Back to Journals » Infection and Drug Resistance » Volume 12
Recent advances in the treatment of C. difficile using biotherapeutic agents
Authors Giau VV ![]() , Lee H, An SSA
, Lee H, An SSA ![]() , Hulme J
, Hulme J
Received 4 March 2019
Accepted for publication 3 May 2019
Published 10 June 2019 Volume 2019:12 Pages 1597—1615
DOI https://doi.org/10.2147/IDR.S207572
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Joachim Wink
Vo Van Giau, 1 Hyon Lee, 2 Seong Soo A An, 1 John Hulme 1
1Department of BioNano Technology, Gachon University, Seongnam-si 461-701, Republic of Korea; 2Department of Neurology, Gachon University Gil Medical Center, Incheon, South Korea
Abstract: Clostridium difficile (C. difficile) is rapidly becoming one of the most prevalent health care–associated bacterial infections in the developed world. The emergence of new, more virulent strains has led to greater morbidity and resistance to standard therapies. The bacterium is readily transmitted between people where it can asymptomatically colonize the gut environment, and clinical manifestations ranging from frequent watery diarrhea to toxic megacolon can arise depending on the age of the individual or their state of gut dysbiosis. Several inexpensive approaches are shown to be effective against virulent C. difficile in research settings such as probiotics, fecal microbiota transfer and immunotherapies. This review aims to highlight the current advantages and limitations of the aforementioned approaches with an emphasis on recent studies.
Keywords: antibiotics, fecal matter transfer, polyclonal adjuvants, C. difficile, biotherapeutic agents
Corrigendum for this paper has been published
Introduction
Approximately 10–35% of all cases of antibiotic-associated diarrhea in developed countries are caused by the gram-positive, spore-forming, toxin-producing anaerobe, Clostridium difficile (C. difficile).1–8 Exposure to antibiotics is recognized as the most important risk factor for C. difficile infection (CDI).9–13 In a recent study, >45% of the CDI patients had taken antibiotics in the 90d prior the development of symptoms, whilst in another case–control study, 48% of the patients were exposed to antibiotics in the 4-week time period to CDI onset.14,15 Fluoroquinolones (FQs), clindamycin (CLI) and cephalosporins (CFs) are the antibiotics commonly associated with CDI.16,17 Resistance to these antibiotics continues to play an important role in the emergence of new C. difficile clones.18 An investigation by Wasels et al showed that in CDI ribotype 27 (RT 027) FQ resistance is associated with a modest fitness cost; a trait linked to the presence of a favorable mutation (Thr82Ile) in the gyrA gene.19 In 2014, Lee et al reported on the emergence of 3 new ribotypes (RT) 014, 017 and 018 in a Korean hospital; all the strains carried the Thr82I1e mutation. Moreover, the same mutation was detected in isolates of some additional ribotypes genetically related or unrelated to RT027.20,21
Increasing age >65 is another known risk factor associated with CDI, accounting for the majority of diarrheal cases in residential facilities.22–26 In the United States alone, near half a million cases have been reported with 29,000 fatalities attributed to CDI.27 Patients in health care settings are particularly susceptible to infection and re-infection with a recurrence rate of over 20% and a mortality rate of over 9% within days of diagnosis. It is also estimated that up to 57% of the long-term care facility residents (LTCF) are asymptomatic carriers of C. difficile. Although CDI often occurs as a secondary infection, it can also occur in healthy adults with similar rates of recurrence.28 An article detailing the various risks (1960–2010) associated with infection can be found in a review by Spigaglia et al.29
Once a patient exhibits symptom, the first step in treatment is the discontinuation of antibiotics associated with CD risk. For the past 40 years, first-line treatments for mild, recurrent and severe CDI have been the drugs metronidazole (MET) and oral vancomycin (VAN). Unfortunately, in 27% of the cases, the drugs do not effectively treat the infection or prevent recurrence. If metronidazole and oral vancomycin treatments are ineffective, fidaxomicin (FDX) can be administered. This RNA polymerase inhibitor has been shown to reduce sporulation and toxin production in hundreds of C. difficile strains.30–33 Although recurrence and relapse rates for FDX are lower compared to VAN, fidaxomicin still fails in approximately 1 out of 8 patients treated with the antibiotics and in clinical trials.34 Moreover, a recent report showed that vancomycin-resistant isolates are >250 times less susceptible to fidaxomicin compared to fidaxomicin-sensitive strains, even though these two antibiotics have different mechanisms of action.35 Failure of FDX in these cases requires the development of novel cost-effective therapies for C. difficile infections, ensuring that new treatments do not promote reduced susceptibility to antibiotics in current use.
One of the most cost-effective alternative therapies to treat C. difficile is FMT. Recent reports suggest that FMT has the potential to dominate recurrent and severe CDI treatments36–38 and in some cases primary CDI as well.39 The impact of FMT and alternative therapies on CDI is yet to be fully realized. In this review, we briefly visit the infection cycle and roles of CDI genes in toxin production, and then discuss several bio-therapeutic options under investigation, highlighting those which have the potential to replace FDX and VAN in the treatment of initial, recurrent and severe CDI. In this regard, in-vivo studies and clinical trials conducted using known bio-therapeutic options are discussed. Finally, we close by looking at the challenges that emerging CDI biotherapeutic treatments currently face.
Infection cycle and the roles of C. difficile genes in toxin production
Transmission of the C. difficile occurs via the fecal-oral route in the form of highly resistant spores. Once passed the acidic pH of the stomach, the spores germinate in the presence of certain bile acids within the intestine. The active cells then progress to the colon where they outcompete the host bacteria for residence in the hypoxic folds and nutrient-rich crypts. As the colonies form and localized resources decline, a quorum threshold is reached initiating toxin production. The amount of toxin produced determines the severity of the infection. Once outside the localized influence of the CD film or crypt, some cells or spores migrate to the anus and are defecated by the host.40 A summary of the CDI cycle is shown in Figure 1.
 | Figure 1 Infection cycle of toxigenic Clostridium difficile in the human gastrointestinal track. As C. difficile is an obligate anaerobic bacterium, transmission occurs primarily via spores. Three sources of infection (health care, animal and community residences) are indicated. Spores and some vegetative cells (most of which are eliminated in the hosts stomach) are ingested. Once past the stomach a range of metabolic factors (primary to secondary bile acid ratio, short chain fatty acids) encourages spore germination in the duodenum. After germination, the cells disseminate to the anaerobic folds of the ileum and cecum, forming colonies (assuming dysbiosis). Once in the colon, some cells enter sporulation, others produce toxins. As toxin levels increase, the epithelial barrier is challenged, this in turn initiates the inflammatory response and upregulates the production of anti-toxin antibodies in the host. |
Most C. difficile (CD) clinical isolates produce two high molecular weight-related toxins, namely TcdA (308 kDa) and TcdB (270 kDa). TcdA and TcdB expression can fluctuate depending on the bacteria’s exposure to various physical (temperature) and chemical (iron and carbon availability) stressors and the types of strains used in trials.41 The proteins are part of the large clostridial toxin (LCT) family which includes C. perfringens cytotoxins, C. sordelli hemorrhagic toxins (TcsL) and (TcsH) as well as C. α-novyi toxin. CD toxin expression is dependent on the regulation of tcdA and tcdB genes located on the pathogenicity locus PaLoc. The PaLoc locus also contains 3 accessory genes tcdC (negatively regulates tcdA and tcdB) tcdE (encodes a putative holing necessary for toxin release) and tcdR (RNA polymerase sigma factor). The roles of tcdC and tcdE remain controversial as toxin production barely differs between tcdC mutant and wild type strains, whilst reports suggest tcdE holio protein may play a role in toxin release.42 Furthermore, several studies show tcdC expression levels do not diminish during the stationary phase of growth, suggesting that tcdC may adopt a modulatory role rather than a repressive one. In addition to tcdA and B, some strains (RT027 type III, RT251) also express a third toxin, binary toxin (CDTa and CDTb). CDT is structurally related to iota toxin and C2 toxins of C. perfingens and C. botulinum and is thought to upregulate TcdA and TcdB production.43–45
The roles of these three toxins in C. difficile disease remain controversial. Over the years, several studies using hamster models and isogenic C. difficile strains have shown that both TcdA and TcdB mutants are capable of causing fulminant disease and death. Recent studies using hamster and mouse models exposed to wild-type, double toxin knockout and isogenic single strains induced innate and pro-inflammatory immune responses. Strains expressing only TcdB resulted in significant weight loss and severe systemic disease in both models, implying the severe aspects of the disease might be attributable to TcdB rather than TcdA.46 Interestingly, the majority of these strains produce a modified form of TcdB (B) toxin that shares enzyme-GTPase substrate site homology with C. sordelli hemorrhagic toxins, allowing it to carry out glucosylation events in the absence of TcdA.47 Furthermore, the observation that A−B+ strains are virulent in infected individuals indicates that B toxin is sufficient for pathology in humans.
However, the role of the CDT toxin in disease pathogenesis remains unclear. Several studies have shown that CDT production in addition to TcdA and B is associated with an elevated risk of recurrence, disease severity and mortality.48 Moreover, reports of blood, inflammation and fluid retention in infected hamster and rabbit models indicate that CDT could be enterotoxic.49 It has also been demonstrated that TcdA/CDT-producing strains are more virulent in hamsters than isogenic TcdA+TcdB−CDT− strains. Additional studies in mice also showed the host eosinophilic response is suppressed in the presence of CDT.50 Recent work by Kaplan et al demonstrated AB toxin production in hypervirulent and non-hypervirulent strains is under the control of a novel thiolactone quorum-signaling peptide which is independent of tcdC-mediated regulation.51 Furthermore, Lyras et al demonstrated that CdtR may act as a global regulator of virulence in epidemic 027 strains and not others, suggesting that each epidemic strain has its own regulatory mechanism.52 These initial experiments highlight the potential roles of CDT and quorum-signaling peptides in pathogenic virulence and toxin production. A schematic depicting the organization of the toxin genes can be found in Figure 2. For the rest of the review, TcdA and TcdB will be used interchangeably with toxin A and toxin B.
