Back to Journals » Pediatric Health, Medicine and Therapeutics » Volume 10
Refractory Kawasaki disease: diagnostic and management challenges
Authors Duignan S, Doyle SL, McMahon CJ
Received 28 December 2018
Accepted for publication 23 May 2019
Published 30 October 2019 Volume 2019:10 Pages 131—139
DOI https://doi.org/10.2147/PHMT.S165935
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Roosy Aulakh
Sophie Duignan,1,2 Sarah L Doyle,2 Colin J McMahon1,2
1Department of Paediatric Cardiology, Our Lady’s Children’s Hospital, Dublin 12, Ireland; 2Department of Immunology, National Children’s Research Centre, Dublin, Ireland
Correspondence: Colin J McMahon
Department of Paediatric Cardiology, Our Lady’s Children’s Hospital, Crumlin, Dublin 12, Ireland
Tel +3 531 428 2854
Fax +3 531 409 6181
Email [email protected]
Abstract: Kawasaki disease (KD), an acute, self-limiting, medium-sized arterial vasculitis, is now the most common cause of acquired heart disease in childhood in the developed world. In this review, we discuss the diagnosis of KD, predicting resistance to traditional therapy and treatment options in refractory or high-risk disease. We also highlight ongoing clinical trials and other potential avenues of research which may prove beneficial in managing children, especially those with resistant KD.
Keywords: aneurysm, arteritis, coronary, inflammation, Kawasaki, vasculitis
Introduction
Kawasaki disease (KD) is an acute, self-limiting inflammatory syndrome of childhood associated with a predominantly medium-sized artery vasculitis with a predilection for the coronary arteries.1 The diagnosis is clinical; there remains no confirmatory laboratory or imaging investigation.
The critical complication of KD is coronary artery abnormalities which can lead to early and late complications including myocardial infarction and death. In developed countries, KD is now the leading cause of acquired heart disease in childhood.1,2 Up to a quarter of children with KD who do not receive treatment will develop coronary artery aneurysms (CAA)3 compared to just 4% of children who receive prompt intravenous immunoglobulin (IVIG) therapy.1,4 Of note, patients under 12 months of age are at particularly high risk of CAA formation if left untreated. This is of particular importance because the presentation is often “incomplete” in this age group and therefore the diagnosis is more difficult and potentially delayed. In untreated cases, there is a 2–3% risk of mortality secondary to coronary complications.2,3,5
For these reasons, there has been an intense interest in the prevention of coronary complications in children with KD, largely based on reducing inflammation early in the disease course. The mainstay of traditional therapy is IVIG and aspirin therapy. However, there is an increasing incidence of patients who do not respond to IVIG therapy. IVIG-resistant or refractory KD is defined as recrudescent or persistent fever at 24–48 hrs following first IVIG infusion and affects 10–20% of patients.1,6–9 Due to the significant morbidity associated with CAA, there has been increasing emphasis on earlier diagnosis and a search for adjunctive and second line therapies in order to decrease treatment failure.
Epidemiology
KD is the second most common vasculitis of childhood after Henoch Schönlein purpura.10 It, reported globally, has a male preponderance and an ethnic bias toward Asian children, particularly Japanese where the incidence is highest in the world and increasing – it reached 308 per 100,000 children in 2014.8,11 Approximately 80% of patients affected by KD are under 5 years of age with peak incidence at 18–24 months.12,13
Etiology
It is hypothesized that KD occurs in genetically predisposed individuals who are exposed to an as yet unidentified, likely infectious, trigger. The concept of an underlying genetic predisposition to KD is based on two key observations. Firstly, there is an increased risk of KD in patients who have a first degree relative with a history of KD.14 Similarly, parents of a child with KD are twice as likely to have a history of KD compared to the general population.15 Secondly, KD has a significantly higher incidence in certain ethnicities, which persists even after their relocation to other regions of the world. This is exemplified by the case of Hawaii where the incidence of KD in those of Japanese descent was 210.5 cases per 100,000 children during 1996–2006.16 This is comparable to the incidence seen in Japan and contrasts dramatically to the incidence reported in white children in Hawaii during the same time period – 13.7 cases per 100,000 children16– which is similar to the incidence rate among white children in the continental USA.12
The concept of an infectious trigger is supported by the symptomatology of KD which resembles common childhood infections, region-specific incidence rates, seasonality, the occurrence of epidemics and the low incidence of recurrence. A study which analyzed KD in 25 countries over more than 40 years found a statistically significant and consistent seasonal fluctuation in KD case numbers with higher numbers seen in winter in the Northern Hemisphere extra-tropics, suggesting a seasonal exposure to an environmental (likely infectious) agent.17 The fact that 80% of KD occurs in those less than five years of age could be due to the immature immune system failing to protect against this agent. Rowley et al have published a series of papers which revealed the presence of IgA plasma cells and an oligoclonal IgA response in arterial tissues from patients with KD,18,19 suggesting an antigen-driven response driven by the entry of a pathogen at a mucosal site likely to be the respiratory tract.
Immune response and pathogenesis
Despite decades of research, the underlying immuno-pathogenic mechanism for KD is not completely understood. The proposed paradigm is an exaggerated immune response (both the innate and adaptive systems) observed in genetically susceptible individuals following an encounter with an (likely viral) infectious agent leading to an imbalance between pro-inflammatory and anti-inflammatory pathways. For example, the IL-1 signaling pathway appears to be key to the pathogenesis of KD; upregulated IL-1 pathway genes and increased IL-1 concentrations have been demonstrated in the peripheral blood of KD patients during the acute phase of the disease.20,21 TNF-alpha has also been found to be significantly increased in peripheral blood monocytes in KD patients compared to healthy controls.21 The self-limited nature of the disease and the low recurrence rate suggest the emergence of T-cell and B-cell memory that is protective. Understanding the exact underlying mechanism is crucial as this informs potential therapeutic targets, for example, anakinra (an IL-1 receptor antagonist) and anti-TNF-alpha agents.
