")
Back to Journals » Biologics: Targets and Therapy » Volume 15
Recent Advances in the Treatment of Hemophilia: A Review
Authors Marchesini E, Morfini M , Valentino L
Received 2 February 2021
Accepted for publication 10 May 2021
Published 15 June 2021 Volume 2021:15 Pages 221—235
DOI https://doi.org/10.2147/BTT.S252580
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Shein-Chung Chow
Emanuela Marchesini,1 Massimo Morfini,2 Leonard Valentino3,4
1Hemophilia Centre, SC Vascular and Emergency Department, University of Perugia, Perugia, Italy; 2Italian Association of Haemophilia Centres (AICE), Naples, Italy; 3National Hemophilia Foundation, New York, NY, USA; 4Rush University, Chicago, IL, USA
Correspondence: Massimo Morfini via Dello Statuto N.1- I-50129, Firenze, Italy
Tel +39 348 2306928
Fax +39 055-4732 18
Email [email protected]
Abstract: Progress in hemophilia therapy has been remarkable in the first 20 years of the third millennium, but the innovation began with the description the fractionation of plasma in 1946. The first concentrates followed the discovery of FVIII in the cryoprecipitate of frozen plasma and FIX in the supernatant in the early 1960s, which led to the initial attempts at replacement therapy. Unfortunately, the lack of screening methods for viral pathogens resulted in people with hemophilia (PWH) receiving concentrates contaminated by hepatitis A virus, hepatitis C virus, and human immunodeficiency virus, as these concentrates were made from large industrial pools of plasma derived from thousands of donors. Fortunately, by 1985, viral screening methods and proper virucidal techniques were developed that made concentrates safe. Increasingly pure products followed the introduction of chromatography steps with monoclonal antibodies in the production process. The problem of immunogenicity of exogenously administered concentrates has not yet had a complete solution. The development of alloantibodies against FVIII in about 25– 35% of PWH is the most serious adverse effect of replacement therapy. The next major advance followed the cloning of the F8 gene and later the F9 genes, which paved the way to produce concentrates of factors obtained by the recombinant DNA technology. The injected FVIII and FIX molecules had a relatively short circulating half-life in the plasma of people with hemophilia A and B, approximately 12 and 18 hours, respectively. The ability to prolong the plasma half-life and extend the interval between injections followed the application of methods to conjugate the factor molecule with the fragment crystallizable of IgG1 or albumin or by adding polyethylene glycol, which has led to an increase in the half-life of concentrates, especially for rFIX. The next frontier in hemophilia therapy is the application of durable and potentially curative therapies such as with gene addition therapy. Experiments in hemophilia B have demonstrated durable responses. Unfortunately, the results with gene therapy for hemophilia A have not been as remarkable and the durability must still be demonstrated. Nonetheless, the long-term safety, predictability, durability, and efficacy of gene therapy for hemophilia A and B remain an open question. At present, only healthy adult PWH have been enrolled in gene therapy clinical trials. The application of gene therapy to children and those with pre-existing antibodies against the delivery vector must also be studied before this therapy becomes widespread.
Keywords: replacement therapy, adverse events, pharmacokinetics, extended half-life concentrates, non-replacement therapy, gene therapy
Introduction
Hemophilia A (HA) and B (HB) are inherited bleeding disorders due to the partial or total deficiency of coagulation factor (F) VIII or FIX, respectively. Hemophilia is due to a gene mutation in the X chromosome, and thus males with an XY genotype are affected while females with XX genotype are carriers of the disease but may also be affected and have bleeding. These women are generally mildly symptomatic, but sometimes, due to non-random inactivation of one of the XX chromosomes (lyonization), they can be more severely affected, especially with menorrhagia. The range of FVIII concentration in the healthy population is quite wide, 64–197 IU/dL, and it also depends on the blood group: 55–150 IU/dL in group O and 71–186 in non-O.1 The FVIII or FIX concentration in plasma of people with hemophilia (PWH) is <1 IU/dL in those with a severe form of the disease, 1–5 IU/dL in those with moderate hemophilia, and 5–40 UI/dL in patients with a mild form. Spontaneous bleeding in PWH is most commonly localized to the joints and skeletal muscles, rarely at skin or mucosal surfaces as occurring in patients affected by thrombocytopenia or those with platelet dysfunction. Bleeding following minor trauma or with surgical procedures may cause severe bleedings at the injury site in PWH. There is a rough correlation between the severity of the disease and the types of bleeding observed in PWH. In severe hemophilia, hemarthrosis, muscle hematomas, CNS hemorrhages, and hematuria are frequent even in the absence of any apparent trauma; in moderately severe hemophilia, moderate trauma or minor surgery may trigger bleeding, and in mild disease, bleeding usually only occurs following severe trauma or major surgery.
Generally, HB is considered less severe than HA,2–4, but there is not complete agreement on this issue.5,6 We observed that arthropathy in Italy was less frequent in HB patients than in HA.7 The difference in genotypes of the two diseases seems to be the reason for this discrepancy. In severe HA, the inversion of intron 22 results in an extreme change of the F8 gene sequence and is the causative mutation in about 50% of the patients. In comparison, in HB, the small and complete deletion of the F9 gene represents only 17%, and missense mutations account for 55% of cases.8 Missense mutations represent about 75% of moderate and 16% of severe HA, respectively. The more severe manifestations of joint and muscle bleeding are cartilage damage and synovial inflammation and hypertrophy developing into a chronic debilitating arthropathy. The muscle and skeletal derangement may soon cause a limitation of movements during daily activities with a consequent decrease in patients’ quality of life.
The Replacement Therapy and Its Adverse Events
After assessing that hemophilia was due to the lack of a plasma clotting factor,9 the disease’s first treatment was introduced in 1948 by the infusion of plasma from healthy blood donors.10 In 1958, Nilsson 1M and Blomback B in Sweden successfully treated HA patients with a globulin fraction of human plasma purified by the Cohn’s fractionation method and contained in the fraction 1-0.11 At about the same time, in 1964, a new era of hemophilia therapy started after the discovery of cryoprecipitate from the plasma of a single blood donor.12,13 Unfortunately, cryoprecipitate’s specific activity was relatively low (FVIII:C 0.2 IU/mg/mL).13,14 The need for production and storage at −20°C in plastic bags made this approach to hemophilia therapy cumbersome and inconvenient. Furthermore, each cryoprecipitate bag contained only about 100 IU of FVIIl. A hypothetical dose of 20–30 IU/kg required the administration of about 14 or 21 bags to a PWH of 70 kg body weight. The pharmaceutical industry soon began producing lyophilized FVIII concentrates, starting from a large pool of plasma. Again, the first step was the cryo-precipitation of the plasma, but purification methods allowed the production process to achieve a remarkably high specific activity, mainly through immunoaffinity chromatography with monoclonal antibodies specific for FVIll. Unfortunately, the concentrates from large plasma pools and the single donor cryoprecipitates were contaminated by the hepatitis viruses15 even though the infection risk was lower for cryoprecipitates.16 Later, HIV transmission occurred in 75% of HA patients treated with commercial clotting factor concentrates17 but only in 20% of those treated with single donor cryoprecipitates,18 possibly since the donors were known to the patient, typically a parent. HIV transmission occurred in more than 50% of the PWH in the United States;19 In Italy, probably due to the large availability of the first Prothrombin Complex Concentrates,20 the prevalence of HIV infection was 47.1% in people with hemophilia B but 26.8% among those with hemophilia A.21 Before the end of the last millennium, three significant advances in the manufacturing process of concentrates impacted the viral safety of products used for replacement therapy:
- The implementation of more accurate screening of blood donors for infection by enveloped viruses.22
- The implementation of several virucidal methods such as pasteurization, vapor heating, and heating in the lyophilized state at 80°C. The addition of solvent/detergent in the manufacturing process’s final steps resulted elimination of lipid enveloped viruses but not on naked viruses such as parvovirus,23,24 HAV25 or transfusion-transmitted virus (TTV).26
- The production of new concentrates through DNA-recombinant biotechnologies.