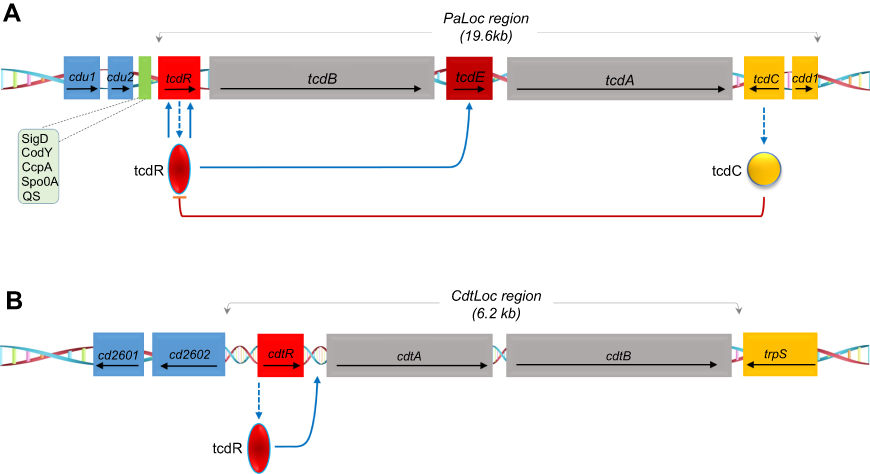 | Figure 2 Schematic representation of the toxin genes and regulatory proteins. (A) Pathogenicity locus (PaLoc) region containing the following genes: tcdR, tcdB, tcdE, tcdC and tcdA. The arrows indicate the direction of transcription. TcdC negatively regulates AB toxin expression. Other regulators Sigma D (SigD), the nutritional repressor CodY (known as GTP-sensing transcriptional pleiotropic repressor CodY), catabolite control protein A (CcpA), Stage 0 sporulation protein A (Spo0A) and quorum sensing (QS)) that affect toxin gene transcription (boxed) mostly act via expression of the tcdR gene. (B) Schematic of the binary toxin locus (CdtLoc) and flanking regions with regulatory interactions. CtdR positively regulates the transcription of cdtA and cdtB. CtdR also regulates the production of AB toxins in various 027 strains but not in ribotypes 078 and 012. |
Probiotics
Research has shown that probiotics can confer a wide range of health benefits especially those directly related to the human gut.53 For example, probiotics can improve your immune system,54 regulate gut microbiota as well as prevent gastrointestinal infections in animals and humans.55 For many years, various species of probiotics have been studied as preventative therapies for CDI, with the most common being within the Bifidobacterium and Lactobacillus genus. Recently, a decade-long study examining the efficacy of a three-strain probiotic mixture involving 45,000 patients was reported.56 The probiotic mixture BioK+® (containing Lactobacillus acidophilus CL1285, Lactobacillus rhamnosus CLR2 and Lactobacillus casei LBC80R) was administered 2–12 hrs after antibiotic treatment and continued for at least 30 days or until treatments were discontinued. In the patients who received the mixture, the CDI rate decreased from 18.0 cases per 10,000 patient days to an average of 2.3 cases per 10,000 patient days. In addition to Bifidobacterium and Lactobacillus, the yeast Saccharomyces has been utilized as preventive treatment for CDI. Of note is the medicinal yeast Saccharomyces boulardii CNCM I-745 which has been approved for the treatment and prevention of diarrhea of various causes.57 Saccharomyces boulardii CNCM I-745 secretes a 54-kDa protease, which is capable of inactivating C. difficile toxins A and B resulting in its efficacy being evaluated in several clinical trials. In 2000, Surawicz et al conducted a randomized, double-blind, placebo-controlled study utilizing S. boulardii for the treatment of recurrent CDI (n=168).58 Patients were given either vancomycin 500 mg daily, vancomycin 2 g daily, or metronidazole 1 g daily for 10 days. On day 7 of the antibiotic course, S. boulardii 500 mg or placebo were administered and continued for a total of 28 days. Results of the study showed that those treated with S. boulardii in addition to vancomycin 2 g daily had a 16.7% recurrence rate versus a 50% recurrence rate in individuals treated with vancomycin 2 g daily and placebo. Unfortunately, a follow-up trial found that S. boulardii was not effective at preventing AAD in elderly patients.59 Additional smaller trials using mixtures of Lactobacillus rhamnosus GG and Lactobacillus plantarum did not demonstrate efficacy of probiotic treatment over the placebo for the prevention of AAD. Similarly, a multicenter, randomized, double-blind, placebo-controlled trial found no benefit to probiotic (mixture of two Lactobacillus and two Bifidobacterium strains) administration in the prevention of CDI in more than 2,941 elderly patients >65 years. Several meta-analyses combining data from studies of different probiotic strains in different patient populations produced results that were largely inconclusive.60 Despite the potential benefit to the host microbiota, long-term safety concerns remain. Among those concerns is the transfer of antibiotic-resistant genes between gut microflora and opportunistic pathogens via mobile genetic elements. Another concern is the resistant profiles of commercial and medicinal probiotics are unavailable.61 Clinical guidelines suggest that for the prevention of AAD, probiotics may be considered based on the evaluation of individual cases. At present, the recommended strain for CDI is S. boulardii CNCM I-745 although the quality of evidence is low.62
Immunotherapies
Given the prevalence of CD within the general populous, the ease with which it is transmitted and the propensity for infection recurrence, a long-term treatment strategy that invigorates the host’s immune response maybe considered the most prudent or cost-effective approach. There are many reviews that document the host’s immune response to Clostridium difficile infection,63–66 but perhaps the most insightful work is by Solomon67 in which the adaptive response to AB toxins, surface-layer proteins (SLPs, Cwp66 and Cwp84) and flagella proteins (FliC and FliD) were addressed. It was found that patients who can generate an enhanced anamnestic systemic immune response to these toxins and proteins are more likely to remain asymptomatic. In addition, symptomatic patients who can mount a rapid immune response early on are less likely to have recurrent CDI. There is now considerable evidence to show that host immune and inflammatory responses contribute in large part to patient outcomes. In the next part of this review, we examine the different types of immunotherapies that can be applied in the treatment and prevention of rCDI and severe CDI.
Traditional vaccines
Over the years, many antibody-based approaches, namely, intravenous (IV) immunoglobulin therapy and polyclonal antibody preparations68–70 have shown efficacy in treating CDI in animals. Commercially pooled polyclonal human intravenous immunoglobulin G (IVIG) is a standard therapeutic preparation made from a plasma pool sourced from 10,000 to 50,000 healthy donors.71 In 1995, Torres et al found that a C. difficile culture filtrate inactivated with formalin was effective in protecting hamsters from CD-induced diarrhea and death.72 Since then, other groups have reported the advantages of using toxoid vaccines to treat CDI and rCDI. Of note is the Sanofi Pasteur (SP) Institute vaccine which has been fast tracked by the food and drug administration (FDA). During phase I trials, the dual toxin (A&B) vaccine induced a complete seroconversion for toxin A at all doses in adults, and at the highest vaccine dose in the elderly. Toxin B seroconversion was lower, both in adults and elderly groups reaching 75%. The antibody response appeared persistent only for toxin A in adult groups, whereas the toxin B response declined 6 months after vaccination,73 suggesting the need for a booster dose. The vaccine also passed phase two trials in which the immunogenicity in adults for primary prevention (NCT01230957) and infected adults for prevention of recurrent disease (NCT00772343) was tested. Phase three trials (NCT01887912) launched by Sanofi in 2013 involved 15,000 people; 10,000 received the vaccine and 5,000 a placebo was terminated in late 2017 when it was determined that the probability of reaching its primary endpoint was low.74 However, phase three trials of a similar competing vaccine created by Pfizer are still currently underway (NCT03090191).
The immunogenicity of vaccines based on polysaccharide (PS) glycans found on the surface of C. difficile cells, namely PSI, PSII, PSIII and lipoteichoic acid-based glycoconjugates has been extensively reported in the literature75,76 and shall not be covered herein. For those readers whose research focus is the development of vaccines against cell-surface components (sortase anchor proteins and cell wall proteins), the recent review by Fagan et al is recommended.77
Recombinant vaccines (RV)
The large-scale production of highly toxic antigens can be a challenging and costly process. Vaccines based on non-toxic fragments of genetically engineered versions of the toxins alleviate some of these concerns including issues of safety.78,79 Karczewski et al investigated the potential of a recombinant vaccine composed of 2 separate fragments of toxin B against C. difficile.80 A combination of toxin B fragments and toxin A were administered to Golden Syrian hamsters. The recombinant vaccine protected animals against a lethal dose of C. difficile spores, with an efficacy equivalent to traditional toxoid vaccines. Other groups have also demonstrated recombinant toxin A and toxin B fragments protect hamsters against C. difficile. The study by Spencer et al noted the glucosyltransferase domain of toxin B induced a greater immune response compared to the binding domain of the whole toxin.81 A similar vaccine preferentially expressing the glucosyltransferase domain of toxin B and the C-terminal receptor binding domain (RBD) of toxin A was reported by Leuzzi et al. The antibodies generated against the glucosyltransferase domain provided more protection in a mouse infection model when used in conjunction with toxin A antibodies.82 While limited protection was observed with some combinations, co-administration of a cell-binding domain fragment of toxin A (toxin A-B1) and the glucosyltransferase moiety of toxin B (toxin B-GT) induced systemic IgGs which neutralized both toxins and protected vaccinated animals from death following challenge with two strains of C. difficile. Further characterization revealed that despite high concentrations of toxin in the gut lumens of vaccinated animals during the acute phase of the disease resulting in minimal toxicological damage.
The size and domain complexity of native recombinant toxins A and B make it a challenge to use them as vaccine candidates. A simple solution is to generate a smaller chimeric vaccine that retains the major neutralizing epitopes from both toxins. In 2012, Wang et al used a non-pathogenic Bacillus megaterium expression system to generate glucosyltransferase-deficient holotoxins.83 The atoxic holotoxins induced potent antitoxin neutralizing antibodies showing little cross-immunogenicity or protection between toxin A and toxin B. The researchers subsequently generated a glucosyltransferase-deficient toxin chimera, cTxAB. Parenteral immunization of mice or hamsters with cTxAB induced rapid and potent neutralizing antibodies against both toxins. Complete and long-lasting disease protection was conferred by cTxAB vaccinations against both laboratory and hypervirulent C. difficile strains.
In 2015, Sun’s group generated a chimeric protein designated mTcd138, comprising the glucosyltransferase and cysteine proteinase domains of toxin B and the receptor-binding domain of toxin A.84 Parental immunizations of mice and hamsters with mTcd138 induced protective antibodies to both toxins and provided protection against infection with the hypervirulent C. difficile strain UK6. Many hypervirulent strains also secrete a binary toxin, namely, CDT. CDT is composed of 2 active components, CDTa and CDTb. Vaccines generated against hypervirulent strains sometimes include attenuated forms of the binary toxin.85 Recently, a novel tetravalent vaccine was generated via a high yield insect-baculovirus system. The immunogenicity of bivalent and tetravalent vaccines was compared in immunized (21days prior spore challenge) hamsters. Investigations revealed that bivalent and tetravalent vaccines induced similar neutralizing antibody titers to toxin A in prototypic strains VPI10463, BI17 and 8864. Only hamsters receiving the tetravalent and binary vaccines alone had elevating neutralizing titers to the binary toxin.86
Monoclonal antibodies
There are many types of antibody-based approaches that have shown efficacy in treating CDI such as intravenous immunoglobulin therapy and polyclonal antibody preparations.87 The toxin pair A/B are the primary targets for therapeutic antibodies against CDI while minor virulence factors such as CDT, surface layer proteins (SLPs) and flagella are sometimes targeted depending on the virulence of the strain under investigation. Initial studies using a mouse rCDI model indicated that the treatment of mice with antitoxin antibodies significantly protects against the morbidity and mortality associated with CDI induced by both historical (VPI 10463, in the case of the toxin challenge models) and hypervirulent strains of C. difficile.88,89
As 85–95% of the clinical isolates test positive for toxin A & B (A+B+), it makes sense to target both toxins. Consequently, a number of antitoxin A/B combinations are already in the initial stages of development.90 In a recent study, more than 20 monoclonal antibodies (mAbs) with neutralizing potential against toxin A and more than 50 with neutralizing potential against toxin B were evaluated.91 Of those 20 mAbs screened, CA 997 was the best at neutralizing toxin A strongly binding the toxin approximately 12 times. A combination of CA1125 and CA1151 mAbs demonstrated a binding valency of 3 for toxin B. Using an established experimental model, individually housed hamsters were then separately dosed on 4 consecutive days with 50 mg/kg of anti-toxin A and 50/kg of anti-toxin B before being orally challenged with C. difficile spores/vegetative cells. A tri-antibody mixture (UCB mAb) offered very high levels of protection (82%) with 9/11 of the hamsters surviving for 28 days. It worth noting that CDA1 exhibited negligible neutralizing activity against toxin A, a finding confirmed in a study by Marozsan et al.92
The intended clinical use of mAb mixtures is for the prevention of recurrent diseases when administered in conjunction with standard-of-care antibiotics. Monoclonal antibody (mAb) and single-domain antibody (sdAb)-based therapies currently dominate the immunotherapeutic pipeline with bezlotoxumab leading the way. Bezlotoxumab (known as 124–1152, MK-3415, CDB1 and MDX-1388) recognizes the C-terminal receptor binding domain of toxin B exhibiting a binding valency of 3. Actoxumab (previously named 3D8, MK-3415, CDA1, MDX-066) is one of the first fully human mAbs to potently neutralize toxin A93. In a landmark study involving 2,655 adults receiving oral standard-of-care antibiotics for primary or recurrent C. difficile infection showed the sustained cure rates (initially clinically cured without recurrence of infection within 12 weeks) with 64% bezlotoxumab alone, 58% with actoxumab-bezlotoxumab and 54% for the placebo group, respectively,.93 The abundances of the Clostridium XIVa clade and Holdemania bacteria in the placebo group prior to treatment were not reported. Akkermansia is another bacterium that is frequently associated with CDI and rCDI as it is thought to contribute to infection by facilitating the access of luminal antigens to the intestinal immune system my mucin degradation.94 Interestingly, a recent CDI study which analyzed the bacterial diversity of the guts of mice under different treatments including MK-3415, vancomycin and vancomycin combined with MK-3415 showed Akkermansia levels to be quite resilient, persisting in high amounts in both vancomycin groups. The authors suggested higher proportions of Lactobacillus and Blautia as well as changes in mucosal composition might attenuate the inflammatory role of Akkermansia.95
To date, PA-50 and PA-41 are two of the most potent mAbs currently under investigation. PA-50 is a humanized anti-toxin A mAb, that targets toxin A RBD at multiple sites and has been shown to neutralize toxin A from a broad range of C. difficile ribotypes.96 The mAb is significantly more potent than actoxumab in-vitro, possibly due to its multivalent interactions with toxin A. PA-41 is a humanized anti-toxin B mAb and is significantly more potent compared to bezlotoxumab. In addition, PA-41 is capable of inhibiting toxins from the same range of C. difficile ribotypes stated previously. In a hamster model for CDI, 95% of the animals treated with a combination of humanized PA-50 and PA-41 showed long-term survival relative to 0% survival of animals treated with standard antibiotic or comparator mAbs.97
Probiotic expression vehicles and single domain antibodies (sdAbs)
The ability to produce antibodies or antibody fragments in a self-limiting manner at the site of infection would be most advantageous in the treatment of CDI. A potential way to accomplish this is to use a probiotic sdAbs expression vehicle.98,99 Of recent note is the work by Andersen et al in which four VHHs (heavy domain only) were expressed on the surface of Lactobacilli.100 Two strains of the probiotic delayed the death of hamsters challenged with AB toxin B and C. difficile spores, with 50% of the hamsters receiving the probiotic surviving until the end of the experiment. More recently, Shkoporov et al expressed two VHHs in Bifidobacterium longum demonstrating toxin A neutralization in vitro.101 The group administered the probiotic bacteria to mice and confirmed the in-vivo expression (secretion) of both single domain antibodies in the guts of mice. In a study by Unger et al,102 recombinant VHHs were generated against the subunit and the binding component. Three out of five CDTa and two from four CDTb specific antibodies were found to neutralize the cytotoxicity of CDTa and CDTb. Surprisingly three of the nanobodies selected for binding to CDTa also indirectly neutralized the binding component (CDTb) by restricting the translocation of CDTa into the cytosol. In other investigations, hamsters immunized with Bacillus subtilis spores expressing a carboxy-terminal segment of toxin A remained resistant to colonization when challenged with C. difficile strain. Anti-toxin A mucosal antibodies obtained following immunization with recombinant B. subtilis spores were able to reduce the adhesion of C. difficile to mucus-producing intestinal cells.103 More recently, Sulea et al utilized the affinity maturation platform “Assisted Design of Antibody and Protein Therapeutics (ADAPT)” to develop a set of mutant sdAbs (camelid sdAb A26.8, a VHH) that bind to toxin A. The designer mutants showed enhanced affinity to toxin A, with the A26.8 double-mutantT56R,T103R neutralizes TcdA cytotoxicity with an IC50 of 12 nM.104 While certainly a consideration for severe CDI, immune-based treatment and prevention of C. difficile infection91,105–112 that have been widely studied (Table 1).
