Back to Journals » Drug Design, Development and Therapy » Volume 14
Recent Progress in the Development of New Antimalarial Drugs with Novel Targets
Authors Belete TM ![]()
Received 1 June 2020
Accepted for publication 9 September 2020
Published 22 September 2020 Volume 2020:14 Pages 3875—3889
DOI https://doi.org/10.2147/DDDT.S265602
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Tin Wui Wong
Tafere Mulaw Belete
Department of Pharmacology, College of Medicine and Health Sciences, University of Gondar, Gondar, Ethiopia
Correspondence: Tafere Mulaw Belete Tel +251 918045943
Email [email protected]
Abstract: Malaria is a major global health problem that causes significant mortality and morbidity annually. The therapeutic options are scarce and massively challenged by the emergence of resistant parasite strains, which causes a major obstacle to malaria control. To prevent a potential public health emergency, there is an urgent need for new antimalarial drugs, with single-dose cures, broad therapeutic potential, and novel mechanism of action. Antimalarial drug development can follow several approaches ranging from modifications of existing agents to the design of novel agents that act against novel targets. Modern advancement in the biology of the parasite and the availability of the different genomic techniques provide a wide range of novel targets in the development of new therapy. Several promising targets for drug intervention have been revealed in recent years. Therefore, this review focuses on the progress made on the latest scientific and technological advances in the discovery and development of novel antimalarial agents. Among the most interesting antimalarial target proteins currently studied are proteases, protein kinases, Plasmodium sugar transporter inhibitor, aquaporin-3 inhibitor, choline transport inhibitor, dihydroorotate dehydrogenase inhibitor, isoprenoid biosynthesis inhibitor, farnesyltransferase inhibitor and enzymes are involved in lipid metabolism and DNA replication. This review summarizes the novel molecular targets and their inhibitors for antimalarial drug development approaches.
Keywords: drug resistance, novel targets, antimalarial agents, mode of action, Plasmodium
Background
Malaria is a devastating parasitic infectious disease, especially in Sub-Saharan African, parts of Asia, and South America. Even though several efforts undertaken, nowadays, it is one of the major causes of morbidity and mortality, mainly in pregnant women and children. According to the World Health Organization (WHO) report in 2018, there were 228 million malaria cases and 405 000 deaths in the world. Almost half the world’s population is at risk of malaria disease, most cases (93%), and deaths (94%) reported in Africa. About 125 million pregnancies are at risk of malaria every year, and 272,000 Children aged less than five years die due to malaria.1 Malaria is also the cause of poverty and a major hindrance to economic development, mainly in Africa.2 The five identified Plasmodium species responsible for inflicting malaria in humans are P. vivax, P. knowlesi, P. ovale, P. malariae, and P. falciparum. Of these, P. falciparum is the most virulent and prevalent Plasmodium species.3
In the absence of an effective vaccine, the therapeutic use of antimalarial agents remains the only method for management and prophylaxis of malarial disease. Several studies showed that the efficacies of most antimalarial agents compromised by the emergency of drug-resistant Plasmodium species.4 Resistance reported for nearly all available antimalarial agents that reinforced the urgent need to develop new antimalarial agents against existing validated targets, as well as to search for novel targets.5 The development of a novel antimalarial agent that acts toward both transmissible gametocytes stages and the intraerythrocytic proliferative asexual, in particular, those of resistant parasite species, is urgently required.6 Several enzymes, ion channels, transporters, interacting molecules in red blood cell (RBC) invasion, and molecules responsible for oxidative stress in the parasite, lipid metabolism, and degradation of hemoglobin are promising novel targets to develop new antimalarial drugs against rapidly mutating malarial parasites.7
The potential of new antimalarial agent judged by several requirements: novel modes of action with no cross-resistance to the current antimalarial agent, single-dose cures, effective against both the asexual blood stages and the gametocytes responsible for transmission. Besides, the new antimalarial agent should have the efficacy to prevent infection (chemoprotective agents), and clear P. vivax hypnozoites from the liver (anti-relapse agents).8
Traditional drug discovery follows many approaches to identifying a new ant-malarial agent to combat malaria disease. These approaches are optimization of current drug regimens and formulations, modifying the existing antimalarial agents, screening of natural products, isolating resistance-reversal agents, utilizing combination chemotherapeutic approaches, and exploiting drugs indicated for other uses.8,9
Besides the traditional drug discovery methods for the identification of new ant-malarial agent, the understanding of Plasmodium cell biology and genome has proved a powerful tool for uncovering mechanisms of resistance, and have great potential to design a novel drugs with both high antimalarial activity and transmission-blocking potential to combat malaria once and for all.10 Genetic screening in P. falciparum identified 2680 genes that are important for asexual blood-stage growth resulting identification of crucial cellular processes, which is vital to develop new a drug.10,11 The new drug should: (i) address drug-resistance issues, (ii) have a rapid onset of action, (iii) be safe, especially in children and pregnant women, and (iv) cure malaria in a single dose.12 The challenge is to find a drug that addresses all of these features. The objective of this review is to have a view on novel targets in the treatment of malaria parasites, which are under investigation by several companies so that readers will have information on previously done works.
Antimalarial Drugs
Currently, most antimalarial agents are targeting the asexual phase of malaria infection that causes symptomatic illness. The pre-erythrocytic (hepatic) stage remains unattractive because clinical symptoms not generated. Antimalarial drugs exhibit considerable stage selectivity of action (see Figure 1).Malarial treatment based on natural products, semi-synthetic, and synthetic compounds developed since the 1940s.13 The existing antimalarial agents grouped into three main classes: quinoline derivative, antifolate, and artemisinin derivatives. There is no single drug has been identified or manufactured that can wipe out all strains of the Plasmodium species. Thus, to fight malarial infection efficiently, the combination of drugs frequently administered at the same time. Quinolines are the most widely used antimalarial agents for the management of malaria. Quinine is an alkaloid isolated from the bark of Cinchona trees, was the first antimalarial agent used for the management of disease in the 17th century. Quinine became the standard therapy for malaria from the mid-19th century to the 1940s.14 The emergence of resistant strains of P. falciparum, in addition to toxicity, has limited the therapeutic use of Quinine. However, Quinine still used against severe malaria, most often combined with a second agent to decrease treatment duration and to minimize the side effects.15,16