Diagnosis
The diagnosis of KD remains a challenge. There is no pathognomonic clinical feature, no confirmatory laboratory or imaging investigation. Clinicians rely, therefore, on the fulfillment of clinical diagnostic criteria. History taking and clinical examination are of paramount importance in the diagnosis, and it should be noted that a clinical feature may have already resolved by the time of presentation. Generally, the American Heart Association guidelines are used in the Western World. The Kawasaki Disease Research Committee guidelines from the Japanese Ministry of Health are used in Asia (see Table 1). To complicate matters further, a cohort present as incomplete KD or atypical KD. This cohort has a higher risk of CAA formation, which may be at least partially due to later treatment onset due to a delay in diagnosis.
 |
Table 1 Kawasaki Disease Research Committee Guidelines (Japanese Ministry of Health) for diagnosis of Kawasaki disease. Copright © 2005. John Wiley and Sons. Adapted from Ayusawa M, Sonobe T, Uemura S, et al. Revision of diagnostic guidelines for Kawasaki disease (the 5th revised edition). Pediatr Int. 2005;47(2):232–234.22 |
Clinical features
The fever of KD is typically high, greater than 39 degrees, and remains for 1–3 weeks without treatment. The principal features of KD tend to occur in sequential order and this can help to differentiate KD from other differential diagnoses.23 A careful history may reveal clinical features which were present during the illness but have resolved by time of presentation. Conjunctival injection, which is bilateral and non-purulent, presents soon after fever onset and is followed by oral changes including erythema and cracking of the lips and a strawberry tongue (erythema and prominent fungiform papillae). Of note, oral ulceration and pharyngeal exudate are not consistent with a diagnosis of KD. Cervical lymphadenopathy then develops and is usually unilateral. The rash usually occurs within 5 days of fever onset and is polymorphous. It is most commonly maculopapular, but may be erythema multiforme-like, scarlatiniform erythroderma or less commonly urticarial or fine micro-pustular eruptions. The rash is usually generalized and characteristically affects the groin creases which may desquamate. Of note, another distinctive dermatological feature is erythema and induration at the BCG (Bacillus Calmette–Guerin) inoculation site. In the case of bullous, petechial or vesicular rashes, an alternative diagnosis should be sought as these are not consistent with KD.1 Finally, extremity changes occur. These are distinctive and often begin with erythematous induration, which may be painful, of the hands and feet. In the subacute phase, 2–3 weeks after fever onset, the fingers and toes may desquamate. As KD is a multi-system inflammatory disorder, a broad range of clinical features may occur.
Laboratory features
During the acute phase of the disease, there is typically an elevation of acute phase reactants including C reactive protein (CRP) and erythrocyte sedimentation rate (ESR) as well as a neutrophil leukocytosis and normocytic normochromic anemia.1,2,24,25 If there are no serum markers of inflammation at early presentation, these should be periodically repeated, and if the inflammatory markers fail to rise, the diagnosis of KD is unlikely. An important point to note is that ESR (but not CRP) may rise following IVIG therapy and therefore it should not be used alone to monitor inflammatory activity.26
Albumin and sodium levels may be low due to increased capillary permeability and indeed this is used as a marker of severity in some prediction scores. Elevation of serum transaminases or gamma glutamyl transpeptidase levels occurs in approximately a third of patients.27 White cells may be found in the urine in up to 80% of children with KD;27 there may also be CSF mononuclear predominant pleocytosis in 30%.28 In the acute phase, thrombocytopenia may occur in the setting of a consumptive coagulopathy with raised D-dimers, while in the subacute phase, thrombocytosis is a characteristic finding and usually begins during the second week following the onset of fever.
Echocardiography
Echocardiography represents the mainstay of cardiac imaging during the acute phase. When abnormal, it is a useful adjunct to aid diagnosis if not all the clinical features are present, or to aid early diagnosis if the fever has not yet lasted 5 days. It can be substituted for a criterion to make a diagnosis of complete KD. However, where echocardiography is normal in the first week of illness, this does not preclude the later development of CAA and it should therefore be repeated at 1–2 weeks and 4–6-week intervals after initial treatment,27 or more frequently, in patients at high risk of CAA, or if CAA are present on the baseline study.
Coronary artery abnormalities occur in 15–25% of untreated patients, and a significant proportion of these will have further cardiovascular manifestations including pericarditis, myocarditis, pericardial effusion, electrocardiographic abnormalities and/or even myocardial infarction (see Table 2).26 Standard therapy with IVIG reduces the incidence of coronary arterial abnormalities to approximately 4%.29 Valvular dysfunction occurs in approximately a quarter of children with KD and most commonly involves the mitral valve.30
 |
Table 2 Coronary artery abnormalities |
Diagnosis
If 4 or more of the principal features of KD are present, experienced clinicians can make the diagnosis with 4 days or rarely even 3 days of fever. This is supported by the presence of the associated laboratory features and, where present, CAA.
Unfortunately, the diagnostic criteria do not correctly identify all children with KD. As untreated KD confers a 4–5 times increased risk of CAA formation, it is important to consider the diagnosis in all children with prolonged unexplained fever, particularly if any of the other principal clinical features are present. The presence of CAAs confirms the diagnosis; however, these are not usually present during the first week of the illness. Infants less than six months of age may present with only prolonged fever and irritability and are also at particularly high risk of CAA.1 The AHA guidelines provide an algorithm to aid the diagnosis of incomplete KD1 adapted from AHA guidelines).