 | Table 1 Immune-based treatment and prevention of Clostridium difficile infection |
Fecal microbiota therapy
The best therapeutic option for recurrent CDI is FMT. The therapy involves the transfer of suspended (saline or water) fecal matter from a healthy donor to a recipient via colonoscopic or nasoduodenal tube and rectal enema.113 Fecal matter is a complex mixture of bacteria, fungi, viruses, human cells, metabolites and more.114 Recommendations state that if there are three or more recurrences of CDI following pulsed vancomycin therapy, FMT should be considered the next therapeutic option.115 In a landmark randomized, open-label, clinical trial (RCT), van Nood and colleagues compared vancomycin alone and vancomycin bowel lavage to vancomycin and bowel lavage with FMT.116 Overall a 94% cure rate was reported for the FMT group while the vancomycin-bowel lavage and vancomycin groups reported cure rates of 23% and 31%, respectively. Further FMT clinical trials have reported similar cure rates.117,118 Recently, Bang et al showed FMT to be a highly effective therapy for refractory and recurrent Clostridium difficile. FMT was performed in nine patients with refractory/recurrent CDI. Bowel movement was normalized within one week after FMT.119 In a randomized double-blind clinical trial, where subjects were treated for rCDI by heterologous FMT (h-FMT) or autologous FMT (a-FMT) as a “placebo”, revealed that, while h-FMT resulted in higher cure rates than a-FMT (90% versus 63%; P=0.019), autologous FMT was, in some cases, successful.120
Several clinical trials have demonstrated the equality of fresh and frozen donor material to cure recurrent C. difficile infection (rCDI)121 More recently, Anand et al showed that the age of the sample donor does not affect the overall microbial diversity of the sample and the clinical efficacy of FMT in rCDI patients.122 All patients receiving FMT from their respective donors had resolution of rCDI symptoms and had a negative C. difficile toxin test 4–12 weeks after FMT. FMT has also been used to treat individuals infected with hypervirulent strains of C. difficile. In the case of a recent CDI outbreak in France, the treating physician adopted a new treatment algorithm by applying FMT in combination with antimicrobial therapy during the first infection episode, mortality of the patients dropped from 64% to 19% with early FMT treatment.123 Tanaka et al also demonstrated that FMT is an effective treatment of new-onset CDI as well.124
Increases in microbial alpha diversity are often reported in FMT recipients with improvements in Bacteroidetes, Clostridium clusters IV and XIVa numbers and a decrease in members of the Enterobacteriaceae family.125 Higher diversity of gut microbiota has been observed in lean individuals when compared to obese individuals, yet diversity is a complex parameter as some recent microbiota studies have shown higher diversity in disease states, such as colon cancer, coeliac disease and Alzheimer’s disease.126 Thus, rather than counting the number of bacterial species, a comprehensive analysis (Source Tracker software program and Bayesian algorithm) of enriched and depleted microbial taxa must be performed and diversity alterations defined for each disease.127 This point was eloquently demonstrated in a recent paper by Staley et al in which a partial engraftment was shown to be sufficient if functionally critical taxa were still present in the subjects following antibiotic therapy.128 Notably subjects cured by a-FMT typically had greater abundances of the Clostridium XIVa clade and Holdemania bacteria prior to treatment, and the relative abundances of these groups increased significantly after FMT compared to heterologous FMT and pre-FMT samples. Provided Clostridium XIVa and IV can be identified in the feces prior aFMT it may be possible to further accelerate the reconstitution of the host flora by supplementing the slurry with phytochemicals (aryl hydrocarbon receptor ligands) thereby boosting colonization resistance.129 Moreover, given aFMTs ability to rapidly improve the post-antibiotic reconstitution of the indigenous fecal microbiome and gut transcriptome in individuals,130,131 it may be prudent to offer the therapy as adjuvant to MET and VAN treatments, as both of these antibiotics are associated with the emergence of potentially pathogenic fungal operational taxonomic units, with predicted bacterial functions enriched for xenobiotic metabolism that could perpetuate the dysbiosis driving CDI (see Figure 3).
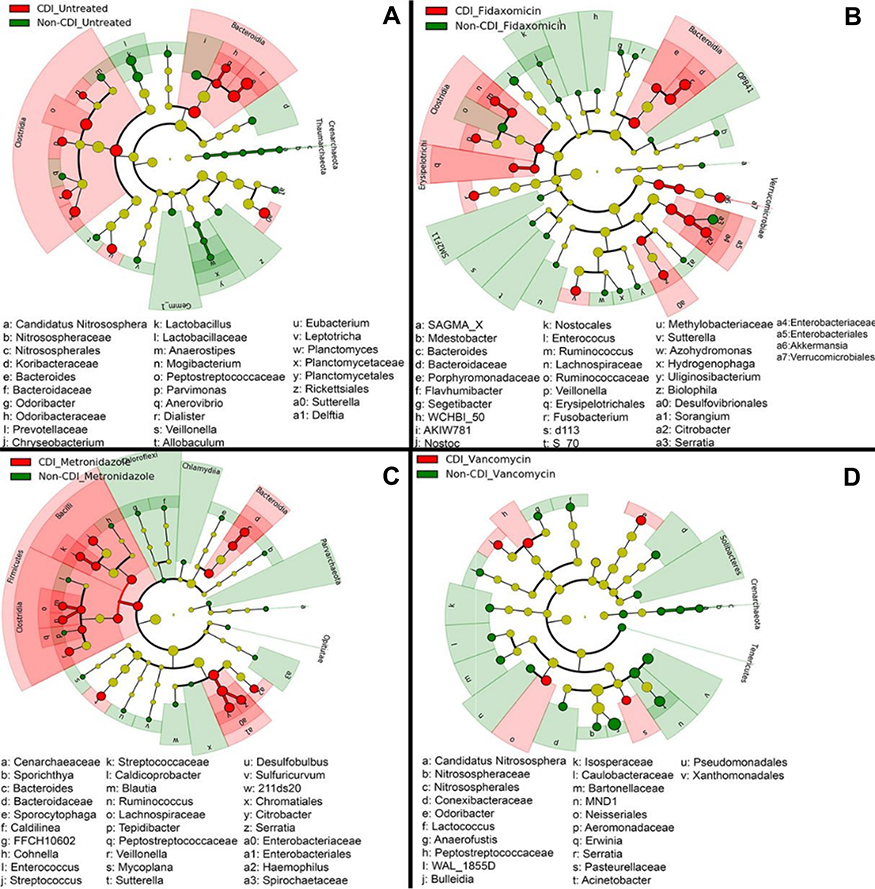 | Figure 3 Cladogram plots were generated in Galaxy to visualize significantly enriched fungal taxa identified in Clostridium difficile infection (CDI) and non-CDI samples considering each treatment cohort separately (A, untreated; B, fidaxomicin; C, metronidazole; D, vancomycin).131 |
CDI is also a common comorbidity of irritable bowel disease (IBD). A recent study by Khortus et al compared the use of FMT in patients with CDI and IBD to those without IBD and found a lower efficacy in clearing the infection in those with IBD after one FMT (74.4% vs 92.1%).132,133 Anderson et al reviewed several case studies in which FMT was used in the treatment of rCDI and refractory CDI infection in IBD.134 A resolution of CDI was found in 11 of the 12 patients and improved response to IBD medication in 6 of 7 patients. In addition, there have been many published case studies showing the positive effects of FMT in IBD of particular note is the work by Moayyedi et al in which the efficacy of FMT in active ulcerative colitis was investigated, remission of IBD was achieved in 24% of the patients.135 A summary of recent FMT trials is shown in Table 2.
 | Table 2 Characteristics of some recent studies concerning fecal microbiota transplantation in C. difficile treatment |
The greatest impediment to the broad dissemination of FMT for the treatment of rCDI and primary cases as well is the uncertainty surrounding its regulation. Regulation of FMT is complicated by the multifarious nature of fecal samples. Ideally, an FMT replacement modality would be safer to use, easy to apply and less expensive than current treatments.