The first-generation recombinant FVIII concentrates were produced in Chinese Hamster Ovary (CHO) cells, transfected with human cDNA encoding for FVIII in a culture medium containing human albumin.27,28 To improve the purity of recombinant FVIll concentrates, albumin was removed from the culture media (second-generation rFVIII concentrates), and finally, albumin was eliminated from the final formulation and a non-protein stabilizer added prior to lyophilization (third-generation rFVIII).29 Soon afterward the introduction of the first rFVIII concentrates, rFIX concentrate was also introduced for HB treatment.30 At about the same time, a modified rFVIII molecule, in which the B domain had been deleted31 or truncated was marketed and showed improved production yield from the cell line and, in the following years, became the standard procedure to produce rFVIII concentrates.32 Unfortunately, after the widespread conversion from plasma-derived FVIII (pdFVIII) to rFVIII concentrates for the therapy of HA, the incidence of neutralizing antibodies directed to the clotting activity of FVIII, so-called “inhibitors” became more evident. This adverse event is very severe because the effect of the infused products can be entirely or partially reduced, according to the titer of inhibitor. The incidence of inhibitors is 24% in HA patients treated with pdFVIII concentrates but 33% and up to 52%33–35 of those treated with rFVIII concentrates, depending on the study. The high incidence of inhibitors against rFVIII in previously untreated patients (PUPs) was also observed in the regulatory clinical trials for approval of all rFVIII concentrates: the incidence was 29.7% for Kogenate® (Bayer), 31.0% for Recombinate® (Baxter), 33.0% for ReFacto® (Pfizer), and 38.0% after the first 18 exposure days to Kogenate®.36,37 The claimed lower immunogenicity of pdFVIII versus rFVIII concentrates was contradicted by a prospective study of PUPs conducted in Germany in which no difference between pdFVIll (36%) and rFVIll (36%) concentrates was observed.38 Other retrospective or observational studies showed discordant results between pdFVIII and rFVIII. A randomized, controlled, open-labeled trial in PUPs with severe hemophilia A (Survey of Inhibitors in Plasma-Product Exposed Toddlers [SIPPET]) was conducted in Europe, Asia and the US in which the incidence of inhibitors after the first exposures to pdFVIII or rFVIII concentrates was compared.39 The cumulative incidence of FVIII inhibitors observed in 29 HA patients treated with pdFVIII was 26.8% vs 44.5% in 47 HA treated with rFVIII concentrates, respectively. The hazard ratio of developing inhibitors was 1.87 (95% Cl: 1.17–2.96) for rFVIII vs pdFVIII concentrates. Similar outcomes were observed from a study conducted in France.40 In conclusion, the immunogenicity of both pdFVIII and rFVIII remains a significant, unsolved adverse event of replacement therapy of HA. After treating HB with pdFIX or rFIX concentrates, FIX inhibitors’ incidence is exceptionally low, about 3%.41 These antibodies can fix the complement, and, in some patients, severe anaphylactic reactions occurred just after the intravenous (iv) infusion of the concentrate.42
The Targets of Replacement Therapy in Hemophilia
The main aim of hemophilia treatment is to prevent life-threatening bleeding and/or treat muscle and joint bleeding. Early, treatment was limited to on-demand therapy, ie, the infusion of concentrate after the occurrence of the bleeding, also allowing the home therapy a prompt and successful treatment.43
Even though this form of therapy can stop bleeding, the damage to joints or the muscles starts a few hours after the onset of bleeding.44,45 The observational Orthopedic Outcome Study demonstrated the advantages of prophylaxis started after the occurrence of bleeding, so-called secondary prophylaxis, compared to on-demand therapy.46
The observation that in hemophilia patients moderate (FVIII/IX: 1–5 IU/dL) or mild disease (FVIII/IX: 5–40 IU/dL), bleeding was uncommon or absent suggested that increasing the factor level with an adequate amount of FVIII/IX concentrate every two or three other days to maintain the FVIII/FIX plasma concentration above 1IU/dL might prevent bleeding. According to a study conducted in the Netherlands, only a baseline FVIII of at least 12 IU/dL was able to entirely abolish the risk of joint bleeding. Another study from the US examined data collected over an 11-year period from males with non-severe hemophilia A or B without inhibitors who were treated on-demand with factor therapy. These authors showed that levels of 20% may be required to prevent all joint bleeding.47 Another study from the Netherlands showed that PWH who have a baseline factor activity of at least 5 IU/dL experience one episode of joint bleeding each year while those with a baseline level of 3 IU/dL experience two episodes yearly.48 Unfortunately, even a single episode of joint bleeding may trigger hemophilia arthropathy.49
Recurrent bleeding results in a progressive, chronic synovitis, cartilage, and bone destruction, and eventually a disabling arthropathy accompanied by muscle atrophy. Iron from degrading red blood cells is responsible for synovial hyperplasia and the release of inflammatory mediators including interleukins (IL)-1, IL-6, and tumour necrosis factor (TNF)-α all result in cartilage destruction.50 Increased production of vascular endothelial growth factor (VEGF) triggers the neo-angiogenesis of synovial vessels.51 Very soon, it was realized that on-demand therapy could not prevent arthropathy or the associated muscle atrophy, with severe side effects on the patients’ movements.52
A randomized trial demonstrated the value of continuous, early prophylaxis started prior to the second episode of joint bleeding (primary prophylaxis)53 on the prevention of joint damage in children with severe hemophilia A:54 the annualized bleeding rate (ABR) was 4.89±3.57 in the on-demand treatment group but only 0.63±1.35 in the prophylaxis group (p<0.01). Similar findings were seen in an Italian study, the ESPRIT in which the bleeding rate was 0.20 episodes/patient/month in those receiving prophylaxis versus 0.52 episodes/patient/month in the group of patients on treated episodically on-demand.55 Even prophylaxis initiated after two or more episodes of joint bleeding but before the onset of joint disease (secondary prophylaxis) decreased the frequency of bleeding and was advantageous to avoid a further deterioration of the joints and muscles damaged from the previous bleeding.56,57 Similar benefits of prophylaxis in adult patients have been demonstrated compared to on-demand treatment.58
Adolescents and young adults may discontinue prophylaxis to avoid the burden of 2–3 weekly infusions, returning to episodic treatment. These individuals experienced a decrement in their quality of life and joint score because of the increase in bleeding frequency.58,59
A Predictor of the Efficacy of Replacement Therapy: Pharmacokinetics
The aim of replacement therapy is to prevent fatal bleeding and the effects of bleeding on the joints and muscles. Some patients can recognize the symptoms forecasting the bleeding, the so-called “aura.” However, prevention of bleeding is better than treating bleeding with on-demand replacement therapy because, in the short-run, joint inflammation leads to new bleeding and starts a vicious circle that ends in severe arthropathy. Therefore, prevention of bleeding, so-called prophylaxis, was hypothesized to result in better outcomes than on-demand therapy, especially if a minimum trough level was maintained in the plasma. Maintaining the trough level of FVIII or FIX above 3–5 IU/dL is sufficient to reduce the number of bleeding events to one or two episodes every year.48
The plasma concentration of standard half-life FVIII or FIX concentrates decreases after intravenous infusion by an exponential decay curve, with a half-life of 12–15 hrs or 25–30 hrs, respectively. There is a large variability among the patients in the response even to the same factor concentrate, and the assessment of pharmacokinetic (PK) characteristics of a particular concentrate in each patient is recommended to predict the dose and the interval between the iv administrations. “One size doesn’t fit all,” and the tailoring of prophylaxis is the best way to assure that each patient has the best and optimal outcome. With a pharmacokinetic study, the maximal concentration (Cmax) of the protein in the plasma just after the infusion and the in vivo recovery (IVR), defined as the ratio between the Cmax and the dose administered in IU/kg can be determined to guide dosing. Other PK parameters are the area under the curve (AUC), the clearance (Cl), the half-life (HL), the volume of distribution (Vd). All of these parameters should be considered to determine the optimal dosing regimen thereby improving the efficacy of the concentrate in preventing bleeding during the patient’s daily life.60 A Cmax near the average normal value can protect the patient during intense activities including some sports; the HL can provide the patients an idea about how long until the next dose is needed. The most critical PK parameter to define the right dose is the Cl, which represents the amount of plasma made free of the concentrate after the infusion. The following formula explains the procedure for an accurate calculation of the dose for repeated treatments, according to three fundamental parameters:
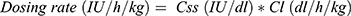

where Tau is the interval between the administrations and CSS is the concentration of infused clotting factor at the steady state. It is also evident that continuous infusion, especially useful during post-surgical treatment, is the least expensive way of delivering therapy, in that Tau is equal to 0.
Another more user-friendly approach to the evaluation of PK parameters is offered by Population PK (PopPK). The Bayesian compromise, on which PopPK is based, provides a rough estimation of PK parameters depending on the performance of the drug in a large population of patients. If the population data are abundant, the patients’ PK parameters will be switched toward the mean of the population (the “shrinkage” effect)62 and vice versa when the population data are sparse: in this case, the data of the patient will have more weight. Even though PopPk has more recently been used in hemophilia, it should be validated versus standard individual PK but it may be useful from a clinical perspective to tailor the treatment’s regimen. It is well known that the inter-patient variability of clotting factor concentrates is extremely large.63 To shed light on this issue, several genetic polymorphisms have recently been evaluated to examine their impact on FVIII individual PK parameters in hemophilia A patients. These polymorphisms include LDLR,64 CLEC4M,65 genetic modifiers of VWF,66 and ASGR2 5ʹ UTR haplotype.67
The Extended Half-Life (EHL) Recombinant FVIII/IX Concentrates (rFVIII/FIX EHL)
The prominent feature limiting the benefit of prophylaxis has been the need for frequent, repeated iv infusions every 2–3 days for FVIII and every 5–7 days for FIX concentrates, respectively. The pharmaceutical industry has developed some biochemical methods to increase the HL of concentrates in an effort to reduce the burden of the treatment on patients, especially for young patients. The ability to prolong the plasma half-life and extend the interval between injections followed the application of methods to conjugate the factor molecule with the fragment crystallizable (Fc) of IgG1 or albumin or by adding polyethylene glycol (PEG), which has led to a significant extension of the half-life of concentrates, especially for rFIX. Modifications were first applied to rFIX:
- pegylation, the linkage of the molecule with PEG (Rebinyn/Refixia®, Novo Nordisk), which had been a successful procedure for many other drugs to extend their HL;
- conjugation with recombinant albumin (Idelvion®-CSL Behring);
- conjugation with Fc fragment (Alprolix®SOBI).
Table 1 provides a summary of the outcomes of the cross-over regulatory studies, comparing the PK of pdFIX or rFIX standard half-life (SHL) with the new rFIX EHL.68–70 Apart from the improvement of terminal HL of rFIX EHL, which increased from 2.43 to 6.28 times for pdFIX or rFIX SHL concentrates, the more outstanding result is the decrease in Cl (1.97–9.85 times). On the contrary, the volumes of the distribution area (VdArea) of both rFIX EHL and rFIX SHL, compared to theoretical plasma volume (40–45 mL/kg), are large (Table 1). In particular, the VdArea of rFIX Fc seems to be exceptionally large, probably due to protein binding to cells expressing the Fc receptor and incomplete recycling of the protein. The Cl is the unique PK parameter valid to design the repeated treatment dosing during prophylaxis or continuous infusion. From the formula to calculate the dosing rate,61 it is evident that the smaller is the Cl, the more extended the dosing interval will be. Therefore, using continuous infusion, where the Tau is null, is the most dose sparing regimen. Consequently, the rFIX EHL concentrates may allow an essential reduction of dosing rate during prophylaxis or an increase of interval between the administrations, an alternative appreciated by the patients to better enable them to tailor their therapy. On the other hand, we must remember that Cl is the ratio between the dose and the AUC. In two comparative PK studies of Phase I/II clinical trials,68,69 the sample timing of pdFIX and rFIX SHL concentrates had been stopped at the post-infusion time 48 hrs, when the FIX concentrations had not reached the patients’ baseline. On the contrary, the samples of rFIX EHL concentrates had been collected up to when the decay curve became very flat (N9-GP up to 168 hrs, Efrenonacog alfa up to 240 hrs, and rIX FP up to 336 hrs). Therefore, the AUC of the comparator concentrates, pdFIX and rFIX SHL, resulted in an underestimation of about 12%69 or 20%68 and consequently the Clearance was overestimated.
