")
Back to Journals » Journal of Inflammation Research » Volume 16
Recent Advances on the Molecular Mechanism and Clinical Trials of Venous Thromboembolism
Authors Huang SL, Xin HY, Wang XY , Feng GG, Wu FQ, Feng ZP, Xing Z , Zhang XH, Xin HW, Luo WY
Received 8 September 2023
Accepted for publication 28 November 2023
Published 14 December 2023 Volume 2023:16 Pages 6167—6178
DOI https://doi.org/10.2147/JIR.S439205
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Tara Strutt
Shao-Li Huang,1– 3,* Hong-Yi Xin,4,5,* Xiao-Yan Wang,4,5,* Guang-Gui Feng,3 Fu-Qing Wu,3 Zhi-Peng Feng,6 Zhou Xing,2 Xi-He Zhang,4,5 Hong-Wu Xin,4,7,8 Wen-Ying Luo1
1Medical Laboratory Center, Affiliated Hospital of Guangdong Medical University, Zhanjiang, Guangdong, 524400, People’s Republic of China; 2First Clinical College, Guangdong Medical University, Guangdong, 524400, People’s Republic of China; 3Clinical laboratory, Lianjiang People’s Hospital, Guangdong, 524400, People’s Republic of China; 4Doctoral Scientific Research Center, Lianjiang People’s Hospital, Guangdong, 524400, People’s Republic of China; 5Guangdong Medical University Affiliated Lianjiang People’s Hospital, Guangdong, 524400, People’s Republic of China; 6Department of Gastroenterology, Yueyang Hospital Affiliated to Hunan Normal University, Yueyang, Hunan, 414000, People’s Republic of China; 7Laboratory of Oncology, Center for Molecular Medicine, School of Basic Medicine, Faculty of Medicine, Yangtze University, Jingzhou, Hubei, 434023, People’s Republic of China; 8Research Centre of Molecular Medicine, Medical College of Chifeng University, Chifeng, Inner Mongolian Autonomous Region, 024000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Wen-Ying Luo, Medical laboratory Center, Affiliated Hospital of Guangdong Medical University, 57th South Renmin Road, Zhanjiang, 524001, People’s Republic of China, Tel +86 18312728748, Email [email protected] Xi-He Zhang, Doctoral Scientific Research Center, Lianjiang People’s Hospital, Guangdong, 524400, People’s Republic of China, Tel +86 19126491230, Email [email protected]
Abstract: Venous thromboembolism is a condition that includes deep vein thrombosis and pulmonary embolism. It is the third most common cardiovascular disease behind acute coronary heart disease and stroke. Over the past few years, growing research suggests that venous thrombosis is also related to the immune system and inflammatory factors have been confirmed to be involved in venous thrombosis. The role of inflammation and inflammation-related biomarkers in cerebrovascular thrombotic disease is the subject of ongoing debate. P-selectin leads to platelet-monocyte aggregation and stimulates vascular inflammation and thrombosis. The dysregulation of miRNAs has also been reported in venous thrombosis, suggesting the involvement of miRNAs in the progression of venous thrombosis. Plasminogen activator inhibitor-1 (PAI-1) is a crucial component of the plasminogen-plasmin system, and elevated levels of PAI-1 in conjunction with advanced age are significant risk factors for thrombosis. In addition, it has been showed that one of the ways that neutrophils promote venous thrombosis is the formation of neutrophil extracellular traps (NETs). In recent years, the role of extracellular vesicles (EVs) in the occurrence and development of VTE has been continuously revealed. With the advancement of research technology, the complex regulatory role of EVs on the coagulation process has been gradually discovered. However, our understanding of the causes and consequences of these changes in venous thrombosis is still limited. Therefore, we review our current understanding the molecular mechanisms of venous thrombosis and the related clinical trials, which is crucial for the future treatment of venous thrombosis.
Keywords: venous thromboembolism, miRNA, neutrophil extracellular traps, plasminogen activator inhibitor-1, EVs, inflammatory factor
Introduction
Venous thromboembolism (VTE) is a multifaceted, potentially fatal event that activates coagulation and fibrinolysis.1 It is a condition that includes pulmonary embolism (PE) and deep vein thrombosis (DVT).2 In addition, it is also the third most prevalent cardiovascular disease after stroke and coronary heart disease (CHD).3 Venous thrombosis (VT) is a leading cause of mortality as well as morbidity worldwide, occurring in approximately one case in 1000 people per year in affluent nations.4 DVT is the thrombosis that blocks the deep venous cavity, disrupting the venous reflux network and resulting in chronic deep venous dysfunction.5,6 Furthermore, DVT is a consequence of a complicated interaction between enzymes and cellular processes, in which the endothelium, platelets, and leukocytes coordinate a pro-inflammatory state that ultimately leads to the clot formation7 with higher mortality rate for acute DVT.8 To date, vascular ultrasound and digital subtraction angiography are the primary techniques for diagnosing DVT. The lower extremity deep venous thrombosis (LEDVT) is diagnosed clinically based on venography, the gold standard.9 Even though both techniques have a high diagnostic value, the latter is quite intrusive and expensive, while the former lacks the ability to diagnose intraperitoneal venous embolism.10 In addition, ultrasound is not a routine and is only performed before discharge for patients with symptoms following major orthopedic surgery.11 The absence of specific clinical signs and non-specific symptoms associated with venous thrombosis may lead to a delayed or inaccurate diagnosis, ultimately resulting in poor patient prognosis.12,13 Both genetic and acquired factors can lead to the formation of venous thrombosis, which is a complex process whose molecular mechanism remains poorly understood. Therefore, exploring the mechanism is essential for effective treatment of venous thrombosis. Recently, there has been a lot of discussion on the mechanisms of venous thrombosis, including inflammatory, immunological, and neutrophil extracellular trap formation aspects, other factors under investigation include age, p-selection, MicroRNA(miRNA), PAI-1-induced venous thrombosis and extracellular vesicles (EVs). We have outlined these relevant molecular mechanisms involved in VTE (Figure 1), which may be an important target for future therapeutic interventions.