Emerging biotherapies
Phages
Phage therapy entails the isolation and inoculation of phages that target and eliminate specific bacteria.136 To date, phage treatments have been successfully developed for Escherichia coli,137 Pseudomonas, Proteus,138,139 Staphylococcus and Streptococcus infections.140 The lack of phage treatments for CDI reflects the technical difficulties (culturing) of working with sporulating anaerobes. The first reported isolation of C. difficile phages was in 1983, since then several phages (mainly temperate) have been described in the literature.141 All known C. difficile phage genomes are double-stranded DNA and belong to the Caudovirales (the order of the tailed phages). CD phages are characterized by their size and morphological type which includes the small myovirus (SMV) ΦMMP02, medium myovirus (MMs) φCD119 and phiCDHM1, long-tailed myoviruses (LTMs) φCD27 and ΦMMP04 and two morphologically distinct siphoviruses (SVs) φCD6356 and φCD38-2.142 In 2016, two novel myoviruses CDKM15 and CDKM9 were isolated and selected for detailed sequence analysis on the basis of their broad host range.143 CDKM15 infected 20/80 strains from 9/20 CD ribotypes, whilst CDKM9 infected 25/80 strains from 12/20 ribotypes. Both phages infected the clinically relevant ribotypes R027 and R001. Genome sequencing analysis of these phages identified new signals for horizontal gene transfer (HGT). The mechanism of DNA packaging for each myovirus could not be classified. Three C. difficile hosts, namely CD105HE1 (Ribotype 076, equine isolate), CD105LC1 (ribotype 027, human isolate) and CD105HS (ribotype 012, environmental isolate) were recently used by Clokie et al to propagate seven phages (6 phiCDHM1-6 and phiCDHS1) producing phase titers ranging from 109 to 1010 PFU/mL.144,145 With the exception of phiCDHS1 the remaining phases were manufactured on a common host (CD105LC1), ensuring any lytic activity was attributable to the specific phage and not due to differences conferred by the host bacterial strain. Using a hamster model, the oral delivery of optimized phage combinations resulted in reduced C. difficile colonization at 36-hr post-infection.
The evolution of bacterial resistance to phages is of genuine concern as recent work suggests CD phages can mediate the horizontal transfer of genetic material via transduction (antibiotic resistant and toxin genes). In a study by Goh et al, the φC2 phage was shown to transduce the antibiotic marker ermB carried on a 13 kbp transposon.146 Moreover, genome sequencing has revealed the presence of defense mechanisms including a clustered regularly interspaced short palindromic repeat (CRISPR)/CAS system147,148 and active type I and type II restriction modification system.
Although hamster CDI models demonstrate various clinical symptoms consistent with those seen in humans, the animals rapidly succumb to the disease. This has resulted in many groups employing artificial gut models, which have revealed many facets of enteric pathogens.149–151 In 2010 and 2013, Meader et al studied C. difficile phage-host interactions using two ex-situ model systems. The first involved studying their dynamics in a batch model and the second in a multi-vessel model (artificial gut model).152,153 Remedial and prophylactic treatments were tested using φCD27, both models exhibited significant reductions in the levels of TcdA and B compared with the controls. The colon model illustrated the potential of phage therapy in treating CDI as well as other factors that could impact treatment.153 Studies by Govind et al showed the single phage φCD119 could lysogenize under the conditions in the mammalian gut and suppress toxin production.154 On the other-hand, Sekulovoic et al and Goh et al demonstrated toxin levels are most likely influenced by strain-phage specific interplay and that considerable variation in the physiological response to phage infection does occur.155,156
More recently, Nale et al utilized Galleria mellonella larvae as an alternative model to study the therapeutic potential of a 4-phage cocktail on CD ribotypes 014/020.157 It was found that multiple phage doses significantly improved the larval remedial regimen with 60% of the larvae surviving until the end of the experiment. The phages were most effective when vancomycin was given prophylactically before bacterial infection resulting in as little as 10 colony forming units (CFU) per larva being recovered. The study demonstrated that multi-phage therapy remains one of the most effective ways of clearing C. difficile and preventing the appearance of resistant/lysogenic clones.
Isolating new CD phages from the terminal part of the gut that exhibits minimal or no temperate activity on the target strain remains challenging.158,159 However, the mouth is a great source of bacteriophages and susceptible bacteria such as E. faecalis which is known to play a role in prolonging dysbiosis. Therefore, it may be suggested that future phage treatments target such species as well as CD, potentially reducing the risk of recurrence and relapse.160 A summary of the challenges CD phage therapy currently face can be found in recent review by Fortier.161
Endolysins
Many bacteriophages isolated from the host environment are not efficient in the rapid eradication of pathogenic hosts, as is the case with φCD27.162 One way to overcome this problem is to clone and express the recombinant version of the endolysin from its phage. Endolysins are produced by many double-stranded DNA bacteriophages to affect the release of new virions from an infected cell by degrading the bacterial cell wall. They have been used to target many well-known infectious bacteria including Streptococcus,163 Staphylococcus,164 Listeria,165 Bacillus166 and Clostridium.167 Unlike CDI,168,169 most infections involve multiple strains requiring the use of broad-spectrum antibiotics. Lysins possess the potential to satisfy this role without the risk of bacterial resistance. Moreover, they have been shown to provide better protection against pathogenic organisms such as C. difficile or E. faecalis compared to their respective phages.170,171 Investigations using φCD27 found that a truncated version of the N-terminus was able lyse all 32 strains of C. difficile tested, as well as less closely related species B. subtilis, Listeria innoculaand B. amyloliquefaciens.172 How the wider activity of the truncated endolysin impacted the broader bacterial community within the GI tract was not reported. In the future, endolysins will undoubtedly play a strategic role in the treatment of systemic (sepsis) and antibiotic resistant infections.
Small molecule inhibitors
The mammalian gut contains hundreds of small molecules whose function is yet to be discovered. Finding molecules that selectively inhibit different stages of the C. difficile life cycle, while sparing the indigenous gut microbiota is important for the development of alternatives to standard antibiotic treatments. 2-aminoimidazole (2-AI) molecules have been shown to overcome the protective mechanisms of multi-drug resistant pathogens such as Staphylococcus aureus and Pseudomonas aeruginosa. Recent work by the group of Theriot et al in which the inhibitory effects of eleven 2-AI molecules on the life cycle of seven strains of C. difficile and an eight-member commensal library of bacteria associated with host colonization resistance were tested. Four of them were found to inhibit toxin production without affecting the growth of both C. difficile strains and the commensal library.169 In addition to 2-AI, there are number of anti-virulence compounds such as Ebselen and benzodiazepinedione that inhibit the glucosyltransferase activity of TcdA and TcdB and have the potential to reduce disease symptoms.170,173 Furthermore, Ebselen has been used in phase II clinical trials, and was recently reported to ameliorate β-amyloid pathology, tau pathology and cognitive impairment in triple-transgenic Alzheimer’s disease mice.174 Whether C. difficile or its toxins should be included in the current “infectious theory” of Alzheimer’s disease is beyond the scope of this review.
An alternative way of inactivating TcdB is by triggering its auto-proteolysis in the gut lumen prior to cell uptake using the allosteric activator inositol hexakisphosphate (IP6). Although IP6 can trigger the auto-processing of TcdB in vitro, the cleavage is abolished if performed in the presence of luminal concentrations (>10 mM) of calcium. In a recent study, Ivarsson et al attempted to address the problem of calcium chelation by synthesizing a series of IP6 analogs where the six phosphate groups were progressively replaced by sulfates culminating in inositol hexasulfate (IS6).175 An optimal balance between allosteric activity and interference by calcium was reached using the phosphate-sulfate hybrid IP2S4. IP2S4 attenuated colitis in CDI mouse models after oral dosing; moreover, a thiol-phosphate form of the analog IT2S4 was shown to rescue mice in a fulminant CDI model. Figure 4 shows in-vivo IP2S4 and IP6 attenuating activities in mice.
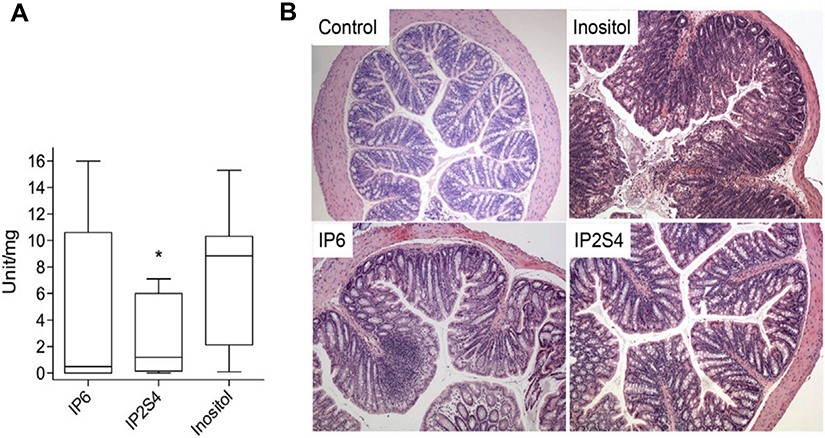 | Figure 4 Swiss mice infected with fecal slur from a patient with recurrent Clostridium difficile infection. (A) Oral administration of IP2S4 but not IP6 significantly reduced the acute inflammatory component of colitis compared with administration of myo-inositol. (B) Histological sections of excised colons. Inositol-treated mice (negative control) displayed overt colonic structural changes characterized by mucosal ulceration and overlying exudate, marked acute and chronic inflammatory infiltrate and submucosal edema. IP2S4- and IP6-treated mice had decreased mucosal damage and inflammatory infiltrate. Copyright ©2018. Reproduced with permission from Elsevier. Ivarsson ME, Durantie E, Huberli C, et al. Small-molecule allosteric triggers of Clostridium difficile toxin B auto-proteolysis as a AQ3 therapeutic strategy. Cell Chem Biol. 2018;26(1):17–26.e13.175 |
Bacteriocins
Bacteriocins are a group of antimicrobial peptides ribosomally produced by Gram-negative and Gram-positive bacteria. A recent study, conducted by Egan et al, explored the role bacteriocins may have in the GIT. In a genome mining project, the authors retrieved 641 genomes (307 whole genomes and 334 draft genomes) from microorganisms in the human gut. The genomes represented 199 bacterial genera, including Lactobacillus, Streptococcus, Clostridium and Bacillus.176
Nisin is a bacteriocin produced by a group of Gram-positive bacteria that belongs to Lactococcus and Streptococcus species. Nisin is classified as a Type A (I) lantibiotic that is synthesized from mRNA and has been used for many years as a food additive. Similar to vancomycin, lanthipeptides such as nisin also targets a cell wall component, in this case lipid II. Recent studies by Fliss et al177 assessed the in vitro efficacy of nisin Z and A on C. difficile cells and spores. Nisin A and Z both inhibited the growth of twenty C. difficile isolates, and minimum inhibitory concentrations (MIC) were estimated at 6.2 μg/mL for nisin Z and 0.8 μg/mL for nisin A. In addition, C. difficile spores were also susceptible to nisin A (25.6 μg/mL), reducing spore viability by 40–50%. The MIC value for nisin A was comparable to the MICs obtained for lacticin 3147. However, when used as standalone therapy resistance to nisin A frequently occurs.178 A simple way to minimize resistance as well as improve therapeutic efficacy is to incorporate a germinator or potentiate the antibiotic/antibiotic peptide with a primary metabolite. The effectiveness of this approach was recently demonstrated by Se-Wook Oh et al in which the synergistic action of nisin and lysozyme (20 nmol/L nisin and 0.2 mmol/L lysozyme) resulted in no viable C. difficile spores being detected after 2 hrs of incubation.179
Lacticin 3147 is another bacteriocin produced by strains of L. lactis180 with potent anti-C. difficile activity with concentrations as low as 18 μg/mL capable of elminating 106 CFU/mL of C. difficile <30 mins, comparable in efficacy to metronidazole and vancomycin in a model fecal environment. An alternative to 3147 is the lantibiotic actagardine. When combined with ramoplanin or metrondiazole it behaves in a partial synergistic/additive fashion against 61.5% and 54.4% of target C. difficile strains investigated.181 In addition, a recent study demonstrated that combinations of the class II bacteriocin, durancin 61A and the broad-spectrum antimicrobial reuterin yielded fractional inhibitory concentration index (FIC) indices of 0.2 against C. difficile, indicating highly synergistic activity.182 But perhaps the bacteriocin with the most therapeutic potential was thuricin. Initial work revealed that thuricin was as effective as metronidazole and vancomycin against C. difficile in a distal colon human model. Moreover, further studies showed thuricin interacted in a partial synergistic manner when combined with ramoplanin against 31% of the target CD strains investigated.183
Conclusions
With the recent emergence of hypervirulent strains in Europe, Australasia45 and North America, there is an urgent need to develop alternative/adjunctive therapeutic options to metronidazole and vancomycin in order to minimize the ongoing problem of recurrence and prevent the spread of vancomycin-resistant enterococci in hospital environments.
