Back to Journals » Drug Design, Development and Therapy » Volume 16
Recent Updates on the Development of Deuterium-Containing Drugs for the Treatment of Cancer
Authors Belete TM ![]()
Received 21 June 2022
Accepted for publication 25 September 2022
Published 4 October 2022 Volume 2022:16 Pages 3465—3472
DOI https://doi.org/10.2147/DDDT.S379496
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Jianbo Sun
Tafere Mulaw Belete
Department of Pharmacology, College of Medicine and Health Sciences, University of Gondar, Gondar, Ethiopia
Correspondence: Tafere Mulaw Belete, Tel +251 918045943, Email [email protected]
Abstract: Cancer is one of the deadliest diseases in the world. In 2020, 19.3 million cancer cases and 10 million deaths were reported in the world. It is supposed that the prevalence of cancer cases will rise to 28.4 million by 2040. Chemotherapy-based regimens have a narrow therapeutic index, severe adverse drug reactions, and lack metabolic stability. Besides, the metabolism of anticancer produces several non-active and toxic metabolites that reduce exposure of the target site to the parent drug. Therefore, developing better-tolerated and effective new anticancer drugs and modification of the existing anticancer drugs to minimize toxicity and increase efficacy has become a very urgent need. Deuterium incorporation reduces the metabolism of certain drugs that are breakdown by pathways involving hydrogen-carbon bond scission. For example, CYP450 mediated oxidative metabolism of drugs that involves the breakdown of a hydrogen-carbon bond affected by deuteration. Deuterium incorporation into the drug increases the half-life and reduces the dose, which provides better safety and efficacy. Deutetrabenazine is the first deuterated form of tetrabenazine approved to treat chorea associated with Huntington’s disease and tardive dyskinesia. The study revealed that Deutetrabenazine has fewer neuropsychiatric side effects with favorable safety than tetrabenazine. The current review highlights the deuterium kinetic isotope effect on drug metabolism, deuterated compound pharmacokinetic property, and safety profile. Besides, this review explains the deuterated anticancer drug development update status.
Keywords: deuterated drug, anticancer drugs, clinical trial, pharmacokinetic property
Background
Cancer is one of the deadliest diseases in the world. In 2020, 19.3 million cancer cases and 10 million deaths were reported in the world. It is supposed that the prevalence of cancer cases will rise to 28.4 million by 2040.1 Radiation, surgery, immunotherapy, and chemotherapy are used in separate or combination to treat cancer. But, these treatment options cause severe adverse reaction. Therefore, developing better-tolerated and effective new anticancer drugs and chemically modify an existing anticancer drugs to minimize toxicity and increase efficacy has become a very urgent need.
Although there is an increased understanding of cancer biology with several new and novel potential targets, the ability to translate these advances into treatment is poor, with a failure rate of about 90%.2 So, drug development needs a large sum of money and a long time because, for every 10,000 compounds, only one becomes a drug.3 Narrow therapeutic index, lack of metabolic stability, and severe adverse drug reaction has been the main problem of the current anticancer drugs.4 Besides, the metabolism of anticancer produces several non-active and toxic metabolites that reduce exposure of the target site to the parent drug.5 Pharmaceutical companies investigate deuterated agents as new chemical entities to bring favorable pharmacokinetic properties. The replacement of hydrogen with deuterium effectively increases the drug’s metabolic stability by prolonging the half-life reduces the dose, which provides better safety and efficacy.6
Deuterium is a stable isotope of hydrogen with similar physical and chemical characteristics. Thus, selective deuteration of a drug keeps the pharmacologic effect. The common protium hydrogen isotope has one proton and one electron. Deuterium, however, has an additional neutron. Deuterium is not radioactive, as is tritium (1 proton and 2 neutrons).7 Deuterium is available naturally at an abundance of about 0.0156%, which help to manufacture enriched D2O (heavy water), which is vital for nuclear reactors. The deuteration reaction is the replacement of covalently bonded hydrogen atom with a deuterium atom. Deuterium gas, high pressure, high temperature, and catalyst-like Pt, Pd, Rh facilitates the replacement of the deuterium atom into the new compound. Considering the shape and size, the deuterated compound has similar to the all hydrogen compound.8,9 However, minor physical character changes were measured in deuterated compounds, including reduced hydrophobicity, decreased acidity of carboxylic acids, and phenol, and increased basicity of the amine. These differences have less effect on the potency or selectivity of the receptor.10,11 The current review highlights the deuterium kinetic isotope effect on drug metabolism, Deuterated agents’ pharmacokinetic property, and safety profile. Besides, this review explains the deuterated anticancer drug development update status.