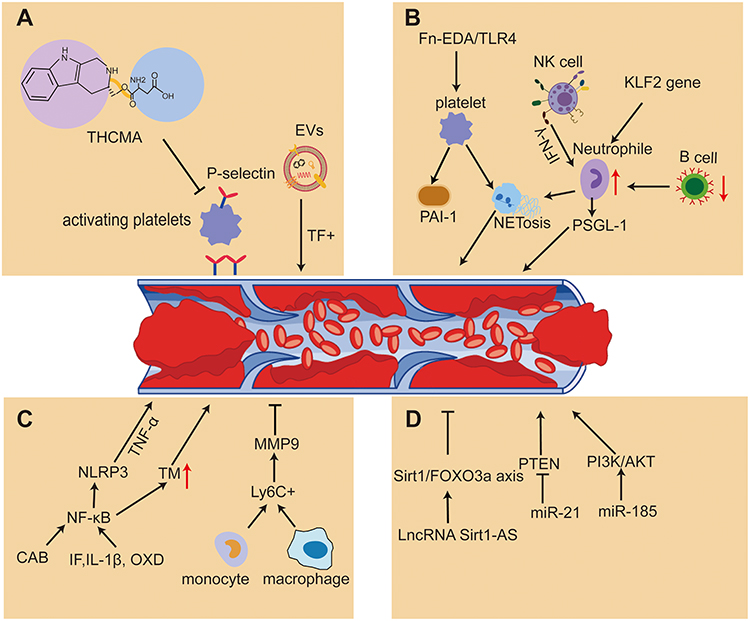 |
Figure 1 Mechanisms of venous thrombosis through PAI-1, inflammatory factors, miRNA, age-related changes, NETs formation, P-selectin activation and immunological processes. (A) THCMA inhibits platelet activation and aggregation by P-selectin to reduce thrombosis and tissue factor-positive (TF+) EVs are associated with VTE (B) Exogenous cellular Fn-EDA stimulated NETosis in neutrophils via TLR4-stimulated thrombin-activated platelets and TLR4 contributes to Fn-EDA-mediated DVT. B-cell deficiency leads to an increase in circulating neutrophils and an increased abundance of NETs within the thrombus. NK cells promote venous thrombosis by activating neutrophils and producing IFN-γ-dependent NETs. (C) Ly6C+ monocytes and macrophages release IL-6 that induce mononuclear cell sources of MMP 9, and MMP 9 participates in the process of thrombolysis. IL-1β, TF, XOD, GAB, and TNF-α regulate venous thrombosis through the NLRP 3 / IL-1 /NF-κB signaling mechanism. (D) Mir-185 inhibits thrombosis by regulating PI3K/Akt signaling pathway, which inhibits inflammation-induced tissue factor expression in deep vein endothelial cells. MiR-21 inhibits the expression of PTEN, increases the proliferation of endothelium and promotes the formation of new blood vessels and venous thrombosis. LncRNA SIRT1-AS reduces the incidence of aging-associated DVT by Sirt1/FOXO3a axis. Abbreviations: THCMA, 3S-1,2,3,4-tetrahydro-β-carboline-3-methyl aspartyl ester; EVs, extracellular vesicles; Fn-EDA, fibronectin containing extra domain A; TLR4, toll-like receptor 4; DVT, deep vein thrombosis; NETs, neutrophil extracellular traps; MMP 9, matrix metalloproteinase 9; TF, tissue factor; XOD, xanthine oxidase; GAB, Grb2-associated binding. |
Plasminogen Activator Inhibitor-1
The main protein of plasminogen-plasminase system, plasminogen activator inhibitor-1 (PAI-1), is a significant inhibitor of tissue-type plasminogen activator (t-PA) and urokinase-type plasminogen activator (U-PA).14 Inflammation stimulates endothelial cells to release tPA and PAI-1 locally. Besides, since platelets are the major circulating pool of PAI-1, activated platelets release a great number of PAI-1, leading to locally high levels of PAI-1 at growing fibrin clot sites,15,16 thus increased PAI-1 is a risk factor for thrombosis. In addition, the polymorphism of RS1799889 in the promoter region of the PAI-1 gene has been detected in patients with thrombosis.17 However, there is no association between elevated levels of PAI-1 and the risk of venous thrombosis in recent study.18 The 5G allele is associated with reduced PAI-1 transcript levels compared with 4G allele, so the presence of the 5G allele may lead to a reduced risk of thrombotic events. However, the risk of thrombosis for the 4G allele carrying the PAI-1 gene is controversial.19 Another study showed that PAI-1-siRNA strengthens the cavity-forming capacity of endothelial progenitor cells (EPCs) and significantly accelerates EPCs homing. After PAI-1 gene silencing, PAI-1 mRNA and protein expression decreased, vascular endothelial growth factor (VEGF) expression increased, and light-like structure enhanced in inferior vena cava tissue. PAI-1 gene silencing could promote VT recanalization by enhancing the lumen-forming capacity of the rat EPCs.20
Inflammatory Factor
There is growing evidence showed that inflammatory factors linked to venous thrombosis. The role of inflammation and inflammation-related biomarkers in cerebrovascular thrombotic disease is a subject of ongoing debate.21,22 Vascular cell adhesion molecule 1 (VCAM-1) plays an important role in leukocyte adherence and migration among vascular endothelial cells and the level are elevated in endothelial cell inflammatory areas.23 Research has revealed that (lymphocyte antigen 6 complex)+Ly6C+ monocytes and macrophages are the primary myeloid cell source of interleukin-6 (IL-6) in lytic thrombus, and IL-6 was known to induce monocyte-derived matrix metalloproteinase 9 (MMP9) production and that MMP9 has a weakened engagement in the process of thrombus lysis. The monocyte-IL6-MMP9 axis reflects a prospective non-anticoagulant target that may promote thrombolysis in individuals with completely occlusive DVT because of its crucial role in the IL-6 signaling pathway, which is essential for venous thrombosis and is influenced by the amount or lack of blood flow around the thrombus.24 Another research also revealed that inflammatory Ly6Chi monocytes regulate the development, proliferation, and lysis of thrombus, which could be treated with transcription factor Nur77 (NR4A1) agonists at any stage of the illness.25 However, investigators were unable to establish any correlation between plasma tissue factor (TF), IL-6, VCAM-1 or D-dimer levels and the development of DVT, but it should be noted that blood samples were only taken at recruitment within three days of injury, which may be too early to detect a prethrombotic status.23 Another study has demonstrated that elevated levels of inflammatory factors interleukin-1β (IL-1β), tissue factor (TF), xanthine oxidase (XOD) and nuclear factor kappa B (NF-κB) may accelerate thrombosis.26 The NLRP3/IL-1/NF-κB signaling mechanisms regulate IL-1 and tumor necrosis factor alpha (TNF-α), which may be essential signs of the prethrombotic condition due to slowed blood flow, impaired vascular endothelium, and elevated tissue factor expression. Therefore, VTE is the result of the coagulation system’s cascade reaction.27–29 Coagulation factor XII (FXII), an essential coagulation factor, was found to be associated with thrombosis, the study revealed that the knockdown of FXII markedly raised superoxide dismutase (SOD) concentrations, reduced the thrombosis and apoptosis, and raised the malondialdehyde (MDA) concentrations in DVT mice. Moreover, TNFα, IL-6, interleukin-8 (IL-8), and phosphatidylinositol 3-kinases (PI3K)/protein kinase B (AKT) signaling activation were all markedly decreased by FXII knockdown. The stimulation of PI3K/AKT signaling by the FXII protein causes an inflammatory response, which in turn leading to DVT. Therefore, focusing on FXII protein may show promise as a DVT therapy strategy.30 As we know, AKT2 is a subtype of AKT, in a mouse model of venous thrombosis, AKT2 could modulate endothelial cell-mediated blood coagulation homeostasis as well as facilitate endothrombotic recanalization and thrombus resolution. Besides, AKT2 could increase the expression of thrombomodulin (TM) and decrease the expression of TF in cultivated endothelial cells.31 According to another research, TM is thought to be a valuable marker for assessing endothelial impairment and plays a crucial role in DVT. The activation of the NF-κB signaling pathway leads to an increase in plasma TM levels and thrombus size.32 As for endothelial cells, study found that prothrombotic procoagulant phospholipids was found on the surfaces of activated endothelial cells.33 The endothelium procoagulant action is supported by phospholipid-disrupting enzymes, TMEM16E and TMEM16F (Ca2+-activated phospholipid-disrupting enzyme), which externalize phosphatidylserine (PS), in the mice model of thrombosis caused by laser damage, PS externalization was inhibited and fibrin production in the vessel wall was decreased without affecting platelets when TMEM16E or TMEM16F were deleted genetically or treated with TMEM16 inhibitors, the results demonstrate the involvement of endothelial TMEM16E in thrombosis and suggest TMEM16E as a possible target for therapeutic intervention to inhibit the development of thrombus.33,34 Besides, another recent research has demonstrated the Grb2-associated binding 2 (GAB2), a signal adapter protein, plays a vital part in the dissemination of