A high index of suspicion is necessary for the diagnosis of KD in certain cases. For example, in the infant of less than six months, in whom prolonged fever and irritability may be the only features and in whom the risk of CAA is particularly high. Similarly, the diagnosis is often more difficult in the older child or teenager, and again, this cohort appears to be at a higher risk of CAAs.
In all of the above scenarios, an expert in KD should be consulted early in the disease course.
Predicting IVIG resistance
Up to 20% of patients will not respond to IVIG, and these patients are at a significantly higher risk of CAA.32 There is evidence that IVIG resistance is increasing.33 The definition of IVIG resistance is different throughout the medical literature with some groups, including the AHA, defining resistance as recrudescence or persistence of fever at 36 hrs following the first IVIG infusion and others using 24 or 48 hrs as the cutoff point. The ability to predict resistance to IVIG and/or risk of CAA formation would allow clinicians to offer more aggressive therapy to high-risk patients earlier in the disease course and potentially decrease the incidence of CAA and resultant morbidity.
Three risk scores based on Asian populations have been created over the past decade (see Table 3). However, when tested on a North American population, the scores demonstrated a sensitivity of 33–42% for predicting IVIG resistance and therefore a low-risk score is not necessarily useful.34 They maintain a high specificity in the 80th percentage and thus have a good positive-predictive value. This also raises the question of whether KD is a slightly different disease in the Western world compared to that seen in Japan and Asia, and if so, whether research in the area is applicable to diverse populations.
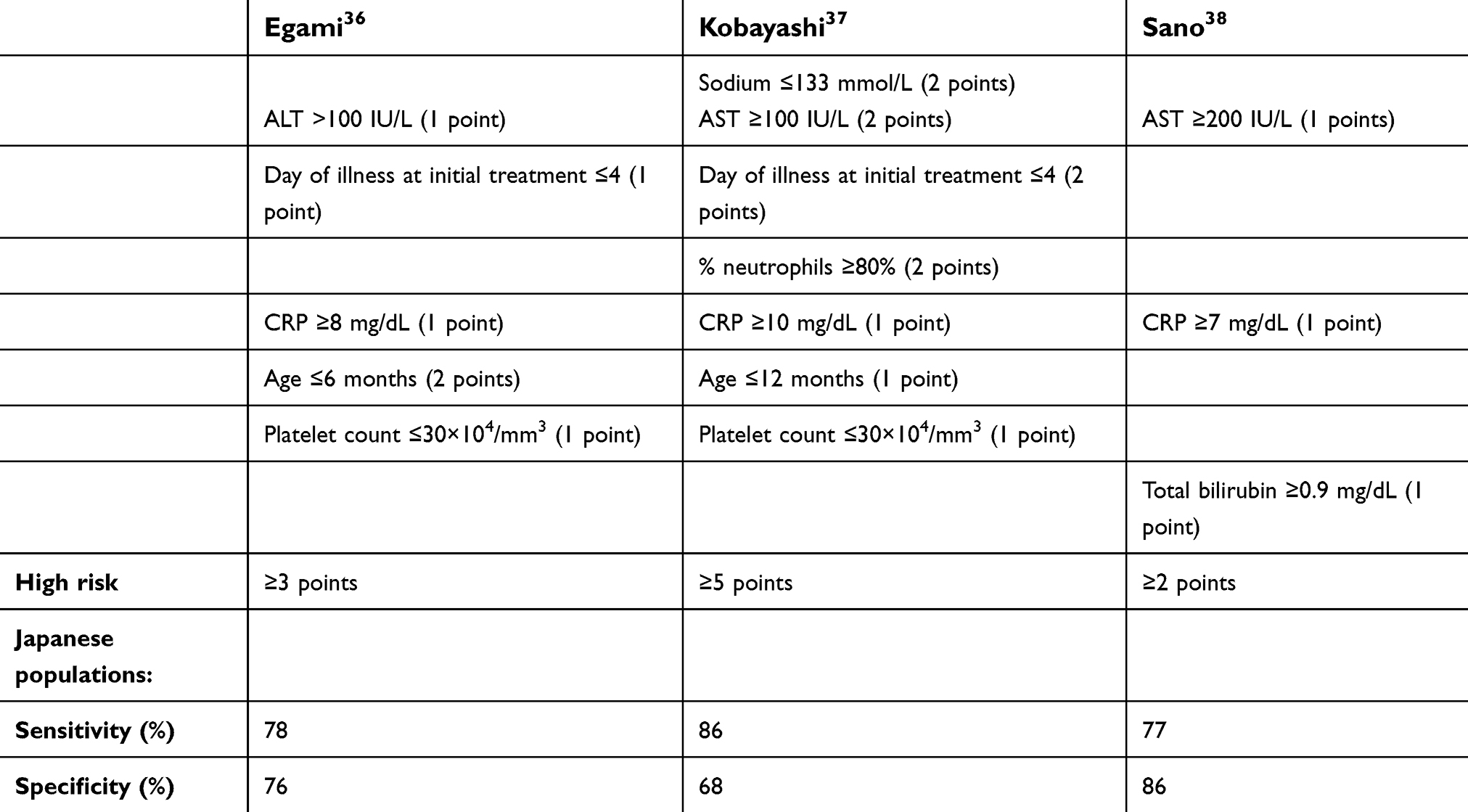 |
Table 3 Scoring systems to predict IVIG resistance |
More recently, baseline coronary artery Z scores have been used to predict the risk of CAA formation in a North American population.35 A baseline maximum Z score ≥2.0 was strongly associated with aneurysm development; it also had an 80% sensitivity and specificity of 74%. The addition of Japanese risk scores to the maximum baseline coronary artery z score did not improve CAA prediction.