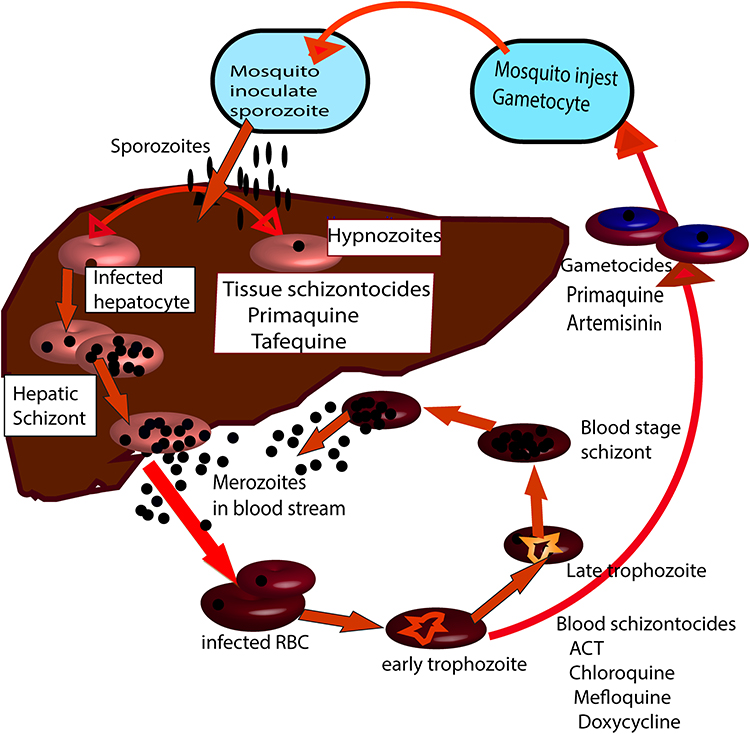 |
Figure 1 The life cycle of Plasmodium parasite in man. Stages and forms of the parasite at which different types of antimalarial drug acts. |
In 1925 the German researchers discovered pamaquine the first synthetic antimalarial agent by modifying methylene blue. Because of limited efficacy and toxicity pamaquine, cannot use for the treatment of malaria. But pamaquine provides lead compounds to develop better antimalarial agents. Mepacrine (quinacrine) is another derivative of methylene blue used to treat malaria during World War II.17
Chloroquine developed during World War II to treat malaria. Chloroquine is a drug of choice to treat malarial due to its efficacy, safety, and low cost. But its irrational use soon led to the emergence of chloroquine-resistant P. falciparum species.18 Primaquine therapeutically used to treat relapsing P. vivax caused by hypnozoites. Primaquine has a potent gametocytocidal effect against P. falciparum. Primaquine causes hemolytic anemia in glucose-6-phosphate dehydrogenase (G6PD) deficient patient. This hemolytic effect intensified the need to obtain new drugs with anti-P. vivax activity.19
New quinoline derivatives were synthesized, giving new drugs such as piperaquine, amodiaquine, and others. After the emergence of chloroquine resistance, amodiaquine, a phenyl-substituted analog of chloroquine, displayed excellent efficacy against chloroquine-resistant strains of P. falciparum.20 Pyronadrine is a Mannich base antimalarial agent developed in 1970 in China. It is effective against drug-resistant P. falciparum strains, P. vivax, P. malariae, and P. ovale. Pyronadrine is now available as ACT with artesunate, which displays excellent efficacy against all malaria parasites.21 Mefloquine developed in the mid-1980s and currently recommended for chemoprophylaxis of malaria caused by all species including chloroquine resistance strains. However, its use associated with several side effects and drug resistance.22 Drugs from Quinolines derived mostly act on blood stages of the parasite, but some antimalarial agents act at the hepatic stage. These drugs inhibit by forming a complex with haem in the parasite food vacuole. So, haem polymerization blocked. As a result, the haem that released during hemoglobin breakdown builds up to toxic levels, thereby killing the parasite with its toxic waste.23
Antifolates are antimalarial agents that inhibit folic acid synthesis, which is crucial for nucleotide and amino acid synthesis. Antifolates block the nuclear division of the Plasmodium species at the schizont stage within the erythrocytes and hepatocytes. Sulfadoxine have similar structures to p-aminobenzoic acid (PABA), a component of folic acid. They inhibit dihydrofolic acid synthesis by inhibiting dihydropteroate synthase, a crucial enzyme for the biosynthesis of nucleic acids.24
Pyrimethamine and Proguanil are schizonticidal antimalarial agents that act at the asexual forms of the Plasmodium species. These drugs inhibit the dihydrofolate reductase (DHFR) enzyme, inhibiting the reduction of dihydrofolate to tetrahydrofolate, which is vital for the biosynthesis of amino acids and nucleic acids. Proguanil is a prodrug that metabolized to cycloguanil. Proguanil is the first antifolate drug developed for malaria treatment. The reason is it destroys the parasites before they invade erythrocytes during their passage into the bloodstream. Besides, proguanil is a safe drug. Pyrimethamine mostly used with other faster-acting drugs. However, its use decreased due to resistance.24,25
Atovaquone is the first approved antimalarial agent that targets the Plasmodium mitochondria. Atovaquone inhibits electron transport by blocking cytochrome b parts of the cytochrome bc1complex, by acting as a ubiquinone analog. When combined with proguanil, atovaquone is safe and effective for pregnant women and children. Atovaquone is effective against the sexual stages of the parasite in the host and the mosquito. So, it inhibits malaria transmission from mosquito to human. A fixed combination with proguanil developed under the trade name Malarone.24,26
In 1972 Artemisinin was extracted from Artemisia annua. Artemisinin and its derivatives including, artemether, dihydroartemisinin, arteether, and artesunate have broad-spectrum activity. Artemisinin inhibits all parasite phases within erythrocytes, particularly at the early phase of their development. It also suppresses gametocyte transmission from humans to mosquitoes.27 Artemisinin and its derivatives are effective against chloroquine and mefloquine-resistant strains. They are safe, potent, and rapid-acting blood schizonticides against all Plasmodium species. However, Artemisinin does not clear the latent liver stages of the parasite. These drugs have short a half-life and poor bioavailability which leads to drug resistance, making them ineffective as a monotherapy. Therefore, Artemisinin derivatives recommended for treatment in combination with other antimalarial agents.28
The antimalarial effect of artemisinins may result from the production of free radicals, produced due to cleavage of the artemisinin endoperoxide bridge in the parasite food vacuole, which inhibits the parasite calcium ATPase and proteasome.29,30 Artemether has a high recrudescence rate when used as a monotherapy. It has rapid oral absorption. The bioavailability doubled when administered in the presence of food. Once in the systemic circulation, artemether hydrolyzed in the gut and liver to dihydroartemisinin.
Artesunate is a semisynthetic derivative due to its rapid antimalarial action, lack of significant resistance and greater water solubility. It is recommended as a first-line drug for severe malaria.31
Tetracyclines and macrolides are slow-acting antimalarial drugs, used as an adjunct to quinine in treating P. falciparum. Doxycycline is also used for chemoprophylaxis in regions with high prevalent drug resistance.32 The current strategy used to fight antimalarial drug resistance is the therapeutic use of drug combinations. This strategy used in the past through the utilization of fixed combinations. WHO recommended artemisinin-based combination therapy (ACT) regime as a first-line to manage uncomplicated P. falciparum. The reason is the combination of the drug decreases the drug resistance and side-effects.33
The ACT contains a potent artemisinin component, which rapidly clears the parasites, plus a longer-acting drug, which eliminates remaining parasites and decreases artemisinin resistance. The ACTs recommended by WHO are artesunate/amodiaquine,artemether/lumefantrine, artesunate/mefloquine, artesunate/pyronaridine, dihydroartemisinin/piperaquine, artesunate/sulfadoxine/pyrimethamine, Arterolane/piperaquine and Artesinin/piperaquine/primaquine. Chloroquine plus primaquine remains the first-line for radical cure of P. vivax. Quinine + tetracycline/doxycycline has a high cure rate but has severe side effects and contraindicated for children and pregnant women.34