the inflammatory signals in endothelial cells induced by IL-1β and other cytokines of inflammation.35 In endothelial cells, GAB2 contributes to the activation of NF-κB and Rho.36 According to the study, greatly reducing IL-1-induced Rho-dependent exocytosis of Von Willebrand factor (VWF) and P-selectin then following adhesion of neutrophils to vascular cells was achieved by either gene silencing of GAB2 or mucosa-associated lymphoid tissue lymphoma translocation protein 1(MALT1), the effector signals molecule in the CBM (CARD recruited membrane-associated protein 3-B cell lymphoma 10 - MALT1) signalosome, or by pharmacologically inhibiting MALT1 with a particular inhibitor, mepazine.36 Additionally, IL-1-induced NF-κB-dependent production of tissue-related factors and VCAM-1 was decreased by MALT1 suppression. Gab2 loss or pharmacological suppression of MALT1 decreased venous thrombosis brought on by inferior vena cava-ligation-induced stenosis or stasis in mice and reduced the concentration of monocytes and neutrophils at the wound area, which was in accordance with the in vitro data.36 Furthermore, the findings of another investigation demonstrated that the neutrophil count, monocyte level, c-reactive protein (CRP) concentration, lymphocyte-to-monocyte ratio (LMR), and neutrophil-to-lymphocyte ratio (NLR) were significantly altered in accordance with the duration of cerebral venous thrombosis (CVT). Patients with CVT exhibit distinct inflammatory patterns throughout the course of their illness: higher levels of NLR and lower levels of LMR during the acute phase; higher levels of LMR and lower levels of CRP during the chronic phase.37
Mechanism of miRNA Leading to Venous Thrombosis
MicroRNA is an endogenous, highly conserved 19–22 short nucleotide fragment of a non-coding RNA molecule.38 As a protein that is directly affected by AKT, glycogen synthase kinase 3 (GSK3) is a crucial part of the PI3K/AKT signaling cascade and has the ability to influence cell survival, death, motility, and migration.39,40 Study has demonstrated that miRNA-185 was relevant with the proliferation and apoptosis of vascular endothelial cells by controlling the late glycation end product receptor (RAGE). MiR-185 can inhibit the expression of tissue factor in rat. The rat deep vein endothelial cells were induced by lipoderma endothelium, which was regulated by PI3K/AKT signaling pathways. MiR-185 inhibits thrombosis by reducing inflammation-induced tissue factor expression.41 In addition, another study found that miR-150 is an important microRNA that plays a key part in all kinds of cell functions. Moreover, miR-150 plays a crucial role in endothelial progenitor cells (EPCs), and its expression is downregulated in EPCs induced by DVT. The upregulation of miR-150 promotes angiogenesis and proliferation of EPCs through the targeting of SRC kinase signaling inhibitor 1 (SRCIN1) both in vitro and in vivo for thrombolysis.42 The Fas ligand (FASLG) gene is a target of miR-21, knockdown of FASLG can impair EPCs function, while the expression of miR-21 may stimulate EPCs proliferation and angiogenesis. In addition, In the EPCs of the DVT model rat, miR-21 expression is diminished. By targeting FASLG, miR-21 may promote the proliferation of endothelial progenitor cells and the creation of new blood vessels, which imply that miR-21 could be a potential indicator of thrombosis.43 In other side, it has been proved that miR-21 is closely related to PTEN (phosphatase and tensin gene), which inhibit cell proliferation and promote apoptosis under normal physiological conditions and therefore plays an important role in thrombosis.44 MiR-21 can increase the proliferation of vascular endothelial cells and promote the formation of new blood vessels by inhibiting the expression of PTEN.45 A new study has uncovered that the function of miR-21 is to increase the rate of angiogenesis and cardiomyocyte survival by target to PTEN in heart failure.46 In summary, miR-21 plays a complex regulatory role in thrombosis, including inhibition of PTEN and FASLG, promotion of endothelial cell proliferation and neovascularization, and contribute to keratinocyte migration, angiogenesis and cardiomyocyte survival in heart failure. Further study should focus on the mechanism of miR-21 in thrombosis and provide a theoretical basis for the development of new treatments.
Mechanisms of Venous Thrombosis Associated with Aging
The risk of venous thrombosis increases with age,47 but the mechanisms underlying the increased risk of thrombosis with age are not well understood. Silent information regulator 1 (Sirt1) is associated with endothelial cell senescence, inflammation, oxidative stress and platelet adhesion. Sirt1antisense (Sirt1-AS) is an antisense long non-coding RNA (lncRNA) of Sirt1, DVT development is related to endothelial cell senescence and low lncRNA expression of Sirt1-AS and Sirt1. Sirt1 delays senescence to reduce the incidence and production of age-related thrombosis, and the Sirt1antisense lncRNA (lncRNA Sirt1-AS) mitigates DVT by modulating the Sirt1/FOXO3a axis. Specifically, it reduces the incidence of senescence-associated DVT by enhancing human vascular endothelial cell (HUVEC) viability and proliferation while decreasing HUVEC apoptosis.48 In a cross-sectional research of mice and humans, researcher found that Deoxyribonuclease 1(DNase 1) could inhibit age-induced increases in endogenous thrombin generation and venous thrombosis. The work revealed that circulating cell-free DNA increased with aging via NETosis-independent processes.49 The NETosis, which involves the release of NETs following the activation of neutrophils in vitro, has been demonstrated to be mediated by hydrogen peroxide (H2O2).50
The Mechanism of Neutrophil Traps Promoting Thrombosis
Vascular Willebrand factor or glycoprotein Iba-dependent platelet “Priming” triggers the activation of integrin αllbβ3, which in turn regulates neutrophil and T cells binding. Neutrophil binding of platelet αllbβ3 to SLC44A2 results in the production of highly prothrombotic NETs.51 Therefore, it is believed that one of ways in which neutrophils encourage venous thrombosis is by forming NETs.52 Additionally, NETs utilize transforming growth factor-β (TGF-β) in thrombi to up-regulate fibroblasts and facilitate fibrotic thrombus remodeling.53 It has long been thought that arterial and venous thrombosis involve different mechanisms,54,55 but recent studies have revealed that the neutrophil play a crucial role in both arterial and venous thrombosis. The transcription factor Kruppel-like Factor 2 (KLF2) is the key regulator of neutrophil activation, which can be triggered by anti-phospholipid antibodies or be lost through KLF2 gene expression. This leads to the aggregation of P-selectin glycoprotein ligand-1 (PSGL-1) via reconstitution of cortical actin, thereby increasing adhesion potential at thrombotic sites.56 Another study also revealed that in the progression of heart failure and myocardial hypertrophy, KLF2 controls thrombosis and activation of neutrophil.57 In addition, recent research demonstrated that under a live microscope, the neutrophil “plucked” an extension of megakaryocytes in blood vessels, known as pre-platelets, to regulate the platelet production. Along with cxcr4-cxcl12-dependent migration to the periatrial megakaryocytes, the neutrophil activate platelets via reactive oxygen species and trigger activation of myosin light chain and extracellular signal-regulated kinases. Through these mechanisms mentioned above, neutrophils accelerate platelet growth and promote stable platelet release. After myocardial infarction, neutrophils lead to excessive release of young reticular platelets and increase the risk of re-ischemia, and on the contrary, ablation neutrophil can normalize platelet and reduce the thrombus burden of recurrent and venous thrombosis after myocardial infarction.58