Acute phase management
The goals of the management of the acute phase of KD are to stop the inflammation as early as possible in order to limit the risk and severity of coronary artery lesions and to prevent coronary artery thrombosis.
Traditional therapy
Intravenous immunoglobulin is initially given as a single dose of 2 g/kg over 8–12 hrs. The exact mechanism of IVIG in KD is poorly understood. Putative mechanisms include immune modulation of T regulatory cells, reduction of pro-inflammatory cytokine production, blockade of adhesion molecules important for inflammatory cell migration to vascular endothelium, provision of anti-cytokine, anti-chemokine and anti-complement antibodies and neutralization of the etiological agent. IVIG reduces the risk of CAA from approximately 25% to less than 5% (using Z score of greater than 2.5 as definition) and less than 1% for giant CAA.39,40 Its effect is dose-dependent and is independent of aspirin. The best results are seen if it is given as soon as possible, ideally within five days of fever onset. However, it is still recommended beyond 10 days if there is persistent fever, coronary artery dilatation or markers of ongoing inflammation, for example, a persistently elevated CRP or ESR.1
Aspirin was traditionally thought to have two roles in the management of KD; an initial anti-inflammatory effect at high dosage and then an anti-thrombotic effect at lower dosage. However, evidence for the efficacy of aspirin to limit CAA formation is lacking. One large retrospective study appears to show no effect on the response rate to IVIG, duration of fever or incidence of CAA.41 One randomized controlled trial comparing IVIG alone to IVIG plus high-dose aspirin therapy found that there was no additional benefit to the IVIG alone group in reducing the incidence of CAA but the confidence intervals in this study were wide.42 A further large RCT is ongoing to elucidate whether the benefit of high-dose aspirin outweighs the risks of initial high-dose aspirin, including that of potentially fatal Reye’s syndrome.43 Currently, an initial dose of 80–100 mg/kg/d into 4 divided doses is used in the United States while a lower dose of 30–50 mg/kg/d tends to be favored in Asia and Western Europe. There is no data to suggest that either dose is superior. There is also practice variation regarding duration of high-dose aspirin therapy. Some centers change to low- dose aspirin once the child has been afebrile for 48–72 hrs; others continue high-dose aspirin until the 14th day of the illness and once the fever has subsided for at least 48–72 hrs. Low-dose aspirin is usually 3–5 mg/kg/day and is continued until the 6–8 weeks after the onset of the illness if the patient has no coronary abnormalities.1 For children who develop coronary artery abnormalities, aspirin (or a different anti-platelet agent) is continued until they regress (transient dilatation may take up to one year to regress) or indefinitely if the CAA remains.
Adjunctive and second-line therapies
Corticosteroids are an established therapy in most vasculitides; however, their role in KD has been more controversial. Over the past decade, the use of adjunctive corticosteroid therapy alongside traditional IVIG and aspirin has been evaluated and the results have been varied. A multi-center prospective randomized, placebo-controlled, double-blinded study found no significant difference in coronary z scores or in duration of fever in those treated with corticosteroids in addition to IVIG.44 This study included 199 patients with a diagnosis of KD regardless of severity. Subsequent Japanese studies have shown the addition of corticosteroids to confer a significantly decreased risk of CAA; however, these studies included only patients deemed at high risk of IVIG resistance based on risk scores which have been shown to have a low sensitivity when used in a North American population.45–48
A recent Cochrane review included 7 randomized trials consisting of 922 patients in total with all severities of KD.49 They found that on pooled analysis, the addition of corticosteroids reduced the subsequent occurrence of CAA by almost a third (odds ratio 0.29) without serious adverse effects.
The current AHA guidelines do not recommend routine use of adjunctive corticosteroids, but rather consideration for high-risk patients.1
To date, there have been no robust clinical trials comparing second-line treatment options for IVIG resistant KD. To further complicate the interpretation of the effect of any given treatment is the fact that in many case reports and case series, multiple second-line options have been used and it is difficult to ascertain which treatment led to the desirable effect.
An appropriate and established initial therapy is a second 2 mg/kg infusion of IVIG. Retrospective cohort studies have shown this to be an effective approach;32 however, it is yet to be evaluated in a randomized controlled prospective trial.
Corticosteroids can be used as a second-line treatment alone or in conjunction with a second dose of IVIG. A recent meta-analysis concluded that steroids were more effective than IVIG in reducing fever but that there was no significant difference in risk of CAA formation.50
Serum TNF-alpha is raised in patients with KD and higher serum levels have been shown to correlate with the development of CAA.51 Infliximab is a chimeric murine/human IgG1 monoclonal antibody which binds specifically to TNF-alpha with high affinity. It has been evaluated as an adjunct to IVIG therapy in a Phase III randomized double-blinded placebo-controlled trial;52 it did not reduce treatment resistance but did reduce duration of fever. In trials comparing rescue infliximab to a second dose of IVIG in IVIG resistant patients, infliximab decreased the duration of fever but the groups had similar coronary arterial outcomes.53,54
The disadvantage of etanercept, another TNF-alpha antagonist, is that it only binds to circulating and not cell-bound TNF-alpha which could potentially impair its efficacy. A prospective open-label trial has demonstrated its safety as an adjunctive treatment to standard therapy.55 Fifteen patients completed the trial and none demonstrated IVIG resistance or CAAs.