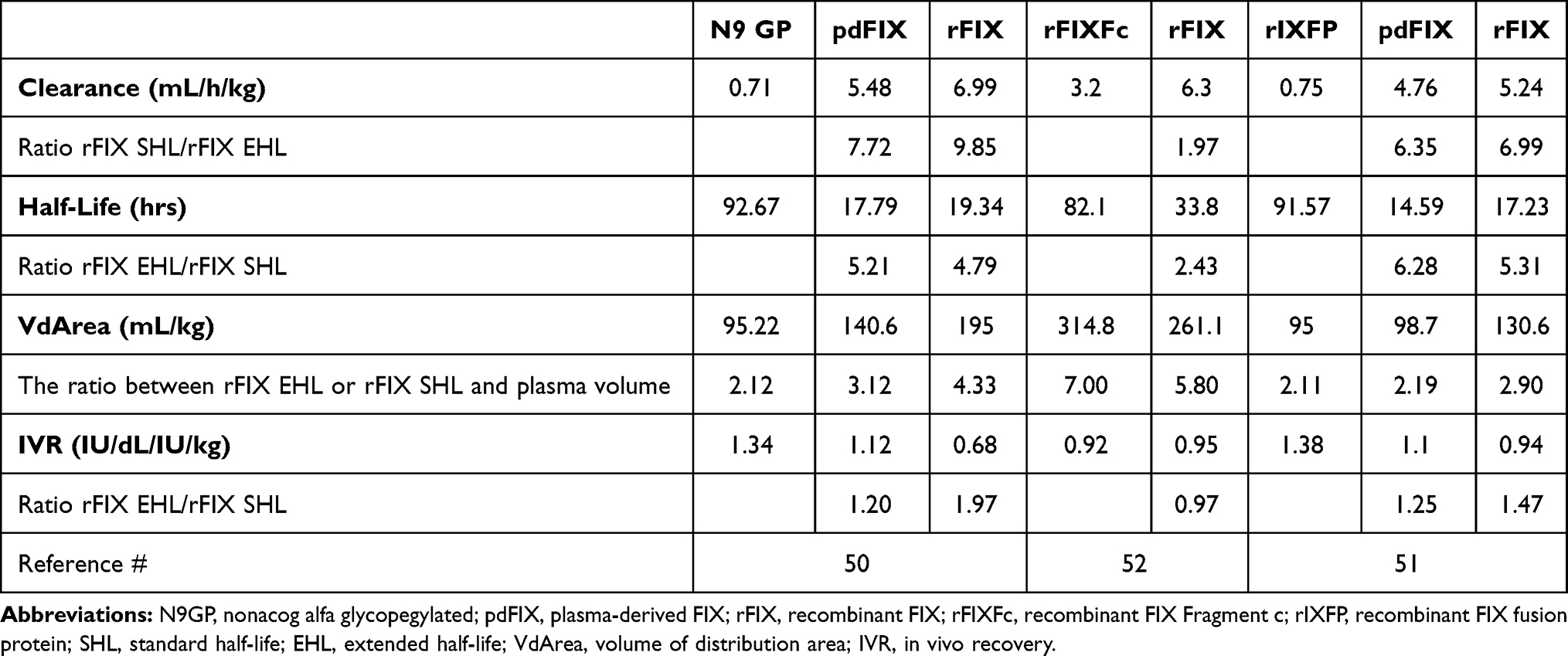 |
Table 1 Ratios Between Some Parameters from the Compared Pharmacokinetic Studies of rFIX EHL versus SHL Concentrates, According to the Outcomes of Phase I/II Clinical Trials |
The improvement of PK parameters of the new rFVIII EHL concentrates has been less evident than those achieved by rFIX EHL (Table 2).71–76 The majority of rFVIII EHL represents a new formulation of older rFVIII SHL after the conjugation with Fc or PEG of different molecular weights and sizes. For many years, pegylation has been used to improve the efficacy of several drugs. To mention some of the first: pegademase to treat the severe combined immunodeficiency syndrome,77 PEG-human growth hormone for acromegaly,78 and PEG interferon-alpha for hepatitis B79 and C.80
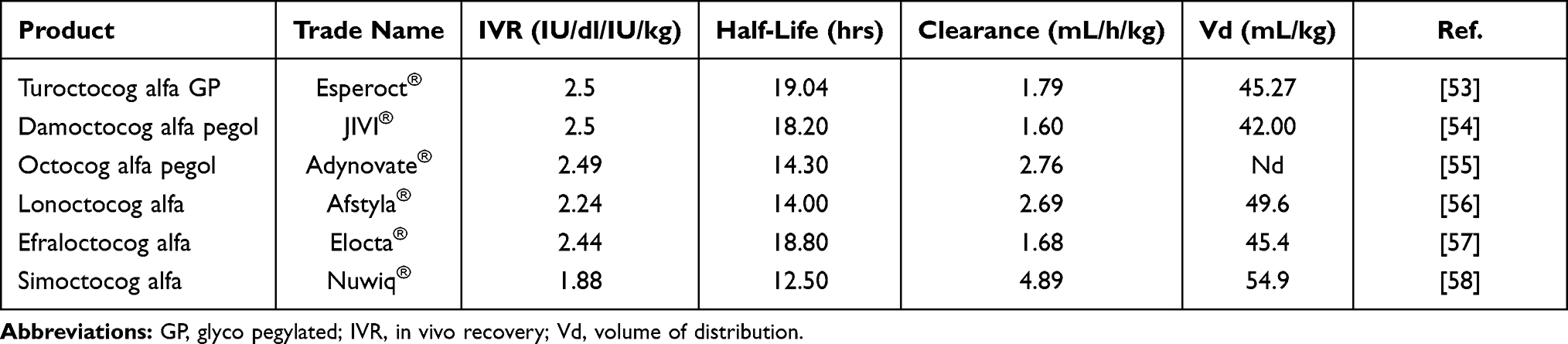 |
Table 2 PK Outcomes of the New rFVIII Concentrates, so Far Licensed |
Recently, several pegylated rFVIII, predominantly B domain deleted, have been approved (Table 2). Unfortunately, the improvement of HL was limited to 2–3 hours compared to the previous SHL formulation. The modest increase in HL allowed for a reduction in the frequency of administration of prophylaxis to twice weekly or every 5 days. Efmoroctocog alfa is B-domain-deleted FVIII covalently attached to the Fc domain of human IgG1. Many human cells express Fc receptors allowing them to bind and take up the FVIII into the cytoplasm through a pinocytosis mechanism and then return it intact to the plasma compartment. This recycling mechanism can increase efmoroctocog alfa HL by about 4–5 hours (Table 2). Additional modifications of the efmoroctocog alfa molecule’s fusion process have been achieved by adding two other proteins, the D’D3 dimer fragment of VWF and a polypeptide chain called XTEN. This new molecule is named BIVV001.81 The D’D3 domain stabilizes FVIII allowing a significant increase of HL in hemophilia mice, up to 37 hrs, about double that of Efmoroctocog Alfa, and resulting in a trough plasma level of 5 IU/dL at 120 hrs after the infusion 25 IU/kg. A non-randomized clinical trial of BIVV001 (NCT04161495), called XTEND-1, is currently ongoing to evaluate two different dosing regimens over a 26-week period: one infusion of BIVV001 at a dose of 50 IU/kg once weekly for prophylaxis and an on-demand regimen of 50 IU/kg as needed to control bleeding.
Non-Replacement Therapies
Despite the progress made with the availability of clotting factors, unmet needs remain for the optimal treatment of PWH. Remarkably, the currently available EHL FVIII/FIX products reduce administration frequency, but treatment is still required to be administered by iv infusion with the attendant challenges associated with venous access, particularly in young children. The problem of immunogenicity of exogenously administered concentrates, the most serious adverse effect of replacement therapy, remains a problem. The incidence of alloantibodies against FVIII and FIX occurs in about 25–35% and 3–5% of the patients with severe HA and HB, respectively. Individuals with inhibitors are poor candidates for prophylaxis, which can only be provided by bypassing agents such as activated prothrombin complex concentrates (APCC) and activated recombinant FVII (rFVIIa), which are very expensive.82 Moreover, inhibitor patients have to infuse bypassing agents frequently, including daily to prevent bleeding resulting in a significant burden of treatment.
