Back to Journals » The Application of Clinical Genetics » Volume 8
Recent advances in gene therapy for lysosomal storage disorders
Authors Rastall D, Amalfitano A
Received 20 February 2015
Accepted for publication 3 April 2015
Published 23 June 2015 Volume 2015:8 Pages 157—169
DOI https://doi.org/10.2147/TACG.S57682
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Prof. Dr. Martin Maurer
David PW Rastall,1 Andrea Amalfitano1,2
1Department of Microbiology and Molecular Genetics, 2Department of Pediatrics, College of Osteopathic Medicine, Michigan State University, East Lansing, MI, USA
Abstract: Lysosomal storage disorders (LSDs) are a group of genetic diseases that result in metabolic derangements of the lysosome. Most LSDs are due to the genetic absence of a single catabolic enzyme, causing accumulation of the enzyme's substrate within the lysosome. Over time, tissue-specific substrate accumulations result in a spectrum of symptoms and disabilities that vary by LSD. LSDs are promising targets for gene therapy because delivery of a single gene into a small percentage of the appropriate target cells may be sufficient to impact the clinical course of the disease. Recently, there have been several significant advancements in the potential for gene therapy of these disorders, including the first human trials. Future clinical trials will build upon these initial attempts, with an improved understanding of immune system responses to gene therapy, the obstacle that the blood–brain barrier poses for neuropathic LSDs, as well other biological barriers that, when overcome, may facilitate gene therapy for LSDs. In this manuscript, we will highlight the recent innovations in gene therapy for LSDs and discuss the clinical limitations that remain to be overcome, with the goal of fostering an understanding and further development of this important field.
Keywords: human trials, clinical trials, gene therapy, lysosomal storage disease, blood-brain barrier, adeno-associated virus, lentivirus, adenovirus
Introduction
Lysosomal storage disorders (LSDs) are a group of over 40 distinct inherited diseases that result in metabolic derangements of the lysosome.1 Clinical features of LSDs vary from disease to disease, but can include cardiomegaly, hepatosplenomegaly, skeletal deformity, cognitive disability, and premature death.2 Most LSDs result from a loss-of-function mutation in a single gene responsible for producing a catalytic lysosomal enzyme.3 As a result of insufficient enzymatic activity, the enzyme’s substrate accumulates over variable amounts of time within the lysosomes of specific tissues, causing the pathophysiology of the respective LSD.1,4
Compared to many other genetic diseases, LSDs are practical targets for gene therapy because of a unique physiologic trait referred to as “cross-correction”. Cross-correction allows specific extracellular LSD enzymes to be taken up and targeted to the lysosomes of otherwise enzyme-deficient cells.5 This phenomenon was exploited to develop the first United States Food and Drug Administration-approved therapies for these diseases, known as enzyme replacement therapies (ERT).6,7 These therapies typically involve repeated (life-long) intravenous administrations of a recombinant LSD enzyme into affected patients. From the bloodstream, the recombinant LSD enzyme can travel to affected tissues throughout the body, enter enzyme-deficient cells, and partially restore deficient lysosomal enzymatic functions. Unfortunately, recombinant enzymes administered intravenously do not easily cross the blood–brain barrier (BBB), so they are unable to resolve the severe neurological manifestations of many LSDs.8,9 Additionally, in some patients, repeated administration of recombinant LSD enzymes can trigger immune reactions to the enzyme that limit the effectiveness of the therapy.10,11
Despite these limitations, the success of ERTs for LSDs demonstrates that improved treatment of LSDs may be achieved by development of gene therapy approaches. This article will focus on recent developments in the field of gene therapy for the treatment of LSDs, including recent successes, as well limitations that remain to be overcome.