Travelers from non-endemic to endemic areas recommended, mefloquine, atovaquone/proguanil, or doxycycline in chemoprophylactic regimens.35 Intermittent preventive treatment advocated in high-risk populations, including sulfadoxine/pyrimethamine during pregnancy and amodiaquine/sulfadoxine-pyrimethamine as seasonal chemoprophylaxis.36 Halofantrine is unsuitable for therapeutic use because of its cardiotoxicity. Dapsone, Mepacrine, amodiaquine, and sulfonamides withdrawn from therapeutic use due to their side effects.36,37 Some of the well known antimalarial agents with their side effects presented in Table 1.
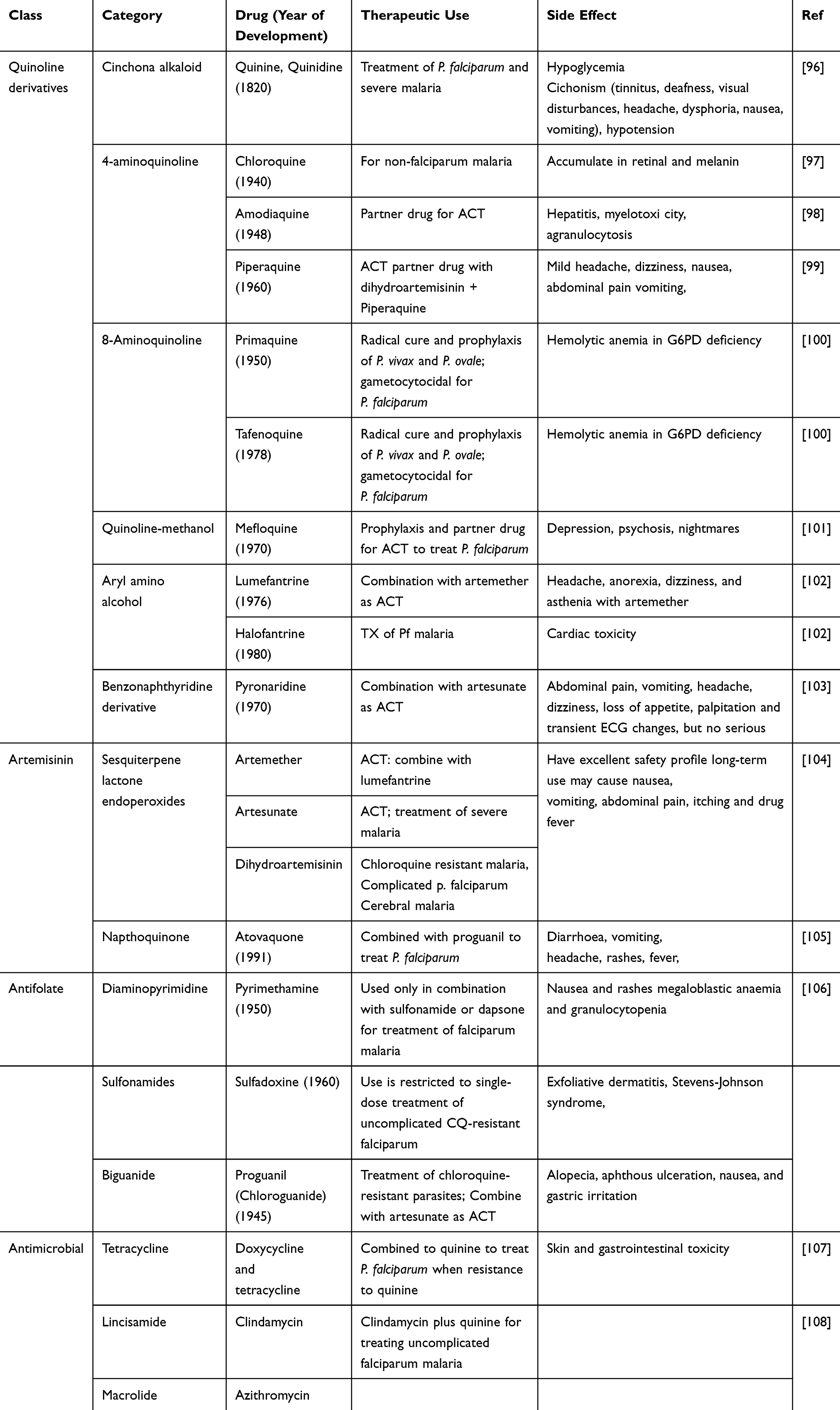 |
Table 1 The Summery of Antimalarial Drugs |
Novel Antimalarial Targets
The current available antimalarial agents identified based on the major metabolic pathway differences of the Plasmodium species with its host. The Major metabolic pathways of the parasite, including heme detoxification, fatty acid synthesis, nucleic acid synthesis, fatty acid synthesis, and oxidative stress are some of the novel sites for drug design.38,39 Although most antimalarial agents used for several years, currently their use limited due to drug resistance. Based on the literature, there is no antimalarial drug identified to inhibit a known drug target.7,40 Instead, most antimalarial drugs identified in vivo animal or in vitro model studies. Therefore, the mode of action for most antimalarial agents remains uncertain. Besides, the mechanism of resistance not well understood for most antimalarial agents.39
Malaria control needs coordinated strategies, like vector control, effective and safe antimalarial agents, and effective vaccines. Considering the high mortality and morbidity due to malaria, the emergency, and the spread of drug resistance, the ineffectiveness of available antimalarial drugs against non-erythrocyte and sexual stages, It is crucial to identify novel antimalarial agent by understanding the basic metabolic pathway of the parasite. To achieve this objective, drug research should direct on validated novel targets to isolate a new lead compound.39,41
The need for identifying novel metabolic targets comes from several reasons. Firstly, except atovaquone and artemisinin derived drugs, most antimalarial agents have no chemical diversity, which may lead to cross-resistance. Secondly, because of the confusing array of putative chemotherapeutic targets, so many have not been validated. If validated it may generate some effective and safe compounds. Identification of novel drug targets and the design of new compounds acting on new targets is nowadays widely used approach all over the world to combat issues raised by the emergence of resistance to existing drugs.40,41 Therefore, investigating inhibitors specific to the new target proteins of the malaria parasite has been exploited for drug target identification. Since the unveiling of the P. falciparum genome, several novel targets for drug intervention have emerged. These potential antimalarial drug targets the crucial metabolite biosynthesis, membrane transport, and signaling system, and the hemoglobin degradation processes.40,42
Malaria Parasite Proteases
Plasmodium proteases are a ubiquitous catalytic and regulatory enzyme that has a key role in the survival of the protozoan parasite and the diseases they cause. It catalyzes the hydrolysis of the peptide bond.43 The effect of Proteases in the pathogenesis malaria disease includes cell/tissue penetration, immune evasion, activation of inflammation, invasion of erythrocyte, hemoglobin and other proteins breakdown, autophagy, and development the parasite.44
Malarial proteases (glutamate aspartate, cysteine, metallo, serine, and threonine) are promising therapeutic targets because disruption of the malaria proteases gene inhibits the degradation of hemoglobin and the development of the parasite in the erythrocyte stages.45
The rupture of erythrocyte and the subsequent invasion by merozoites require malarial protease enzyme. A Synthetic peptide (GlcA-Val-Leu-Gly-Lys-NHC2H5) inhibits P. falciparum schizont cysteine protease Pf 68. It inhibits erythrocyte invasion and parasite development. This suggests that a protease plays a key role in erythrocyte invasion by the parasites. So, protease is a promising target for antimalarial drugs development.46
In P. falciparum food vacuole several aspartic proteases (plasmepsin I, II, III, IV) and cysteine proteases (falcipain-1, falcipain-2/, falcipain-3) have been isolated which used for degradation of hemoglobin as displayed in Figure 2.
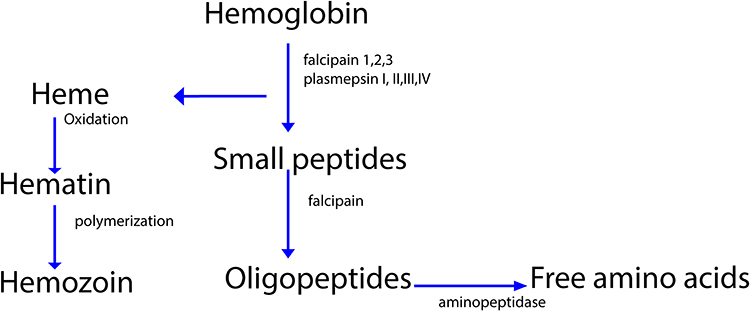 |
Figure 2 Degradation of hemoglobin by protease. |
Incubation of cultured P. falciparum parasites with the protease inhibitor leupeptin and E-64 causes accumulation of undegraded globin. Leupeptin inhibits both cysteine and some serine proteases, but E-64 specifically inhibit cysteine protease.47,48 After parasites incubation with aspartic protease inhibitor pepstatin, globin did not accumulate. Several studies showed that cysteine protease inhibitors not only inhibited globin degradation but also inhibited earlier steps in hemoglobin breakdown, like denaturation of the hemoglobin, the release of heme from globin, and production of hemozoin.49 These results suggest that cysteine protease is required for initial steps in hemoglobin degradation by P. falciparum. Both E-64 and pepstatin blocked P. falciparum development synergistically. However, only E-64 blocked globin hydrolysis.48,49 Several cysteine protease inhibitors like fluoromethyl ketones and vinyl sulfones inhibited P. falciparum growth and hemoglobin degradation. In the malaria animal model, fluoromethyl ketone inhibits P. vinckei protease activity and cured 80% of murine malaria infections. Thus, protease inhibitors are promising candidate antimalarial drugs. Subsequent works identified biologically active falcipain inhibitors, including chalcones, phenothiazines that block parasite metabolism and development.50
Serine protease involved in both schizont rupture and erythrocyte reinvasion in the P. falciparum life cycle. It can be blocked by several serine protease inhibitors and the best choice because no human enzyme homolog is available. The protease inhibitor LK3 isolated from Streptomyces species degrades serine protease of malaria.51 Maslinic acid is a natural pentacyclic triterpene that inhibits the maturation of the parasite from the ring stage to the schizont stage, which terminate the release of merozoites and its invasion. A series of highly potent 2-pyrimidine carbonitrile inhibit falcipain-2 and falcipain-3.52 Inhibition of plasmepsins by statine and allophenylnorstatin-based inhibitors block hemoglobin degradation and kill the parasites. There are several cysteine protease blockers available including, Epoxomicin, lactacystin, MG132, WEHI-842, WEHI-916, and chymostatin.