The alternative therapies discussed each have their advantages, vaccination and monoclonal antibodies are probably the most cost effective in the long term.131 On the other hand, they do not reduce the bacterial load nor prevent C. difficile colonization or potential spore transmission. Moreover, challenges to vaccination strategy will arise from a patient’s inability to generate a rapid, long-lasting and protective response. However, it is pleasing to note that many anti-toxin therapies are on the cusp of approval and when combined with other biotherapeutic options such as FMT or tailored spore formulations individual therapeutic solutions will become more available.
In addition to FMT and immunotherapies, multi strain-phage treatments are one of the most promising emerging therapies. However, numerous obstacles persist regarding the isolation and therapeutic application of C. difficile phages. Of particular concern is how different combinations or the same combination can affect toxin production in different hosts. Moreover, it is important know the exact phage and antibiotic resistance patterns of C. difficile strains in order to minimize the risk of recurrence. As of yet, no experimental models have investigated the use of multiple bacterial phages in the treatment of rCDI and dysbiosis.
Although the biotherapies discussed herein have the potential to improve patient outcomes, the most difficult step is translating these discoveries into therapeutics that are safe for humans. This review has not covered other treatment options, such as alternative antibiotics and antimicrobial agents.184–187 Future treatments will undoubtedly include a combination of these therapies with the aim of reducing rCDI, and the number of antibiotic resistant genes in C. difficile patients.188
Acknowledgments
This research was supported by the National Research Foundation of Korea (NRF) Grants awarded by the Korean government (MEST, No 2017R1A2B4012636) and the Gachon University Research Fund GCU-2018-0682.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Vindigni SM, Broussard EK, Surawicz CM. Alteration of the intestinal microbiome: fecal microbiota transplant and probiotics for Clostridium difficile and beyond. Expert Rev Gastroenterol Hepatol. 2013;7(7):615–628. doi:10.1586/17474124.2013.832501
2. Tsutsumi LS, Owusu YB, Hurdle JG, Sun D. Progress in the discovery of treatments for C. difficile infection: a clinical and medicinal chemistry review. Curr Top Med Chem. 2014;14(1):152–175.
3. Kim PK, Huh HC, Cohen HW, et al. Intracolonic vancomycin for severe Clostridium difficile colitis. Surg Infect (Larchmt). 2013;14(6):532–539. doi:10.1089/sur.2012.158
4. Mulcahy-O’Grady H, Workentine ML, Challenge T. Potential of metagenomics in the clinic. Front Immunol. 2016;7:29. doi:10.3389/fimmu.2016.00029
5. Khan FY, Elzouki AN. Clostridium difficile infection: a review of the literature. Asian Pac J Trop Med. 2014;7s1:S6–S13. doi:10.1016/S1995-7645(14)60197-8
6. McCarville JL, Caminero A, Verdu EF. Novel perspectives on therapeutic modulation of the gut microbiota. Therap Adv Gastroenterol. 2016;9(4):580–593. doi:10.1177/1756283X16637819
7. Lau CS, Chamberlain RS. Probiotics are effective at preventing Clostridium difficile-associated diarrhea: a systematic review and meta-analysis. Int J Gen Med. 2016;9:27–37. doi:10.2147/IJGM.S98280
8. Di Bella S, Ascenzi P, Siarakas S, Petrosillo N, Di Masi A. Clostridium difficile Toxins A and B: insights into pathogenic properties and extraintestinal effects. Toxins. 2016;8(5):134. doi:10.3390/toxins8050134
9. Sofia MA, Rubin DT. The impact of therapeutic antibodies on the management of digestive diseases: history, current practice, and future directions. Dig Dis Sci. 2017;62(4):833–842. doi:10.1007/s10620-017-4479-0
10. Baines SD, Wilcox MH. Antimicrobial resistance and reduced susceptibility in Clostridium difficile: potential consequences for induction, treatment, and recurrence of C. difficile infection. Antibiotics. 2015;4(3):267–298. doi:10.3390/antibiotics4030267
11. Ofosu A. Clostridium difficile infection: a review of current and emerging therapies. Ann Gastroenterol. 2016;29(2):147–154. doi:10.20524/aog.2016.0006
12. von Müller L. Aktuelles zu Clostridium-difficile-infektionen. Dtsch Med Wochenschr. 2016;141(16):1144–1147. doi:10.1055/s-0042-107443
13. Lübbert C, John E, von Müller L. Clostridium Difficile infection: guideline-based diagnosis and treatment. Dtsch Arztebl Int. 2014;111(43):723–731. doi:10.3238/arztebl.2014.0723
14. Dial S, Kezouh A, Dascal A, Barkun A, Suissa S. Patterns of antibiotic use and risk of hospital admission because of Clostridium difficile infection. CMAJ. 2008;179(8):767–772. doi:10.1503/cmaj.071812
15. Wilcox MH, Mooney L, Bendall R, Settle CD, Fawley WN. A case-control study of community-associated Clostridium difficile infection. J Antimicrob Chemother. 2008;62(2):388–396. doi:10.1093/jac/dkn163
16. Tenover FC, Tickler IA, Persing DH. Antimicrobial-resistant strains of Clostridium difficile from North America. Antimicrob Agents Chemother. 2012;56(6):2929–2932. doi:10.1128/AAC.00220-12
17. Fuzi M. Dissimilar fitness associated with resistance to fluoroquinolones influences clonal dynamics of various multiresistant bacteria. Front Microbiol. 2016;7:1017. doi:10.3389/fmicb.2016.01017
18. Abdulgader SM, Shittu AO, Nicol MP, Kaba M. Molecular epidemiology of Methicillin-resistant Staphylococcus aureus in Africa: a systematic review. Front Microbiol. 2015;6:348. doi:10.3389/fmicb.2015.00348
19. Wasels F, Kuehne SA, Cartman ST, et al. Fluoroquinolone resistance does not impose a cost on the fitness of Clostridium difficile in vitro. Antimicrob Agents Chemother. 2015;59(3):1794–1796. doi:10.1128/AAC.04503-14
20. Valiente E, Cairns MD, Wren BW. The Clostridium difficile PCR ribotype 027 lineage: a pathogen on the move. Clin Microbiol Infect. 2014;20(5):396–404. doi:10.1111/1469-0691.12619
21. Lee JH, Lee Y, Lee K, Riley TV, Kim H. The changes of PCR ribotype and antimicrobial resistance of Clostridium difficile in a tertiary care hospital over 10 years. J Med Microbiol. 2014;63(Pt 6):819–823. doi:10.1099/jmm.0.072082-0
22. Kouzegaran S, Ganjifard M, Tanha AS. Detection, ribotyping and antimicrobial resistance properties of Clostridium difficile strains isolated from the Cases of Diarrhea. Mater Sociomed. 2016;28(5):324–328. doi:10.5455/msm.2016.28.324-328
23. Gupta A, Khanna S. Community-acquired Clostridium difficile infection: an increasing public health threat. Infect Drug Resist. 2014;7:63–72. doi:10.2147/IDR.S46780
24. Lambert PJ, Dyck M, Thompson LH, Hammond GW. Population-based surveillance of Clostridium difficile infection in Manitoba, Canada, by using interim surveillance definitions. Infect Control Hosp Epidemiol. 2009;30(10):945–951. doi:10.1086/605719
25. Bloomfield LE, Riley TV. Epidemiology and risk factors for community-associated Clostridium difficile infection: a narrative review. Infect Dis Ther. 2016;5(3):231–251. doi:10.1007/s40121-016-0117-y
26. Martinez FJ, Leffler DA, Kelly CP. Clostridium difficile outbreaks: prevention and treatment strategies. Risk Manag Healthc Policy. 2012;5:55–64. doi:10.2147/RMHP.S13053
27. Lessa FC, Mu Y, Bamberg WM, et al. Burden of Clostridium difficile infection in the United States. N Engl J Med. 2015;372(9):825–834. doi:10.1056/NEJMoa1408913
28. Ozaki E, Kato H, Kita H, et al. Clostridium difficile colonization in healthy adults: transient colonization and correlation with enterococcal colonization. J Med Microbiol. 2004;53(Pt 2):167–172. doi:10.1099/jmm.0.05376-0
29. Spigaglia P. Recent advances in the understanding of antibiotic resistance in Clostridium difficile infection. Ther Adv Infect Dis. 2016;3(1):23–42. doi:10.1177/2049936115622891
30. Chaparro-Rojas F, Mullane KM. Emerging therapies for Clostridium difficile infection - focus on fidaxomicin. Infect Drug Resist. 2013;6:41–53. doi:10.2147/IDR.S24434
31. Chandrasekaran R, Lacy DB. The role of toxins in Clostridium difficile infection. FEMS Microbiol Rev. 2017;41(6):723–750. doi:10.1093/femsre/fux048
32. Aktories K, Papatheodorou P, Schwan C. Binary Clostridium difficile toxin (CDT) - A virulence factor disturbing the cytoskeleton. Anaerobe. 2018. doi:10.1016/j.anaerobe.2018.03.001
33. Samarkos M, Mastrogianni E, Kampouropoulou O. The role of gut microbiota in Clostridium difficile infection. Eur J Intern Med. 2018;50:28–32. doi:10.1016/j.ejim.2018.02.006
34. Crook DW, Walker AS, Kean Y, et al. Fidaxomicin versus vancomycin for Clostridium difficile infection: meta-analysis of pivotal randomized controlled trials. Clin Infect Dis. 2012;55 Suppl 2:S93–S103. doi:10.1093/cid/cis499
35. Wolfe C, Pagano P, Pillar CM, Shinabarger DL, Boulos RA. Comparison of the in vitro antibacterial activity of Ramizol, fidaxomicin, vancomycin, and metronidazole against 100 clinical isolates of Clostridium difficile by broth microdilution. Diagn Microbiol Infect Dis. 2018;92(3):250–252. doi:10.1016/j.diagmicrobio.2018.06.002
36. Cornely OA, Watt M, McCrea C, Goldenberg SD, De Nigris E. Extended-pulsed fidaxomicin versus vancomycin for Clostridium difficile infection in patients aged >/=60 years (EXTEND): analysis of cost-effectiveness. J Antimicrob Chemother. 2018;73(9):2529–2539. doi:10.1093/jac/dky184
37. Baro E, Galperine T, Denies F, et al. Cost-effectiveness analysis of five competing strategies for the management of multiple recurrent community-onset Clostridium difficile infection in France. PLoS One. 2017;12(1):e0170258. doi:10.1371/journal.pone.0170258
38. Jiang M, N-H L, Ip M, You JHS. Cost-effectiveness analysis of ribotype-guided fecal microbiota transplantation in Chinese patients with severe Clostridium difficile infection. PLoS One. 2018;13(7):e0201539. doi:10.1371/journal.pone.0201539
39. Le P, Nghiem VT, Mullen PD, Deshpande A. Cost-effectiveness of competing treatment strategies for Clostridium difficile infection: a systematic review. Infect Control Hosp Epidemiol. 2018;39(4):412–424. doi:10.1017/ice.2017.303
40. Sunenshine RH, McDonald LC. Clostridium difficile-associated disease: new challenges from an established pathogen. Cleve Clin J Med. 2006;73(2):187–197.