Cellular fibronectin containing extra domain A (Fn-EDA) is a toll-like receptor 4 (TLR4) endogenous ligand that promotes thrombotic inflammation.59–63 Overweight and obesity are known to increase the chance of developing VTE.64,65 In a diet-induced obesity mouse model, researchers found increased plasma levels of Fn-EDA in high-fat (HF) fed animals.59 Additionally, individuals with VTE had high plasma levels of Fn-EDA and were linked to body mass index (BMI). Under co-morbid diet-induced obesity circumstances, genetic ablation of Fn-EDA decreased susceptibility to DVT. Besides, the research revealed that exogenous cellular Fn-EDA stimulated NETosis in neutrophils via TLR4-stimulated thrombin-activated platelets and that TLR4 contributes to Fn-EDA-mediated DVT. Therefore, Fn-EDA/TLR4 axis may be involved in NETosis and the development of DVT in mice. The elevated levels of Fn-EDA in plasma may be an important mechanism for promoting DVT in the context of diet-induced obesity.59
Defibrotide, a heterogeneous mixture of polyanionic oligonucleotides, has been currently approved for the treatment of transplant-related venous occlusive disease. Recently, researchers have conducted in vitro experiments and mouse models to determine the mechanism by which defibrotide inhibits NET formation and venous thrombosis in antiphospholipid syndrome (APS). This study revealed the role of NETs in the thrombotic complications of APS. At a dose of 1–10 μg mL−1, defibrin significantly inhibited NET formation in IgG-stimulated control neutrophils isolated from APS patients.66 Defibrotide elevates intracellular cyclic AMP levels in neutrophils, thereby mitigating the inhibition of NET formation through blockade of adenosine A2A receptors or suppression of cyclic AMP-dependent kinases. In a model where antiphospholipid antibodies accelerated thrombosis, defibrin at doses ranging from 15–150 mg/kg/day inhibited both NET formation and venous thrombosis and the effect was diminished in adenosine A2A receptor knockout mice. This study has demonstrated the mechanism by which defibrin can counteract thrombotic inflammation mediated by neutrophils in APS.66
Selection
P-selectin is an adhesion molecule expressed on the surface of activated platelets and endothelial cells,67,68 leading to platelet-monocyte aggregation and stimulating vascular inflammation and thrombosis.69 Therefore, inhibiting the expression of p-selectin is a good option for reducing the thrombosis. 3S-1,2,3,4-tetrahydro-β-carboline-3-methyl aspartyl ester (THCMA) is a new small molecule inhibitor of p-selectin, which can remarkably restrict platelet aggregation in vitro and down-regulate serum p-selectin and TNFα expression levels in vivo. THCMA has been successfully developed as a nanomedicine and is 100-fold more effective in inhibiting arterial and venous thrombosis and 10-fold more effective in suppressing inflammation than PSI-697, a drug in clinical trials.68,69 The doses of THCMA that inhibit thrombosis do not produce clotting disorders and no risk of bleeding, the drug significantly improves oral efficacy, which could be used for oral therapy of arterial and venous thrombosis, cancer-related thrombosis and inflammation.68
Immunological Mechanism of Venous Thrombosis
Over the past few years, growing research suggest that the production of venous thrombosis also involves the immune system. As thrombosis originates from severe hypoxia in deep venous environment, endothelial cells are subjected to oxidative stress. This encourages the binding of additional pattern recognition molecules or mannose-binding lectin (MBL) to the surface of endothelial cell via the lectin pathway, which in turn activates mannose-binding lectin-associated serine protease 2 (Masp-2), then Masp-2 can cleave prothrombin to thrombin and forms fibrin.70–74 According to the study by Damoah et al, having elevated levels of the complement-activating enzyme Masp-2 raises the chance for developing venous thromboembolism in the future.75 Furthermore, the absence of B cells in mice indirectly contributes to venous thrombosis by elevating neutrophil counts and increasing fibrinogen levels. The inferior vena cava (IVC) stenosis model demonstrated an augmented incidence of venous thrombosis due to B-cell deficiency, which was hypothesized by investigators to be caused by a rise in circulating neutrophils leading to an increased abundance of NETs within the thrombus and upregulation of fibrinogen production.76 Through the production of Interferon-γ (IFN-γ)-dependent NETs, natural killer (NK) cells contribute to the development of venous thrombosis, which DVT decreases with NK cell depletion.77 Moreover, the activation of innate effect-memory T cells plays a crucial role in regulating venous wall inflammation and thrombus lysis after thrombosis.78 Besides, a study demonstrated that regulatory T cells (Treg) clustered in venous clots of blood, where they formed the stromal cell protein SPARC (secreted protein acidic and rich in cysteine) which promoted the MMP activity of monocytes. Treg thrombolysis is through the control of monocyte recruitment, differentiation, and regulation of the activity of MMP, which is possible to use clot Tregs therapeutically to speed up thrombus clearance.79 Mast cell protease-4 (MMCP-4) is a chymase-type particle-localized protease that has been discovered to be crucial in the development of DVT, in the deep vein thrombosis-affected mice, chymase decreased the activity of plasmin within thrombus, the inhibition of chymase could eliminate and avoid deep vein thrombosis without lengthening the bleeding duration, which reduce chymase activity without disrupting the coagulation cascade, these findings offer a potential pharmaceutical approach to treat or prevent DVT.80
Immobilization is known to be an important risk factor for the VTE development, but a protracted state of inactivity of paralyzed spinal cord injury (SCI) patients and free-ranging hibernating brown bears is protected from VTE. Thienel et al has demonstrated that mass spectrometry-based proteomics can identify antithrombotic properties in the platelets of hibernating brown bears, with heat shock protein 47(HSP47) being most markedly diminished. The rationale behind this is that the downregulation or ablation of HSP47 leads to a decrease in immune cell activation and neutrophil trap formation, thereby promoting thromboprotection in patients with spinal cord injury and bears and mice. This cross-species conservation of platelet characteristics may provide antithrombotic therapy as well as prognostic indicators.81
Immune thrombosis refers to the innate immune response triggered by the formation of blood clots in blood vessels and serves as a framework for the interaction between the immune system (innate and adaptive) and endothelial dysregulation-mediated thrombotic response caused by inflammation. The immune cells support the related molecules and produce specific intravascular scaffolds that promote pathogen recognition, containment, and destruction. These mechanisms preserve the integrity of the host without resulting in serious side effects.82,83 The previous study showed IL-6, IL-8, and monocyte chemoattractant protein (MCP-1) were the independent predictors of accelerated VTE development and they concluded that systemic inflammation is a key driver of VTE risk after major trauma.84 Future work will need to further determine the importance of immunothrombosis for host protection and characterize in more detail the host molecules involved in this process, without causing significant collateral damage to the host.