Phosphatidylinositol 4‑kinase (PfPI4K) Inhibitor
Phosphoinositide lipid kinases (PIKs) are ubiquitous enzymes that phosphorylate lipids to regulate proliferation, survival, trafficking, and intracellular signaling.53 The most widely studied classes of PIKs in the parasite are phosphoinositide 3-kinase (PI3K) and phosphatidylinositol 4-kinase (PI4K). Inhibition of these enzymes identified as a potential target for the development of antimalarial drugs with an ideal activity profile for the prevention, treatment, and elimination of malaria.54 UCT943, Imidazopyrazines (KAF156), and aminopyridine are a new antimalarial compound class that targets the PI(4)K and inhibits the intracellular development of multiple Plasmodium species at each stage of infection in the host. Therefore, targeting (PI3K) and PI(4)K may open up new avenues of target-based drug discovery to identify novel antimalaria drugs. KAF156 is currently in Phase II clinical trial.55,56 MMV048 is a compound that has good prophylactic activity against P.cynomolgi in vivo and has the promising potential to act as a transmission-blocking drug. MMV048 is currently in Phase IIa clinical trials in Ethiopia.11
Targeting Transporters
For rapid growth in infected erythrocyte, the Plasmodium species need enough amounts of substrates to fuel their vigorous metabolism. Hence, the parasite prepares the host erythrocyte by inducing specialized transporter, which is significantly different from the host cell transporter for the uptake and removal of metabolites. So, transporters like carrier proteins and channels are potential targets due to their major role as in the transport of metabolites, electrolytes, and nutrients.57 These are the plasmodial surface anion channel (PSAC) and parasitophorous vacuolar membrane (PVM), which provide a sequential diffusive pathway for nutrient entry to the intracellular parasite.58
PSAC is the most promising target due to its critical role in different types of nutrient acquisition (hypoxanthine, cysteine, glutamine, glutamate, isoleucine, methionine, proline, tyrosine, pantothenic acid, and choline) into the intracellular parasite. PSAC has no clear homology with known host channel genes.58,59 Phlorizin, Dantrolene, furosemide, and niflumate are effective anion transporter blockers. Drugs like glibenclamide, meglitinide, and tolbutamide inhibit the influx of choline into parasite-infected erythrocytes.60,61
Plasmodium Sugar Transporter Inhibitor
Blood forms of P. falciparum almost entirely depend on glycolysis for energy production, without energy stores; rely on constant uptake of glucose. The parasite converts Pyruvate to lactate to produce ATP, which require for replicating in the intraerythrocytic site.62 Glucose first transported from the blood into the parasitized erythrocyte via a combination of the host cell’s glucose transporter called GLUT1, which is abundant in the erythrocyte membrane and the parasite-induced ‘New Permeability Pathways’.63 The glucose transported into the parasite via P. Falciparium Hexose transporter (PFHT). PFHT has some of the typical sugar transporter features. GLUT1 is selective for D-glucose, whereas PFHT can transport both D-glucose and D-fructose. Therefore, the differences between GLUT1 and PFHT in terms of their interaction with substrates, proposed that selective inhibition of PFHT is a promising novel target to develop a new antimalarial drug.64 A long chain O‐3‐hexose derivative (Compound 3361) inhibits the uptake of glucose and fructose by PFHT but, it does not inhibit hexose transport by the major mammalian glucose and fructose transporters (GLUT1 and 5). Compound 3361 also inhibits glucose uptake by P. vivax of PFHT. In previous studies, Compound 3361 kills’ P. falciparum in culture and reduces the multiplication of P. berghei in the mouse model.65
Parasite’s Lactate Transporter Inhibitor
Blood form of the malaria parasite relies primarily on anaerobic glycolysis for growth and development.60 Glucose is uptaken by the parasite-infected erythrocytes up to 100 times faster than uninfected erythrocytes. The parasite metabolizes glucose by glycolysis into lactic acid, which is exported from the parasite by lactate: H+ symport mechanism into the external environment.66 Both lactate export and glucose uptake are critical for keeping energy needs, intracellular Ph, and osmotic stability of the parasite. The lactate: H+ symport transport system inhibition is a promising novel target to develop a new drug. Several compounds like MMV007839 and MMV000972, kill asexual blood-stage P. falciparum parasites via inhibition of the lactate: H+ transporter.67
P‑Type Na+ATPase Inhibitor (PfATP4) Inhibitor
Like other cell types, erythrocytes maintain a low internal Na+ level. However, the parasite increases the permeability of the erythrocyte cell membrane, facilitates the entrance of Na+ that causes the erythrocyte’s cytoplasm Na+ concentration increases to a level of the extracellular medium. Thus, despite the parasite exist in the intracellular site, the parasite finds itself in a high-Na+ medium and must efflux Na+ ions across its plasma membrane to keep a low cytoplasmic Na+ level for survival. In this case, the parasite’s influx of Na+ regulated by using a P-type ATPase transporter (PfATP4) that serves as the parasite’s primary Na+-efflux pump mechanism as shown in Figure 3.68 Inhibition of this transporter causes increased Na+ amount inside the parasite, ultimately leading to the death of the malaria parasite. Several compounds including, cipargamin, which is in Phase 2, (+)-SJ733, which is in Phase1 and KAE609, which is in phase 2 target PfATP4 as their Mechanism of action.67,69
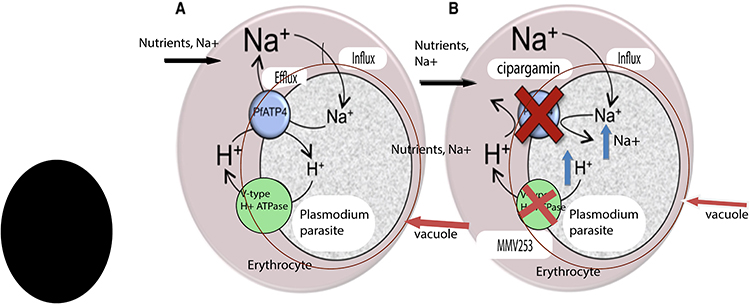 |
Figure 3 Proposed mechanism of the parasite-induced PfATP4 and V‑type H +–ATPase in the death of infected erythrocytes following inhibition by cipargamin. |
V‑Type H +–ATPase Inhibitor
The Plasmodium species control its Na+ level by using a P-type ATPase transporter. It also imports H + by a similar pathway. To regulate the increasing H + concentration and keep an intracellular pH of 7.3, the malaria parasite uses a complementary V-type ATPase transporter to efflux H +. It is a promising target to develop a novel drug. MMV253 inhibits the V-type H + ATPase as its mechanism of action through the mutant selection and whole-genome sequencing.70,71
Aquaporin-3 Inhibitor
Aquaporin-3 (AQP3) is an aquaglyceroporin that facilitates the movement of both water and glycerol in mammalian cells. AQP3 induced in human hepatocytes in response to parasite infection, which has a significant role in the parasite replication. AQP3 provides the pathway for the entry of glycerol into P. berghei and contributes to the replication of the parasite during the asexual intraerythrocytic stages.72 Genetic depletion of AQP3 significantly suppresses in P. berghei liver stage of the parasite load. Furthermore, treatment with auphen, an AQP3 inhibitor, reduces both P. berghei in hepatocytes and P. falciparum parasitemia load in erythrocytes, proposing that the host protein has a crucial role in different life stages of the parasite.73 The most interesting is the genetic disruption of AQP3 in mice is not lethal, proposing the host protein has the potential therapeutic novel target. This work increases our understanding of host liver processes influenced by Plasmodium infection and highlights the potential of targeting these processes for future antimalarial drug development.71,72
Choline Transport Inhibitor
Phospholipids play a key role in the intra-erythrocytic life cycle of P. falciparum, both as structural components of the membranes and as regulatory molecules that regulate several enzymatic activities. These molecules are vital for parasite multiplication inside RBC. After the invasion of RBC, the level of phospholipids increases, with phosphatidylcholine as the major lipid of its cell membranes component. The parasites synthesize phosphatidylcholine de novo using choline as a precursor. This de novo pathway is essential for parasite growth and survival. Inhibition of choline transport into the parasite, inhibit phosphatidylcholine biosynthesis resulting in parasite death.74 Albitiazolium is a drug that has reached Phase II trials, acts primarily by inhibiting the transport of choline into the parasite. Albitiazolium accumulates in the Plasmodium up to 1000-fold and inhibits parasite growth without recrudescence. It is active effective in severe conditions. Notably, a single injection is curative at high parasitemia levels.75,76
Cholinephosphate cytidylyl transferase is a rate-limiting step in phosphatidylcholine de novo biosynthesis.77 The bisquaternary ammonium compound G25 and bis-cationic compound T3 inhibits phosphatidylcholine synthesis in the parasite. G25 is 1000-fold less toxic to mammalian cell lines. These agents are crucial lead compounds for antimalarial agent discovery and development.78,79