The coagulation system is a complex mechanism in which procoagulant and anticoagulant proteins determine the correct coagulation balance. The hemostatic status could be altered, increasing a bleeding or thrombotic tendency when changes affect the coagulation cascade’s balance. The bleeding phenotype of hemophilia patients is determined by several factors that influence the natural hemostatic balance83,84 based on the equilibrium between natural procoagulant and anticoagulant factors. Considering the above observations, novel molecules to rebalance the hemostatic system in PWH have been developed to be less burdensome and more effective. These novel agents act to rebalance the coagulation system by enhancing coagulation (emicizumab)85 or inhibiting anticoagulant pathways (eg, fitusiran, concizumab).86,87
Enhancing Coagulation with Hemlibra® (INN Emicizumab, ACE910)
Emicizumab-KYWH is a new humanized antibody specific for both FIXa and FX, generated by a complex biomolecular process: specific monoclonal antibodies for FIXa and FX were produced by CHO or human embryonic kidney (HEK-293) cells, afterward transfected with plasmids carrying the genes for heavy and light chains of IgG.85 Emicizumab acts as an FVIIIa cofactor mimetic. This molecule binds FIXa and FX and assumes a three-dimensional structure in the tenase complex. The weak binding affinities guarantee the balance of activation coagulation and FX activation for downstream reactions.88
The target activity of emicizumab are sites of bleeding because its function depends on phospholipids. Emicizumab has both similarities and dissimilarities to activated FVIII.85 Subcutaneous (sq) administration of emicizumab at a loading dose of 3.0 mg/kg initially, and 1.5 mg/kg weekly afterward for 4 weeks completely prevented the bleedings. The PK profile of emicizumab is linear with an HL of 4–5 weeks.89 The first trial was conducted in 64 healthy subjects, divided into five groups, six subjects in each: escalating doses of emicizumab (0.001, 0.01, 0.1, 0.3, and 1 µg/kg) were administered sq and two subjects received a placebo as a control in the part A of the study. After assessing tolerability and safety, part B study was performed with three similar groups of subjects receiving 0.1, 0.3, and 1.0 mg/kg of emicizumab sq. Plasma emicizumab concentrations increased soon after the sq injection achieving a Cmax proportional to the doses, followed by a monophasic decay and returning to the baseline value in about 84–168 days. The HL ranged from 28.3 to 34.4 days. During the first 24 days after administration of emicizumab, a sharp decrease of aPTT and an increase of peak height of thrombin generation (TG) were observed in FVIII-neutralized plasma without a rise in hyper-coagulability markers (D-Dimer, TAT, PT-INR, or platelet count) neither occurrence of any adverse event.89 A global Phase III open-label clinical trial program called HAVEN was performed to assess once weekly, once each fortnight, and monthly sq prophylaxis with emicizumab in HA patients with and without FVIII inhibitors. The model-based simulation determined dosing regimens without dose escalation. The ABR, defined as bleeding events requiring additional treatment, (HAVEN 1 – patients >12 yrs) was 2.9 (95% CI 1.7–5.0) in those receiving emicizumab prophylaxis and 23.3 (95% CI.:12.3–43.9) in the control group, resulting in an 87% decrease in the ABR in the emicizumab-treated patients, among which 63% experienced zero bleeds. Thrombotic microangiopathy (TMA) occurred in 2 patients, and cavernous sinus thrombosis in one patient treated contemporaneously with APCC. Another patient experienced TMA soon after starting emicizumab therapy; due to severe rectal bleedings, he was treated even with APCC without success, and the recurrent bleeding was fatal. The concomitant use of emicizumab with rFVIIa did not cause similar adverse events in any patients. The efficacy of emicizumab was demonstrated in 1- to 15-year-old hemophilia patients with inhibitors in which 86% of patients reported no bleeding. In patients without inhibitors to FVIII, the percentage of patients who did not experience any bleedings was approximately 60%. A summary of the HAVEN clinical trial has been reported in Table 3.
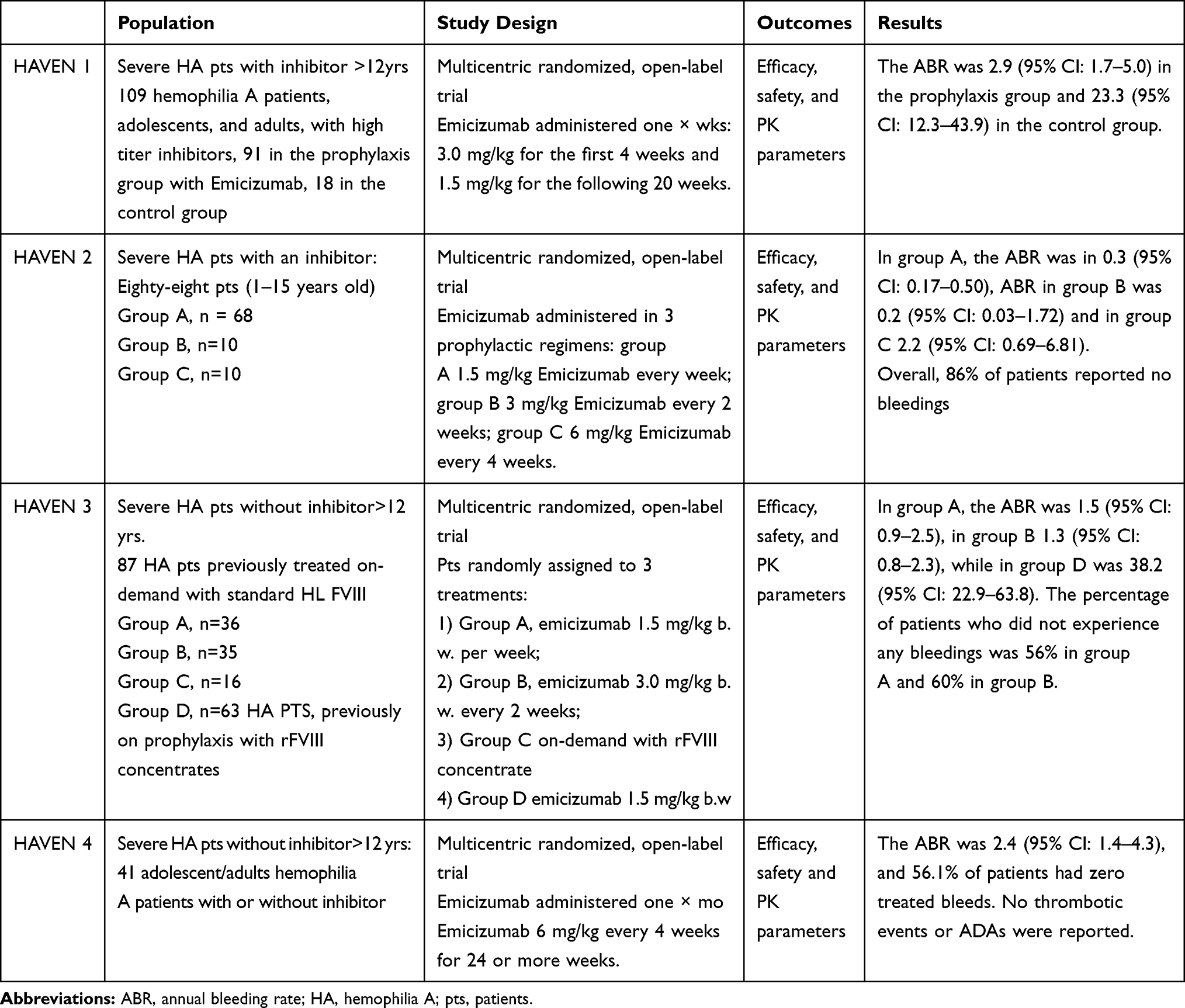 |
Table 3 Summary of HAVEN 1, 2, 3, and 4 Studies |
Dampening the Anti-Coagulant Pathway Towards Equilibrium
Fitusiran
Fitusiran is an antithrombin RNA interference molecule (ALN-AT3; fitusiran, (Alnylam/Sanofi) which decreases antithrombin (AT) messenger RNA expression in the liver.90,91 AT is a natural anticoagulant which inactivates FXa and thrombin. Deficiency of AT results in a prothrombotic state due to excess TG. Fitusiran decreases the plasma concentration of AT and improves thrombin generation, reducing the bleeding tendency, and increasing the survival of HA mice following a hemostatic challenge.92 The administration of fitusiran in animal models showed a significant and prolonged reduction of AT levels, with an improvement in TG that appears to be dose-dependent and persistent. The first trial with fitusiran was conducted in 4 healthy volunteers and 25 HA or HB patients without inhibitors. The participants received a single weekly subcutaneous dose that was escalated until progressively lower plasma levels of AT were observed. After monthly administration, the AT levels showed a reduction of 70% to 90% and a parallel increase of TG in both hemophilia A and B patients. During the Phase 2 study, fitusiran was administered at 50 and 80 mg once-monthly in 14 and 19 patients with and without inhibitor, respectively. The enrolled patients showed a decrease in AT levels of 80% of normal. The overall median ABR in fitusiran-treated patients without inhibitor was one in comparison to 12 in the on-demand factor replacement group and two in the prophylaxis group before the study entry. In treated patients, the median ABR was 0 compared to 38 before study entry. All breakthrough bleeds were managed with FVIII, FIX products, APCC, or rFVIIa, without complications or thrombotic events. The Phase 3 studies to evaluate patient safety and efficacy (with or without inhibitors) are ongoing in three clinical trials. These studies are providing data about fitusiran administration in prophylaxis at 80 mg once monthly.
Recently, the fitusiran trials have been temporarily suspended due to an occurrence of sinus vein thrombosis in a non-inhibitor HA. After evaluating clinical risk and safety and implementing a change in the dosing regimens with respect to fitusiran and any supplemental therapies administered for breakthrough bleeding, dosing has resumed in these trials.