Extracellular Vesicles (EVs) in the Coagulation Mechanism of VTE
Many clinical cases of VTE have unknown causes and lack typical clinical symptoms, so there is an urgent need to develop reliable biomarkers for assisting in the prediction and diagnosis of VTE. Extracellular vesicles (EVs) produced by activated, damaged, or apoptotic cells carry a variety of bioactive substances and play diverse physiological roles while participating in the pathological processes of numerous diseases. The procoagulant specificity of EVs is also associated with the expression of TF. The previous study showed tumor cells constitutively release EVs that may contribute to thrombosis in cancer patients. Clinical studies have found that levels of circulating tumor–derived, tissue factor-positive (TF+) EVs in pancreatic cancer patients are associated with VTE.85 Besides, the activity of PS and TF expressed by EVs is much higher than that of the mother cell, which can significantly shorten mice’s bleeding time and promote thrombosis formation.85 The researchers discovered that inhibiting CD36 reduced the binding of endothelial, monocyte, and platelet EVs to resting platelets.86 The binding of EVs to platelets was also reduced by annexin V or an antibody to PS, suggesting that EV PS plays a role in the binding process.86 In addition, EV also carries P-selectin glycoprotein ligand-1 and glycoprotein GpIb, which respectively mediate EVs activation and platelet activation. Endothelial cells and vWF interact to rapidly deposit TF-associated EVs at the site of thrombus formation, thus more effectively promoting initiation and amplification of the coagulation cascade.87
In addition to directly promoting coagulation, EVs can also promote thrombosis through indirect mechanisms. For example, activated platelets can transfer “cargo” such as arachidonic acid via EVs, thereby inducing the activation of other platelets.88 There is a close relationship between inflammation and thrombosis, and it has recently been discovered that EVs can indirectly promote coagulation by influencing the inflammatory system. High-level EVs can significantly up-regulate the expression of pro-inflammatory signaling molecules, activate systemic coagulation response and induce inflammation and apoptosis.89
Summary and Future Perspectives
In this review, we provide a comprehensive overview of the mechanisms underlying PAI-1, inflammatory factors, miRNA, age-related changes, neutrophil extracellular traps formation, P-selectin activation, immunological processes and extracellular vesicles (EVs) in relation to venous thrombosis (Figure 1). We reviewed venous thrombosis biomarkers from ongoing clinical trials, as shown in Table 1, as well as published clinical trials related to venous thrombosis as shown in Table 2. With regard to venous thrombosis, we have outlined the important points as follows: firstly, increased PAI-1 is regarded as a thrombosis risk factor, although there is debatable evidence about the thrombosis risk association with the 4G allele in the PAI-1 gene.19 Besides, vein thrombosis is influenced by inflammatory factors, such as IL6, IL-1, and TNF-α, as well as other factors mediated by various signaling pathways like VCAM-1, TF, TM, VWF, and P-selectin. Venous thrombosis is also connected to the immune system and miRNA. Importantly, although immobility is a known risk factor for venous thrombosis, the downregulation or ablation of HSP47 in patients with SCI and bears leads to a decrease in immune cell activation and neutrophil trap formation, thereby promoting thromboprotection in SCI patients and bears. This offers a crucial concept for the management of venous thrombosis. The connection between inflammation and the immune system in venous thrombosis warrants more research as well. Additionally, NETosis is also involved in the mechanism of venous thrombosis caused by aging, which may share similar linked mechanism. Notably, age-related and immunological venous thrombosis have been linked to neutrophil trap development; hence, one potential treatment for venous thrombosis might be to inhibit neutrophil trap formation. Besides, EVs involved in the pathological process of various clinical VTE‑related diseases, and has potential applications in indicating the risk of VTE and aiding in the diagnosis and treatment of VTE.
 |
Table 1 Ongoing Clinical Trials Involving Biomarker in VTE |
 |
Table 2 Published Literature Involving Venous Thrombosis |
However, venous thrombosis is a complex illness, there are still unknown mechanisms. Therefore, inactive venous thrombosis may not be predicted even after testing all available indicators. It is critical to continue to explore of the underlying mechanisms of venous thrombosis. The more clearly the mechanisms of venous thrombosis are understood, the greater therapeutic and diagnostic targets for venous thrombosis are identified.
Data Sharing Statement
The authors declare that the submitted data is available. This paper does not contain any other individual or collective published or written works data except those specifically annotated and cited in the paper.
Consent for Publication
We declare that all authors agreed to publish the manuscript at this journal based and followed publication ethics.
Funding
This study was supported by the Affiliated Hospital of Guangdong Medical University “Clinical Medicine+” CnTech Co-construction Platform (no. CLP2021B004), the Discipline Construction Project of Guangdong Medical University (no.4SG21279P), the Discipline Construction Project of Guangdong Medical University (no. 4SG21276P), the Basic and Applied Basic Research Foundation of Guangdong Province Regional Joint Fund Project (The Key Project) (no. 2020B1515120021), Zhanjiang city science and technology development special fund competitive allocation project (no. 2020A01037), and basic and applied basic research funding committee of Guangdong province, China (no. 2023A1515010798), and the open projects of the key laboratory of human genetic disease research of Inner Mongolia (no. YC202201).