Suitability of LSDs as targets for gene therapy
Gene therapy is based upon a simple concept – namely, a working copy of an appropriate gene is provided to a patient to either stabilize or reverse a clinical disease state. There are several caveats that must be considered when evaluating a gene therapy for a LSD, including: 1) can the existing pathology be reversed or merely slowed in its rate of progression?; 2) can the appropriate tissues or cells affected by the respective LSD be targeted by the respective gene therapy approach?; and 3) will the patient’s immune system perceive the protein produced by the gene therapy treatment as foreign? Despite these caveats, LSDs provide three advantages that allow them to be practical targets for gene therapy. Firstly, each LSD is a single-gene recessive disorder and the pathophysiology of the diseases is relatively well understood. The development of several animal models that mimic the pathophysiology of various LSDs furthers accurate preclinical evaluations of various forms of gene therapy.12,13 Secondly, clinical studies of residual LSD enzyme activities in more mildly affected LSD patients have shown that even small improvements in enzyme activity can be associated with significant impacts on the clinical course of the disease. Studies in both Pompe disease (glycogen storage disease type II [GSDII] knockout [KO])14 and metachromatic leukodystrophy (MLD)15,16 have demonstrated that the most severe infantile-onset forms of the disease correlate with <1% enzyme activity, and that adult onset forms correlate with <10%–15% of normal enzyme activity. A similar pattern has been observed in Fabry disease, Niemann–Pick disease, and Gaucher’s disease.17,18 These results suggest that if gene therapy is able to produce even a relatively small amount of a respective LSD enzyme, it may have a large clinical impact upon the course of the disease. Finally, the most promising feature of LSDs as targets for the successful implementation of gene therapy is the “cross-correction” phenomenon. Lysosomal enzymes are secreted in small amounts to the extracellular space where they can be taken up and targeted to lysosomes of neighboring cells.5 If supraphysiologic amounts of a respective LSD enzyme can be provided via gene therapy, secreted enzymes may enter the bloodstream and be taken up by distant organs, providing clinical benefits analogous or superior to ERT approaches. The most successful gene therapy approaches can exploit this pathway by expressing high concentrations of enzyme from the liver or other target organs and allowing that organ to secrete enzyme for “systemic cross-correction” (Figure 1).
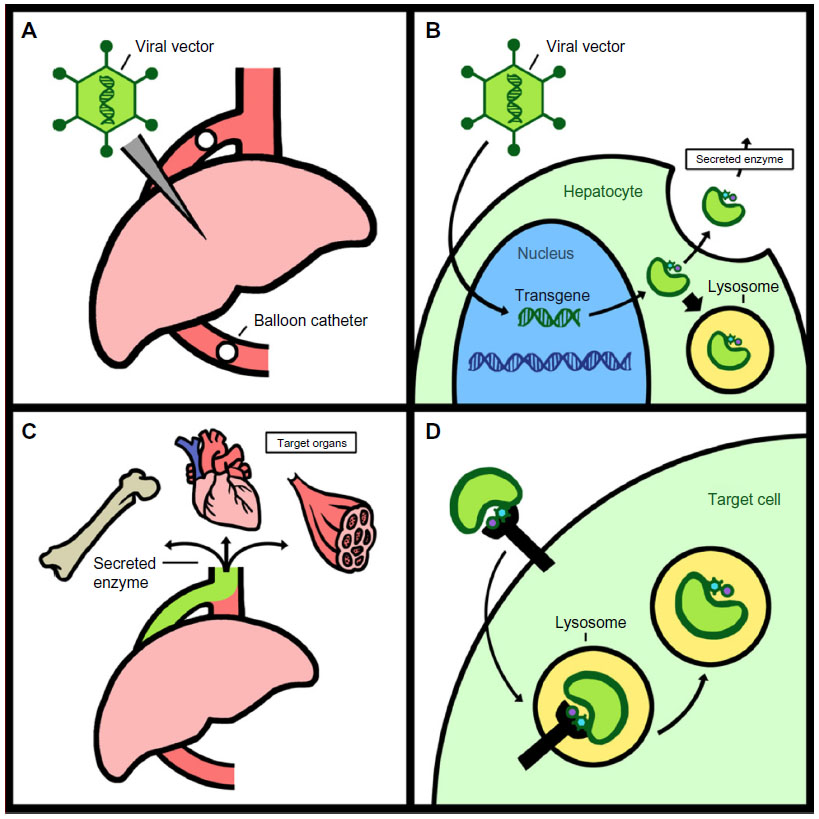 | Figure 1 Systemic cross-correction. |
Gene therapy vectors
There have been many different gene transfer “vectors” produced for the purpose of gene therapy; however, the vast majority of animal studies focus on the use of recombinant, virus-based vectors due to their high probability for allowing robust gene transfer and expression into a variety of cells and tissue types (Figure 2). Under the right circumstances, each of these vectors have demonstrated long-term efficacy in animal models of LSDs, and each are viable candidates for human gene therapy of various LSDs.19–24,31
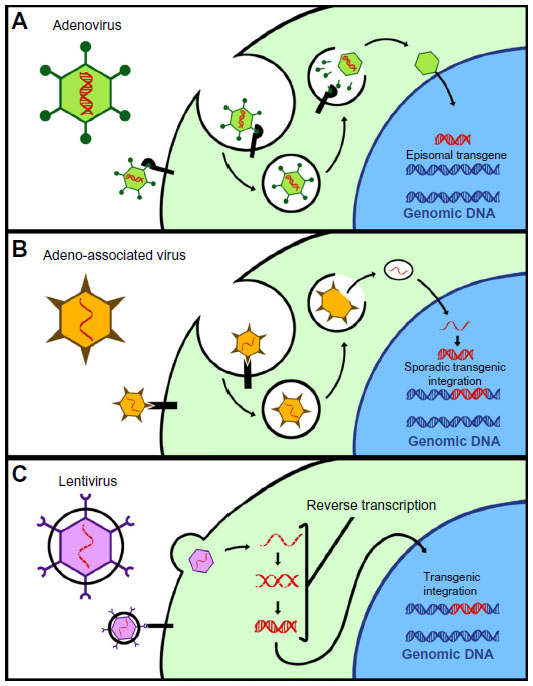 | Figure 2 Commonly used viral vectors. |
