 2018;496(4):1357–1361. doi:10.1016/j.bbrc.2018.02.022
96. Im JH, Kang KW, Kim SY, et al. GPR119 agonist enhances gefitinib responsiveness through lactate-mediated inhibition of autophagy. J Exp Clin Cancer Res. 2018;37(1):295. doi:10.1186/s13046-018-0949-2
97. Hara T, Takamura A, Kishi C, et al. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol. 2008;181(3):497–510.
98. Xia H, Wang W, Crespo J, et al. Suppression of FIP200 and autophagy by tumor-derived lactate promotes naïve T cell apoptosis and affects tumor immunity. Sci Immunol. 2017;2:17. doi:10.1126/sciimmunol.aan4631
99. Kolesnik DL, Pyaskovskaya ON, Solyanik GI. Impact of lactic acidosis on the survival of Lewis lung carcinoma cells. Exp Oncol. 2017;39(2):112–116. doi:10.31768/2312-8852.2017.39(2):112-116
100. Payen VL, Mina E, Van Hée VF, Porporato PE, Sonveaux P. Monocarboxylate transporters in cancer. Molecular Metabolism. 2020;33:48–66. doi:10.1016/j.molmet.2019.07.006
101. Jin P, Jiang J, Xie N, et al. MCT1 relieves osimertinib-induced CRC suppression by promoting autophagy through the LKB1/AMPK signaling. Cell Death Dis. 2019;10(8):615. doi:10.1038/s41419-019-1844-2
102. Long Y, Gao Z, Hu X, et al. Downregulation of MCT4 for lactate exchange promotes the cytotoxicity of NK cells in breast carcinoma. Cancer Med. 2018;7(9):4690–4700. doi:10.1002/cam4.1713
103. Ray R, Chen G, Vande Velde C, et al. BNIP3 heterodimerizes with Bcl-2/Bcl-X (L) and induces cell death independent of a Bcl-2 homology 3 (BH3) domain at both mitochondrial and nonmitochondrial sites. J Biol Chem. 2000;275(2):1439–1448. doi:10.1074/jbc.275.2.1439
104. Zou YM, Hu GY, Zhao XQ, et al. Hypoxia-induced autophagy contributes to radioresistance via c-Jun-mediated Beclin1 expression in lung cancer cells. J Huazhong Univ Sci Technol Med Sci. 2014;34(5):761–767. doi:10.1007/s11596-014-1349-2
105. Mazure NM, Pouysségur J. Hypoxia-induced autophagy: cell death or cell survival? Curr Opin Cell Biol. 2010;22(2):177–180. doi:10.1016/j.ceb.2009.11.015
106. Lee S, Hallis SP, Jung KA, Ryu D, Kwak MK. Impairment of HIF-1α-mediated metabolic adaption by NRF2-silencing in breast cancer cells. Redox Biol. 2019;24:101210. doi:10.1016/j.redox.2019.101210
107. Zhao C, Chen J, Cheng L, Xu K, Yang Y, Su X. Deficiency of HIF-1α enhances influenza A virus replication by promoting autophagy in alveolar type II epithelial cells. Emerging Microbes Infections. 2020;9(1):691–706. doi:10.1080/22221751.2020.1742585
108. Galluzzi L, Pietrocola F, Levine B, Kroemer G. Metabolic Control of Autophagy. Cell. 2014;159(6):1263–1276. doi:10.1016/j.cell.2014.11.006
109. White E, Mehnert JM, Chan CS. Autophagy, Metabolism, and Cancer. Cold Spring Harb Symp Quant Biol. 2017;21(22):5037.
110. Fan Q, Yang L, Zhang X, et al. Autophagy promotes metastasis and glycolysis by upregulating MCT1 expression and Wnt/β-catenin signaling pathway activation in hepatocellular carcinoma cells. J Exp Clin Cancer Res. 2018;37(1):9. doi:10.1186/s13046-018-0673-y
111. Roy S, Leidal AM, Ye J, Ronen SM, Debnath J. Autophagy-dependent shuttling of TBC1D5 controls plasma membrane translocation of GLUT1 and glucose uptake. Mol Cell. 2017;67(1):S109727651730360X. doi:10.1016/j.molcel.2017.05.020
112. Jiao L, Zhang HL, Li DD, et al. Regulation of glycolytic metabolism by autophagy in liver cancer involves selective autophagic degradation of HK2 (hexokinase 2). Autophagy. 2018;14(4):671–684. doi:10.1080/15548627.2017.1381804
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.