Dihydroorotate Dehydrogenase Inhibitor
A key-step for the spreading of Plasmodium species in the human host is the extensive and rapid division of parasite DNA, which depends on the availability of essential metabolites, such as pyrimidines. In the malaria parasite, pyrimidine nucleotide has a vital role in the synthesis of DNA, phospholipids, and glycoproteins. Nucleotide synthesis follows two major pathways: the salvage pathway and the de novo pathway. Dihydroorotate dehydrogenase (DHODH) is an important enzyme that catalyzes the oxidation of dihydroorotate to orotate, a rate-limiting step in the de novo pyrimidine synthesis. So, DHODH represents a potentially promising target for antimalarial drug development.80 A human cell access pyrimidines by salvage of already formed pyrimidine or by de novo synthesis. If the de novo biosynthesis pathway inhibited the cell will depend on the salvage pathway, and the cell will not die. However, inhibition of de novo pyrimidine biosynthesis in the parasite causes the death of those cells because the malaria parasite lacks the pyrimidine salvage pathway, which makes the parasite vulnerable to inhibition of DHODH.81 DSM190 and DSM265 are a selective inhibitor of the parasite DHODH enzyme, the later is currently in phase 2 clinical trials. P218 is a DHODH inhibitor effective against all pyrimethamine-resistant strains currently in phase 1. KAF156 (Ganaplacide) is currently in Phase 2b clinical trials with lumefantrine.82
Isoprenoid Biosynthesis Inhibitor
Isoprenoids are necessary for post-translational lipid modification of proteins and asexual replication of P. falciparum. Isoprenoid synthesized from five-carbon precursor isopentyl diphosphate (IPP) or its isomer dimethylallyl diphosphate (DMAPP) synthesized by one of the two independent pathways. The mevalonate pathway and the 2C-methyl-D-erythritol 4-phosphate (MEP) pathway. The two pathways are mutually exclusive in most microorganisms. Bacteria and P. falciparum rely exclusively on the MEP pathway, but humans do not. So, Enzymes in the MEP pathway explored as potential therapeutic novel targets. The rate-limiting step in the MEP pathways catalyzed by P. falciparum 1-deoxy-Dxylulose- 5-phosphate reductoisomerase (pfDxr), making this parasite enzyme a promising target to develop a novel antimalarial agent.83,84 PfDXR inhibitor inhibited P. falciparum growth, without being toxic to human cells. PfDXR is a potential novel target in antimalarial drug development.83 Fosmidomycin, MMV019313, and MMV008138, inhibits DOXP reductoisomerase, a key enzyme of the DOXP pathway which is absent in human. Because inhibition of protein prenylation in the malaria parasite disrupts asexual parasite growth and which is a potential antimalarial target.85
Farnesyltransferase Inhibitor
Prenylated protein has a vital role in several cellular processes including vesicular trafficking, signal transduction, regulation of DNA replication, and cell division. Such post-translational modifications help the binding of intracellular proteins to membranes and promote protein-protein interactions. Farnesyl transferase catalyzes the transfer of the farnesyl group, a 15-carbon isoprenoid lipid unit, from farnesyl pyrophosphate to the C-terminus of proteins containing the CaaX motif. Farnesyltransferase enzyme is a promising novel target to develop an antimalaria drug because its inhibition kills the parasite.86
Previously, resistance evolution of the parasite with the farnesyltransferase inhibitor BMS-388,891 a tetrahydroquinoline, showed mutations in the protein at the peptide substrate-binding domain. In selections with BMS-339,941 another tetrahydroquinoline, mutation identified in the farnesyl pyrophosphate binding pocket. In another study, mutations found in the beta subunit of the farnesyltransferase in P. falciparum strains resistant to MMV019066. Modeling studies revealed that the mutations distort the critical interaction site of the small molecule with the farnesylation active site, thereby conferring resistance.87
P. falciparum Translational Elongation Factor 2 Inhibitor
One of the promising targets for developing a new drug is the blocking P. falciparum ribosome, and other parts of the translational machinery responsible for protein synthesis. Plasmodium species possess three genomes: nuclear, mitochondrial, and apicoplast (from a relic chloroplast). All genomes require translational machinery to function. Protein syntheses Inhibitors have major clinical success as potent antibiotics. Doxycycline, clindamycin, and azithromycin, have an antimalarial therapeutic use, because they inhibit the ribosomes within the Plasmodium specie’s mitochondria and apicoplast, leading to loss of effect of these organelles.88 Interestingly, the P. falciparum ribosomes occupy an evolutionary middle ground between prokaryotic and eukaryotic, differentiating it significantly from the human ribosome to give an important promising novel target. P. falciparum elongation factor 2 (pfEF2) is a component of a ribosome that catalyzes the GTP-dependent translocation of the ribosome along messenger RNA, and is crucial for protein synthesis in eukaryotes. PfEF2 isolated as a novel target for antimalarial drug development.87,89
Inhibition of protein synthesis exemplified in the discovery of the sordarin, a natural product which selectively blocks fungal protein synthesis by inhibiting the yeast eukaryotic elongation factor 2. Similarly, M5717 (previously DDD107498), a selective inhibitor of the 80S ribosome interacting PfEF2, is currently in phase 1 study, validating the potential of the PfEF2as an effective target for antimalarial drugs.88,90
Anti-Adhesive Polysaccharide
The main features of severe malaria are the sequestration of parasite-infected erythrocytes, inflammation, and Microvascular obstruction. The falciparum parasite uses heparan sulfate during attachment to the endothelium and other blood cells causing obstructions of blood flow. Inhibition of these abnormal cells and pathogen interactions with drugs restore hampered blood flow and affect the parasite growth.91
Several studies showed that sevuparin is an antiadhesive polysaccharide agent manufactured from heparin with eliminated antithrombin effect. Sevuparin inhibits invasion of merozoites into erythrocytes, binding of infected erythrocytes to uninfected and infected erythrocytes and binding to vascular endothelial cells. Besides, sevuparin binds to the N-terminal extracellular, heparan sulfate binding structure of P. falciparum erythrocyte membrane protein 1, the Duffy-binding like domain 1α (DBL1α), known as a vital contributor to sequestration of infected erythrocytes.92,93 Some of the clinical trials that are undergoing in different phases summarized in Table 2.
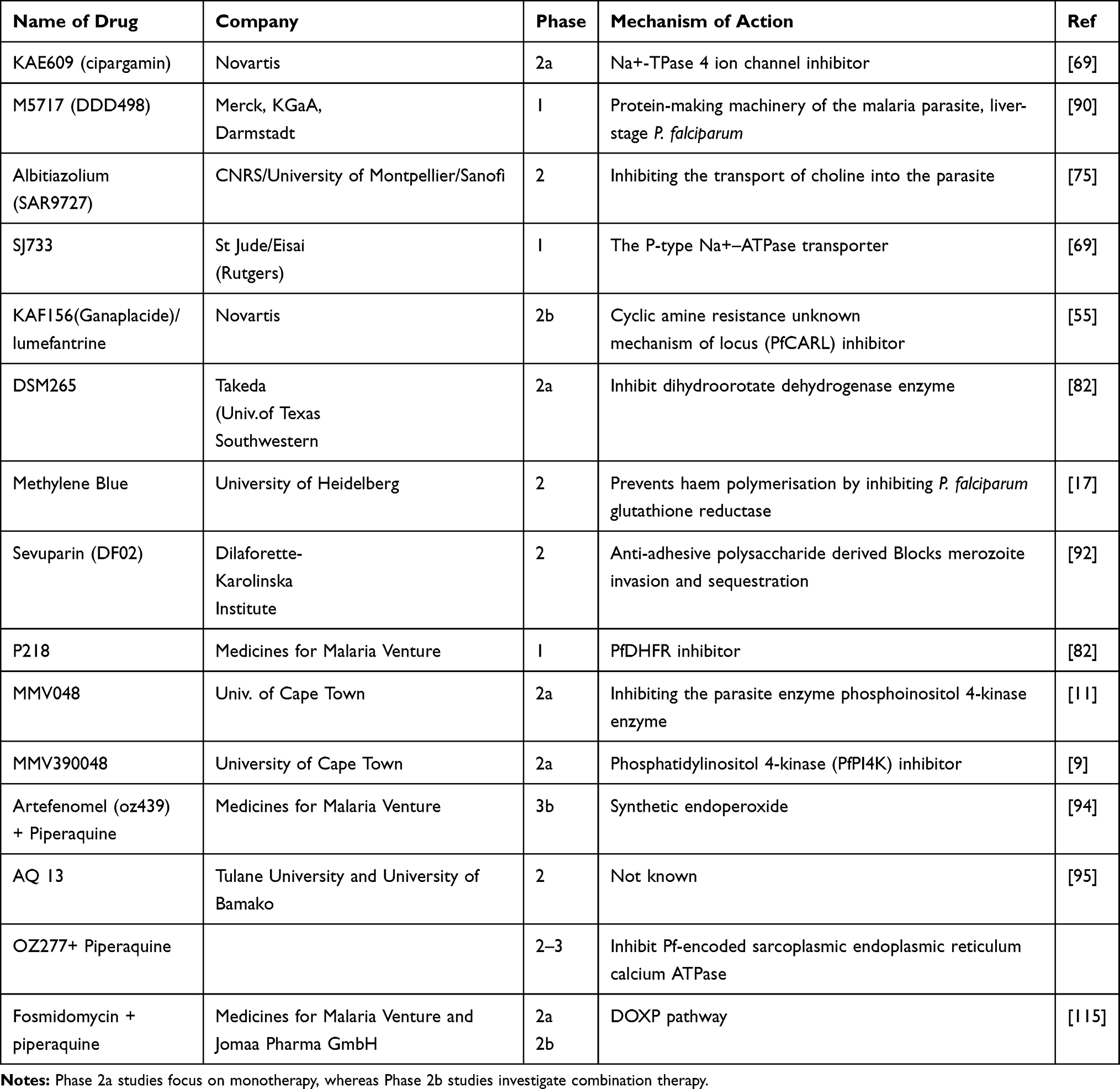 |
Table 2 Overview of Some of the Ongoing Clinical Trial Performed in Malaria Drug Development |
Besides, new drug discovery effective vaccine development is crucial to control malaria. Several vaccines developed in the past targeting the different parasitic life cycle stages. Transmission blocker like PpPf S25, targets the parasite stages in the mosquito vector. Vaccines targeting blood-stage forms, such as the apical membrane antigen-1 (AMA-1) and merozoite surface protein 1 (MSP1) showed disappointing results due to the complex metabolic pathways of erythrocytes and merozoites. Vaccines targeting the pre-erythrocytic sporozoite stage remain the most promising approach. RTS, S/AS02A vaccine induces an anti-circumsporozoite antibody that has completed Phase III efficacy trials.94.95
Conclusion
It is very concerning that the recent emergence of resistance of malaria parasites to available drugs continues to grow, increasingly limiting our ability to control this serious disease. However, it is reassuring that many new approaches to antimalarial drug discovery are now under evaluation, as reviewed here. Recent increases in the pace of progress in this area suggest that, if support for antimalarial drug discovery is adequate, new approaches should lead to the development of new antimalarials that can act through novel mechanisms of action soon. Over the past few years, high-throughput screens have identified several novel chemotypes that developed into highly promising antimalarial candidates. The pace of research progress is high; meaning updates to reviews such as this will always be needed. Apart from efficacy, toxic side effects, pharmacokinetic compatibility, and the potential to develop resistance would all be the major parameters in the eventual development of a successful drug. An understanding of the mechanisms underlying antimalarial drug resistance should also help us to circumvent the emergence of resistance to new generations of antimalarials soon.