Concizumab
Concizumab is a humanized IgG4 antibody monoclonal selective for the second Kunitz domain of tissue factor pathway inhibitor (TFPI). TFPI is the primary regulator of the tissue factor pathway and inhibits the initiation phase of the coagulation system-binding factor Xa and TF- FVIIa due to the high affinity of TFPI for these serine proteases. This chemical binding inactivates TFPI and reduces the function of this natural anticoagulant. Concizumab restores TG by mitigating the effect of TFPI on the coagulation system. Its effects are achieved by reducing TFPI levels and in turn, TG.93 In a Phase 1 clinical study with anti-TFPI antibodies, a dose-dependent effect was observed with a decreased bleeding tendency.94 After a single infusion, the bioavailability of concizumab was 93%, and the HL was 72 h. The safety, PK, and pharmacodynamics of different doses of concizumab (0.25, 0.5, or 0.8 mg/kg every 4 days) administered subcutaneously to HA patients were evaluated in a phase 1 trial. No safety events were observed, and the PK and pharmacodynamics of the concizumab dose in respect to the TFPI plasma concentration and TG levels confirmed the previous data: 100 ng/mL of concizumab reduced the bleeding tendency. A linear increase in D-dimer and Fragment 1+2 of prothrombin was observed among the laboratory parameters, but no anti-drug antibodies were detected. A multicenter, randomized, double-blinded phase 2 study evaluated the efficacy of daily subcutaneously administered concizumab in HA and HB patients with or without inhibitors. Concizumab was detected in blood samples 43 days after the administration. The profiles of PK performed in healthy volunteers, and hemophilia subjects were similar. A linear relationship between concizumab, D-dimer, and prothrombin Fragment 1+2 levels was observed. The D-dimer response in HA patients and healthy volunteers was similar when they received a dose of concizumab 36-fold higher than volunteers.95
The Explorer 3 trial was a placebo-controlled, multiple-dose, dose-escalation study where concizumab was administered subcutaneously.96 The authors observed a dose-dependent decrease in free and total TFPI and a procoagulant effect. The evaluation of safety and efficacy of concizumab was performed in the Explorer trial program; particularly, the Explorer 4 and 5 trials evaluated the efficacy of concizumab in severe HA and HB patients with and without inhibitors. The design of the trial was a dose-escalation, with an initial dose 0.15 mg/kg (150 mg/kg/day) escalated to a maximum of 0.25 mg/kg (250 mg/kg/day). Patients were observed for 6 months, followed by an addition 12 months of observation in the extension phase. The inhibitor trial Explorer 4 was a multicenter, open label, randomized controlled trial. The non-inhibitor trial Explorer 5 was a multicenter, single-arm, open-label phase 2 trial.97 Concizumab 0.5 mg/kg was administered initially in the Explore 4. The dose was escalated to find a minimal effective and safe dose for each patient. Patients were randomized to prophylaxis with concizumab or on-demand treatment with rFVIIa. Median ABR was 4.5 for concizumab and 19.7 for rFVIIa. In the concizumab group, no significant differences between spontaneous and traumatic bleeds were observed, and the ABR for spontaneous bleeding was 2.3 (95% CI: 1.4–3.6). Seventy treated bleeding episodes were observed in the trial in 23 patients (63.9%), and the median ABR was 4.5. In general, the treatment with concizumab appeared safe and well tolerated. A few AEs were observed without severe AEs and withdrawals for AE. Notably, no thromboembolic events and no deaths were observed.97 Moreover, on March 16, 2020, Novo Nordisk temporarily suspended the clinical trials of Concizumab due to the occurrence of thrombosis in three patients.98 After a modification of a protocol for the treatment of breakthrough bleeding, the phase 3 trial was resumed on August 13, 2020.99
The Gene Therapy
Significant advances in therapy for HA and HB over the past four decades have been achieved, as described in the preceding sections. Despite these innovations, gaps still exist in terms of efficacy, safety, and patient administration ease.100 Breakthrough bleeding continues to occur with EHL products to treat HA and HB patients despite optimized regimens using PK to tailor the treatment regimen and with novel non-factor products, albeit to a lesser degree. The long-term safety of drug conjugates designed to extend the HL of FVIII/FIX products remains a concern for some, as do the non-factor products, which are sometimes criticized as “not natural FVIII or FIX.” All these products require repeated administrations either by the intravenous or subcutaneous route, and adherence to a treatment schedule is necessary. Besides, patients frequently voice their concerns and dissatisfaction with the available treatments for their hemophilia due to the imposed limitations in career and educational choices for the patient and employment choices for patients and caregivers, inability for patients to participate in recreational activities fully, and potential limitations in family life as well as the persistent concern regarding health insurance status. People with hemophilia may experience the inability to live with spontaneity in their life due to their hemophilia.101
Gene therapy offers the possibility to eliminate the need for repeated administration of a medication to prevent bleeding, enhance the quality of life, and provide the spontaneity people with hemophilia lack due to its single administration paradigm. This transformative effect has been realized by people with hemophilia who have undergone a liver transplant for other reasons (eg, HCV-related liver failure).
The ultimate goal for hemophilia treatments should be a “functional cure” and “health equity”.101 Gene therapy research has explored various mechanisms for transgene delivery, including non-viral techniques including chemical, electroporation, and polymer-based procedures; however, viral-mediated delivery (eg, lentiviral, adeno-associated, or gene editing) of a transgene encoding the information necessary for the recipient cells to express the FVIII or FIX protein to potentially cure the patients’ hemophilia has been most commonly used. Recently, efforts have focused on the r-adeno-associated virus (rAAV) as a preferred delivery method, although three trials currently underway will utilize lentiviral gene delivery combined with autologous stem cell transplantation for patients with HA (NCT04418414, NCT03818763; and NCT03217032) and one trial for HB (NCT3961243). One trial for patients with HB is underway using a gene-editing approach (NCT3961243). The Tables 4 and 5 summarize the prior and current, ongoing trials using an rAAV to deliver the F8 or F9 transgene for people with HA or HB, respectively.
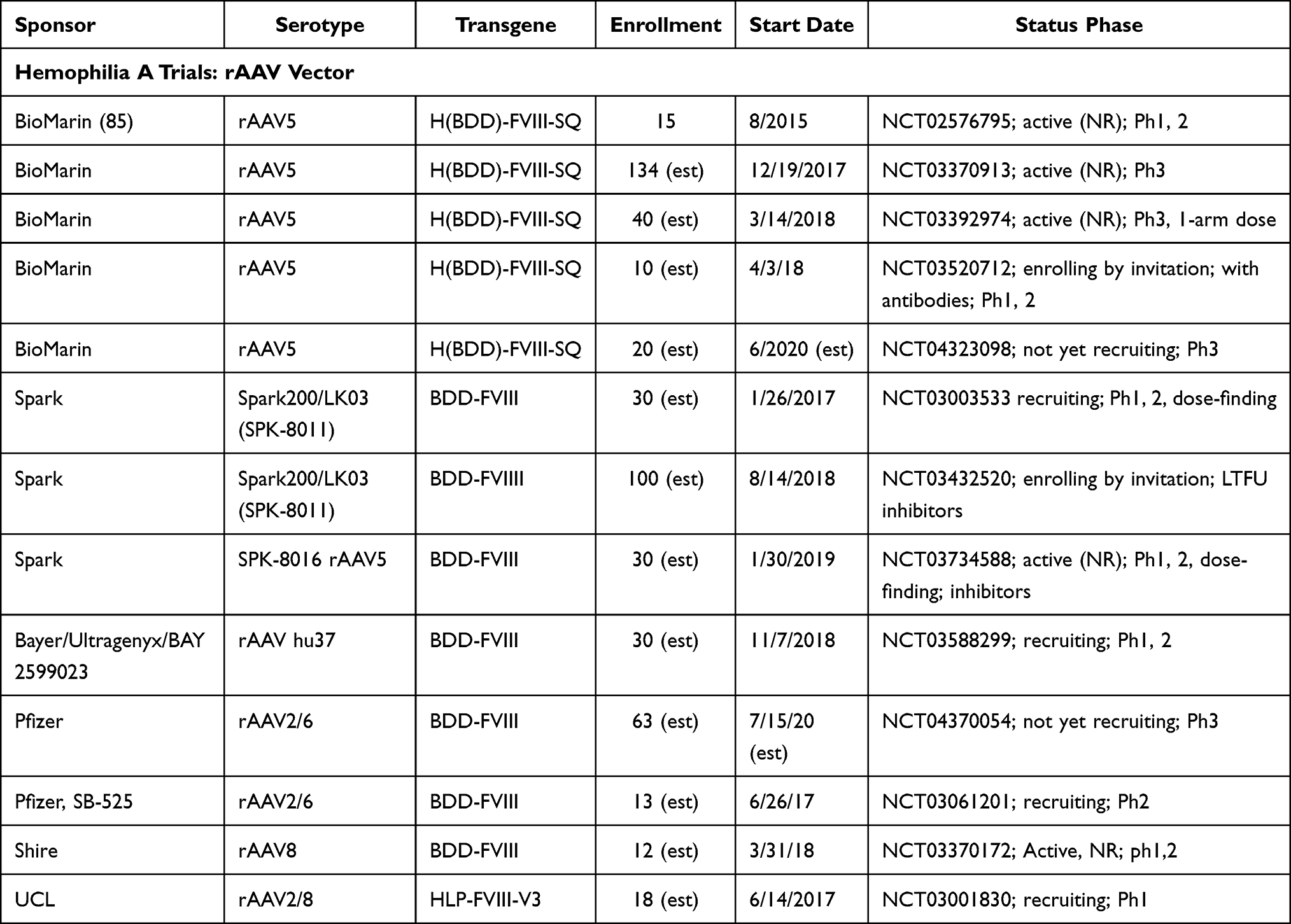 |
Table 4 Completed and ongoing Gene Therapy Trials in Hemophilia A |
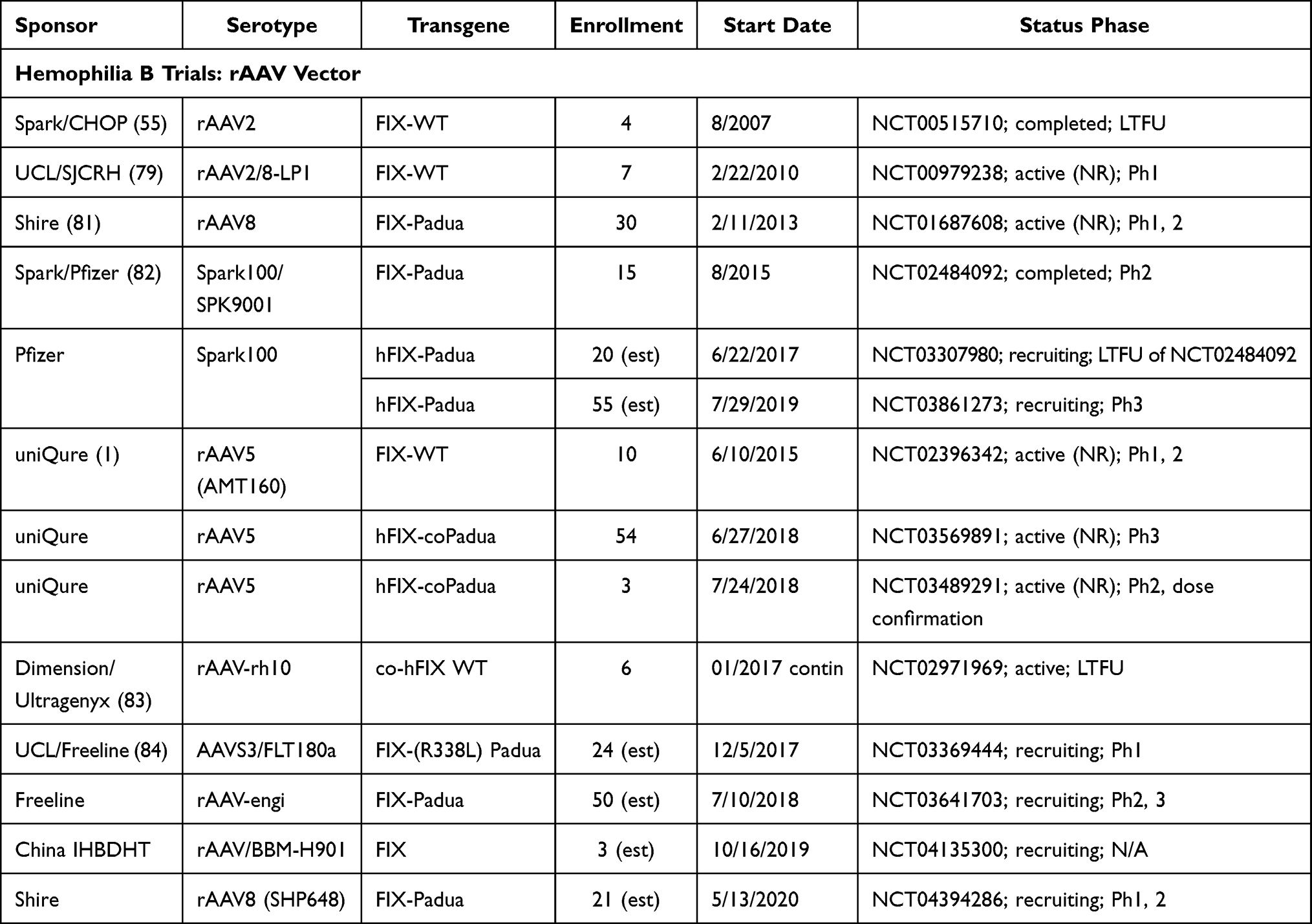 |
Table 5 Completed and Ongoing Gene Therapy Trials in Hemophilia B |
Any gene therapy’s key attributes must focus on safety, effectiveness, predictability, durability, and ultimately, providing a meaningful impact for patients. A core set of outcome measures was de scribed by Iorio et al102 and included the frequency of bleeding events and the achieved FVIII or FIX level, the duration of transgene expression, alleviation of chronic pain utilization of healthcare resources, and mental health. To date, the data from the completed and ongoing clinical trials of gene therapy in HA and HB have demonstrated benefit in terms of reductions in bleeding frequency and the need for supplemental infusions of FVIII or FIX concentrates to treat breakthrough bleeding events; however, data to support the benefit in the other core outcome parameters are lacking.103
In terms of safety, open questions regarding the cause of and long-term implications of elevations in liver enzymes and the risks of insertional mutagenesis remain for clinicians and patients alike.