Adenovirus
Generally speaking, adenovirus-based gene therapy vectors have been the most widely used vector in human clinical trials to date, with 23.3% of all gene therapy clinical trials utilizing adenoviruses.25 Adenoviruses are double-stranded DNA viruses that do not integrate into the host genome, can infect nonreplicating cells, and can transfer up to 37 kb of genetic information.
Early generation vectors based on adenoviruses were potent activators of the innate immune system, activating TLR9,26 TLR2,26 complement,27 and the NLRP3 inflammasome.28 New strategies have been developed to limit immunity with adenoviruses, including the preemptive administration of medications to prevent innate immune responses to the vector,29 the use of liver targeting veno-occlusive systems diminishing systemic exposure to high doses of recombinant adenovirus vectors,30,31 and the development of advanced generation viruses that lack viral genes, such as helper-dependent adenoviruses (HD-Ads) that lack all viral genes.30–33 HD-Ads have decreased immunity and sustained expression of transgenes in mice and nonhuman primates. Although HD-Ads are still capable of activating innate immunity, as would any virus-based vector,34 the mechanism and degree of innate immune activation can be comparable to nonviral methods for gene therapy.35 Recently, HD-Ads have been demonstrated to persist in nonhuman primates for up to 7 years, demonstrating their capacity for durable expression in immune-tolerant animals.31
Additional advances in adenovirus-based gene transfer vectors include the development of a variety of alternative serotype-based vectors that evade pre-existing immunity, and have other interesting properties. For instance, canine adenovirus serotype 2 has been observed to be less immunogenic than classic adenoviral vectors,36 and to have high tropism for neurons,37 allowing it to be used for the improved treatment of neonatal mucopolysaccharisosis mice.38
Retroviruses and lentivirus
Retroviruses (RVs) and lentiviruses (LVs) are enveloped single-stranded RNA viruses. RVs and LVs integrate directly into the host’s genome. This ability to integrate into the host’s DNA is both a great advantage and a liability. On the one hand, integration allows the genes delivered by such vectors to potentially be permanent, persisting indefinitely within the host cell despite repeated cell division, such as in hematopoietic stem cells (HSCs).39 On the other hand, multiple integrations can also promote insertional mutagenesis. For instance, in gene therapy trials using γ-RVs to treat SCID-X1 immunodeficiency, multiple patients developed T-cell acute lymphoblastic leukemia that was directly attributable to the RV randomly integrating next to, and activating the LMO-2 oncogene.40,41 Recent work has been aimed at improving the safety profile of RVs and LVs by targeting or limiting their ability to randomly integrate, or to include, “transcriptional insulators” that decrease the chance to transcriptionally activate bystander genes.42,43 In gene therapy for LSDs, RVs and LVs are frequently used to transfect cells ex vivo. These cells can then be reinfused into the same patient that they were harvested from, and theoretically supply enzyme to neighboring cells in the body through cross-correction.