Deuterium Safety
Several studies showed that deuterium has very low systemic toxicity. Single-celled organisms can grow in conditions of full deuteration. Lower organisms including fish and tadpoles can survive in about 30% D2O. Mice and dogs can tolerate long-term replacement of 10–15% of body fluid hydrogen with deuterium; however, toxicity was reported with chronic exposure above 25–32%.8,12 In humans, toxicity was not reported upon acute exposure of 15–23% deuterium replacement in the whole body plasma. Besides, excess deuterated water was administered to healthy participants, including pregnant women and neonates without displaying side effects.11,13
Deuterated water excretion is via the urine with a half-life of about 10 days similar to that of H2O.14 Although the amount of deuterium in the drug is inconsequential, the effect of deuteration on a drug is less predictable, and PK changes due to deuteration may cause unexpected adverse drug effects. For example, shunting the elimination pathway of a drug may produce a favorable PK but may induce new toxicities. Deuterated metabolites may be hard to eliminate than expected.15 JNJ-38877605 is a curative an antitumor agent that inhibits c-Met tyrosine kinase. However, JNJ-38877605 clinical trial was terminated because the formation of insolubility metabolites. Deuteration of JNJ38877605 decreases toxic metabolites formation and renal toxicity. Besides, deuterated JNJ38877605 showed better antitumor effect, oral exposure, and metabolic stability than the JNJ38877605.16 It is important to note that several deuterated molecules have been developed over the years, but deutetrabenazine is the only drug to be approved for clinical use. The earlier deuterated drugs were abandoned due to unexpected adverse reactions.15,17
Deuterium Kinetic Isotope Effect on Drug Pharmacokinetic Property
Deuterium-carbon bond is shorter and 6−10 times more stable than the hydrogen-carbon bond, which makes it hard to break. Thus, the slow rate of bond breakdown results from the kinetic isotope effect (KIE). The KIE is the change of chemical reaction rate when one of the atoms in a compound is substituted by its isotopes.18 Due to the higher molecular weight of deuterium, C-D bonds have a lower vibrational frequency and zero-point energy and needs higher activation energy that decreases the rate (k) of C-D bond cleavage. This rate effect is the deuterium isotope effect (DIE) and is expressed as kH/kD the ratio of the rate of C-H vs C-D bond cleavage. The DIE affects the pharmacokinetic property of several drugs that are metabolized by pathways involving C-H bond breakage. However, minor shifts in physical properties such as reduced hydrophobicity and altered pKa for acids and bases are insignificant to affect the potency or selectivity of the targets.18,19
Deuteration of drugs reduces the metabolic rate (especially oxidative) in the gut wall or liver, which causes the parent drug to enter into the systemic circulation or increases the bioavailability of the parent drug. In most cases, the systemic clearance rate is unaffected. Deuterated drugs with this property may need a reduced dose regime and produce less metabolite. Since gastrointestinal irritation relates to the amount of dosed drug than the concentration of the drug in the blood, this effect may help to increase tolerability.20 Generally, deuterium incorporation reduces the metabolism of certain drugs that are breakdown by pathways involving hydrogen-carbon bond scission.21 For example, CYP450 mediated oxidative metabolism of drugs that involves the breakdown of a hydrogen-carbon bond affected by deuteration. Such as deuteration of paroxetine decreases the inactivation of CYP2D6 and reduces drug–drug interactions for favoring the metabolic profile.19,21
However, DIE on the drug’s pks usually masked by competing effects in-vivo, such as alternate metabolic pathways and different rate-limiting steps in enzymatic reactions and biological sequestration. Because of the large active site cavity of CYP enzymes, which oxidize at a non-deuterated site compensates for reduced metabolism at the deuterated site resulting in the loss of an isotope effect on the intrinsic clearance and redistribution of the relative abundance of metabolites.21 So, the deuteration of drug affect CYP450 metabolism is difficult to predict and varies from drug to drug. Besides, deuterium incorporation may change the ratio of parent drugs to metabolites that change the amounts of metabolites formed. The metabolites formed are similar to those from the non-deuterated compound, except for the presence of deuterium. But, decreased metabolism reduces the exposure of the target site to undesirable metabolites or increases exposure to the target site.7,19,21
Deuteration of a drug not only reduces drug metabolism, but also may add novel effect relative to the parental drug. For example, the deuterated analog of phenylbutyrate significantly induced apoptosis and inhibited colon cancer cell proliferation as compared with phenylbutyrate. The main effect of deuteration is to decrease the rate of drug clearance, which increases the half-life and duration of action. The clinical advantage is to maintain similar systemic exposure with increased trough level and decreased peak levels. This helps administer lower doses or less frequently, leading to increased efficacy and decreased adverse effects.7,19,22 The isolation of the beneficial enantiomer from the undesired ones helps in the development of drugs such as esomeprazole magnesium, escitalopram, eszopiclone, and levalbuterol. However, drugs with hydrogen at the chiral center are found in racemate form due to their unstable stereocenters that make enantiomers interconvert in vivo. This switching can stabilize through deuterium substitution.23,24 Deuterium substitution at the chiral center may decrease atom abstraction rate and stabilize the configuration in favor of the preferred enantiomer as revealed in CC-122, a thalidomide derivative compound used for the treatment of hematological cancers and solid tumors.24 In this case, the (S)-deuterated enantiomer is antitumorigenic, whereas the other enantiomer does not affect tumor growth.7
Deuterated Anticancer Drug Development Update
Deutetrabenazine is the leading deuterated drug approved by the FDA for the management of chorea and tardive dyskinesia due to Huntington’s disease. The success of deutetrabenazine triggered pharmaceutical companies to invest in the discovery of deuterated agents.