Furthermore, pre-existing antibodies to AAV represent a significant hurdle to overcome in approximately half of the patients, and dosing in children and the need for repeat dosing are also open questions. Nonetheless, many patients are eager to realize gene therapy’s full potential to achieve a “functional cure” and health equity.
Conclusions
Progress in hemophilia therapy has been remarkable in the first 20 years of the third millennium, but the innovation started with the description the fractionation of plasma in 1946. Over the past 75 years, the life expectancy and quality of life of people living with hemophilia has improved dramatically, and their life is approaching that of their unaffected peers. Screening methods for viral pathogens contaminating blood and blood products combined with virucidal techniques to eliminate contaminating pathogens has made concentrates safe. The cloning of the F8 gene and later the F9 gene paved the way to factor concentrates developed using recombinant DNA technology. The short circulating plasma half-life of the injected FVIII and FIX molecules has been extended with innovative technologies. The availability of rFIX EHL concentrates has improved the adherence of people with hemophilia B to comply with the prophylaxis regimen; however, the advantages of rFVIII EHL concentrates are so far less evident but well accepted by the patients. The goal for replacement therapy is to prevent bleeding and the development of arthropathy but remains an ambition for the community leading to the use of individual PK or PopPK to tailor the prophylaxis regimen due to the large inter-patient variability. Despite these advances, bleeding and adherence to the treatment regimen continues to be a problem for many patients. A new era of prevention of bleeding began with the introduction of a bi-specific monoclonal antibody that mimics the effects FVIII in the tenase complex. The excellent efficacy, subcutaneous route of administration and long duration of effect have won favour with many PWH. Other non-replacement therapies are also in development, but the thrombotic risk of these novel agents mandates careful evaluation. Several gene therapy trials are currently underway in PWH, but the long-term safety and efficacy remains to be established. The high cost of hemophilia treatments, including replacement therapy and non-factor therapies and the potential very high costs for gene therapy, limit access to people living in countries with high gross national income. Worldwide, about 70% of PWH do not have access to replacement therapy. Their life expectancy is reduced and the consequences of joint and other bleeding leads to severe reductions in their motor skills and the ability to execute activities of daily living. The World Federation of Hemophilia distributes coagulation factor concentrates and emicizumab in low- and low–middle-income countries through a humanitarian aid program which has improved life of a limited number of PWH in the developing world.
Disclosure
Dr Massimo Morfini reports grants from Pfizer, personal fees from Bayer, personal fees from Kedrion, personal fees from Bioverativ, personal fees from Novo Nordisk, personal fees from Sanofi, outside the submitted work. The authors do not report any other conflicts of interest regarding this paper.
References
1. Jang JH, Seo JY, Bang SH, et al. Establishment of reference intervals for von Willebrand factor antigen and eight coagulation factors in a Korean population following the Clinical and Laboratory Standards Institute guidelines. Blood Coagul Fibrinolysis. 2010;21:251–255. doi:10.1097/MBC.0b013e3283367931
2. Mannucci PM, Franchini M. Is haemophilia B less severe than haemophilia A? Haemophilia. 2013;19:499–502. doi:10.1111/hae.12133
3. Melchiorre D, Linari S, Manetti M, et al. Clinical, instrumental, serological and histological findings suggest that hemophilia B may be less severe than hemophilia A. Haematologica. 2016;101:219–225. doi:10.3324/haematol.2015.133462
4. Franchini M, Mannucci PM. Haemophilia B is clinically less severe than haemophilia A: further evidence. Blood Transfus. 2018;16:121–122. doi:10.2450/2016.0158-16
5. Santagostino E, Fasulo MR. Hemophilia a and hemophilia B: different types of diseases? Semin Thromb Hemost. 2013;39:697–701. Review. doi:10.1055/s-0033-1353996
6. van Miert JH, Hop H, Borjas-Howard JF, Meijer K. A milder clinical course for severe hemophilia B: a true or biased effect? Haematologica. 2016;101:e266. doi:10.3324/haematol.2016.147298
7. Tagariello G, Iorio A, Santagostino E, et al. Comparison of the rates of joint arthroplasty in patients with severe factor VIII and IX deficiency: an index of different clinical severity of the 2 coagulation disorders. Blood. 2009;114:779–784. doi:10.1182/blood-2009-01-195313
8. Belvini D, Salviato R, Radossi P, et al. Molecular genotyping of the Italian cohort of patients with hemophilia B. Haematologica. 2005;90:635–642.
9. Lane S. Haemorrhagic diathesis. Successful transfusion of blood. Lancet. 1840;35:185–188. doi:10.1016/S0140-6736(00)40031-0
10. Ingram GI. The history of haemophilia. Haemophilia. 1997;3(Suppl 1):5–15. doi:10.1111/j.1365-2516.1997.tb00168.x
11. Bloamback B, Bloamback M, Nilsson IM. Note on the purification of human antihemophilic globulin. Acta Chem Scand. 1958;12:1878. doi:10.3891/acta.chem.scand.12-1878
12. Pool JG, Gershgold EJ, Pappenhagen AR. High-potency antihaemophilic factor concentrate prepared from cryoglobulin precipitate. Nature. 1964;203:312. doi:10.1038/203312a0
13. Pool JG, Shannon AE. Production of high-potency concentrates of antihemophilic globulin in a closed-bag system. N Engl J Med. 1965;273:1443–1447. doi:10.1056/NEJM196512302732701
14. Morfini M. In vitro characteristics of highly purified Factor VIII concentrates. Ann Hematol. 1991;63:123–125. doi:10.1007/BF01703241
15. Kasper CK, Kipnis SA. Hepatitis and clotting-factor concentrates. JAMA. 1972;221:510. doi:10.1001/jama.1972.03200180052024
16. Morfini M, Rafanelli D, Longo G, et al. Hepatitis-free interval after clotting factor therapy in first infused haemophiliacs. Thromb Haemost. 1986;56:268–270. doi:10.1055/s-0038-1661664
17. Evatt BL, Gomperts ED, McDougal JS, Ramsey RB. The coincidental appearance of LAV/HTLV-III antibodies in hemophiliacs and the onset of the AIDS epidemic. N Engl J Med. 1985;312:483–486. doi:10.1056/NEJM198502213120805
18. Ragni MV, Tegtmeier GE, Levy JA, et al. AIDS retrovirus antibodies in hemophiliacs treated with factor VIII or factor IX concentrates, cryoprecipitate, or fresh frozen plasma: prevalence, seroconversion rate, and clinical correlations. Blood. 1986;67:592–595. doi:10.1182/blood.V67.3.592.592
19. Evatt BL. The tragic history of AIDS in the hemophilia population, 1982–1984. J Thromb Haemost. 2006;4:2295–2301. doi:10.1111/j.1538-7836.2006.02213.x
20. Morfini M, Mannucci PM, Mariani G, et al. Evaluation of prophylactic replacement therapy in haemophilia B. Scand J Haematol. 1976;16:41–47. doi:10.1111/j.1600-0609.1976.tb01115.x
21. Ghirardini A, Schinaia N, Chiarotti F, et al. Epidemiology of hemophilia and of HIV infection in Italy. GICC. Gruppo Italiano Coagulopatie Congenite. J Clin Epidemiol. 1994;47:1297–1306. doi:10.1016/0895-4356(94)90135-X
22. Mannucci PM. Clinical evaluation of viral safety of coagulation factor VIII and IX concentrates. Vox Sang. 1993;64:197–203. doi:10.1111/j.1423-0410.1993.tb03055.x
23. Bartolomei Corsi O, Azzi A, Morfini M, et al. Human parvovirus infection in haemophiliacs first infused with treated clotting factor concentrates. J Med Virol. 1988;25:165–170. doi:10.1002/jmv.1890250206
24. Santagostino E, Mannucci PM, Gringeri A, et al. Transmission of parvovirus B19 by coagulation factor concentrates exposed to 100 degrees C heat after lyophilization. Transfusion. 1997;37:517–522. doi:10.1046/j.1537-2995.1997.37597293884.x
25. Mannucci PM, Gdovin S, Gringeri A, et al. Transmission of hepatitis A to patients with hemophilia by factor VIII concentrates treated with organic solvent and detergent to inactivate viruses. The Italian Collaborative Group. Ann Intern Med. 1994;120:1–7. doi:10.7326/0003-4819-120-1-199401010-00001
26. Azzi A, De Santis R, Morfini M, et al. TT virus contaminates first-generation recombinant factor VIII concentrates. Blood. 2001;98:2571–2573. doi:10.1182/blood.V98.8.2571
27. Gomperts E, Lundblad R, Adamson R. The manufacturing process of recombinant factor VIII, Recombinate. Transfus Med Rev. 1992;6:247–251. doi:10.1016/S0887-7963(92)70175-8
28. Bray GL, Gomperts ED, Courter S, et al. A multicenter study of recombinant factor VIII (Recombinate): safety, efficacy, and inhibitor risk in previously untreated patients with hemophilia A. The Recombinate Study Group. Blood. 1994;83:2428–2435.