The nuclear factor activated T-cells)–calcineurin pathway is implicated in the pathogenesis of KD. Thus, cyclosporine, a calcineurin inhibitor, has been studied as both an adjunctive therapy and as a rescue therapy for KD. One retrospective case series reported defervescence and resolution of inflammation in all 9 patients treated with cyclosporine following failure of multiple other therapies.56 Importantly, in mouse models that were induced with a coronary arteritis with similar histopathological features to those of the coronary artery lesions seen in KD, the administration of calcineurin inhibitors led to exacerbation of the coronary arteritis, and thus the effect on CAA needs to be examined in larger studies.57 The first randomized, open-label, blinded-end point trial to evaluate its efficacy as an adjunctive therapy is underway.58
In a KD mouse model, IL-1 receptor antagonists successfully inhibited the development of vasculitis, and IL-1 receptor-deficient mice were protected from induced coronary lesions.59 Retrospective case series and case reports60,61 of anakinra have been promising.62 The largest case series to date reported on 11 patients who had failed at least 2 different treatments for KD prior to anakinra.63 All patients showed rapid and sustained improvement in clinical and biochemical inflammation measured by resolution of fever and decrease in CRP. Overall, coronary artery dimensions decreased in all but one patient, but this was not a rapid occurrence and it is not clear whether this was as a result of the anakinra.
Statins lower low-density lipoprotein cholesterol, improve endothelial function, reduce oxidative stress and inhibit inflammation.64 Several observations have led to an interest in atorvastatin as a potential adjunctive therapy to inhibit the progression of aneurysms in KD. Genetic studies have shown an association between polymorphisms in matrix metalloproteinase (MMP) genes and both KD susceptibility and aneurysm formation.65 In a murine model, atorvastatin inhibited MMP-9 secretion in the blood vessel wall.66 It has also been shown to attenuate MMP-9 gene expression in vitro in human endothelial cells.67 Atorvastatin has been shown to increase the number and suppressive function of regulatory T cells in adults with rheumatoid arthritis.68 The safety of atorvastatin use in the context of CAA following KD has been evaluated in 20 patients. The group reported excellent compliance at a median of 2.5 years of treatment and apparent safety of the drug in young children (the youngest in the study was 10 months of age at treatment initiation); however, studies with longer follow-up are required.
Cyclophosphamide, methotrexate and plasma exchange have also been used in treatment-resistant cases of KD.69–73 Cyclophosphamide carries high-risk toxicity and therefore should only be considered in severe refractory cases. Methotrexate was used at a relatively low dose in the largest case series to date and there were therefore no adverse reactions reported. This suggests it should be further evaluated, potentially as an adjunctive therapy for high-risk patients. Plasma exchange is a high-risk procedure and should therefore only be considered in extreme cases of refractory KD. The largest series reported to date included 125 patients who were resistant to IVIG and treated with plasma exchange.74 Of those children with normal coronary arteries prior to plasma exchange, no child went on to develop later coronary arterial abnormalities.
Conclusion
The diagnosis of KD remains a challenge and it should always be considered in cases of prolonged fever, particularly in the infant with irritability. Expert opinion should be sought early, and if possible, echocardiography should be performed. Identifying patients at high risk of refractory KD and thus CAAs is a research priority in order to identify high-risk patients early in the disease course who may benefit from more aggressive treatment. As nearly half of infants under 6 months of age treated for KD within 10 days of illness go on to develop CAA, perhaps this cohort should be treated more aggressively regardless of other clinical predictors.75 Currently, as there is no validated risk score for Western populations, a combination of features including baseline bloods and coronary Z-score should be used to determine disease severity. In the future, a risk score may include genetic testing for high-risk SNPs. There is currently a dearth of evidence for choosing adjunctive therapy or second-line treatment in refractory KD. Results of several randomized-controlled and open-label trials are awaited and may guide the future management of KD patients.
Disclosure
The authors report no conflicts of interest in this work.
References
1. McCrindle BW, Rowley AH, Newburger JW, et al. Diagnosis, treatment, and long-term management of kawasaki disease: a scientific statement for health professionals from the American Heart Association. Circulation. 2017;135(17):e927–99. doi:10.1161/CIRCULATIONAHA.117.027305
2. Brogan PA, Bose A, Burgner D, et al. Kawasaki disease: an evidence based approach to diagnosis, treatment, and proposals for future research. Arch Dis Child. 2002;86(4):286–290. doi:10.1136/adc.86.4.286
3. Kato H, Akagi T, Sugimura T, et al. Kawasaki disease. Coron Artery Dis. 1995;6(3):194–206.
4. Burns JC, Shike H, Gordon JB, Malhotra A, Schoenwetter M, Kawasaki T. Sequelae of Kawasaki disease in adolescents and young adults. J Am Coll Cardiol. 1996;28(1):253–257.
5. Dajani AS, Taubert KA, Gerber MA, et al. Diagnosis and therapy of Kawasaki disease in children. Circulation. 1993;87(5):1776–1780.
6. Durongpisitkul K, Soongswang J, Laohaprasitiporn D, Nana A, Prachuabmoh C, Kangkagate C. Immunoglobulin failure and retreatment in Kawasaki disease. Pediatr Cardiol. 2003;24(2):145–148. doi:10.1007/s00246-002-0216-2
7. Han RK, Silverman ED, Newman A, McCrindle BW. Management and outcome of persistent or recurrent fever after initial intravenous gamma globulin therapy in acute Kawasaki disease. Arch Pediatr Adolesc Med. 2000;154(7):694–699. doi:10.1001/archpedi.154.7.694
8. Makino N, Nakamura Y, Yashiro M, et al. Epidemiological observations of Kawasaki disease in Japan, 2013-2014. Pediatr Int. 2018;60(6):581–587. doi:10.1111/ped.13544
9. Kim GB, Park S, Eun LY, et al. Epidemiology and clinical features of Kawasaki disease in South Korea, 2012-2014. Pediatr Infect Dis J. 2017;36(5):482–485. doi:10.1097/INF.0000000000001474
10. Gardner-Medwin JMM, Dolezalova P, Cummins C, Southwood TR. Incidence of Henoch-Schönlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins. Lancet. 2002;360(9341):1197–1202.