Donafenib, a deuterium derivative of sorafenib, is a novel multikinase inhibitor such as platelet-derived growth factor receptor and vascular endothelial growth factor receptor. Donafenib was developed for the management of different cancers, such as hepatocellular carcinoma, colorectal cancer, and thyroid cancer.19 In 2021, donafenib was approved by the National Medical Products Administration of China for the management of unresectable hepatocellular carcinoma. Currently, several deuterated drugs are on trial for the management of different disorders. Among these compounds HC-1119, BMS-986165, AVP-786, RT001, ALK-001, and donafenib reached Phase III clinical trials.19,25
Hc-1119: A Deuterated Enzalutamide
Enzalutamide is an androgen receptor inhibitor approved for the management of metastatic castration-resistant prostate cancer after chemotherapy. However, its dose-dependent central nervous system (CNS)-related toxicities hamper its clinical use. So, deuteration helps to improve enzalutamide’s PK properties and decrease side effects.26 enzalutamide metabolized to the active metabolite N-demethylenzalutamide mainly by CYP2C8 and CYP3A4/5. enzalutamide is also metabolized to the inactive metabolite enzalutamide carboxylic acid by the carboxylesterase enzyme and contributes about 75% of the drug exposure. The deuteration of the N-CH3 moiety enzalutamide to the N-CD3 decreases the N-demethylation pathway. So, HC-1119, a deuterated form of enzalutamide developed for the management of metastatic castration-resistant prostate cancer.26,27
Due to the kinetic isotope effect, HC-1119 showed slow metabolism and increased drug exposure, which help to decrease the dose of HC-1119 to get similar efficacy with fewer side effects.27 HC-1119 showed higher drug concentrations and a better antitumor effect than the enzalutamide in vivo model. The AUC of HC-1119 in mice, rats, and dogs was higher than that of enzalutamide.28 Besides, studies revealed that HC-1119 has a lower CNS concentration than enzalutamide at similar plasma concentration levels.27 Thus, the differentiation of HC-1119 from enzalutamide gives warrants clinical development. At the steady-state, 80 mg HC-1119 achieved an effective plasma concentration equivalent to 160 mg enzalutamide. This favorable PK profile makes HC-1119 may offer a reduced dosage and higher safety margin compared to enzalutamide.28,29 As the HC-1119 cannot cross the blood–brain barrier, which reduces the central nervous system adverse effects of enzalutamide, and increase its safety.
Currently, five clinical trials for HC-1119 are undertaken on Clinicaltrials.gov. Three of these are in Phase I (NCT03776968, NCT03774056, and NCT03778047). The remaining two studies are Phase III (NCT03851640, NCT03850795).
Dosimertinib: A Deuterated Osimertinib
The epidermal growth factor receptor (EGFR) is a tyrosine kinase receptor that has a vital role in different biological processes such as cell survival, proliferation, migration, and differentiation. EGFR tyrosine kinase inhibitors such as gefitinib, erlotinib, afatinib, dacomitinib, rociletinib, and osimertinib provide a significant clinical benefit.30 Particularly, osimertinib show significant clinical outcome with fewer adverse reactions and is a standard drug to manage advanced EGFR mutation-positive non-small-cell lung cancer.31
Several studies show that osimertinib metabolism is undertaken by CYP450 to produce two major active metabolites: AZ5104 and AZ7550, but AZ5104 has reduced selectivity and causes severe toxicities. Therefore, improving favorable PK profiles of osimertinib may be feasible to decrease the toxicity and achieve an excellent clinical outcome.32 Dosimertinib deuteraterated analog of osimertinib, has better pk properties that increase the concentration of the drug in plasma, achieving better clinical benefits for the safety and excretion of the drugs. Several studies showed that dosimertinib has robust in vivo antitumor efficacy and better PK profiles, with lesser toxicity than osimertinib.33 Currently, Dosimertinib is in Phase I trial (CXHL2000060 and CXHL2000061).
MBRI-001: A Deuterated Plinabulin
Plinabulin is a derivative of the marine natural bioactive compound “diketopiperazine phenylahistin”, which showed potent depolymerization effects on microtubules. Plinabulin also acts its effect via the anti-angiogenesis, interruption of tumor blood flow, and induction of cancer apoptosis via the c-Jun N-terminal kinase (JNK) pathway. Currently, the combination of plinabulin and docetaxel is under Phase 3 trial for the management of lung cancers.34 But, plinabulin Showed low efficacy due to its poor PKs character. So, MBRI-001, the deuterated analog of plinabulin was developed with better pk property and antitumor effects. MBRI-001 has rapid distribution in various tissues, especially in the lung which has a significant concentration difference from other tissues. MBRI-001 showed better stability in rat and liver microsomes than plinabulin in vitro. MBRI-001 showed greater activity on microtubule hepatocellular carcinoma than that of plinabulin. MBRI-001 showed better efficacy against cancer growth with lower toxicity in mice than docetaxel.35,36 The combination of MBRI-001 with gefitinib showed a significant antitumor effect than monotherapy.35 The combination of MBRI-001 and sorafenib showed a better antitumor activity that may provide a new approach for treating HCC in the future.36 Therefore, MBRI-001 could be the most promising anti-cancer agent in the future.