Disclosure
The authors declare that they have no known competing commercial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. Alirezaei T, Sattari H, Irilouzadian R. Significant decrease in plasma D-dimer levels and mean platelet volume after a 3-month treatment with rosuvastatin in patients with venous thromboembolism. Clin Cardiol. 2022;45(7):717–722. doi:10.1002/clc.23833
2. Lutsey PL, Zakai NA. Epidemiology and prevention of venous thromboembolism. Nat Rev Cardiol. 2023;20(4):248–262. doi:10.1038/s41569-022-00787-6
3. Zoller B, Svensson PJ, Dahlback B, Lind-Hallden C, Hallden C, Elf J. Genetic risk factors for venous thromboembolism. Expert Rev Hematol. 2020;13(9):971–981. doi:10.1080/17474086.2020.1804354
4. Næss IA, Christiansen SC, Romundstad P, Cannegieter SC, Rosendaal FR, Hammerstrøm J. Incidence and mortality of venous thrombosis: a population‐based study. J Thromb Haemost. 2007;5(4):692–699. doi:10.1111/j.1538-7836.2007.02450.x
5. Liang J, Mei S, Qiao X, et al. A botanical medicine dragon’s blood exhibited clinical antithrombosis efficacy similar to low molecular weight heparin. Sci China Life Sci. 2021;64(10):1691–1701. doi:10.1007/s11427-020-1848-8
6. Koitabashi N, Niwamae N, Taguchi T, Ohyama Y, Takama N, Kurabayashi M. Remarkable regression of massive deep vein thrombosis in response to intensive oral rivaroxaban treatment. Thromb J. 2015;13(1):13. doi:10.1186/s12959-015-0045-1
7. Momi S, Canino J, Vismara M, et al. Proline-rich tyrosine kinase Pyk2 regulates deep vein thrombosis. Haematologica. 2022;107(6):1374–1383. doi:10.3324/haematol.2021.279703
8. Tang JJ, Meng QY, Cai ZX, Li XQ. Corrigendum to “Transplantation of VEGFl65-overexpressing vascular endothelial progenitor cells relieves endothelial injury after deep vein thrombectomy” [Thromb. Res. 137 (2016) 41–45]. Thromb Res. 2017;154:107. doi:10.1016/j.thromres.2017.04.026
9. Jin T, Jiang L, Zhang X. Influence of Lower Extremity Deep Venous Thrombosis in Cerebral Infarction on Coagulation Index and Thromboelastogram and Its Risk Factors. J Healthc Eng. 2022;2022:1–6.
10. Han Y, Bai X, Wang X. Exosomal myeloperoxidase as a biomarker of deep venous thrombosis. Ann Transl Med. 2022;10(1):9. doi:10.21037/atm-21-5583
11. Akpinar EE, Hosgun D, Akan B, Ates C, Gulhan M. Does thromboprophylaxis prevent venous thromboembolism after major orthopedic surgery? J Bras Pneumol. 2013;39(3):280–286. doi:10.1590/S1806-37132013000300004
12. Torres-Macho J, Mancebo-Plaza AB, Crespo-Gimenez A, et al. Clinical features of patients inappropriately undiagnosed of pulmonary embolism. Am J Emerg Med. 2013;31(12):1646–1650. doi:10.1016/j.ajem.2013.08.037
13. Elliott CG, Goldhaber SZ, Jensen RL. Delays in diagnosis of deep vein thrombosis and pulmonary embolism. Chest. 2005;128(5):3372–3376. doi:10.1378/chest.128.5.3372
14. Declerck PJ, Gils A. Three decades of research on plasminogen activator inhibitor-1: a multifaceted serpin. Semin Thromb Hemost. 2013;39(04):356–364. doi:10.1055/s-0033-1334487
15. Nougier C, Benoit R, Simon M, et al. Hypofibrinolytic state and high thrombin generation may play a major role in SARS‐COV2 associated thrombosis. J Thromb Haemost. 2020;18(9):2215–2219. doi:10.1111/jth.15016
16. Meltzer ME, Lisman T, de Groot PG, et al. Venous thrombosis risk associated with plasma hypofibrinolysis is explained by elevated plasma levels of TAFI and PAI-1. Blood. 2010;116(1):113–121. doi:10.1182/blood-2010-02-267740
17. Erkal B, Kalayci Yigin A, Palanduz S, Dasdemir S, Seven M. The Effect of PAI-1 Gene Variants and PAI-1 Plasma Levels on Development of Thrombophilia in Patients With Klinefelter Syndrome. Am J Men’s Health. 2018;12(6):2152–2156. doi:10.1177/1557988318801158
18. Vuckovic BA, Djeric MJ, Tomic BV, Djordjevic VJ, Bajkin BV, Mitic GP. Influence of decreased fibrinolytic activity and plasminogen activator inhibitor-1 4G/5G polymorphism on the risk of venous thrombosis. Blood Coagul Fibrin. 2018;29(1):19–24. doi:10.1097/MBC.0000000000000656
19. Sundquist K, Wang X, Svensson PJ, et al. Plasminogen activator inhibitor-1 4G/5G polymorphism, factor V Leiden, prothrombin mutations and the risk of VTE recurrence. Thromb Haemostasis. 2015;114(12):1156–1164. doi:10.1160/TH15-01-0031
20. Li H, Zhang B, Lu S, et al. siRNA-mediated silencing of PAI-1 gene acts as a promoter over the recanalization of endothelial progenitor cells in rats with venous thrombosis. J Cell Physiol. 2019;234(11):19921–19932. doi:10.1002/jcp.28590
21. Shi K, Tian DC, Li ZG, Ducruet AF, Lawton MT, Shi FD. Global brain inflammation in stroke. Lancet Neurol. 2019;18(11):1058–1066. doi:10.1016/S1474-4422(19)30078-X
22. Atkinson AJ, Colburn WA, DeGruttola VG, et al; Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001;69(3):89–95. doi:10.1067/mcp.2001.113989
23. Hickey BA, Cleves A, Alikhan R, Pugh N, Nokes L, Perera A. Can we use biomarkers of coagulation to predict which patients with foot and ankle injury will develop deep vein thrombosis? Foot Ankle Surg. 2019;25(1):59–62. doi:10.1016/j.fas.2017.08.002
24. Obi AT, Sharma SB, Elfline MA, et al. Experimental venous thrombus resolution is driven by IL-6 mediated monocyte actions. Sci Rep. 2023;13(1):3253.
25. Shahneh F, Christian Probst H, Wiesmann SC, et al. Inflammatory monocyte counts determine venous blood clot formation and resolution. Arterioscler Thromb Vasc Biol. 2022;42(2):145–155. doi:10.1161/ATVBAHA.121.317176
26. Pai RZ, Fang Q, Tian G, Zhu B, Ge X. Expression and role of interleukin-1beta and associated biomarkers in deep vein thrombosis. Exp Ther Med. 2021;22(6):1366. doi:10.3892/etm.2021.10800
27. Mukhopadhyay S, Johnson TA, Duru N, et al. Fibrinolysis and inflammation in venous thrombus resolution. Front Immunol. 2019;10:1348. doi:10.3389/fimmu.2019.01348
28. Deguchi H, Elias DJ, Navarro S, Espana F, Griffin JH. Elevated serum amyloid A is associated with venous thromboembolism. Thromb Haemostasis. 2013;109(02):358–359. doi:10.1160/TH12-10-0722
29. Abuduhalike R, Abudouwayiti A, Juan S, MaheMuti A. Study on the Mechanism of NLRP3/IL-1/ NF-κB signaling pathway and macrophage polarization in the occurrence and development of VTE. Ann Vasc Surg. 2023;89:280–292. doi:10.1016/j.avsg.2022.09.056
30. Meng Y, Yin Q, Ma Q, et al. FXII regulates the formation of deep vein thrombosis via the PI3K/AKT signaling pathway in mice. Int J Mol Med. 2021;47(5):1–3.