Abbreviations
AQP3, Aquaporin-3; pfEF2, P. falciparum elongation factor 2; WHO, World Health Organization; DHODH, Dihydroorotate dehydrogenase.
Data Sharing Statement
All data are provided in the manuscript or found from published papers as cited.
Acknowledgments
I would like to acknowledge the School of Pharmacy (University of Gondar) for providing the resources.
Disclosure
The author declares that they have no competing interests for this work.
References
1. World Health Organization. World Malaria Report. 2019.
2. Mulaw T, Wubetu M, Dessie B, Demeke G, Molla Y. Evaluation of antimalarial activity of the 80% methanolic stem bark extract of combretum molle against Plasmodium berghei in mice. J Evid Based Integr Med. 2019;29(24):2515690X19890866.
3. Conroy AL, Datta D, John CC. What causes severe malaria and its complications in children? Lessons learned over the past 15 years. BMC Med. 2019;17(1):52. doi:10.1186/s12916-019-1291-z
4. Patel P, Bharti PK, Bansal D, et al. Prevalence of mutations linked to antimalarial resistance in Plasmodium falciparum from Chhattisgarh, Central India: a malaria elimination point of view. Sci Rep. 2017;7(1):1–8. doi:10.1038/s41598-017-16866-5
5. Menard D, Dondorp A. Antimalarial drug resistance: a threat to malaria elimination. Cold Spring Harb Perspect Med. 2017;7(7):a025619. doi:10.1101/cshperspect.a025619
6. Mishra M, Mishra VK, Kashaw V, Iyer AK, Kashaw SK. Comprehensive review on various strategies for antimalarial drug discovery. Eur J Med Chem. 2017;5(125):1300–1320. doi:10.1016/j.ejmech.2016.11.025
7. Sahu NK, Sahu S, Kohli DV. Novel molecular targets for antimalarial drug development. Chem Biol Drug Des. 2008;71(4):287–297. doi:10.1111/j.1747-0285.2008.00640.x
8. Diagana TT. Supporting malaria elimination with 21st century antimalarial agent drug discovery. Drug Discov Today. 2015;20(10):1265–1270. doi:10.1016/j.drudis.2015.06.009
9. Rosenthal PJ. Antimalarial drug discovery: old and new approaches. J Exp Biol. 2003;206(21):3735–3744. doi:10.1242/jeb.00589
10. Cortopassi WA, Celmar Costa Franca T, Krettli AU. A systems biology approach to antimalarial drug discovery. Expert Opin Drug Discov. 2018;13(7):617–626. doi:10.1080/17460441.2018.1471056
11. Mathews ES, John AR. Tackling resistance: emerging antimalarials and new parasite targets in the era of elimination. F1000Research. 2018;7:1170. doi:10.12688/f1000research.14874.1
12. Burrows JN, Duparc S, Gutteridge WE, et al. New developments in anti-malarial target candidate and product profiles. Malar J. 2017;16(1):26. doi:10.1186/s12936-016-1675-x
13. Burrows JN, Burlot E, Campo B, et al. Antimalarial drug discovery–the path towards eradication. Parasitology. 2014;141(1):128–139. doi:10.1017/S0031182013000826
14. Gachelin G, Garner P, Ferroni E, Tröhler U, Chalmers I. Evaluating cinchona bark and quinine for treating and preventing malaria. J R Soc Med. 2017;110(1):31–40. doi:10.1177/0141076816681421
15. Thu AM, Phyo AP, Landier J, Parker DM, Nosten FH. Combating multidrug-resistant Plasmodium falciparum malaria. FEBS J. 2017;284(16):2569–2578. doi:10.1111/febs.14127
16. Hokkanen M. Quinine, malarial fevers and mobility: a biography of a ‘European fetish’, c. 1859–c. 1940. In: Medicine, Mobility and the Empire. Manchester University Press; 2017.
17. Pinheiro L, Feitosa LM, Silveira FF, Boechat N. Current antimalarial therapies and advances in the development of semi-synthetic artemisinin derivatives. An Acad Bras Ciênc. 2018;90(1):1251–1271. doi:10.1590/0001-3765201820170830
18. Solomon VR, Lee H. Chloroquine and its analogs: a new promise of an old drug for effective and safe cancer therapies. Eur J Pharmacol. 2009;625(1–3):220–233. doi:10.1016/j.ejphar.2009.06.063
19. John GK, Douglas NM, Von Seidlein L, et al. Primaquine radical cure of Plasmodium vivax: a critical review of the literature. Malar J. 2012;11(1):280. doi:10.1186/1475-2875-11-280
20. Parhizgar AR, Tahghighi A. Introducing new antimalarial analogues of chloroquine and amodiaquine: a narrative review. Iran J Med Sci. 2017;42(2):115.
21. Kumar S, Kumari R, Pandey R. New insight-guided approaches to detect, cure, prevent and eliminate malaria. Protoplasma. 2015;252(3):717–753.
22. González R, Pons‐Duran C, Piqueras M, Aponte JJ, Ter Kuile FO, Menéndez C. Mefloquine for preventing malaria in pregnant women. Cochrane Database Syst Rev. 2018;(3). doi:10.1002/14651858.CD011444.pub3.
23. Singh K, Kaur T. Pyrimidine-based antimalarials: design strategies and antiplasmodial effects. MedChemComm. 2016;7(5):749–768. doi:10.1039/C6MD00084C
24. Antony HA, Parija SC. Antimalarial drug resistance: an overview. Trop Parasitol. 2016;6(1):30. doi:10.4103/2229-5070.175081
25. Singh S, Singh A, Singh M, et al. Modern advancement in the area of antimalarial drug development. Indian J Heterocycl Chem. 2018;28(2):185–193.
26. Goodman CD, Buchanan HD, McFadden GI. Is the mitochondrion a good malaria drug target? Trends Parasitol. 2017;33(3):185–193. doi:10.1016/j.pt.2016.10.002
27. Aderibigbe BA. Design of drug delivery systems containing artemisinin and its derivatives. Molecules. 2017;22(2):323. doi:10.3390/molecules22020323
28. Liu CX. Discovery and development of artemisinin and related compounds. Chin Herb Med. 2017;9(2):101–114. doi:10.1016/S1674-6384(17)60084-4
29. Bridgford JL, Xie SC, Cobbold SA, et al. Artemisinin kills malaria parasites by damaging proteins and inhibiting the proteasome. Nat Commun. 2018;9(1):1–9. doi:10.1038/s41467-018-06221-1
30. Krishna S, Pulcini S, Moore CM, Staines HM, Staines HM. Pumped up: reflections on PfATP6 as the target for artemisinins. Trends Pharmacol Sci. 2014;35(1):4–11. doi:10.1016/j.tips.2013.10.007
31. Tayyab Ansari M, Saeed Saify Z, Sultana N, et al. Malaria and artemisinin derivatives: an updated review. Mini Rev Med Chem. 2013;13(13):1879–1902. doi:10.2174/13895575113136660097
32. Gaillard T, Madamet M, Tsombeng FF, Dormoi J, Pradines B. Antibiotics in malaria therapy: which antibiotics except tetracyclines and macrolides may be used against malaria? Malar J. 2016;15(1):556.
33. Ogbonna A, Uneke CJ. Artemisinin-based combination therapy for uncomplicated malaria in Sub-Saharan Africa: the efficacy, safety, resistance and policy implementation since Abuja 2000. Trans R Soc Trop Med Hyg. 2008;102(7):621–627. doi:10.1016/j.trstmh.2008.03.024
34. Charunwatthana P, Pukrittayakamee S. Combination anti-malarial therapy and WHO recommendations. J R Inst Thai. 2010;100–106.