Hc-1144: A Deuterated Tivozanib
The phosphorylation of vascular endothelial growth factor receptors (VEGFR) leads to develop new tumors. Tivozanib is a potent and selective VEFGR inhibitor. Besides, the half-life of tivozanib is four days, and once-daily administration keeps effective serum concentrations. Tivozanib provided favorable progression-free and overall survival for patients than sorafenib. Tivozanib inhibits angiogenesis, vascular permeability in tumor tissues, and tumor growth. However, Tivozanib causes hypertension (34–44%) and dysphonia (5–21%) compared to sorafenib, a less effective VEFGR inhibitor.37,38 The deuterated form of Tivozanib (HC-1144) was as effective in vitro as Tivozanib itself. But, HC-1144 had a larger half-life, larger max concentration (Cmax, meaning smaller doses can be given), 1.5 times larger AUC (meaning the drug stayed in the body for a long time), and a similar peak time (meaning the release of the drug was not further delayed).39
CTP-221: A Deuterated (S)-Lenalidomide
Lenalidomide is a derivative of thalidomide with pleiotropic effects in human malignancies. lenalidomide has a plethora of anti-cancer characteristics including anti-proliferative, anti-angiogenic, immunomodulatory, and pro-erythropoietic properties. Lenalidomide in combination with dexamethasone approved for the management of myelodysplastic syndromes-associated anemia, and in relapsed/refractory multiple myeloma. Lenalidomide in combination with rituximab is approved for the management of relapsed mantle cell lymphoma, previously treated follicular lymphoma, and marginal zone lymphoma. In 2020, the lenalidomide combination with tafasitamab a cytolytic CD19 targeting monoclonal antibody approved to manage relapsed diffuse large B cell lymphoma.40
Lenalidomide has a short half-life (3–4 h). lenalidomide is a mixture of R and S-enantiomers that undergoes rapid chiral inversion through epimerization. The S-enantiomer possesses a potent antitumor effect, whereas the R-deuterated enantiomer does not affect tumor growth. The administration of S-enantiomer lenalidomide is less effective, because it rapidly converts in vivo to the R/S mixture.41,42 CTP-221 is a deuterated S-enantiomer of lenalidomide at key positions, which decreases the interconversion of enantiomers in-vivo. This helps to administer the potent S-enantiomer with less exposure to R-enantiomer. The preclinical studies showed that CTP-221 has more potency than racemic lenalidomide.17,19 The rates of epimerization of CTP-221 and S-lenalidomide were compared and CTP-221 was 2–3 times stable to epimerization than S-lenalidomide.43
Deuterated Tamoxifen
Tamoxifen is a nonsteroidal antiestrogen used as an adjunct chemotherapeutic agent to treat breast cancer. However, tamoxifen increases endometrial cancer risk and hepatic tumor. Tamoxifen induces and promotes tumors via its estrogenic activity. The proposed metabolic pathway of tamoxifen to a reactive electrophile involves allylic α-carbon oxidation that produces a reactive quinone methide.44 The deuterium replacement reduces (5-times) covalent bond formation to hepatic proteins in both mice. It also decreased binding with CYP3A4 enzymes. Genotoxicity of tamoxifen decreased 2 to 3 fold in-vivo in rats and in vitro in an MCL-5 human cell line that retains CYP450 activity by using d5-tamoxifen in which deuterium is substituted for hydrogen in the allylic ethyl group. These and other results suggest that liver carcinogenicity in rats caused by tamoxifen involves allylic α-carbon oxidation that may generate a reactive quinone methide.45,46
Brp800: A Deuterated Dasatinib
The pan-Src family kinase inhibitor dasatinib is approved for the treatment of chronic myeloid leukemia but has less effect in the treatment of lung cancer.47 BRP800 is a deuterated form of dasatinib Showed a novel activity, such as inhibition of cell proliferation by inhibiting cell cycle signals via targeting the cyclin D-CDK4/6-pRb-E2F pathway. Deuterium substitution keeps inhibition of c-Src activity and showed better anti-non-small cell lung cancer effect.