31. Xie W, Zhang L, Luo W, Zhai Z, Wang C, Shen YH. AKT2 regulates endothelial-mediated coagulation homeostasis and promotes intrathrombotic recanalization and thrombus resolution in a mouse model of venous thrombosis. J Thromb Thrombolys. 2020;50(1):98–111. doi:10.1007/s11239-020-02112-9
32. Cheng X, Sun B, Liu S, Li D, Yang X, Zhang Y. Identification of thrombomodulin as a dynamic monitoring biomarker for deep venous thrombosis evolution. Exp Ther Med. 2021;21(2):142. doi:10.3892/etm.2020.9574
33. Schmaier AA, Anderson PF, Chen SM, et al. TMEM16E regulates endothelial cell procoagulant activity and thrombosis. J Clin Invest. 2023;133(11). doi:10.1172/JCI163808
34. Filep JG. Two to tango: endothelial cell TMEM16 scramblases drive coagulation and thrombosis. J Clin Invest. 2023;133(11). doi:10.1172/JCI170643
35. Kondreddy V, Magisetty J, Keshava S, Rao LVM, Pendurthi UR. Gab2 (Grb2-Associated Binder2) plays a crucial role in inflammatory signaling and endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2021;41(6):1987–2005. doi:10.1161/ATVBAHA.121.316153
36. Kondreddy V, Keshava S, Das K, Magisetty J, Rao L, Pendurthi UR. The Gab2-MALT1 axis regulates thromboinflammation and deep vein thrombosis. Blood. 2022;140(13):1549–1564. doi:10.1182/blood.2022016424
37. Dias L, Pinto MJ, Castro P, Carvalho M. Inflammatory biomarkers correlate with time evolution in cerebral venous thrombosis. J Stroke Cerebrovascular Dis. 2021;30(7):105844. doi:10.1016/j.jstrokecerebrovasdis.2021.105844
38. Abe H, Kunita A, Otake Y, et al. Virus-host interactions in carcinogenesis of Epstein-Barr virus-associated gastric carcinoma: potential roles of lost ARID1A expression in its early stage. PLoS One. 2021;16(9):e256440. doi:10.1371/journal.pone.0256440
39. Beurel E, Grieco SF, Jope RS. Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Therapeut. 2015;148:114–131.
40. Kaidanovich-Beilin O, Woodgett JR. GSK-3: functional insights from cell biology and animal models. Front Mol Neurosci. 2011;4. doi:10.3389/fnmol.2011.00040
41. Lu R, Zhu W, Sun H, et al. Study on the effect and mechanism of miR-185 on lower extremity deep venous thrombosis. Mol Biotechnol. 2022;64(3):330–337. doi:10.1007/s12033-021-00412-w
42. Wang W, Zhu X, Du X, et al. MiR-150 promotes angiogensis and proliferation of endothelial progenitor cells in deep venous thrombosis by targeting SRCIN1. Microvasc Res. 2019;123:35–41. doi:10.1016/j.mvr.2018.10.003
43. Du X, Hong L, Sun L, et al. miR-21 induces endothelial progenitor cells proliferation and angiogenesis via targeting FASLG and is a potential prognostic marker in deep venous thrombosis. J Transl Med. 2019;17(1). doi:10.1186/s12967-019-2015-z
44. Laurent P, Severin S, Gratacap M, Payrastre B. Class I PI 3-kinases signaling in platelet activation and thrombosis: PDK1/Akt/GSK3 axis and impact of PTEN and SHIP1. Adv Bio Regul 2014;54:162–174. doi:10.1016/j.jbior.2013.09.006
45. Xie J, Wu W, Zheng L, et al. Roles of MicroRNA-21 in skin wound healing: a comprehensive review. Front Pharmacol. 2022;13:828627. doi:10.3389/fphar.2022.828627
46. Surina S, Fontanella RA, Scisciola L, Marfella R, Paolisso G, Barbieri M. miR-21 in Human Cardiomyopathies. Front Cardiovasc Med. 2021;8:767064. doi:10.3389/fcvm.2021.767064
47. Rosendaal FR. Thrombosis in the young: epidemiology and risk factors. a focus on venous thrombosis. Thromb Haemostasis. 2018;78:1–6.
48. Lou Z, Zhu J, Li X, et al. LncRNA Sirt1-AS upregulates Sirt1 to attenuate aging related deep venous thrombosis. Aging. 2021;13(5):6918–6935. doi:10.18632/aging.202550
49. Kumar R, Sonkar VK, Swamy J, et al. DNase 1 protects from increased thrombin generation and venous thrombosis during aging: cross‐sectional study in mice and humans. J Am Heart Assoc. 2022;11(2). doi:10.1161/JAHA.121.021188
50. Fuchs TA, Abed U, Goosmann C, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176(2):231–241. doi:10.1083/jcb.200606027
51. Constantinescu-Bercu A, Grassi L, Frontini M, Salles-Crawley II, Woollard K, Crawley JT. Activated αIIbβ3 on platelets mediates flow-dependent NETosis via SLC44A2. Elife. 2020;9. doi:10.7554/eLife.53353
52. von Bruhl ML, Stark K, Steinhart A, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209(4):819–835. doi:10.1084/jem.20112322
53. Sharma S, Hofbauer TM, Ondracek AS, et al. Neutrophil extracellular traps promote fibrous vascular occlusions in chronic thrombosis. Blood. 2021;137(8):1104–1116. doi:10.1182/blood.2020005861