41. Dostal A, Lacroix C, Bircher L, et al. Iron modulates butyrate production by a child gut microbiota In Vitro. mBio. 2015;6:6. doi:10.1128/mBio.01453-15
42. Chilton CH, Crowther GS, Spiewak K, et al. Potential of lactoferrin to prevent antibiotic-induced Clostridium difficile infection. J Antimicrob Chemother. 2016;71(4):975–985. doi:10.1093/jac/dkv452
43. Zanella Terrier MC, Simonet ML, Bichard P, Frossard JL. Recurrent Clostridium difficile infections: the importance of the intestinal microbiota. World J Gastroenterol. 2014;20(23):7416–7423. doi:10.3748/wjg.v20.i23.7416
44. Perez-Cobas AE, Moya A, Gosalbes MJ, Latorre A. Colonization resistance of the gut microbiota against Clostridium difficile. Antibiotics (Basel). 2015;4(3):337–357. doi:10.3390/antibiotics4030337
45. Wehrhahn MC, Keighley C, Kurtovic J, et al. A series of three cases of severe Clostridium difficile infection in Australia associated with a binary toxin producing clade 2 ribotype 251 strain. Anaerobe. 2018;55:117–123. doi:10.1016/j.anaerobe.2018.11.009.
46. King AM, Mackin KE, Lyras D. Emergence of toxin A-negative, toxin B-positive Clostridium difficile strains: epidemiological and clinical considerations. Future Microbiol. 2015;10(1):1–4. doi:10.2217/fmb.14.115
47. Chaves-Olarte E, Freer E, Parra A, Guzman-Verri C, Moreno E, Thelestam M. R-Ras glucosylation and transient RhoA activation determine the cytopathic effect produced by toxin B variants from toxin A-negative strains of Clostridium difficile. J Biol Chem. 2003;278(10):7956–7963. doi:10.1074/jbc.M209244200
48. Stewart DB, Berg A, Hegarty J. Predicting recurrence of C. difficile colitis using bacterial virulence factors: binary toxin is the key. J Gastrointest Surg. 2013;17(1):
49. Hutton ML, Mackin KE, Chakravorty A, Lyras D. Small animal models for the study of Clostridium difficile disease pathogenesis. FEMS Microbiol Lett. 2014;352(2):140–149. doi:10.1111/1574-6968.12367
50. Cowardin CA, Buonomo EL, Saleh MM, et al. The binary toxin CDT enhances Clostridium difficile virulence by suppressing protective colonic eosinophilia. Nat Microbiol. 2016;1(8):16108. doi:10.1038/nmicrobiol.2016.108
51. Darkoh C, DuPont HL, Norris SJ, Kaplan HB. Toxin synthesis by Clostridium difficile is regulated through quorum signaling. MBio. 2015;6(2):e02569. doi:10.1128/mBio.02569-14
52. Lyon SA, Hutton ML, Rood JI, Cheung JK, Lyras D. CdtR regulates TcdA and TcdB production in Clostridium difficile. PLoS Pathog. 2016;12(7):e1005758. doi:10.1371/journal.ppat.1005758
53. Culligan EP, Hill C, Sleator RD. Probiotics and gastrointestinal disease: successes, problems and future prospects. Gut Pathog. 2009;1(1):19. doi:10.1186/1757-4749-1-19
54. Wong A, Ngu DYS, Dan LA, Ooi A, Lim RLH. Detection of antibiotic resistance in probiotics of dietary supplements. Nutr J. 2015;14(1):95. doi:10.1186/s12937-015-0084-2
55. Zheng M, Zhang R, Tian X, Zhou X, Pan X, Wong A. Assessing the risk of probiotic dietary supplements in the context of antibiotic resistance. Front Microbiol. 2017;8:908–908. doi:10.3389/fmicb.2017.00908
56. Maziade PJ, Pereira P, Goldstein EJ. A decade of experience in primary prevention of Clostridium difficile infection at a community hospital using the probiotic combination Lactobacillus acidophilus CL1285, Lactobacillus casei LBC80R, and Lactobacillus rhamnosus CLR2 (Bio-K+). Clin Infect Dis. 2015;60 Suppl 2:S144–147. doi:10.1093/cid/civ178
57. More MI, Vandenplas Y. Saccharomyces boulardii CNCM I-745 improves intestinal enzyme function: a trophic effects review. Clin Med Insights Gastroenterol. 2018;11:1179552217752679. doi:10.1177/1179552217752679
58. Surawicz CM, McFarland LV, Greenberg RN, et al. The search for a better treatment for recurrent Clostridium difficile disease: use of high-dose vancomycin combined with Saccharomyces boulardii. Clin Infect Dis. 2000;31(4):1012–1017. doi:10.1086/318130
59. Pozzoni P, Riva A, Bellatorre AG, et al. Saccharomyces boulardii for the prevention of antibiotic-associated diarrhea in adult hospitalized patients: a single-center, randomized, double-blind, placebo-controlled trial. Am J Gastroenterol. 2012;107(6):922–931. doi:10.1038/ajg.2012.56
60. Wullt M, Hagslatt ML, Odenholt I. Lactobacillus plantarum 299v for the treatment of recurrent Clostridium difficile-associated diarrhoea: a double-blind, placebo-controlled trial. Scand J Infect Dis. 2003;35(6–7):365–367. doi:10.1080/00365540310010985
61. Gueimonde M, Sanchez B, GdLR-G C, Margolles A. Antibiotic resistance in probiotic bacteria. Front Microbiol. 2013;4:202. doi:10.3389/fmicb.2013.00077
62. Cameron D, Hock QS, Kadim M, et al. Probiotics for gastrointestinal disorders: proposed recommendations for children of the Asia-Pacific region. World J Gastroenterol. 2017;23(45):7952–7964. doi:10.3748/wjg.v23.i45.7952
63. Julliard W, De Wolfe TJ, Fechner JH, Safdar N, Agni R, Mezrich JD. Amelioration of Clostridium difficile infection in mice by dietary supplementation with indole-3-carbinol. Ann Surg. 2017;265(6):1183–1191. doi:10.1097/SLA.0000000000001830
64. Hryckowian AJ, Pruss KM, Sonnenburg JL. The emerging metabolic view of Clostridium difficile pathogenesis. Curr Opin Microbiol. 2017;35:42–47. doi:10.1016/j.mib.2016.11.006
65. Dolan KT, Chang EB. Diet, gut microbes, and the pathogenesis of inflammatory bowel diseases. Mol Nutr Food Res. 2017;61(1). doi:10.1002/mnfr.201600129
66. Kelly CP, Kyne L. The host immune response to Clostridium difficile. J Med Microbiol. 2011;60(Pt 8):1070–1079. doi:10.1099/jmm.0.030015-0
67. Solomon K. The host immune response to Clostridium difficile infection. Ther Adv Infect Dis. 2013;1(1):19–35. doi:10.1177/2049936112472173
68. Mathur H, Rea MC, Cotter PD, Ross RP, Hill C. The potential for emerging therapeutic options for Clostridium difficile infection. Gut Microbes. 2014;5(6):696–710. doi:10.4161/19490976.2014.983768
69. Simon M, Chervin S, Brown S, et al. Polyclonal Antibody Therapies for Clostridium difficile Infection. Antibodies. 2014;3:272–288. doi:10.3390/antib3040272
70. Hurez V, Kazatchkine MD, Vassilev T, et al. Pooled normal human polyspecific IgM contains neutralizing anti-idiotypes to IgG autoantibodies of autoimmune patients and protects from experimental autoimmune disease. Blood. 1997;90(10):4004–4013.
71. Humphreys DP, Wilcox MH. Antibodies for treatment of Clostridium difficile infection. Clin Vaccine Immunol. 2014;21(7):913–923. doi:10.1128/CVI.00116-14
72. Torres JF, Lyerly DM, Hill JE, Monath TP. Evaluation of formalin-inactivated Clostridium difficile vaccines administered by parenteral and mucosal routes of immunization in hamsters. Infect Immun. 1995;63(12):4619–4627.
73. Greenberg RN, Marbury TC, Foglia G, Warny M. Phase I dose finding studies of an adjuvanted Clostridium difficile toxoid vaccine. Vaccine. 2012;30(13):2245–2249. doi:10.1016/j.vaccine.2012.01.065
74. Feher C, Mensa J. A comparison of current guidelines of five international societies on Clostridium difficile infection management. Infect Dis Ther. 2016;5(3):207–230. doi:10.1007/s40121-016-0122-1
75. Feher C, Soriano A, Mensa J. A review of experimental and off-label therapies for Clostridium difficile infection. Infect Dis Ther. 2017;6(1):1–35. doi:10.1007/s40121-016-0140-z
76. Broecker F, Hanske J, Martin CE, et al. Multivalent display of minimal Clostridium difficile glycan epitopes mimics antigenic properties of larger glycans. Nat Commun. 2016;7:11224. doi:10.1038/ncomms11224
77. Kirk JA, Banerji O, Fagan RP. Characteristics of the Clostridium difficile cell envelope and its importance in therapeutics. Microb Biotechnol. 2017;10(1):76–90. doi:10.1111/1751-7915.12372
78. Hussack G, Tanha J. An update on antibody-based immunotherapies for Clostridium difficile infection. Clin Exp Gastroenterol. 2016;9:209–224. doi:10.2147/CEG.S84017
79. Sun X, Hirota SA. The roles of host and pathogen factors and the innate immune response in the pathogenesis of Clostridium difficile infection. Mol Immunol. 2015;63(2):193–202. doi:10.1016/j.molimm.2014.09.005
80. Karczewski J, Zorman J, Wang S, et al. Development of a recombinant toxin fragment vaccine for Clostridium difficile infection. Vaccine. 2014;32(24):2812–2818. doi:10.1016/j.vaccine.2014.02.026
81. Spencer J, Leuzzi R, Buckley A, et al. Vaccination against Clostridium difficile using toxin fragments: observations and analysis in animal models. Gut Microbes. 2014;5(2):225–232. doi:10.4161/gmic.27712
82. Leuzzi R, Spencer J, Buckley A, et al. Protective efficacy induced by recombinant Clostridium difficile toxin fragments. Infect Immun. 2013;81(8):2851–2860. doi:10.1128/IAI.01341-12
83. Wang H, Sun X, Zhang Y, et al. A chimeric toxin vaccine protects against primary and recurrent Clostridium difficile infection. Infect Immun. 2012;80(8):2678–2688. doi:10.1128/IAI.00215-12
84. Wang YK, Yan YX, Kim HB, et al. A chimeric protein comprising the glucosyltransferase and cysteine proteinase domains of toxin B and the receptor binding domain of toxin A induces protective immunity against Clostridium difficile infection in mice and hamsters. Hum Vaccin Immunother. 2015;11(9):2215–2222. doi:10.1080/21645515.2015.1052352
85. Secore S, Wang S, Doughtry J, et al. Development of a novel vaccine containing binary toxin for the prevention of Clostridium difficile disease with enhanced efficacy against NAP1 strains. PLoS One. 2017;12(1):e0170640. doi:10.1371/journal.pone.0170640
86. Leuzzi R, Adamo R, Scarselli M. Vaccines against Clostridium difficile. Hum Vaccin Immunother. 2014;10(6):1466–1477. doi:10.4161/hv.28428
87. Yang Z, Ramsey J, Hamza T, et al. Mechanisms of protection against Clostridium difficile infection by the monoclonal antitoxin antibodies actoxumab and bezlotoxumab. Infect Immun. 2015;83(2):822–831. doi:10.1128/IAI.02897-14
88. Anosova NG, Cole LE, Li L, et al. A combination of three fully human toxin A- and toxin B-specific monoclonal antibodies protects against challenge with highly virulent epidemic strains of Clostridium difficile in the hamster model. Clin Vaccine Immunol. 2015;22(7):711–725. doi:10.1128/CVI.00763-14
89. Morrison C. Antibacterial antibodies gain traction. Nat Rev Drug Discov. 2015;14(11):737–738. doi:10.1038/nrd4770
90. Qiu H, Cassan R, Johnstone D, et al. Novel Clostridium difficile anti-toxin (TcdA and TcdB) humanized monoclonal antibodies demonstrate In Vitro neutralization across a broad spectrum of clinical strains and In Vivo potency in a hamster spore challenge model. PLoS One. 2016;11(6):e0157970. doi:10.1371/journal.pone.0157970
91. Davies NL, Compson JE, Mackenzie B, et al. A mixture of functionally oligoclonal humanized monoclonal antibodies that neutralize Clostridium difficile TcdA and TcdB with high levels of in vitro potency shows in vivo protection in a hamster infection model. Clin Vaccine Immunol. 2013;20(3):377–390. doi:10.1128/CVI.00625-12
92. Marozsan AJ, Ma D, Nagashima KA, et al. Protection against Clostridium difficile infection with broadly neutralizing antitoxin monoclonal antibodies. J Infect Dis. 2012;206(5):706–713. doi:10.1093/infdis/jis416
93. Hernandez LD, Kroh HK, Hsieh E, et al. Epitopes and mechanism of action of the Clostridium difficile toxin A-neutralizing antibody actoxumab. J Mol Biol. 2017;429(7):1030–1044. doi:10.1016/j.jmb.2017.02.010
94. Milani C, Ticinesi A, Gerritsen J, et al. Gut microbiota composition and Clostridium difficile infection in hospitalized elderly individuals: a metagenomic study. Sci Rep. 2016;6:25945. doi:10.1038/srep25945
95. Martinez I, Lattimer JM, Hubach KL, et al. Gut microbiome composition is linked to whole grain-induced immunological improvements. ISME J. 2013;7(2):269–280. doi:10.1038/ismej.2012.104
96. Dzunkova M, D’Auria G, Xu H, et al. The monoclonal antitoxin antibodies (Actoxumab-Bezlotoxumab) treatment facilitates normalization of the gut microbiota of mice with Clostridium difficile infection. Front Cell Infect Microbiol. 2016;6:119. doi:10.3389/fcimb.2016.00119
97. Ritter AS, Petri WA
98. Hussack G, Arbabi-Ghahroudi M, Mackenzie CR, Tanha J. Isolation and characterization of Clostridium difficile toxin-specific single-domain antibodies. Methods Mol Biol. 2012;911:211–239.