Adeno-associated virus
Adeno-associated virus (AAV) is a 4.7 kb nonenveloped single-stranded DNA parvovirus. AAVs infect cells by interacting with specific receptors that differ between serotypes. For this reason, AAV tropism is serotype-dependent.44 Gene transfer vectors based on AAVs are capable of attaining sustained transgene expression in a wide variety of cells. Like adenoviruses and LVs, AAVs can also trigger innate or adaptive immune responses against the vector and the transgenes they express in a variety of settings.45–52 Because they are capable of integration, another risk of AAV-based vectors is insertional mutagenesis and genotoxicity.53,54 A unique feature of AAV vectors is the potential ability of certain serotypes to cross the BBB and enter the central nervous system (CNS), making them attractive vectors for the therapy of neuropathic LSDs.55 For this reason, AAV9 has been used in a variety of attempts to achieve CNS-detected gene therapy for neuropathic LSDs.56,57 The greatest limitation of AAV-based gene transfer vectors may be their potential for scalable production. Recently, a practical alternative method using a cell suspension system for AAV production has been developed.58 This system has allowed for AAV to be produced cost effectively in current good manufacturing practices-compliance and has translated into the first approved gene therapy drug available in Europe, Glybera® (alipogene tiparvovec).59
Advances in gene therapy for LSDs
Over the past few years, many advances have been made in the construction and administration of gene therapy vectors. These advances either improved the safety or increased the potential efficacy of the vectors by overcoming limitations that were experienced in earlier gene therapy attempts.62,66
Overcoming adaptive immune responses
Overcoming the adaptive immune responses (cellular or humoral) to gene transfer is a critical issue for the successful treatment of LSDs in humans. Adaptive immune responses are a major limitation in ERT,10,11 and adaptive immune responses can develop against the viral vector or against the transgene, and they have been reported with adenovirus,60 AAV,45,47,48,50 and LV.61,62 Bypassing adaptive immunity was the main limitation in early attempts to treat LSDs with gene therapy; however, recent advances have allowed for the evasion of immune responses, and even the induction of tolerance, demonstrating an interesting potential advantage of gene therapy for the treatment of LSDs.66,69
These concepts are illustrated well in Pompe disease (GSDII). Pompe disease is caused by mutations in the GAA gene that result in insufficient acid α-glucosidase (GAA) production and the accumulation of glycogen within the lysosomes of affected tissues.1 Infantile-onset Pompe disease presents at <1 year of age with musculoskeletal weakness, cardiomegaly, and progressive respiratory insufficiency. Without treatment, patients can succumb to cardiac or respiratory complications by 9–24 months of age. The current treatment for GSDII is ERT, in which recombinant GAA is administered intravenously once every 2 weeks. For most patients, this therapy is quite efficacious; however, some patients develop potent immune reactions against the enzyme that can significantly limit the long-term efficacy of the therapy.10,11
A similar problem was encountered in the first attempts to treat GSDII with gene therapy in animal models. Early attempts to introduce a functional GAA gene into GSDII KO mice63 and GSDII KO quails64 confirmed that robust levels of enzyme expression could be achieved in vivo, but these levels of expression gradually tapered over time. This problem was ameliorated when similar attempts were made in immune-deficient GSDII KO animals, demonstrating that adaptive immunity was leading to the loss of GAA activities over time.65 Two potentially synergistic strategies have been developed for reducing immunity induced by gene therapy for Pompe disease: one is the development of liver-targeted therapies; and the other is immunosuppression/immunomodulation.
Liver-directed gene therapy
One strategy to avoid both innate and adaptive immune responses is liver-directed gene therapy. To target transgene expression to the liver, and to avoid expression in immune cells, liver-specific promoters have been employed. This has proved effective at minimizing adaptive immunity in GSDII KO mice. Specifically, GSDII KO mice were injected with AAV2/8 vectors expressing GAA driven by either a liver-specific promoter or a ubiquitously expressed promoter derived from the cytomegalovirus early region enhancer and promoter elements.66 GSDII KO mice receiving the vector with the ubiquitously expressed promoter developed antibody and T-cell responses against the transgene that limited the expression of GAA, and ultimately failed to correct the phenotype. In contrast, use of the liver-specific promoter prevented the formation of neutralizing antibodies and cytotoxic T-cells against the GAA transgene in the same animals, and it allowed for sustained plasma secretion of GAA, ultimately correcting glycogen storage and supplying GAA to muscle via systemic cross-correction.66 Some have suggested that liver-specific promoters avoid immunity by preventing transgene expression within antigen-presenting cells.67 While this may be the case, it appears that liver-directed gene therapy is capable of actively inducing tolerance as well.68 This was demonstrated in the same murine model of Pompe disease. In GSDII KO mice, AAV2/8 expressing GAA with a liver-specific promoter avoided anti-GAA immunity when injected on its own; it also allowed for protection from the immune responses induced by the ubiquitously expressed vector, suggesting the induction of tolerance.69 As immune responses are the major limiting factor for ERT, induction of tolerance suggests a role for gene therapy in patients where immune responses to the transgene are predicted to be limiting.8,9 Similarly, liver-directed gene therapy approaches have been successfully employed to treat mucopolysaccharidosis (MPS) type I (MPSI) cats,70 MPSIIA mice,71 GSDII KO mice,72 Von Gierke disease mice,73 and Fabry mice.74,75
Early attempts at liver-directed gene therapy introduced the vector into the hepatic bloodstream, but they required high doses of virus to achieve significant levels of hepatocyte transduction.76 Unfortunately, the doses required also induced dose-related toxicity.77,78 To address these issues, a procedure was developed that utilized minimally invasive, hydrodynamic occlusion of the liver and allowed for substantially higher levels of hepatocyte transduction with substantially lower viral doses to be utilized.79 For LSDs that can be treated with liver-directed therapy, this technique is a major breakthrough and will likely benefit future trials in large animals and humans.