BRP800 mainly showed an antiproliferative but not proapoptotic effect. BRP800 has no significant effect on antiapoptotic gene expressions such as Bcl-2 and Mcl1, or the induction of apoptotic enzymes such as caspase 3, 8, or 9. But, BRP800 decreased the induction of cell cycle promoting genes such as cyclins D1, D3, E, A, and CDK4 and induced cell cycle negative regulators including p21, p27, and p53. Based on these findings, BRP800 arrest cells division at the G0/G1 phase in a dose-dependent manner, and G0/G1 fraction was increased from 64% in control to 85% in BRP800-treated cells. Compared with docetaxel, BRP800 is potent with a similar antitumor effect but less adverse reaction. Based on these finding, BRP800 is an antiproliferative agent that block c-Src and cell cycle progression.48
Donafenib: A Deuterated Sorafenib
Hepatocellular carcinoma (HCC) is the most prevalent kind of liver cancer. The incidence of liver cancer is increasing and greater than half of the newly diagnosed patients are at advanced stage that is illegible for surgical procedures. Advanced HCC is resistant to conventional chemotherapy. However, the development of specific target-acting drugs such as tyrosine kinase inhibitors increased the survival of patients with HCC.47 Sorafenib is the drug of choice for the treatment of advanced HCC patient. However, sorafenib has a limited effect due to tumor heterogeneity, tumor escape, and the lack of biomarkers for the response to the treatment. Besides, it has adverse effects such as fatigue, hypertension, rash, abdominal pain hand-foot skin reaction, and diarrhea.49,50
Donafenib is a derivative of sorafenib with a trideuterated N-methyl group that improves the pharmacokinetic and safety profile. Preclinical, phase Ia and Ib trials have shown favorable efficacy and safety profile.51 A phase II/III clinical trial revealed that donafenib (200mg twice daily) had better efficacy than sorafenib (400mg twice daily) in patients with metastatic HCC. Besides, due to the lower dose of the drug, serious side effects were reported in the donafenib group than in the sorafenib group. Currently, donafenib used to treat advanced HCC and trials are being undertaken on its efficacy in several solid tumor types.52,53 In June 2021, donafenib approved to manage patient with unresectable HCC who have not previously received systemic treatment.54
Conclusion