29. Morfini M, Rapisarda CAP. Safety of recombinant coagulation factors in treating hemophilia. Expert Opin Drug Saf. 2019;18:75–85. doi:10.1080/14740338.2019.1574743
30. White G
31. Pittman DD, Alderman EM, Tomkinson KN, et al. Biochemical, immunological, and in vivo functional characterization of B-domain-deleted factor VIII. Blood. 1993;81:2925. doi:10.1182/blood.V81.11.2925.2925
32. Morfini M, Zanon E. Emerging drugs for the treatment of hemophilia A and B. Expert Opin Emerg Drugs. 2016;21:301–313. doi:10.1080/14728214.2016.1220536
33. Schwarzinger I, Pabinger I, Korninger C, et al. Incidence of inhibitors in patients with severe and moderate hemophilia A treated with factor VIII concentrates. Am J Hematol. 1987;24:241–245. doi:10.1002/ajh.2830240303
34. Ehrenforth S, Kreuz W, Scharrer I, Kornhuber B. Factor VIII inhibitors in haemophiliacs. Lancet. 1992;340:253. doi:10.1016/0140-6736(92)90530-G
35. Ehrenforth S, Kreuz W, Scharrer I, et al. Incidence of development of factor VIII and factor IX inhibitors in haemophiliacs. Lancet. 1992;339:594–598. doi:10.1016/0140-6736(92)90874-3
36. Lusher JM. First and second-generation recombinant factor VIII concentrates in previously untreated patients: recovery, safety, efficacy, and inhibitor development. Semin Thromb Hemost. 2002;28:273–276. doi:10.1055/s-2002-32662
37. Lusher JM, Arkin S, Abildgaard CF, Schwartz RS. Recombinant factor VIII for the treatment of previously untreated patients with hemophilia A. Safety, efficacy, and development of inhibitors. Kogenate Previously Untreated Patient Study Group. N Engl J Med. 1993;328:453–459. doi:10.1056/NEJM199302183280701
38. Kreuz W, Ettingshausen CE, Zyschka A, et al. Inhibitor development in previously untreated patients with hemophilia A: a prospective long-term follow-up comparing plasma-derived and recombinant products. Semin Thromb Hemost. 2002;28:285–290. doi:10.1055/s-2002-32664
39. Peyvandi F, Mannucci PM, Garagiola I, et al. A randomized trial of factor VIII and neutralizing antibodies in hemophilia A. N Engl J Med. 2016;374:2054–2064. doi:10.1056/NEJMoa1516437
40. Calvez T, Chambost H, d’Oiron R, et al. Analyses of the France Coag cohort support differences in immunogenicity among one plasma-derived and two recombinant factor VIII brands in boys with severe hemophilia A. Haematologica. 2018;103:179–189. doi:10.3324/haematol.2017.174706
41. Ljung R, Auerswald G, Benson G, et al. Inhibitors in haemophilia A and B: management of bleeds, inhibitor eradication and strategies for difficult-to-treat patients. Eur J Haematol. 2019;102:111–122. doi:10.1111/ejh.13193
42. Thorland EC, Drost JB, Lusher JM, et al. Anaphylactic response to factor IX replacement therapy in haemophilia B patients: complete gene deletions confer the highest risk. Haemophilia. 1999;5:101–105. doi:10.1046/j.1365-2516.1999.00303.x
43. Jones P, Fearns M, Forbes C, Stuart J. Haemophilia A home therapy in the United Kingdom 1975–6. Br Med J. 1978;1:1447–1450. doi:10.1136/bmj.1.6125.1447
44. van Meegeren ME, Roosendaal G, Jansen NW, et al. Blood-induced joint damage: the devastating effects of acute joint bleeds versus micro-bleeds. Cartilage. 2013;4:313–320. doi:10.1177/1947603513497569
45. van Vulpen LF, van Meegeren ME, Roosendaal G, et al. Biochemical markers of joint tissue damage increase shortly after a joint bleed; an explorative human and canine in vivo study. Osteoarthritis Cartilage. 2015;23:63–69. doi:10.1016/j.joca.2014.09.008
46. Aledort LM, Haschmeyer RH, Pettersson HJ. A longitudinal study of orthopedic outcomes for severe factor-VIII-deficient haemophiliacs. The Orthopaedic Outcome Study Group. Intern Med. 1994;236:391–399.
47. Soucie JM, Monahan PE, Kulkarni R, et al. The frequency of joint hemorrhages and procedures in nonsevere hemophilia A vs B. Blood Adv. 2018;2:2136–2144. doi:10.1182/bloodadvances.2018020552
48. den Uijl IEM IEM, Fischer K, Van Der Bom JG, et al. Plug analysis of low frequency bleeding data: the association of joint bleeds according to baseline FVIII activity levels. Haemophilia. 2011;17:41–44. doi:10.1111/j.1365-2516.2010.02383.x
49. Gringeri A, Ewenstein B, Reininger A. The burden of bleeding in haemophilia: is one bleed too many? Haemophilia. 2014;20:459–463. doi:10.1111/hae.12375
50. Valentino LA, Hakobyan N, Enockson C. Blood–induced joint disease: the confluence of dysregulated oncogenes, inflammatory signals, and angiogenic cues. Semin Hematol. 2008;45(Suppl.1):S50–7. doi:10.1053/j.seminhematol.2008.03.017
51. Zetterberg E, Palmblad J, Wallensten R, et al. Angiogenesis is increased in advanced haemophilic joint disease and characterised by normal pericyte coverage. Eur J Haematol. 2014;92:256–262. doi:10.1111/ejh.12227
52. Van Creveld S. Prophylaxis of joint hemorrhages in hemophilia. Acta Haematol. 1969;41:206–214. doi:10.1159/000208852
53. Blancette VS, Key NS, Ljung LR, et al. Subcommittee on Factor VIII, Factor IX and Rare Coagulation Disorders of the Scientific and Standardization Committee of the International Society on Thrombosis and Hemostasis. J Thromb Haemost. 2014;12:1935–1939. doi:10.1111/jth.12672
54. Manco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357:535–544. doi:10.1056/NEJMoa067659
55. Gringeri A, Lundin B, von Mackensen S, et al.; ESPRIT Study Group. A randomized clinical trial of prophylaxis in children with hemophilia A (the ESPRIT Study). J Thromb Haemost. 9;2011:700–710. doi:10.1111/j.1538-7836.2011.04214.x
56. Tagliaferri A, Feola G, Molinari AC, et al.; POTTER Study Group. Benefits of prophylaxis versus on-demand treatment in adolescents and adults with severe haemophilia A: the POTTER study. Thromb Haemost. 114;2015:35–45. doi:10.1160/TH14-05-0407
57. Manco-Johnson MJ, Lundin B, Funk S, et al. Effect of late prophylaxis in hemophilia on joint status: a randomized trial. Thromb Haemost. 2017;15:2115–2124. doi:10.1111/jth.13811
58. Manco-Johnson MJ, Sanders J, Ewing N, et al.; TEEN/TWEN Study Group. Consequences of switching from prophylactic treatment to on-demand treatment in late teens and early adults with severe haemophilia A: the TEEN/TWEN study. Haemophilia. 19;2013:727–735. doi:10.1111/hae.12177
59. Nijdam A, Foppen W, De Kleijn P, et al. Discontinuing early prophylaxis in severe haemophilia leads to deterioration of joint status despite low bleeding rates. Thromb Haemost. 2016;115:931–938. doi:10.1160/TH15-08-0637
60. Valentino LA, Pipe SW, Collins PW, et al. Association of peak factor VIII levels and area under the curve with bleeding in patients with haemophilia A on every third day pharmacokinetic-guided prophylaxis. Haemophilia. 2016;22:514–520. doi:10.1111/hae.12905
61. Gabrielsson J, Weiner D. Pharmacokinetic and Pharmacodynamic Data Analysis: Concepts and Applications.
62. Riviere JE. Comparative Pharmacokinetics: Principles, Techniques & Application.
63. Bjorkman S, Oh M, Spotts G, et al. Population pharmacokinetics of recombinant factor VIII: the relationships of pharmacokinetics to age and body weight. Blood. 2012;119:612–618. doi:10.1182/blood-2011-07-360594
64. Lunghi B, Bernardi F, Martinelli N, et al. Functional polymorphisms in the LDLR and pharmacokinetics of Factor VIII concentrates. J Thromb Haemost. 2019;17:1288–1296. doi:10.1111/jth.14473
65. Garcia-Martínez I, Borràs N, Martorell M, et al. Common genetic variants in ABO and CLEC4M modulate the pharmacokinetics of recombinant FVIII in severe hemophilia A patients. Thromb Haemost. 2020;120:1395–1406. doi:10.1055/s-0040-1714214
66. Ogiwara K, Swystun LL, Paine AS, et al. Factor VIII pharmacokinetics associates with genetic modifiers of VWF and FVIII clearance in an adult hemophilia A population. J Thromb Haemost. 2021;19:654–663. doi:10.1111/jth.15183
67. Lunghi B, Morfini M, Martinelli N, et al. Polymorphisms in the asialoglycoprotein receptor minor subunit gene (ASGR2) contribute to pharmacokinetics of factor VIII concentrates in Hemophilia A; March 3, 2021.