35. Toovey S, Nieforth K, Smith P, et al. Comparative benefit of malaria chemoprophylaxis modelled in United Kingdom travellers. Travel Med Infect Dis. 2014;12(6):726–732. doi:10.1016/j.tmaid.2014.08.005
36. Owusu-Boateng I, Anto F. Intermittent preventive treatment of malaria in pregnancy: a cross-sectional survey to assess uptake of the new sulfadoxine–pyrimethamine five dose policy in Ghana. Malar J. 2017;16(1):323. doi:10.1186/s12936-017-1969-7
37. AlKadi HO. Antimalarial drug toxicity: a review. Chemotherapy. 2007;53(6):385–391. doi:10.1159/000109767
38. Oyelade J, Isewon I, Aromolaran O, et al. Computational identification of metabolic pathways of Plasmodium falciparum using the-shortest path algorithm. Int J Genomics. 2019;2019.
39. Fidock DA, Rosenthal PJ, Croft SL, Brun R, Nwaka S. Antimalarial drug discovery: efficacy models for compound screening. Nat Rev Drug Discov. 2004;3(6):509–520. doi:10.1038/nrd1416
40. Comer E, Beaudoin JA, Kato N, et al. Diversity-oriented synthesis-facilitated medicinal chemistry: toward the development of novel antimalarial agents. J Med Chem. 2014;57(20):8496–8502. doi:10.1021/jm500994n
41. Aide P, Candrinho B, Galatas B, et al. Setting the scene and generating evidence for malaria elimination in Southern Mozambique. Malar J. 2019;18(1):190. doi:10.1186/s12936-019-2832-9
42. Deu E. Proteases as antimalarial targets: strategies for genetic, chemical, and therapeutic validation. FEBS J. 2017;284(16):2604–2628. doi:10.1111/febs.14130
43. Teixeira C, Gomes RB, Gomes P. Falcipains, Plasmodium falciparum cysteine proteases as key drug targets against malaria. Curr Med Chem. 2011;18(10):1555–1572.
44. Roy KK. Targeting the active sites of malarial proteases for antimalarial drug discovery: approaches, progress and challenges. Int J Antimicrob Agents. 2017;50(3):287–302. doi:10.1016/j.ijantimicag.2017.04.006
45. Verma S, Dixit R, Pandey KC. Cysteine proteases: modes of activation and future prospects as pharmacological targets. Front Pharmacol. 2016;25(7):107.
46. Rosenthal PJ. Proteases of malaria parasites: new targets for chemotherapy. Emerg Infect Dis. 1998;4(1):49. doi:10.3201/eid0401.980107
47. Raj R, Kumar V. Anti-malarial drug discovery: new enzyme inhibitors. Nat Prod Targeting Clin Relevant Enzyme. 2017;2:277–296.
48. Glushakova S, Mazar J, Hohmann‐Marriott MF, Hama E, Zimmerberg J. Irreversible effect of cysteine protease inhibitors on the release of malaria parasites from infected erythrocytes. Cell Microbiol. 2009;11(1):95–105. doi:10.1111/j.1462-5822.2008.01242.x
49. Istvan ES, Mallari JP, Corey VC, et al. Esterase mutation is a mechanism of resistance to antimalarial compounds. Nat Commun. 2017;8(1):1–8. doi:10.1038/ncomms14240
50. Na BK, Shenai BR, Sijwali PS, et al. Identification and biochemical characterization of vivapains, cysteine proteases of the malaria parasite Plasmodium vivax. Biochem J. 2004;378(2):529–538. doi:10.1042/bj20031487
51. Nigussie D, Beyene T, Shah NA, Belew S. New targets in malaria parasite chemotherapy: a review. Malaria Contr Elimination. 2015;1:S1–007.
52. Mavondo GA, Mkhwananzi BN, Mabandla MV. Pre-infection administration of asiatic acid retards parasitaemia induction in Plasmodium berghei murine malaria infected sprague-dawley rats. Malar J. 2016;15(1):226. doi:10.1186/s12936-016-1278-6
53. Kandepedu N, Gonzàlez Cabrera D, Eedubilli S, et al. Identification, characterization, and optimization of 2, 8-disubstituted-1, 5-naphthyridines as novel Plasmodium falciparum phosphatidylinositol-4-kinase inhibitors with in vivo efficacy in a humanized mouse model of malaria. J Med Chem. 2018;61(13):5692–5703. doi:10.1021/acs.jmedchem.8b00648
54. McNamara CW, Lee MC, Lim CS, et al. Targeting Plasmodium PI (4) K to eliminate malaria. Nature. 2013;504(7479):248–253. doi:10.1038/nature12782
55. Blascod DL, Wittyd MJ, Doninid C, et al. UCT943, A Next Generation Plasmodium Falciparum PI4K Inhibitor Preclinical Candidate for the Treatment of Malaria 2. 2018.
56. Bhagavathula AS, Elnour AA, Shehab A. Alternatives to currently used antimalarial drugs: in search of a magic bullet. Infect Dis Poverty. 2016;5(1):103. doi:10.1186/s40249-016-0196-8
57. Krishna S, Eckstein-Ludwig U, Joët T, et al. Transport processes in Plasmodium falciparum-infected erythrocytes: potential as new drug targets. Int J Parasitol. 2002;32(13):1567–1573. doi:10.1016/S0020-7519(02)00185-6
58. Desai SA. Targeting ion channels of Plasmodium falciparum-infected human erythrocytes for antimalarial development. Curr Drug Targets Infect Disord. 2004;4(1):79–86. doi:10.2174/1568005043480934
59. Haldar K, Samuel BU, Mohandas N, Harrison T, Hiller NL. Transport mechanisms in Plasmodium-infected erythrocytes: lipid rafts and a tubovesicular network. Int J Parasitol. 2001;31(12):1393–1401. doi:10.1016/S0020-7519(01)00251-X
60. Nguitragool W, Bokhari AA, Pillai AD, et al. Malaria parasite clag3 genes determine channel-mediated nutrient uptake by infected red blood cells. Cell. 2011;145(5):665–677. doi:10.1016/j.cell.2011.05.002
61. Lisk G, Kang M, Cohn JV, Desai SA. Specific inhibition of the plasmodial surface anion channel by dantrolene. Eukaryot Cell. 2006;5(11):1882–1893.
62. Tilley L, Dixon MW, The KK. Plasmodium falciparum-infected red blood cell. Int J Biochem Cell Biol. 2011;43(6):839–842. doi:10.1016/j.biocel.2011.03.012
63. Dickerman BK, Elsworth B, Cobbold SA, et al. Identification of inhibitors that dually target the new permeability pathway and dihydroorotate dehydrogenase in the blood stage of Plasmodium falciparum. Sci Rep. 2016;22(6):37502. doi:10.1038/srep37502
64. Heitmeier MR, Hresko RC, Edwards RL, et al. Identification of druggable small molecule antagonists of the Plasmodium falciparum hexose transporter PfHT and assessment of ligand access to the glucose permeation pathway via FLAG-mediated protein engineering. PLoS One. 2019;14(5):e0216457. doi:10.1371/journal.pone.0216457
65. Meireles P, Sales‐Dias J, Andrade CM, et al. GLUT1–mediated glucose uptake plays a crucial role during Plasmodium hepatic infection. Cell Microbiol. 2017;19(2):e12646. doi:10.1111/cmi.12646
66. Marchetti RV, Lehane AM, Shafik SH, Winterberg M, Martin RE, Kirk K. A lactate and formate transporter in the intraerythrocytic malaria parasite, Plasmodium falciparum. Nat Commun. 2015;6(1):1–7. doi:10.1038/ncomms7721
67. Rosling JE, Ridgway MC, Summers RL, Lehane AM. Biochemical characterization and chemical inhibition of PfATP4-associated Na+-ATPase activity in Plasmodium falciparum membranes. J Biol Chem. 2018;293(34):13327–13337. doi:10.1074/jbc.RA118.003640
68. Crawford ED, Quan J, Horst JA, Ebert D, Wu W, DeRisi JL. Plasmid-free CRISPR/Cas9 genome editing in Plasmodium falciparum confirms mutations conferring resistance to the dihydroisoquinolone clinical candidate SJ733. PLoS One. 2017;12(5):e0178163. doi:10.1371/journal.pone.0178163
69. Zhang R, Suwanarusk R, Malleret B, et al. A basis for rapid clearance of circulating ring-stage malaria parasites by the spiroindolone KAE609. J Infect Dis. 2016;213(1):100–104. doi:10.1093/infdis/jiv358
70. Yadav BS, Chaturvedi N, Marina N. Recent advances in system based study for anti-malarial drug development process. Curr Pharm Des. 2019;25(31):3367–3377. doi:10.2174/1381612825666190902162105