11. Makino N, Nakamura Y, Yashiro M, et al. Descriptive epidemiology of Kawasaki disease in Japan, 2011–2012: from the results of the 22nd nationwide survey. J Epidemiol. 2015;25(3):239–245. doi:10.2188/jea.JE20140089
12. Holman RC, Belay ED, Christensen KY, Folkema AM, Steiner CA, Schonberger LB. Hospitalizations for Kawasaki syndrome among children in the United States, 1997–2007. Pediatr Infect Dis J. 2010;29(6):483–488.
13. Lynch M, Holman RC, Mulligan A, Belay ED, Schonberger LB. Kawasaki syndrome hospitalizations in Ireland, 1996 through 2000. Pediatr Infect Dis J. 2003;22(11):959–963. doi:10.1097/01.inf.0000095194.83814.ee
14. Fujita Y, Nakamura Y, Sakata K, et al. Kawasaki disease in families. Pediatrics. 1989;84(4):666–669.
15. Uehara R, Yashiro M, Nakamura Y, Yanagawa H. Kawasaki disease in parents and children. Acta Paediatr. 2003;92(6):694–697. doi:10.1111/j.1651-2227.2003.tb00602.x
16. Holman RC, Christensen KY, Belay ED, et al. Racial/ethnic differences in the incidence of Kawasaki syndrome among children in Hawaii. Hawaii Med J. 2010;69(8):194–197.
17. Burns JC, Herzog L, Fabri O, et al. Seasonality of Kawasaki disease: a global perspective. PLoS One. 2013;8(9):e74529. doi:10.1371/journal.pone.0074529
18. Rowley AH, Eckerley CA, Jäck HM, Shulman ST, Baker SC. IgA plasma cells in vascular tissue of patients with Kawasaki syndrome. J Immunol. 1997;159(12):5946–5955.
19. Rowley AH, Shulman ST, Spike BT, Mask CA, Baker SC. Oligoclonal IgA response in the vascular wall in acute Kawasaki disease. J Immunol. 2001;166(2):1334–1343. doi:10.4049/jimmunol.166.2.1334
20. Hoang LT, Shimizu C, Ling L, et al. Global gene expression profiling identifies new therapeutic targets in acute Kawasaki disease. Genome Med. 2014;6(11):541. doi:10.1186/s13073-014-0102-6
21. Suzuki H, Uemura S, Tone S, et al. Effects of immunoglobulin and gamma-interferon on the production of tumour necrosis factor-alpha and interleukin-1 beta by peripheral blood monocytes in the acute phase of Kawasaki disease. Eur J Pediatr. 1996;155(4):291–296. doi:10.1007/BF02002715
22. Ayusawa M, Sonobe T, Uemura S, et al. Revision of diagnostic guidelines for Kawasaki disease (the 5th revised edition). Pediatr Int. 2005;47(2):232–234. doi:10.1111/j.1442-200x.2005.02033.x
23. Saguil A, Fargo M, Grogan S. Diagnosis and management of Kawasaki disease. Am Fam Physician. 2015;91(6):365–371.
24. Newburger JW, Takahashi M, Gerber MA, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on rheumatic fever, endocarditis, and Kawasaki disease, council on cardiovascular disease in the young, American Heart Association. Pediatrics. 2004;114(6):1708–1733. doi:10.1542/peds.2004-2182
25. Tremoulet AH, Jain S, Chandrasekar D, Sun X, Sato Y, Burns JC. Evolution of laboratory values in patients with Kawasaki disease. Pediatr Infect Dis J. 2011;30(12):1022–1026. doi:10.1097/INF.0b013e31822d4f56
26. Newburger JW, Takahashi M, Gerber MA, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the committee on rheumatic fever, endocarditis and Kawasaki disease, council on cardiovascular disease in the Young, American Heart Association. Circulation. 2004;110(17):2747–2771. doi:10.1161/01.CIR.0000145143.19711.78
27. Newburger JW, Takahashi M, Burns JC. Kawasaki disease. J Am Coll Cardiol. 2016;67(14):1738–1749. doi:10.1016/j.jacc.2015.12.073
28. Dengler LD, Capparelli EV, Bastian JF, et al. Cerebrospinal fluid profile in patients with acute Kawasaki disease. Pediatr Infect Dis J. 1998;17(6):478–481. doi:10.1097/00006454-199806000-00008
29. Newburger JW, Takahashi M, Beiser AS, et al. A single intravenous infusion of gamma globulin as compared with four infusions in the treatment of acute Kawasaki syndrome. N Engl J Med. 1991;324(23):1633–1639. doi:10.1056/NEJM199106063242305
30. Printz BF, Sleeper LA, Newburger JW, et al. Noncoronary cardiac abnormalities are associated with coronary artery dilation and with laboratory inflammatory markers in acute Kawasaki disease. J Am Coll Cardiol. 2011;57(1):86–92. doi:10.1016/j.jacc.2010.08.619
31. JCS Joint Working Group. Guidelines for diagnosis and management of cardiovascular sequelae in Kawasaki disease (JCS 2013). Digest version. Circ J. 2014;78(10):2521–2562. doi:10.1253/circj.CJ-66-0096