Transient immunosuppression/immunomodulation
A second method for avoiding immunity is the use of transient immunosuppression. This method has been explored in a GSDII KO mouse model.80 In that model, AAV expressing GAA with a ubiquitous promoter generated potent humoral immunity that limited the efficacy of the gene therapy.80 However, pretreatment of the animals with nondepleting anti-CD4 antibodies transiently inhibited the function of CD4+ T-cells and prevented the formation of anti-GAA antibodies. This greatly improved the efficacy of the gene therapy attempt, resulting in decreased glycogen storage in the heart and skeletal muscles of the treated animals.80
The importance of transient immunosuppression has also been observed in the gene therapy of MPSI cats. MPSI is a neuropathic LSD that presents with musculoskeletal malformation, severe cognitive declines, joint pain, and hepatosplenomegaly. When a retroviral vector was used in a feline model of MPSI, potent cytotoxic T-lymphocyte (CTL) responses eliminated transfected cells and prevented long-term efficacy of the therapy.62 In this same model, CTL responses could be avoided by transiently suppressing the animal’s immune system by treatments with the use of the T-cell coreceptor-binding drug, CTLA4-Ig. This resulted in the sustained efficacy and subsequent normalization of substrate accumulations within the liver.62 Transient immunosuppression has also been shown to block downstream antibody responses to adenoviral vectors, allowing for their repeated administration.81,82 This approach has also shown success in a nonhuman primate model with AAV.83 Importantly, transient immunosuppression has been demonstrated to be safe in a human clinical trial for MPSIIIA, suggesting a future for this practice in human gene therapy approaches for many LSDs.84
Gene therapy for neuropathic LSDs
There is mounting evidence for the potential use of gene therapy to treat neuropathic LSDs. Over 50% of LSDs have neurologic involvement,1 and gene therapy is particularly important for this class of LSDs, as currently no therapies exist for many of these diseases. The principal obstacle in treating these diseases is the BBB. ERT is ineffective at crossing the BBB, and most viral vectors (adenoviruses, LVs, and most AAVs) will not cross the fully matured BBB after intravenous administration.8,9,85,86 This obstacle has led to a number of promising innovations that are each at different stages of translation.
Intracranial injections
Several strategies have been developed to bypass the BBB for the treatment of LSDs (Figure 3). The most direct and developed strategy has been to anatomically bypass the BBB with intracranial injections of the respective gene transfer vector. This approach has been utilized extensively in models of infantile neuronal ceroid lipofuscinoses (INCL) and MPSIIIA.84,92 Both are fatal neuropathic LSDs that develop in early childhood. INCL is caused by mutations in the PPT1 (also called CLN2) gene, resulting in deficiency of palmitoyl protein thioesterase-1 (PPT1). INCL presents at around 18 months with symptoms of visual defects, cognitive defects, seizures, and results in premature death.87 MPSIIIA is caused by mutations in the SGSH gene that result in the deficiency of N-sulfoglucosamine sulfohydrolase (SGSH) and the accumulation of heparin sulfate glycosaminoglycans within the CNS. MSP IIIA patients present with cognitive delay at age 3 years, lose the ability to walk independently by age 10 years, and die by age 15 years.88 There are currently no therapies approved for either of these fatal disorders.