99. Hussack G, Arbabi-Ghahroudi M, van Faassen H, et al. Neutralization of Clostridium difficile toxin A with single-domain antibodies targeting the cell receptor binding domain. J Biol Chem. 2011;286(11):8961–8976. doi:10.1074/jbc.M110.198754
100. Andersen KK, Strokappe NM, Hultberg A, et al. Neutralization of Clostridium difficile toxin B mediated by engineered Lactobacilli that produce single-domain antibodies. Infect Immun. 2016;84(2):395–406. doi:10.1128/IAI.00870-15
101. Shkoporov AN, Khokhlova EV, Savochkin KA, Kafarskaia LI, Efimov BA. Production of biologically active scFv and VHH antibody fragments in Bifidobacterium longum. FEMS Microbiol Lett. 2015;362(12):fnv083. doi:10.1093/femsle/fnv083
102. Unger M, Eichhoff AM, Schumacher L, et al. Selection of nanobodies that block the enzymatic and cytotoxic activities of the binary Clostridium difficile toxin CDT. Sci Rep. 2015;5:7850. doi:10.1038/srep07850
103. Hong HA, Hitri K, Hosseini S, et al. Mucosal antibodies to the C terminus of toxin A prevent colonization of Clostridium difficile. Infect Immun. 2017;85(4):e01060-16. doi:10.1128/IAI.01060-16.
104. Sulea T, Hussack G, Ryan S, Tanha J, Purisima EO. Application of Assisted Design of Antibody and Protein Therapeutics (ADAPT) improves efficacy of a Clostridium difficile toxin A single-domain antibody. Sci Rep. 2018;8(1):2260. doi:10.1038/s41598-018-20599-4
105. Lyerly DM, Phelps CJ, Toth J, Wilkins TD. Characterization of toxins A and B of Clostridium difficile with monoclonal antibodies. Infect Immun. 1986;54(1):70–76.
106. Roberts A, McGlashan J, Al-Abdulla I, et al. Development and evaluation of an ovine antibody-based platform for treatment of Clostridium difficile infection. Infect Immun. 2012;80(2):875–882. doi:10.1128/IAI.05684-11
107. van Dissel JT, de Groot N, Hensgens CM, et al. Bovine antibody-enriched whey to aid in the prevention of a relapse of Clostridium difficile-associated diarrhoea: preclinical and preliminary clinical data. J Med Microbiol. 2005;54(Pt 2):197–205. doi:10.1099/jmm.0.45773-0
108. Numan SC, Veldkamp P, Kuijper EJ, van Den Berg RJ, van Dissel JT. Clostridium difficile-associated diarrhoea: bovine anti-Clostridium difficile whey protein to help aid the prevention of relapses. Gut. 2007;56(6):888–889. doi:10.1136/gut.2006.119016
109. Mattila E, Anttila VJ, Broas M, et al. A randomized, double-blind study comparing Clostridium difficile immune whey and metronidazole for recurrent Clostridium difficile-associated diarrhoea: efficacy and safety data of a prematurely interrupted trial. Scand J Infect Dis. 2008;40(9):702–708. doi:10.1080/00365540801964960
110. Hutton ML, Cunningham BA, Mackin KE, et al. Bovine antibodies targeting primary and recurrent Clostridium difficile disease are a potent antibiotic alternative. Sci Rep. 2017;7(1):3665. doi:10.1038/s41598-017-03982-5
111. O’Brien JB, McCabe MS, Athie-Morales V, McDonald GS, Ni Eidhin DB, Kelleher DP. Passive immunisation of hamsters against Clostridium difficile infection using antibodies to surface layer proteins. FEMS Microbiol Lett. 2005;246(2):199–205. doi:10.1016/j.femsle.2005.04.005
112. Ghose C, Eugenis I, Sun X, et al. Immunogenicity and protective efficacy of recombinant Clostridium difficile flagellar protein FliC. Emerg Microbes Infect. 2016;5:e8. doi:10.1038/emi.2016.8
113. Bojanova DP, Bordenstein SR. Fecal transplants: what is being transferred? PLoS Biol. 2016;14(7):e1002503. doi:10.1371/journal.pbio.1002503
114. Clatworthy AE, Pierson E, Hung DT. Targeting virulence: a new paradigm for antimicrobial therapy. Nat Chem Biol. 2007;3(9):541–548. doi:10.1038/nchembio.2007.24
115. Surawicz CM, Brandt LJ, Binion DG, et al. Guidelines for diagnosis, treatment, and prevention of Clostridium difficile infections. Am J Gastroenterol. 2013;108(4):
116. van Nood E, Vrieze A, Nieuwdorp M, et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N Engl J Med. 2013;368(5):407–415. doi:10.1056/NEJMoa1205037
117. Pecere S, Sabatelli M, Fantoni M, Ianiro G, Gasbarrini A, Cammarota G. Letter: faecal microbiota transplantation in combination with fidaxomicin to treat severe complicated recurrent Clostridium difficile infection. Aliment Pharmacol Ther. 2015;42(8):1030. doi:10.1111/apt.13362
118. Gough E, Shaikh H, Manges AR. Systematic review of intestinal microbiota transplantation (fecal bacteriotherapy) for recurrent Clostridium difficile infection. Clin Infect Dis. 2011;53(10):994–1002. doi:10.1093/cid/cir632
119. Bang BW, Park JS, Kim HK, et al. Fecal microbiota transplantation for refractory and recurrent Clostridium difficile infection: a case series of nine patients. Korean J Gastroenterol. 2017;69(4):226–231. doi:10.4166/kjg.2017.69.4.226
120. Kelly CR, Khoruts A, Staley C, et al. Effect of fecal microbiota transplantation on recurrence in multiply recurrent Clostridium difficile infection: a randomized trial. Ann Intern Med. 2016;165(9):609–616. doi:10.7326/M16-0271
121. Carlucci C, Petrof EO, Allen-Vercoe E. Fecal microbiota-based therapeutics for recurrent Clostridium difficile infection, ulcerative colitis and obesity. EBioMedicine. 2016;13:37–45. doi:10.1016/j.ebiom.2016.09.029
122. Anand R, Song Y, Garg S, et al. Effect of aging on the composition of fecal microbiota in donors for FMT and its impact on clinical outcomes. Dig Dis Sci. 2017;62(4):1002–1008. doi:10.1007/s10620-017-4449-6
123. Lagier JC, Delord M, Million M, et al. Dramatic reduction in Clostridium difficile ribotype 027-associated mortality with early fecal transplantation by the nasogastric route: a preliminary report. Eur J Clin Microbiol Infect Dis. 2015;34(8):1597–1601. doi:10.1007/s10096-015-2394-x
124. Tanaka T, Kato H, Fujimoto T. Successful fecal microbiota transplantation as an initial therapy for Clostridium difficile infection on an outpatient basis. Intern Med. 2016;55(8):999–1000. doi:10.2169/internalmedicine.55.5701
125. Kang DW, Adams JB, Gregory AC, et al. Microbiota Transfer Therapy alters gut ecosystem and improves gastrointestinal and autism symptoms: an open-label study. Microbiome. 2017;5(1):10. doi:10.1186/s40168-016-0225-7
126. Giau VV, Wu SY, Jamerlan A, An SSA, Kim SY, Hulme J. Gut microbiota and their neuroinflammatory implications in Alzheimer’s disease. Nutrients. 2018;10(11):1765. doi:10.3390/nu10111765.