32. Burns JC, Capparelli EV, Brown JA, Newburger JW, Glode MP. Intravenous gamma-globulin treatment and retreatment in Kawasaki disease. US/Canadian Kawasaki Syndrome Study Group. Pediatr Infect Dis J. 1998;17(12):1144–1148. doi:10.1097/00006454-199812000-00009
33. Kibata T, Suzuki Y, Hasegawa S, et al. Coronary artery lesions and the increasing incidence of Kawasaki disease resistant to initial immunoglobulin. Int J Cardiol. 2016;1(214):209–215. doi:10.1016/j.ijcard.2016.03.017
34. Sleeper LA, Minich LL, McCrindle BM, et al. Evaluation of Kawasaki disease risk-scoring systems for intravenous immunoglobulin resistance. J Pediatr. 2011;158(5):831–835.e3. doi:10.1016/j.jpeds.2010.10.031
35. Son MBF, Gauvreau K, Kim S, et al. Predicting coronary artery aneurysms in Kawasaki disease at a North American Center: an assessment of baseline z scores. J Am Heart Assoc. 2017;6:6. doi:10.1161/JAHA.116.005378
36. Egami K, Muta H, Ishii M, et al. Prediction of resistance to intravenous immunoglobulin treatment in patients with Kawasaki disease. J Pediatr. 2006;149(2):237–240. doi:10.1016/j.jpeds.2006.07.024
37. Kobayashi T, Inoue Y, Takeuchi K, et al. Prediction of intravenous immunoglobulin unresponsiveness in patients with Kawasaki disease. Circulation. 2006;113(22):2606–2612. doi:10.1161/CIRCULATIONAHA.105.592865
38. Sano T, Kurotobi S, Matsuzaki K, et al. Prediction of non-responsiveness to standard high-dose gamma-globulin therapy in patients with acute Kawasaki disease before starting initial treatment. Eur J Pediatr. 2007;166(2):131–137. doi:10.1007/s00431-006-0223-z
39. Newburger JW, Takahashi M, Burns JC, et al. The treatment of Kawasaki syndrome with intravenous gamma globulin. N Engl J Med. 1986;315(6):341–347. doi:10.1056/NEJM198608073150601
40. Terai M, Shulman ST. Prevalence of coronary artery abnormalities in Kawasaki disease is highly dependent on gamma globulin dose but independent of salicylate dose. J Pediatr. 1997;131(6):888–893. doi:10.1016/S0022-3476(97)70038-6
41. Hsieh K-S, Weng K-P, Lin -C-C, Huang T-C, Lee C-L, Huang S-M. Treatment of acute Kawasaki disease: aspirin’s role in the febrile stage revisited. Pediatrics. 2004;114(6):e689–e693. doi:10.1542/peds.2004-1037
42. Furusho K, Kamiya T, Nakano H, et al. Intravenous gamma-globulin for Kawasaki disease. Acta Paediatr Jpn. 1991;33(6):799–804. doi:10.1111/j.1442-200X.1991.tb02611.x
43. Kuo H-C, Guo MM-H, Lo M-H, Hsieh K-S, Huang Y-H. Effectiveness of intravenous immunoglobulin alone and intravenous immunoglobulin combined with high-dose aspirin in the acute stage of Kawasaki disease: study protocol for a randomized controlled trial. BMC Pediatr. 2018;18(1):200. doi:10.1186/s12887-018-0993-2
44. Newburger JW, Sleeper LA, McCrindle BW, et al. Randomized trial of pulsed corticosteroid therapy for primary treatment of Kawasaki disease. N Engl J Med. 2007;356(7):663–675. doi:10.1056/NEJMc063190
45. Ogata S, Ogihara Y, Honda T, Kon S, Akiyama K, Ishii M. Corticosteroid pulse combination therapy for refractory Kawasaki disease: a randomized trial. Pediatrics. 2012;129(1):e17–e23. doi:10.1542/peds.2011-0148
46. Okada K, Hara J, Maki I, et al. Pulse methylprednisolone with gammaglobulin as an initial treatment for acute Kawasaki disease. Eur J Pediatr. 2009;168(2):181–185. doi:10.1007/s00431-008-0753-7
47. Kobayashi T, Saji T, Otani T, et al. Efficacy of immunoglobulin plus prednisolone for prevention of coronary artery abnormalities in severe Kawasaki disease (RAISE study): a randomised, open-label, blinded-endpoints trial. Lancet. 2012;379(9826):1613–1620. doi:10.1016/S0140-6736(11)61930-2
48. Miyata K, Kaneko T, Morikawa Y, et al. Efficacy and safety of intravenous immunoglobulin plus prednisolone therapy in patients with Kawasaki disease (Post RAISE): a multicentre, prospective cohort study. Lancet Child Adolesc Health. 2018. doi:10.1016/S2352-4642(18)30293-1
49. Wardle AJ, Connolly GM, Seager MJ, Tulloh RM. Corticosteroids for the treatment of Kawasaki disease in children. Cochrane Database Syst Rev. 2017;27(1):CD011188.