Deuterium kinetic isotope effect decreases the rate of metabolism that increases the plasma half-life the drug. In case of toxic metabolites production, deuteration of the drug may help to reduced toxic metabolites production and increase the exposure of the parent drug for the target site. It is difficult to determine the effects of deuterium replacement on PKs character of a drug. Improving the PKs and toxicological profile of the parent drugs by deuteration is promising for the treatment of cancer. Most drug development failure related to three main reasons: these are poor PKs property, lack of efficacy and severe adverse reaction. Deuterated drugs may improve these limitations of the parent drug. Since most trials under taken on non-deuterated versions, there is great hope that this will enable faster, smarter and cheaper trials of the deuterated version. Therefore, the advancement of the deuterated compounds in clinical trials is highly promising and list compounds listed in Table 1.
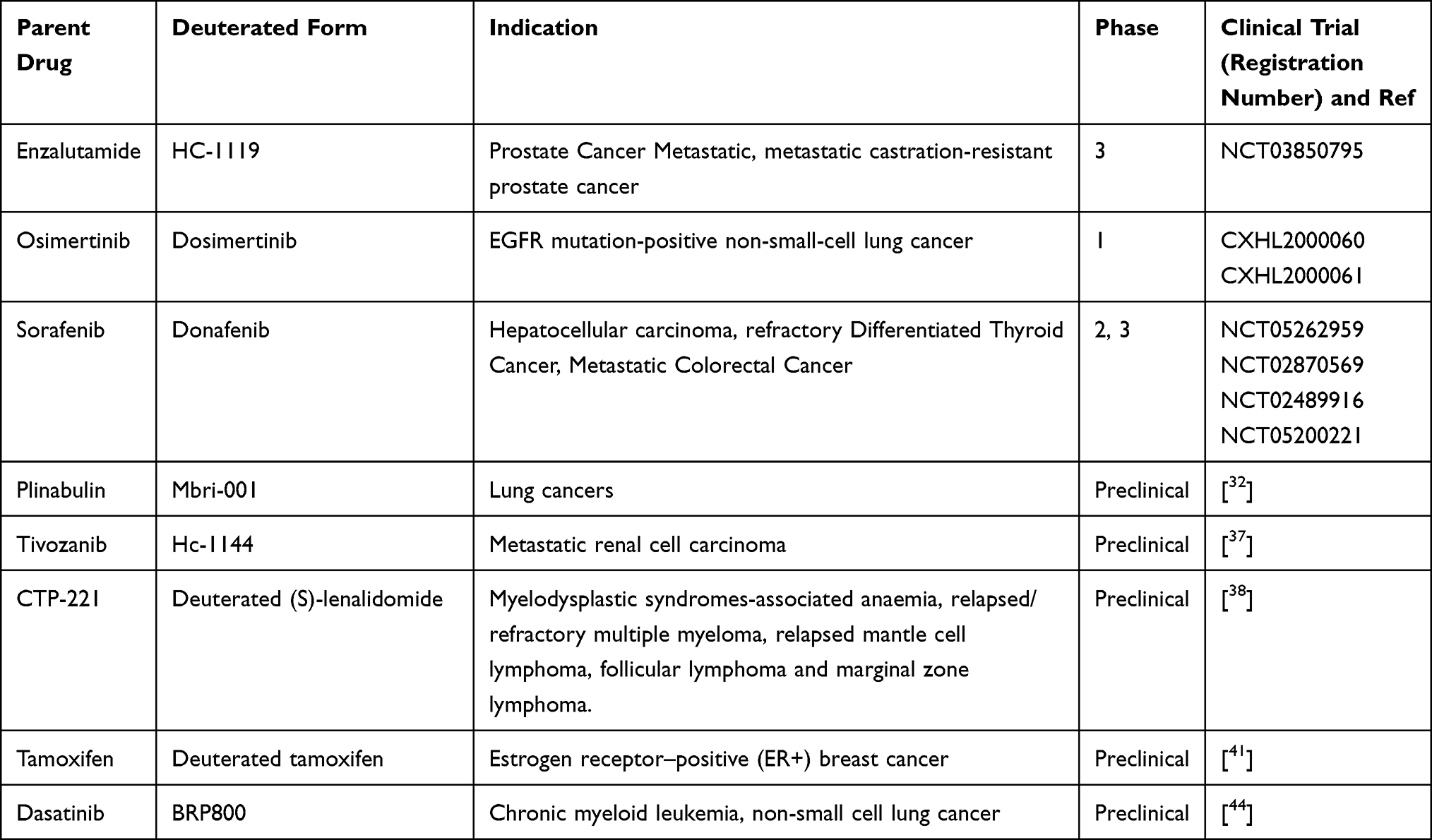 |
Table 1 List of Deuterated Anticancer Drug in Preclinical and Clinical Trial |
Abbreviations
AUC, area under the curve; PK, pharmacokinetic; KIE, kinetic isotope effect; H, hydrogen; D, deuterium; EGFR, epidermal growth factor receptor; VEGFR, vascular endothelial growth factor receptors; HCC, hepatocellular carcinoma.
Data Sharing Statement
The data supporting the findings of the article is available in this article.
Acknowledgments
I would like to acknowledge Mrs. Fasika Abu for editing the paper.
Disclosure
The authors declare no conflicts of interest, financial or otherwise.
References
1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
2. Adams DJ. The Valley of Death in anticancer drug development: a reassessment. Trends Pharmacol Sci. 2012;33(4):173–180. doi:10.1016/j.tips.2012.02.001
3. Moridani M, Harirforoosh S. Drug development and discovery: challenges and opportunities. Drug Discov Today. 2014;19(11):1679. doi:10.1016/j.drudis.2014.06.003
4. Sunny S, Thampi A, Shetty N, Babasahib SK, Chacko C. Assessment of adverse effects of most commonly prescribed anticancer drugs in a Tertiary Care Teaching Hospital. Indian J Pharm Pract. 2017;10(4). doi:10.5530/ijopp.10.4.55
5. Słoczyńska K, Gunia-Krzyżak A, Koczurkiewicz P, et al. Metabolic stability and its role in the discovery of new chemical entities. Acta Pharmaceutica. 2019;69(3):345–361. doi:10.2478/acph-2019-0024
6. Gajula SN, Nadimpalli N, Sonti R. Drug metabolic stability in early drug discovery to develop potential lead compounds. Drug Metab Rev. 2021;53(3):459–477. doi:10.1080/03602532.2021.1970178
7. Kaur S, Gupta M. Deuteration as a tool for optimization of metabolic stability and toxicity of drugs. Glob J Pharmaceu Sci. 2017;1:555566.
8. Harbeson SL, Tung RD. Deuterium medicinal chemistry: a new approach to drug discovery and development. Med Chem News. 2014;24(2):8–22.
9. Perrin CL, Ohta BK, Kuperman J, Liberman J, Erdélyi M. Stereochemistry of β-deuterium isotope effects on amine basicity. J Am Chem Soc. 2005;127(26):9641–9647. doi:10.1021/ja0511927