71. Ashley EA, Phyo AP. Drugs in development for malaria. Drugs. 2018;78(9):861–879.
72. Bietz S, Montilla I, Külzer S, Przyborski JM, Lingelbach K. Recruitment of human aquaporin 3 to internal membranes in the Plasmodium falciparum infected erythrocyte. Mol Biochem Parasitol. 2009;167(1):48–53. doi:10.1016/j.molbiopara.2009.04.006
73. Posfai D, Sylvester K, Reddy A, et al. Plasmodium parasite exploits host aquaporin-3 during liver stage malaria infection. PLoS Pathog. 2018;14(5):e1007057. doi:10.1371/journal.ppat.1007057
74. Penarete-Vargas DM, Boisson A, Urbach S, et al. A chemical proteomics approach for the search of pharmacological targets of the antimalarial clinical candidate albitiazolium in Plasmodium falciparum using photocrosslinking and click chemistry. PLoS One. 2014;9(12):12. doi:10.1371/journal.pone.0113918
75. Schiafino-Ortega S, Baglioni E, Pérez-Moreno G, et al. 1, 2-Diphenoxiethane salts as potent antiplasmodial agents. Bioorg Med Chem Lett. 2018;28(14):2485–2489. doi:10.1016/j.bmcl.2018.05.060
76. Wein S, Van Ba CT, Maynadier M, et al. New insight into the mechanism of accumulation and intraerythrocytic compartmentation of albitiazolium, a new type of antimalarial. Antimicrob Agents Chemother. 2014;58(9):5519–5527. doi:10.1128/AAC.00040-14
77. Déchamps S, Wengelnik K, Berry-Sterkers L, Cerdan R, Vial HJ, Gannoun-Zaki L. The kennedy phospholipid biosynthesis pathways are refractory to genetic disruption in Plasmodium berghei and therefore appear essential in blood stages. Mol Biochem Parasitol. 2010;173(2):69–80. doi:10.1016/j.molbiopara.2010.05.006
78. Peyrottes S, Caldarelli S, Wein S, Perigaud C, Pellet A, Vial H. Choline analogues in malaria chemotherapy. Curr Pharm Des. 2012;18(24):3454–3466.
79. Mesa-Vanegas AM. Potential antiplasmodial and antimalarial agents of natural and synthetic origin. Rev Colomb Cienc Quim Farm. 2018;47(3):375–399. doi:10.15446/rcciquifa.v47n3.77371
80. Xu M, Zhu J, Diao Y, et al. Novel selective and potent inhibitors of malaria parasite dihydroorotate dehydrogenase: discovery and optimization of dihydrothiophenone derivatives. J Med Chem. 2013;56(20):7911–7924. doi:10.1021/jm400938g
81. Llanos-Cuentas A, Casapia M, Chuquiyauri R, et al. Antimalarial activity of single-dose DSM265, a novel plasmodium dihydroorotate dehydrogenase inhibitor, in patients with uncomplicated Plasmodium falciparum or Plasmodium vivax malaria infection: a proof-of-concept, open-label, phase 2a study. Lancet Infect Dis. 2018;18(8):874–883. doi:10.1016/S1473-3099(18)30309-8
82. Reis RA, Calil FA, Feliciano PR, Pinheiro MP, Nonato MC. The dihydroorotate dehydrogenases: past and present. Arch Biochem Biophys. 2017;15(632):175–191. doi:10.1016/j.abb.2017.06.019
83. Belete TM. Novel targets to develop new antibacterial agents and novel alternatives to antibacterial agents. Human Microbiome J. 2019;11(1):1–13. doi:10.1016/j.humic.2019.01.001
84. Goble JL, Adendorff MR, de Beer TA, Stephens LL, Blatch GL. The malarial drug target Plasmodium falciparum 1-deoxy-d-xylulose-5-phosphate reductoisomerase (PfDXR): development of a 3-D model for identification of novel, structural and functional features and for inhibitor screening (supplementary information). Protein Pept Lett. 2010;17(1):109–120. doi:10.2174/092986610789909548
85. Gisselberg JE, Herrera Z, Orchard LM, Llinás M, Yeh E. Specific inhibition of the bifunctional farnesyl/geranylgeranyl diphosphate synthase in malaria parasites via a new small-molecule binding site. Cell Chem Biol. 2018;25(2):185–193. doi:10.1016/j.chembiol.2017.11.010
86. Grellier P, Depoix D, Schrével J, Florent I. Discovery of New Targets for Antimalarial Chemotherapy. 2008:219–225.
87. Eastman RT, White J, Hucke O, et al. Resistance mutations at the lipid substrate binding site of Plasmodium falciparum protein farnesyltransferase. Mol Biochem Parasitol. 2007;152(1):66–71. doi:10.1016/j.molbiopara.2006.11.012
88. Sheridan CM, Garcia VE, Ahyong V, DeRisi JL. The Plasmodium falciparum cytoplasmic translation apparatus: a promising therapeutic target not yet exploited by clinically approved anti-malarials. Malar J. 2018;17(1):465. doi:10.1186/s12936-018-2616-7
89. Jackson KE, Habib S, Frugier M, et al. Protein translation in Plasmodium parasites. Trends Parasitol. 2011;27(10):467–476. doi:10.1016/j.pt.2011.05.005
90. Rottmann M, Jonat B, Gumpp C, et al. Preclinical antimalarial combination Study of M5717, a Plasmodium falciparum elongation factor 2 inhibitor, and pyronaridine, a hemozoin formation inhibitor. Antimicrob Agents Chemother. 2020;64(4). doi:10.1128/AAC.02181-19
91. Armistead JS, Wilson IB, Van Kuppevelt TH, Dinglasan RR. A role for heparan sulfate proteoglycans in Plasmodium falciparum sporozoite invasion of anopheline mosquito salivary glands. Biochem J. 2011;438(3):475–83107. doi:10.1042/BJ20110694
92. Batchvarova M, Shan S, Zennadi R, et al. Sevuparin Reduces Adhesion of Both Sickle Red Cells and Leukocytes to Endothelial Cells in vitro and Inhibits Vaso-Occlusion in vivo. 2013:182.
93. Vogt AM, Barragan A, Chen Q, Kironde F, Spillmann D, Wahlgren M. Heparan sulfate on endothelial cells mediates the binding of Plasmodium falciparum–infected erythrocytes via the DBL1α domain of PfEMP1. J Am Soc Hematol. 2003;101(6):2405–2411.
94. Ogeto T, Ndubi F, Murithi M, et al. Malaria vaccines targeting the pre-erythrocytic stage: a scoping review. F1000Research. 2020;9(680):680. doi:10.12688/f1000research.24320.1
95. Ewer KJ, Sierra-Davidson K, Salman AM, et al. Progress with viral vectored malaria vaccines: a multi-stage approach involving “unnatural immunity”. Vaccine. 2015;33(52):7444–7451. doi:10.1016/j.vaccine.2015.09.094
96. Shanks GD. Historical review: problematic malaria prophylaxis with quinine. Am J Trop Med Hyg. 2016;95(2):269–272. doi:10.4269/ajtmh.16-0138
97. Lacava AC. Ocular complications of chloroquine and derivatives therapy. Arq Bras Oftalmol. 2010;73(4):384–389. doi:10.1590/S0004-27492010000400019
98. Niu YR, Wei B, Chen B, et al. Amodiaquine‐induced reproductive toxicity in adult male rats. Mol Reprod Dev. 2016;83(2):174–182. doi:10.1002/mrd.22603
99. Davis TM, Hung TY, Sim K, Karunajeewa HA, Ilett KF. Piperaquine. Drugs. 2005;65(1):75–87. doi:10.2165/00003495-200565010-00004
100. Lu KY, Derbyshire ER. Tafenoquine: a step toward malaria elimination. Biochemistry. 2020;59(8):911–920. doi:10.1021/acs.biochem.9b01105
101. González R, Hellgren U, Greenwood B, Menéndez C. Mefloquine safety and tolerability in pregnancy: a systematic literature review. Malar J. 2014;13(1):75. doi:10.1186/1475-2875-13-75
102. Bouchaud O, Imbert P, Touze JE, Dodoo AN, Danis M, Legros F. Fatal cardiotoxicity related to halofantrine: a review based on a worldwide safety data base. Malar J. 2009;8(1):289. doi:10.1186/1475-2875-8-289
103. Croft SL, Duparc S, Arbe-Barnes SJ, et al. Review of pyronaridine anti-malarial properties and product characteristics. Malar J. 2012;11(1):270. doi:10.1186/1475-2875-11-270
104. Meshnick SR. Artemisinin: mechanisms of action, resistance and toxicity. Int J Parasitol. 2002;32(13):1655–1660. doi:10.1016/S0020-7519(02)00194-7
105. Nixon GL, Moss DM, Shone AE, et al. Antimalarial pharmacology and therapeutics of atovaquone. J Antimicrob Chemother. 2013;68(5):977–985. doi:10.1093/jac/dks504
106. Nzila A. The past, present and future of antifolates in the treatment of Plasmodium falciparum infection. J Antimicrob Chemother. 2006;57(6):1043–1054. doi:10.1093/jac/dkl104
107. Gaillard T, Madamet M, Pradines B. Tetracyclines in malaria. Malar J. 2015;14(1):445.
108. Gaillard T, Dormoi J, Madamet M, Pradines B. Macrolides and associated antibiotics based on similar mechanism of action like lincosamides in malaria. Malar J. 2016;15(1):85. doi:10.1186/s12936-016-1114-z
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.