50. Yang X, Liu G, Huang Y, Chen S, Du J, Jin H. A meta-analysis of re-treatment for intravenous immunoglobulin-resistant Kawasaki disease. Cardiol Young. 2015;25(6):1182–1190. doi:10.1017/S1047951114002601
51. Matsubara T, Furukawa S, Yabuta K. Serum levels of tumor necrosis factor, interleukin 2 receptor, and interferon-gamma in Kawasaki disease involved coronary-artery lesions. Clin Immunol Immunopathol. 1990;56(1):29–36. doi:10.1016/0090-1229(90)90166-N
52. Tremoulet AH, Jain S, Jaggi P, et al. Infliximab for intensification of primary therapy for Kawasaki disease: a phase 3 randomised, double-blind, placebo-controlled trial. Lancet. 2014;383(9930):1731–1738. doi:10.1016/S0140-6736(13)62298-9
53. Son MB, Gauvreau K, Burns JC, et al. Infliximab for intravenous immunoglobulin resistance in Kawasaki disease: a retrospective study. J Pediatr. 2011;158(4):644–649.e1. doi:10.1016/j.jpeds.2010.10.012
54. Mori M, Hara T, Kikuchi M, et al. Infliximab versus intravenous immunoglobulin for refractory Kawasaki disease: a phase 3, randomized, open-label, active-controlled, parallel-group, multicenter trial. Sci Rep. 2018;8(1):1994. doi:10.1038/s41598-017-18387-7
55. Choueiter NF, Olson AK, Shen DD, Portman MA. Prospective open-label trial of etanercept as adjunctive therapy for Kawasaki disease. J Pediatr. 2010;157(6):960–966.e1. doi:10.1016/j.jpeds.2010.06.014
56. Tremoulet AH, Pancoast P, Franco A, et al. Calcineurin inhibitor treatment of intravenous immunoglobulin-resistant Kawasaki disease. J Pediatr. 2012;161(3):506–512.e1. doi:10.1016/j.jpeds.2012.02.048
57. Murata K, Motomura Y, Tanaka T, et al. Calcineurin inhibitors exacerbate coronary arteritis via the MyD88 signalling pathway in a murine model of Kawasaki disease. Clin Exp Immunol. 2017;190(1):54–67. doi:10.1111/cei.13002
58. Aoyagi R, Hamada H, Sato Y, et al. Study protocol for a phase III multicentre, randomised, open-label, blinded-end point trial to evaluate the efficacy and safety of immunoglobulin plus cyclosporin A in patients with severe Kawasaki disease (KAICA trial). BMJ Open. 2015;5(12):e009562. doi:10.1136/bmjopen-2015-009562
59. Lee Y, Schulte DJ, Shimada K, et al. Interleukin-1β is crucial for the induction of coronary artery inflammation in a mouse model of Kawasaki disease. Circulation. 2012;125(12):1542–1550. doi:10.1161/CIRCULATIONAHA.111.072769
60. Guillaume M-P, Reumaux H, Dubos F. Usefulness and safety of anakinra in refractory Kawasaki disease complicated by coronary artery aneurysm. Cardiol Young. 2018;28(5):739–742. doi:10.1017/S1047951117002864
61. Sánchez-Manubens J, Gelman A, Franch N, et al. A child with resistant Kawasaki disease successfully treated with anakinra: a case report. BMC Pediatr. 2017;17(1):102. doi:10.1186/s12887-017-0969-7
62. Shafferman A, Birmingham JD, Cron RQ. High dose Anakinra for treatment of severe neonatal Kawasaki disease: a case report. Pediatr Rheumatol Online J. 2014;12:26. doi:10.1186/1546-0096-12-26
63. Kone-Paut I, Cimaz R, Herberg J, et al. The use of interleukin 1 receptor antagonist (anakinra) in Kawasaki disease: a retrospective cases series. Autoimmun Rev. 2018;17(8):768–774. doi:10.1016/j.autrev.2018.01.024
64. Liao JK, Laufs U. Pleiotropic effects of statins. Annu Rev Pharmacol Toxicol. 2005;45:89–118. doi:10.1146/annurev.pharmtox.45.120403.095748
65. Shimizu C, Matsubara T, Onouchi Y, et al. Matrix metalloproteinase haplotypes associated with coronary artery aneurysm formation in patients with Kawasaki disease. J Hum Genet. 2010;55(12):779–784. doi:10.1038/jhg.2010.109
66. Blankier S, McCrindle BW, Ito S, Yeung RSM. The role of atorvastatin in regulating the immune response leading to vascular damage in a model of Kawasaki disease. Clin Exp Immunol. 2011;164(2):193–201. doi:10.1111/j.1365-2249.2011.04331.x
67. Izidoro-Toledo TC, Guimaraes DA, Belo VA, Gerlach RF, Tanus-Santos JE. Effects of statins on matrix metalloproteinases and their endogenous inhibitors in human endothelial cells. Naunyn Schmiedebergs Arch Pharmacol. 2011;383(6):547–554. doi:10.1007/s00210-011-0623-0
68. Tang TT, Song Y, Ding Y-J, et al. Atorvastatin upregulates regulatory T cells and reduces clinical disease activity in patients with rheumatoid arthritis. J Lipid Res. 2011;52(5):1023–1032. doi:10.1194/jlr.M010876
69. Lee MS, An SY, Jang GC, Kim DS. A case of intravenous immunoglobulin-resistant Kawasaki disease treated with methotrexate. Yonsei Med J. 2002;43(4):527–532. doi:10.3349/ymj.2002.43.5.613
70. Wallace CA, French JW, Kahn SJ, Sherry DD. Initial intravenous gammaglobulin treatment failure in Kawasaki disease. Pediatrics. 2000;105(6):E78. doi:10.1542/peds.105.6.e78
71. Lee TJ, Kim KH, Chun J-K, Kim DS. Low-dose methotrexate therapy for intravenous immunoglobulin-resistant Kawasaki disease. Yonsei Med J. 2008;49(5):714–718. doi:10.3349/ymj.2008.49.5.714
72. Ahn SY, Kim DS. Treatment of intravenous immunoglobulin-resistant Kawasaki disease with methotrexate. Scand J Rheumatol. 2005;34(2):136–139.
73. Mori M, Imagawa T, Katakura S, et al. Efficacy of plasma exchange therapy for Kawasaki disease intractable to intravenous gamma-globulin. Mod Rheumatol. 2004;14(1):43–47. doi:10.3109/s10165-003-0264-3
74. Hokosaki T, Mori M, Nishizawa T, et al. Long-term efficacy of plasma exchange treatment for refractory Kawasaki disease. Pediatr Int. 2012;54(1):99–103. doi:10.1111/j.1442-200X.2011.03487.x
75. Dominguez SR, Anderson MS, El-Adawy M, Glodé MP. Preventing coronary artery abnormalities: a need for earlier diagnosis and treatment of Kawasaki disease. Pediatr Infect Dis J. 2012;31(12):1217–1220. doi:10.1097/INF.0b013e318266bcf9
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.