10. Harbeson SL, Tung RD. Deuterium in drug discovery and development. Annu Rep Med Chem. 2011;46(1):403–417.
11. Blake MI, Crespi HL, Katz JJ. Studies with deuterated drugs. J Pharm Sci. 1975;64(3):367–391. doi:10.1002/jps.2600640306
12. DeWitt SH, Maryanoff BE. Deuterated drug molecules: focus on FDA-approved deutetrabenazine: published as part of the biochemistry series “biochemistry to bedside”. Biochemistry. 2018;57(5):472–473. doi:10.1021/acs.biochem.7b00765
13. Paton DM. Deutetrabenazine: treatment of hyperkinetic aspects of Huntington’s disease, tardive dyskinesia and Tourette syndrome. Drugs Today. 2017;53(2):89–102. doi:10.1358/dot.2017.53.2.2589164
14. Fusch CH, Hungerland E, Scharrer B, Moeller H. Water turnover of healthy children measured by deuterated water elimination. Eur J Pediatr. 1993;152(2):110–114. doi:10.1007/BF02072485
15. Russak EM, Bednarczyk EM. Impact of deuterium substitution on the pharmacokinetics of pharmaceuticals. Ann Pharmacother. 2019;53(2):211–216. doi:10.1177/1060028018797110
16. Zhan Z, Peng X, Sun Y, Ai J, Duan W. Evaluation of deuterium-labeled JNJ38877605: pharmacokinetic, metabolic, and in vivo antitumor profiles. Chem Res Toxicol. 2018;31(11):1213–1218. doi:10.1021/acs.chemrestox.8b00191
17. Dean M, Sung VW. Review of deutetrabenazine: a novel treatment for chorea associated with Huntington’s disease. Drug Des Devel Ther. 2018;12:313. doi:10.2147/DDDT.S138828
18. Rao N, Kini R, Kad P. Deuterated Drugs. Pharma Chem J. 2022;55(12):1372–1377. doi:10.1007/s11094-022-02584-4
19. Sharma R, Strelevitz TJ, Gao H, et al. Deuterium isotope effects on drug pharmacokinetics. I. System-dependent effects of specific deuteration with aldehyde oxidase cleared drugs. Drug Metabol Disposition. 2012;40(3):625–634. doi:10.1124/dmd.111.042770
20. Pirali T, Serafini M, Cargnin S, Genazzani AA. Applications of deuterium in medicinal chemistry. J Med Chem. 2019;62(11):5276–5297. doi:10.1021/acs.jmedchem.8b01808
21. Hok L, Mavri J, Vianello R. The effect of deuteration on the H2 receptor histamine binding profile: a computational insight into modified hydrogen bonding interactions. Molecules. 2020;25(24):6017. doi:10.3390/molecules25246017
22. Kržan M, Keuschler J, Mavri J, Vianello R. Relevance of hydrogen bonds for the histamine H2 receptor-ligand interactions: a lesson from deuteration. Biomolecules. 2020;10(2):196. doi:10.3390/biom10020196
23. Leek H, Thunberg L, Jonson AC, Öhlén K, Klarqvist M. Strategy for large-scale isolation of enantiomers in drug discovery. Drug Discov Today. 2017;22(1):133–139. doi:10.1016/j.drudis.2016.09.018
24. Seebach D, Sting AR, Hoffmann M. Self‐regeneration of stereocenters (SRS)—applications, limitations, and abandonment of a synthetic principle. Angewandte Chemie Int Edition. 1996;35(2324):2708–2748. doi:10.1002/anie.199627081
25. Cargnin S, Serafini M, Pirali T. A primer of deuterium in drug design. Future Med Chem. 2019;11(16):2039–2042. doi:10.4155/fmc-2019-0183
26. Pang X, Peng L, Chen Y. Effect of N‐methyl deuteration on pharmacokinetics and pharmacodynamics of enzalutamide. J Labelled Comp Radiopharm. 2017;60(9):401–409. doi:10.1002/jlcr.3516
27. Li X, Cheng K, Li X, et al. Phase I clinical trial of HC‐1119: a deuterated form of enzalutamide. Int J Cancer. 2021;149(7):1473–1482. doi:10.1002/ijc.33706
28. Wu X, Feng W, Yang M, et al. HC-1119, a deuterated enzalutamide, inhibits migration, invasion and metastasis of the AR-positive triple-negative breast cancer cells. Mol Biol Rep. 2022;8(49). doi:10.1007/s11033-022-07749-8
29. Ma H, Xu W, Ni J, et al. Phase I clinical trial of HC‐1119 soft capsule in Chinese healthy adult male subjects: pharmacokinetics and safety of single‐dose proportionality and effects of food. Prostate. 2022;82(2):276–285. doi:10.1002/pros.24271
30. Hojjat-Farsangi M. Small-molecule inhibitors of the receptor tyrosine kinases: promising tools for targeted cancer therapies. Int J Mol Sci. 2014;15(8):13768–13801. doi:10.3390/ijms150813768
31. Yang Z, Yang N, Ou Q, et al. Investigating novel resistance mechanisms to third-generation EGFR tyrosine kinase inhibitor osimertinib in non–small cell lung cancer patients. Clin Cancer Res. 2018;24(13):3097–3107. doi:10.1158/1078-0432.CCR-17-2310
32. Ishikawa E, Yokoyama Y, Chishima H, et al. Development and validation of a new liquid chromatography-tandem mass spectrometry assay for the simultaneous quantification of Afatinib, dacomitinib, osimertinib, and the active metabolites of osimertinib in human serum. J Chromatogr B. 2022;1199:123245. doi:10.1016/j.jchromb.2022.123245