54. Secomb TW. Hemodynamics. Compr Physiol. 2016;6:975–1003.
55. Aird WC. Phenotypic heterogeneity of the endothelium: i. Structure, function, and mechanisms. Circ Res. 2007;100(2):158–173. doi:10.1161/01.RES.0000255691.76142.4a
56. Nayak L, Sweet DR, Thomas A, et al. A targetable pathway in neutrophils mitigates both arterial and venous thrombosis. Sci Transl Med. 2022;14(660):j7465. doi:10.1126/scitranslmed.abj7465
57. Tang X, Wang P, Zhang R, et al. KLF2 regulates neutrophil activation and thrombosis in cardiac hypertrophy and heart failure progression. J Clin Invest. 2022;132(3). doi:10.1172/JCI147191
58. Petzold T, Zhang Z, Ballesteros I, et al. Neutrophil “plucking” on megakaryocytes drives platelet production and boosts cardiovascular disease. Immunity. 2022;55(12):2285–2299. doi:10.1016/j.immuni.2022.10.001
59. Dhanesha N, Jain M, Doddapattar P, Undas A, Chauhan AK. Cellular fibronectin promotes deep vein thrombosis in diet‐induced obese mice. J Thromb Haemost. 2021;19(3):814–821. doi:10.1111/jth.15206
60. Dhanesha N, Ahmad A, Prakash P, Doddapattar P, Lentz SR, Chauhan AK. Genetic ablation of extra domain A of fibronectin in hypercholesterolemic mice improves stroke outcome by reducing thrombo-inflammation. Circulation. 2015;132(23):2237–2247. doi:10.1161/CIRCULATIONAHA.115.016540
61. Dhanesha N, Chorawala MR, Jain M, et al. Fn-EDA (Fibronectin Containing Extra Domain A) in the plasma, but not endothelial cells, exacerbates stroke outcome by promoting thrombo-inflammation. Stroke. 2019;50(5):1201–1209. doi:10.1161/STROKEAHA.118.023697
62. Chorawala MR, Prakash P, Doddapattar P, Jain M, Dhanesha N, Chauhan AK. Deletion of extra Domain A of fibronectin reduces acute myocardial ischaemia/reperfusion injury in hyperlipidaemic mice by limiting thrombo-inflammation. Thromb Haemostasis. 2018;118(08):1450–1460. doi:10.1055/s-0038-1661353
63. Doddapattar P, Jain M, Dhanesha N, Lentz SR, Chauhan AK. Fibronectin containing extra domain A induces plaque destabilization in the innominate artery of aged apolipoprotein E-deficient mice. Arterioscl Throm Vas. 2018;38(3):500–508. doi:10.1161/ATVBAHA.117.310345
64. Stein PD, Beemath A, Olson RE. Obesity as a risk factor in venous thromboembolism. Am J Med. 2005;118(9):978–980. doi:10.1016/j.amjmed.2005.03.012
65. Klovaite J, Benn M, Nordestgaard BG. Obesity as a causal risk factor for deep venous thrombosis: a M endelian randomization study. J Intern Med. 2015;277(5):573–584. doi:10.1111/joim.12299
66. Ali RA, Estes SK, Gandhi AA, et al. Defibrotide inhibits antiphospholipid antibody-mediated neutrophil extracellular trap formation and venous thrombosis. Arthritis Rheumatol. 2022;74(5):902–907. doi:10.1002/art.42017
67. Neri T, Nieri D, Celi A. P-selectin blockade in COVID-19-related ARDS. Am J Physiol Lung C. 2020;318(6):L1237–L1238. doi:10.1152/ajplung.00202.2020
68. Feng Q, Wang M, Muhtar E, Wang Y, Zhu H. Nanoparticles of a new small-molecule P-selectin inhibitor attenuate thrombosis, inflammation, and tumor growth in two animal models. Int J Nanomed. 2021;16:5777–5795. doi:10.2147/IJN.S316863
69. Japp AG, Chelliah R, Tattersall L, et al. Effect of PSI‐697, a novel P‐selectin inhibitor, on platelet–monocyte aggregate formation in humans. J Am Heart Assoc. 2013;2(1):e6007. doi:10.1161/JAHA.112.006007
70. Reitsma PH, Versteeg HH, Middeldorp S. Mechanistic view of risk factors for venous thromboembolism. Arterioscl Throm Vas. 2012;32(3):563–568. doi:10.1161/ATVBAHA.111.242818
71. Bovill EG, van der Vliet A. Venous valvular stasis-associated hypoxia and thrombosis: what is the link? Annu Rev Physiol. 2011;73(1):527–545. doi:10.1146/annurev-physiol-012110-142305
72. Collard CD, Montalto MC, Reenstra WR, Buras JA, Stahl GL. Endothelial oxidative stress activates the lectin complement pathway: role of cytokeratin 1. Am J Pathol. 2001;159(3):1045–1054. doi:10.1016/S0002-9440(10)61779-8
73. Kataoka H, Hamilton JR, McKemy DD, et al. Protease-activated receptors 1 and 4 mediate thrombin signaling in endothelial cells. Blood. 2003;102(9):3224–3231. doi:10.1182/blood-2003-04-1130
74. Durigutto P, Macor P, Pozzi N, et al. Complement activation and thrombin generation by MBL bound to beta2-Glycoprotein I. J Immunol. 2020;205(5):1385–1392. doi:10.4049/jimmunol.2000570
75. Damoah CE, Snir O, Hindberg K, et al. High levels of complement activating enzyme MASP-2 are associated with the risk of future incident venous thromboembolism. Arterioscler Thromb Vasc Biol. 2022;42(9):1186–1197. doi:10.1161/ATVBAHA.122.317746
76. Hasselwander S, Xia N, Mimmler M, et al. B lymphocyte-deficiency in mice promotes venous thrombosis. Heliyon. 2022;8(11):e11740. doi:10.1016/j.heliyon.2022.e11740
77. Bertin FR, Rys RN, Mathieu C, Laurance S, Lemarié CA, Blostein MD. Natural killer cells induce neutrophil extracellular trap formation in venous thrombosis. J Thromb Haemost. 2019;17(2):403–414. doi:10.1111/jth.14339
78. Luther N, Shahneh F, Brahler M, et al. Innate effector-memory T-cell activation regulates post-thrombotic vein wall inflammation and thrombus resolution. Circ Res. 2016;119(12):1286–1295. doi:10.1161/CIRCRESAHA.116.309301
79. Shahneh F, Grill A, Klein M, et al. Specialized regulatory T cells control venous blood clot resolution through SPARC. Blood. 2021;137(11):1517–1526. doi:10.1182/blood.2020005407
80. Lapointe C, Vincent L, Giguère H, et al. Chymase Inhibition resolves and prevents deep vein thrombosis without increasing bleeding time in the mouse model. J Am Heart Assoc. 2023;12(4). doi:10.1161/JAHA.122.028056
81. Thienel M, Muller-Reif JB, Zhang Z, et al. Immobility-associated thromboprotection is conserved across mammalian species from bear to human. Science. 2023;380(6641):178–187. doi:10.1126/science.abo5044
82. Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol. 2013;13(1):34–45. doi:10.1038/nri3345
83. Vazquez-Garza E, Jerjes-Sanchez C, Navarrete A, Joya-Harrison J, Rodriguez D. Venous thromboembolism: thrombosis, inflammation, and immunothrombosis for clinicians. J Thromb Thrombolys. 2017;44(3):377–385. doi:10.1007/s11239-017-1528-7
84. Mankame AR, Sanders KE, Cardenas JC. Time-dependent changes in proinflammatory mediators are associated with trauma-related venous thromboembolism. Shock. 2023;60(5):637–645. doi:10.1097/SHK.0000000000002216
85. Geddings JE, Hisada Y, Boulaftali Y, et al. Tissue factor-positive tumor microvesicles activate platelets and enhance thrombosis in mice. J Thromb Haemost. 2016;14(1):153–166. doi:10.1111/jth.13181
86. Ghosh A, Li W, Febbraio M, et al. Platelet CD36 mediates interactions with endothelial cell–derived microparticles and contributes to thrombosis in mice. J Clin Invest. 2008;118(5):1934–1943.
87. Falati S, Liu Q, Gross P, et al. Accumulation of tissue factor into developing thrombi in vivo is dependent upon microparticle P-selectin glycoprotein ligand 1 and platelet P-selectin. J Exp Med. 2003;197(11):1585–1598. doi:10.1084/jem.20021868
88. Rossaint J, Kuhne K, Skupski J, et al. Directed transport of neutrophil-derived extracellular vesicles enables platelet-mediated innate immune response. Nat Commun. 2016;7(1):13464. doi:10.1038/ncomms13464
89. Lin H, Chen H, Qi B, et al. Brain-derived extracellular vesicles mediated coagulopathy, inflammation and apoptosis after sepsis. Thromb Res. 2021;207:85–95. doi:10.1016/j.thromres.2021.09.014
90. Edelmann B, Gupta N, Schnoeder TM, et al. JAK2-V617F promotes venous thrombosis through β1/β2 integrin activation. J Clin Invest. 2018;128(10):4359–4371. doi:10.1172/JCI90312
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.