127. Baumler AJ, Sperandio V. Interactions between the microbiota and pathogenic bacteria in the gut. Nature. 2016;535(7610):85–93. doi:10.1038/nature18849
128. Staley C, Kelly CR, Brandt LJ, Khoruts A, Sadowsky MJ. Complete microbiota engraftment is not essential for recovery from recurrent Clostridium difficile infection following fecal microbiota transplantation. mBio. 2016;7(6). doi:10.1128/mBio.01965-16
129. Julliard W, Fechner JH, Owens L, et al. Modeling the effect of the aryl hydrocarbon receptor on transplant immunity. Transplant Direct. 2017;3(5):e157. doi:10.1097/TXD.0000000000000666
130. Suez J, Zmora N, Zilberman-Schapira G, et al. Post-antibiotic gut mucosal microbiome reconstitution is impaired by probiotics and improved by autologous FMT. Cell. 2018;174(6):1406–1423.e1416. doi:10.1016/j.cell.2018.08.047
131. Lamendella R, Wright JR, Hackman J, et al. Antibiotic treatments for Clostridium difficile infection are associated with distinct bacterial and fungal community structures. mSphere. 2018;3(1). doi:10.1128/mSphere.00572-17
132. Khoruts A, Rank KM, Newman KM, et al. Inflammatory bowel disease affects the outcome of fecal microbiota transplantation for recurrent Clostridium difficile infection. Clin Gastroenterol Hepatol. 2016;14(10):1433–1438. doi:10.1016/j.cgh.2016.02.018
133. Lamas B, Richard ML, Leducq V, et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat Med. 2016;22(6):598–605. doi:10.1038/nm.4102
134. Anderson JL, Edney RJ, Whelan K. Systematic review: faecal microbiota transplantation in the management of inflammatory bowel disease. Aliment Pharmacol Ther. 2012;36(6):503–516. doi:10.1111/j.1365-2036.2012.05220.x
135. Moayyedi P, Surette MG, Kim PT, et al. Fecal microbiota transplantation induces remission in patients with active ulcerative colitis in a randomized controlled trial. Gastroenterology. 2015;149(1):102–109.e106. doi:10.1053/j.gastro.2015.04.001
136. Abedon ST. Phage therapy: eco-physiological pharmacology. Scientifica. 2014;2014:581639. doi:10.1155/2014/726179
137. Sybesma W, Zbinden R, Chanishvili N, et al. Bacteriophages as potential treatment for urinary tract infections. Front Microbiol. 2016;7:465. doi:10.3389/fmicb.2016.00465
138. Khawaldeh A, Morales S, Dillon B, et al. Bacteriophage therapy for refractory Pseudomonas aeruginosa urinary tract infection. J Med Microbiol. 2011;60(Pt 11):1697–1700. doi:10.1099/jmm.0.029744-0
139. Melo LDR, Veiga P, Cerca N, et al. Development of a phage cocktail to control proteus mirabilis catheter-associated urinary tract infections. Front Microbiol. 2016;7:1024. doi:10.3389/fmicb.2016.01024
140. Merabishvili M, Vervaet C, Pirnay JP, et al. Stability of Staphylococcus aureus phage ISP after freeze-drying (lyophilization). PLoS One. 2013;8(7):e68797. doi:10.1371/journal.pone.0068797
141. Hargreaves KR, Clokie MR. Clostridium difficile phages: still difficult? Front Microbiol. 2014;5:184. doi:10.3389/fmicb.2014.00547
142. Sullivan MJ, Petty NK, Beatson SA. Easyfig: a genome comparison visualizer. Bioinformatics. 2011;27(7):1009–1010. doi:10.1093/bioinformatics/btr039
143. Rashid S, Barylski J, Hargreaves K, Millard A, Vinner G, Clokie M. Two novel myoviruses from the north of Iraq reveal insights into Clostridium difficile phage diversity and biology. Viruses. 2016;8(11):310. doi:10.3390/v8110310
144. Meessen-Pinard M, Sekulovic O, Fortier LC. Evidence of in vivo prophage induction during Clostridium difficile infection. Appl Environ Microbiol. 2012;78(21):7662–7670. doi:10.1128/AEM.02275-12
145. Nale JY, Spencer J, Hargreaves KR, et al. Bacteriophage combinations significantly reduce Clostridium difficile growth in vitro and proliferation in vivo. Antimicrob Agents Chemother. 2016;60(2):968–981. doi:10.1128/AAC.01774-15
146. Goh S, Ong PF, Song KP, Riley TV, Chang BJ. The complete genome sequence of Clostridium difficile phage phiC2 and comparisons to phiCD119 and inducible prophages of CD630. Microbiology. 2007;153(Pt 3):676–685. doi:10.1099/mic.0.2006/002436-0
147. Sebaihia M, Wren BW, Mullany P, et al. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat Genet. 2006;38(7):779–786. doi:10.1038/ng1830
148. Zhang Q, Widmer G, Tzipori S. A pig model of the human gastrointestinal tract. Gut Microbes. 2013;4(3):193–200. doi:10.4161/gmic.23867
149. Zackular JP, Moore JL, Jordan AT, et al. Dietary zinc alters the microbiota and decreases resistance to Clostridium difficile infection. Nat Med. 2016;22(11):1330–1334. doi:10.1038/nm.4174
150. Seekatz AM, Young VB. Clostridium difficile and the microbiota. J Clin Invest. 2014;124(10):4182–4189. doi:10.1172/JCI72336
151. Merril CR, Scholl D, Adhya SL. The prospect for bacteriophage therapy in Western medicine. Nat Rev Drug Discov. 2003;2(6):489–497. doi:10.1038/nrd1111
152. Meader E, Mayer MJ, Gasson MJ, Steverding D, Carding SR, Narbad A. Bacteriophage treatment significantly reduces viable Clostridium difficile and prevents toxin production in an in vitro model system. Anaerobe. 2010;16(6):549–554. doi:10.1016/j.anaerobe.2010.08.006
153. Meader E, Mayer MJ, Steverding D, Carding SR, Narbad A. Evaluation of bacteriophage therapy to control Clostridium difficile and toxin production in an in vitro human colon model system. Anaerobe. 2013;22:25–30. doi:10.1016/j.anaerobe.2013.05.001
154. Govind R, Fralick JA, Rolfe RD. In vivo lysogenization of a Clostridium difficile bacteriophage ΦCD119. Anaerobe. 2011;17(3):125–129. doi:10.1016/j.anaerobe.2011.05.012
155. Sekulovic O, Garneau JR, Neron A, Fortier LC. Characterization of temperate phages infecting Clostridium difficile isolates of human and animal origins. Appl Environ Microbiol. 2014;80(8):2555–2563. doi:10.1128/AEM.00237-14
156. Goh S, Hussain H, Chang BJ, Emmett W, Riley TV, Mullany P. Phage ϕC2 mediates transduction of Tn6215, encoding erythromycin resistance, between Clostridium difficile strains. mBio. 2013;4(6). doi:10.1128/mBio.00840-13
157. Nale JY, Chutia M, Carr P, Hickenbotham PT, Clokie MR. ‘Get in Early’; Biofilm and Wax Moth (Galleria mellonella) models reveal new insights into the therapeutic potential of Clostridium difficile bacteriophages. Front Microbiol. 2016;7:1383. doi:10.3389/fmicb.2016.01383
158. Sekulovic O, Meessen-Pinard M, Fortier LC. Prophage-stimulated toxin production in Clostridium difficile NAP1/027 lysogens. J Bacteriol. 2011;193(11):2726–2734. doi:10.1128/JB.00787-10
159. Nagy E, Foldes J. Inactivation of metronidazole by Enterococcus faecalis. J Antimicrob Chemother. 1991;27(1):63–70. doi:10.1093/jac/27.1.63
160. Anonye BO. Commentary: bacteriophage transfer during faecal microbiota transplantation in Clostridium difficile infection is associated with treatment outcome. Front Cell Infect Microbiol. 2018;8:104. doi:10.3389/fcimb.2018.00026
161. Fortier LC. Bacteriophages contribute to shaping clostridioides (Clostridium) difficile species. Front Microbiol. 2018;9:2033. doi:10.3389/fmicb.2018.02033
162. Abaev I, Foster-Frey J, Korobova O, et al. Staphylococcal phage 2638A endolysin is lytic for Staphylococcus aureus and harbors an inter-lytic-domain secondary translational start site. Appl Microbiol Biotechnol. 2013;97(8):3449–3456. doi:10.1007/s00253-012-4252-4
163. Gaeng S, Scherer S, Neve H, Loessner MJ. Gene cloning and expression and secretion of Listeria monocytogenes bacteriophage-lytic enzymes in Lactococcus lactis. Appl Environ Microbiol. 2000;66(7):2951–2958. doi:10.1128/aem.66.7.2951-2958.2000
164. Schuch R, Nelson D, Fischetti VA. A bacteriolytic agent that detects and kills Bacillus anthracis. Nature. 2002;418(6900):884–889.
165. Bai J, Kim YT, Ryu S, Lee JH. Biocontrol and rapid detection of food-borne pathogens using bacteriophages and endolysins. Front Microbiol. 2016;7:474. doi:10.3389/fmicb.2016.00474
166. Eyre DW, Walker AS, Griffiths D, et al. Clostridium difficile mixed infection and reinfection. J Clin Microbiol. 2012;50(1):142–144. doi:10.1128/JCM.05177-11
167. He M, Miyajima F, Roberts P, et al. Emergence and global spread of epidemic healthcare-associated Clostridium difficile. Nat Genet. 2013;45(1):109–113. doi:10.1038/ng.2478
168. Ajuebor J, McAuliffe O, O’Mahony J, Ross RP, Hill C, Coffey A. Bacteriophage endolysins and their applications. Sci Prog. 2016;99(2):183–199. doi:10.3184/003685016X14627913637705
169. Thanissery R, Zeng D, Doyle RG, Theriot CM. A small molecule-screening pipeline to evaluate the therapeutic potential of 2-aminoimidazole molecules against Clostridium difficile. Front Microbiol. 2018;9:1206. doi:10.3389/fmicb.2018.01206
170. Letourneau JJ, Stroke IL, Hilbert DW, et al. Synthesis and SAR studies of novel benzodiazepinedione-based inhibitors of Clostridium difficile (C. difficile) toxin B (TcdB). Bioorg Med Chem Lett. 2018;28(23-24):3601–3605. doi: 10.1016/j.bmcl.2018.10.047.
171. Letourneau JJ, Stroke IL, Hilbert DW, et al. Identification and initial optimization of inhibitors of Clostridium difficile (C. difficile) toxin B (TcdB). Bioorg Med Chem Lett. 2018;28(4):756–761. doi:10.1016/j.bmcl.2018.01.005
172. Mayer MJ, Narbad A, Gasson MJ. Molecular characterization of a Clostridium difficile bacteriophage and its cloned biologically active endolysin. J Bacteriol. 2008;190(20):6734–6740. doi:10.1128/JB.00686-08
173. Beilhartz GL, Tam J, Zhang Z, Melnyk RA. Comment on “A small-molecule antivirulence agent for treating Clostridium difficile infection”. Sci Transl Med. 2016;8(370):370tc372. doi:10.1126/scitranslmed.aaf0746
174. Xie Y, Tan Y, Zheng Y, Du X, Liu Q. Ebselen ameliorates beta-amyloid pathology, tau pathology, and cognitive impairment in triple-transgenic Alzheimer’s disease mice. J Biol Inorg Chem. 2017;22(6):851–865. doi:10.1007/s00775-017-1463-2
175. Ivarsson ME, Durantie E, Huberli C, et al. Small-molecule allosteric triggers of Clostridium difficile toxin B auto-proteolysis as a therapeutic strategy. Cell Chem Biol. 2018;26(1):17–26.e13.
176. Egan K, Field D, Ross RP, Cotter PD, Hill C. In silico prediction and exploration of potential bacteriocin gene clusters within the bacterial genus geobacillus. Front Microbiol. 2018;9:2116. doi:10.3389/fmicb.2018.02116
177. Le Lay C, Dridi L, Bergeron MG, Ouellette M, Fliss IL. Nisin is an effective inhibitor of Clostridium difficile vegetative cells and spore germination. J Med Microbiol. 2016;65(2):169–175. doi:10.1099/jmm.0.000202
178. Khosa S, Lagedroste M, Smits SH. Protein defense systems against the lantibiotic nisin: function of the immunity protein nisi and the resistance protein NSR. Front Microbiol. 2016;7:504. doi:10.3389/fmicb.2016.00504
179. Chai C, Lee KS, Imm GS, Kim YS, Oh SW. Inactivation of Clostridium difficile spore outgrowth by synergistic effects of nisin and lysozyme. Can J Microbiol. 2017;63(7):638–643. doi:10.1139/cjm-2016-0550
180. Rea MC, O’Sullivan O, Shanahan F, et al. Clostridium difficile carriage in elderly subjects and associated changes in the intestinal microbiota. J Clin Microbiol. 2012;50(3):867–875. doi:10.1128/JCM.05176-11
181. Mathur H, Field D, Rea MC, Cotter PD, Hill C, Ross RP. Bacteriocin-antimicrobial synergy: a medical and food perspective. Front Microbiol. 2017;8:1205. doi:10.3389/fmicb.2017.01205
182. Hanchi H, Hammami R, Gingras H, et al. Inhibition of MRSA and of Clostridium difficile by durancin 61A: synergy with bacteriocins and antibiotics. Future Microbiol. 2017;12:205–212. doi:10.2217/fmb-2016-0113
183. Rea MC, Sit CS, Clayton E, et al. Thuricin CD, a posttranslationally modified bacteriocin with a narrow spectrum of activity against Clostridium difficile. Proc Natl Acad Sci. 2010;107(20):9352–9357. doi:10.1073/pnas.0913554107
184. Li C, Teng P, Peng Z, Sang P, Sun X, Cai J. Bis-cyclic guanidines as a novel class of compounds potent against Clostridium difficile. ChemMedChem. 2018;13(14):1414–1420. doi:10.1002/cmdc.201800240
185. Petrosillo N, Granata G, Cataldo MA. Novel antimicrobials for the treatment of Clostridium difficile infection. Front Med. 2018;5:96. doi:10.3389/fmed.2018.00096
186. Tyrrell KL, Citron DM, Merriam CV, Leoncio E, Goldstein EJC. In vitro activity of DS-2969b and comparator antimicrobial agents against Clostridioides (Clostridium) difficile, methicillin-resistant Staphylococcus aureus, and other anaerobic bacteria. Anaerobe. 2018;54:39–41. doi:10.1016/j.anaerobe.2018.04.010
187. Van Giau V, An SSA, Hulme J. Recent advances in the treatment of pathogenic infections using antibiotics and nano-drug delivery vehicles. Drug Des Devel Ther. 2019;13:327–343. doi:10.2147/DDDT.S190577
188. Millan B, Park H, Hotte N, et al. Fecal microbial transplants reduce antibiotic-resistant genes in patients with recurrent Clostridium difficile infection. Clin Infect Dis. 2016;62(12):1479–1486. doi:10.1093/cid/ciw185
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.