33. Meng Y, Yu B, Huang H, et al. Discovery of dosimertinib, a highly potent, selective, and orally efficacious deuterated EGFR targeting clinical candidate for the treatment of non-small-cell lung cancer. J Med Chem. 2021;64(2):925–937. doi:10.1021/acs.jmedchem.0c02005
34. Heist RS, Aren OR, Mita AC, et al. Randomized Phase 2 trial of plinabulin (NPI-2358) plus docetaxel in patients with advanced non-small cell lung cancer (NSCLC). J Clin Oncol. 2014;32(15_suppl):8054. doi:10.1200/jco.2014.32.15_suppl.8054
35. Ma M, Zhao J, Cheng H, et al. In vitro and in vivo pharmacokinetic and pharmacodynamic study of MBRI-001, a deuterium-substituted plinabulin derivative as a potent anti-cancer agent. Bioorg Med Chem. 2018;26(16):4687–4692. doi:10.1016/j.bmc.2018.08.009
36. Deng M, Li L, Zhao J, Yuan S, Li W. Antitumor activity of the microtubule inhibitor MBRI-001 against human hepatocellular carcinoma as monotherapy or in combination with sorafenib. Cancer Chemother Pharmacol. 2018;81(5):853–862. doi:10.1007/s00280-018-3547-2
37. Jamil MO, Hathaway A, Mehta A. Tivozanib: status of development. Curr Oncol Rep. 2015;17(6):1–7. doi:10.1007/s11912-015-0451-3
38. Molina AM, Hutson TE, Nosov D, et al. Efficacy of tivozanib treatment after sorafenib in patients with advanced renal cell carcinoma: crossover of a phase 3 study. Eur J Cancer. 2018;94:87–94. doi:10.1016/j.ejca.2018.02.009
39. Guo S, Pang X, Peng L, et al. Design, synthesis and biological evaluation of deuterated Tivozanib for improving pharmacokinetic properties. Bioorg Med Chem Lett. 2015;25(11):2425–2428. doi:10.1016/j.bmcl.2015.03.088
40. Ioannou N, Jain K, Ramsay AG. Immunomodulatory drugs for the treatment of B cell malignancies. Int J Mol Sci. 2021;22(16):8572. doi:10.3390/ijms22168572
41. Chen N, Zhou S, Palmisano M. Clinical pharmacokinetics and pharmacodynamics of lenalidomide. Clin Pharmacokinet. 2017;56(2):139–152. doi:10.1007/s40262-016-0432-1
42. Hanashima Y, Sano E, Sumi K, et al. Antitumor effect of lenalidomide in malignant glioma cell lines. Oncol Rep. 2020;43(5):1580–1590. doi:10.3892/or.2020.7543
43. Uttamsingh V, Gallegos R, Cheng C, et al. CTP-221, a deuterated S-enantiomer of lenalidomide, is greatly stabilized to epimerization and results in a more desirable pharmacokinetic profile than racemic lenalidomide. Cancer Res. 2013;73(8):3357. doi:10.1158/1538-7445.AM2013-3357
44. Garrido J, Garrido MP, Maria Oliveira-Brett A, Borges F. An electrochemical outlook on tamoxifen biotransformation: current and future prospects. Curr Drug Metab. 2011;12(4):372–382. doi:10.2174/138920011795202965
45. Jarman M, Poon GK, Rowlands MG, et al. The deuterium isotope effect for the α-hydroxylation of tamoxifen by rat liver microsomes accounts for the reduced genotoxicity of [D5-ethyl] tamoxifen. Carcinogenesis. 1995;16(4):683–688. doi:10.1093/carcin/16.4.683
46. Gjerde J, Kisanga ER, Hauglid M, Holm PI, Mellgren G, Lien EA. Identification and quantification of tamoxifen and four metabolites in serum by liquid chromatography–tandem mass spectrometry. J Chromatogr A. 2005;1082(1):6–14. doi:10.1016/j.chroma.2005.01.004
47. Rupniewska E, Roy R, Mauri FA, et al. Targeting autophagy sensitises lung cancer cells to Src family kinase inhibitors. Oncotarget. 2018;9(44):27346. doi:10.18632/oncotarget.25213
48. Ling C, Chen G, Chen G, et al. A deuterated analog of dasatinib disrupts cell cycle progression and displays anti‐non‐small cell lung cancer activity in vitro and in vivo. Int J Cancer. 2012;131(10):2411–2419. doi:10.1002/ijc.27504
49. Miller RE, Brough R, Bajrami I, et al. Synthetic lethal targeting of ARID1A-mutant ovarian clear cell tumors with dasatinib. Mol Cancer Ther. 2016;15(7):1472–1484. doi:10.1158/1535-7163.MCT-15-0554
50. Luo XY, Wu KM, He XX. Advances in drug development for hepatocellular carcinoma: clinical trials and potential therapeutic targets. J Exp Clin Cancer Res. 2021;40(1):1–23. doi:10.1186/s13046-021-01968-w
51. Negri F, Porta C. Donafenib in Chinese patients with advanced hepatocellular carcinoma (HCC): really a new standard of care, or should we change paradigm for drug development in HCC? Oncol Rev. 2021;15(2). doi:10.4081/oncol.2021.564
52. Li Q, Zhu H. Donafenib treatment for hepatocellular carcinoma: a case report. Medicine. 2021;100(25):e26373.
53. Qin S, Bi F, Gu S, et al. Donafenib versus sorafenib in first-line treatment of unresectable or metastatic hepatocellular carcinoma: a randomized, open-label, parallel-controlled phase II-III trial. J Clin Oncol. 2021;39(27):3002–3011. doi:10.1200/JCO.21.00163
54. Keam SJ, Duggan S. Donafenib: first approval. Drugs. 2021;81(16):1915–1920. doi:10.1007/s40265-021-01603-0
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.