68. Negrier C, Knobe K, Tiede A, et al. Enhanced pharmacokinetic properties of a glyco-PEGylated recombinant factor IX: a first human dose trial in patients with hemophilia B. Blood. 2011;118:2695–2701. doi:10.1182/blood-2011-02-335596
69. Santagostino E, Negrier C, Klamroth R, et al. Safety and pharmacokinetics of a novel recombinant fusion protein linking coagulation factor IX with albumin (rIX-FP) in hemophilia B patients. Blood. 2012;120:2405–2411. doi:10.1182/blood-2012-05-429688
70. Powell JS, Pasi KJ, Ragni MV, et al. Phase 3 study of recombinant factor IX Fc fusion protein in hemophilia B. N Engl J Med. 2013;369:2313–2323. doi:10.1056/NEJMoa1305074
71. Tiede A, Brand B, Fischer R, et al. Enhancing the pharmacokinetic properties of recombinant factor VIII: first-in-human trial of glycoPEGylated recombinant factor VIII in patients with hemophilia A. J Thromb Haemost. 2013;11:670–678. doi:10.1111/jth.12161
72. Coyle TE, Reding MT, Lin JC, et al. Phase I study of BAY 94-9027, a PEGylated B-domain-deleted recombinant factor VIII with an extended half-life, in subjects with hemophilia A. J Thromb Haemost. 2014;12:488–496. doi:10.1111/jth.12506
73. Konkle BA, Stasyshyn O, Chowdary P, et al. Pegylated, full-length, recombinant factor VIII for prophylactic and on-demand treatment of severe hemophilia A. Blood. 2015;126:1078–1085. doi:10.1182/blood-2015-03-630897
74. Klamroth R, Simpson M, von Depka-prondzinski M, et al. Comparative pharmacokinetics of rVIII-SingleChain and octocog alfa (Advate®) in patients with severe haemophilia A. Haemophilia. 2016;22:730–738. doi:10.1111/hae.12985
75. Powell JS, Josephson NC, Quon D, et al. Safety and prolonged activity of recombinant factor VIII Fc fusion protein in hemophilia A patients. Blood. 2012;119:3031–3037. doi:10.1182/blood-2011-09-382846
76. Klukowska A, Szczepański T, Vdovin V, et al. Long-term tolerability, immunogenicity and efficacy of Nuwiq® (human-cl rhFVIII) in children with severe haemophilia A. Haemophilia. 2018;24:595–603. doi:10.1111/hae.13460
77. Hershfield MS, Buckley RH, Greenberg ML, et al. Treatment of adenosine deaminase deficiency with polyethylene glycol-modified adenosine deaminase. N Engl J Med. 1987;316(10):589–596. doi:10.1056/NEJM198703053161005
78. Clark R, Olson K, Fuh G, et al. Long-acting growth hormones produced by conjugation with polyethylene glycol. J Biol Chem. 1996;271:21969–21977. doi:10.1074/jbc.271.36.21969
79. Cooksley WG, Piratvisuth T, Lee SD, et al. Peginterferon alpha-2a (40 kDa): an advance in the treatment of hepatitis B e antigen-positive chronic hepatitis B. J Viral Hepat. 2003;10:298–305. doi:10.1046/j.1365-2893.2003.00450.x
80. Palumbo E. PEG-interferon in acute and chronic hepatitis C: a review. Am J Ther. 2009;16:573–578. doi:10.1097/MJT.0b013e3181960819
81. Yee A, Gildersleeve RD, Gu S, et al. A von Willebrand factor fragment containing the D’D3 domains is sufficient to stabilize coagulation factor VIII in mice. Blood. 2014;124:445–452. doi:10.1182/blood-2013-11-540534
82. Gringeri A, Mantovani LG, Scalone L, Mannucci PM; COCIS Study Group. Cost of care and quality of life for patients with hemophilia complicated by inhibitors: the COCIS Study. Blood. 2003;102:2358–2363. doi:10.1182/blood-2003-03-0941
83. Nogami K, Shima M. Phenotypic heterogeneity of hemostasis in severe hemophilia. Semin Thromb Hemost. 2015;41:826–831. doi:10.1055/s-0034-1395349
84. Jayandharan GR, Srivastava A. The phenotypic heterogeneity of severe hemophilia. Semin Thromb Hemost. 2008;34:128–141. doi:10.1055/s-2008-1066024
85. Kitazawa T, Igawa T, Sampei Z, et al. A bis- pecific antibody to factors IXa and X restores factor VIII hemostatic activity in a hemophilia A model. Nat Med. 2012;18:1570–1574. doi:10.1038/nm.2942
86. Sehgal A, Barros S, Ivanciu L, et al. An RNAi therapeutic targeting antithrombin to rebalance the coagulation system and promote hemostasis in hemophilia. Nat Med. 2015;21:492–497. doi:10.1038/nm.3847
87. Hilden I, Lauritzen B, Sørensen BB, et al. Hemostatic effect of a monoclonal antibody mAb 2021 blocking the interaction between FXa and TFPI in a rabbit hemophilia model. Blood. 2012;119:5871–5878. doi:10.1182/blood-2012-01-401620
88. Lenting PJ, Denis CV, Christophe OD. Emicizumab, a bispecific antibody recognizing coagulation factors IX and X: how does it compare to factor VIII? Blood. 2017;130:2463–2468. doi:10.1182/blood-2017-08-801662
89. Uchida N, Sambe T, Yoneyama K, et al. A first-in-human phase 1 study of ACE910, a novel factor VIII-mimetic bispecific antibody, in healthy subjects. Blood. 2016;127:1633–1641. doi:10.1182/blood-2015-06-650226
90. Ragni MV. Targeting antithrombin to treat hemophilia. N Engl J Med. 2015;373:389–391. doi:10.1056/NEJMcibr1505657
91. Muczynski V, Christophe OD, Denis CV, Lenting PJ. Emerging therapeutic strategies in the treatment of hemophilia A. Semin Thromb Hemost. 2017;43:581–590. doi:10.1055/s-0037-1604053
92. Bolliger D, Szlam F, Suzuki N, et al. Heterozygous antithrombin deficiency improves in vivo hemostasis in factor VIII-deficient mice. Thromb Haemost. 2010;103:1233–1238. doi:10.1160/TH09-10-0732
93. Broze GJ, Warren LA, Novotny WF, et al. The lipoprotein-associated coagulation inhibitor that inhibits the factor VII-tissue factor complex also inhibits factor Xa: insight into its possible mechanism of action. Blood. 1988;71:335–343. doi:10.1182/blood.V71.2.335.335
94. Pasi KJ, Rangarajan S, Georgiev P, et al. Targeting of antithrombin in hemophilia A or B with RNAi therapy. N Engl J Med. 2017;377:819–828. doi:10.1056/NEJMoa1616569
95. Chowdary P, Lethagen S, Friedrich U, et al. Safety and pharmacokinetics of anti-TFPI antibody (concizumab) in healthy volunteers and patients with hemophilia: a randomized first human dose trial. J Thromb Haemost. 2015;13:743–754. doi:10.1111/jth.12864
96. Eichler H, Angchaisuksiri P, Kavakli K, et al. A randomized trial of safety, pharmacokinetics, and pharmacodynamics of concizumab in people with hemophilia A. J Thromb Haemost. 2018;16:2184–2195. doi:10.1111/jth.14272
97. Shapiro AD, Angchaisuksiri P, Astermark J, et al. Subcutaneous concizumab prophylaxis in hemophilia A and hemophilia A/B with inhibitors: phase 2 trial results. Blood. 2019;134:1973–1982. doi:10.1182/blood.2019001542
98. Figueiredo M. Novo Nordisk Pauses 3 Clinical Trials of Concizumab Amid Safety Concerns. Hemophilia News Today. March 18, 2020. Available from: https://hemophilianewstoday.com/2020/08/14/novo-nordisk-resumes-phase-3-trials-of-concizumab-in-hemophilia-a-and-b/. Accessed May 22, 2021.
99. Ray F. Novo Nordisk Resumes Phase 3 Trials of Concizumab in Hemophilia A and B. Hemophilia News Today. August 14, 2020. Available from: https://hemophilianewstoday.com/2020/08/14/novo-nordisk-resumes-phase-3-trials-of-concizumab-in-hemophilia-a-and-b/. Accessed May 22, 2021.
100. Valentino LA, Khair K. Prophylaxis for hemophilia A without inhibitors: treatment options and considerations. Expert Rev Hematol. 2020;13:731–743. doi:10.1080/17474086.2020.1775576
101. Skinner MW, Nugent D, Wilton P, et al. Achieving the unimaginable: health equity in haemophilia. Haemophilia. 2020;26:17–24. Review. doi:10.1111/hae.13862
102. Iorio A, Skinner MW, Clearfield E, et al. Core outcome set for gene therapy in haemophilia: results of the coreHEM multistakeholder project. Haemophilia. 2018;24:e167–e172. doi:10.1111/hae.13504
103. Sidonio RF
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.