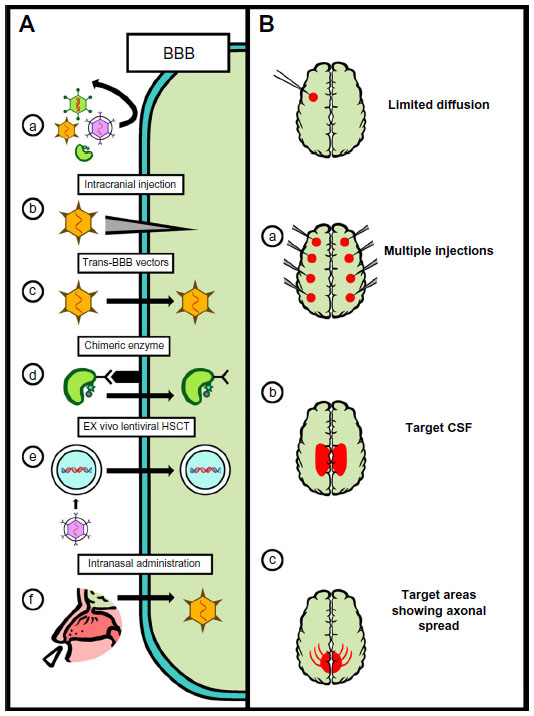 | Figure 3 Strategies for CNS-directed gene therapy. |
Preclinical studies for INCL suggested that intracranial injections of AAV2 expressing PPT1 cleared accumulated storage material from PPT1 KO mice.89 The same vector was shown to broadly transduce the brain of nonhuman primates.91 The vector successfully produced tripetidyl peptidase (TTP)-1 within the neurons of the injected regions, and although the injections themselves caused minor physical damage, no histological damage could be attributed to the vector.90,91 This work was translated into the first gene therapy trial for a LSD.92 Ten children with INCL received 12 intracranial injections of an AAV2 vector expressing TTP-1. A modified Hamburg neurologic rating scale demonstrated that the subjects that received gene therapy exhibited a slower cognitive decline compared to untreated controls and historical data. Unfortunately, it was also demonstrated that humoral immune responses developed in four of the ten subjects, which may have limited the overall effectiveness of the therapy. Similar humoral immunity developed in a clinical study of Canavan disease following intracranial administration of AAV2 vectors expressing aspartoacylase.51 These early studies demonstrated that gene therapy could be potentially effective for neurological disorders, and they also highlighted the importance of the immune system, even within immune-privileged sites like the CNS.
Lessons learned from this trial were incorporated into a clinical trial in MPSIIIA that employed transient immunosuppression alongside intracranial injections.84 Four MPSIIIA patients aged 2–6 years were injected intracranially with an AAV2/10 vector expressing SGSH and sulfatase modifying factor 1. They received 12 simultaneous intracranial injections into the white matter of the basal ganglia. Importantly, patients in this study received immunosuppressive agents (mycophenolate, mofetil, and tacrolimus) beginning 15 days prior to surgery and extending for 8 weeks after the procedure. No adverse outcomes were associated with immunosuppression or administration of the viral vector, demonstrating a markedly improved safety profile over the INCL trial.92 Though the study design limits conclusions about efficacy, the patients may have had a decreased decline in cognitive abilities, and the youngest patient showed improvement in several cognitive scores, including motor skills and independent thought. The behavioral data correlated with magnetic resonance imaging data that showed decreased brain atrophy in two patients and no atrophy in the other two patients. These promising results suggest that intracranial gene therapy may be a viable option for certain neuropathic LSDs.
Trans-BBB vectors for neuropathic LSDs
Another strategy to bypass the BBB is to use alternative serotypes of viruses that may be capable of enhanced entry into the CNS directly from the bloodstream. Early work demonstrated that AAV955 and other AAV serotypes93 crossed the BBB, as performed in neonatal mice. However, the large injection volumes that were used in these studies potentially could have mechanically disrupted the delicate intracerebral vasculature of the neonatal mice, complicating interpretation of these results.94 Subsequent work demonstrated that AAV9 was capable of crossing the BBB in adult nonhuman primates,95 and this approach has been explored as an alternative to intracranial injections in models of MPS. In MPSIIIB mice, intravenous administration of AAV9 was able to correct the pathology in the CNS and the periphery.56 As a preclinical study, the same vector was administered intravenously into adult cynomolgus monkeys; the use of AAV9 was able to drive the expression of α-N-acetylglucosaminidase in both the CNS and the periphery, demonstrating that this approach may be viable for a clinical trial.96
In addition to AAV9, other AAV vectors have been investigated for their ability to cross the BBB. Serotypes AAVrh8 and AAVrh10 were shown to cross the BBB in adult mice, with AAVrh10 also shown to cross the BBB in adult marmosets, although with variable results.97 Another approach has involved identifying new xenobiotic AAVs that may be capable of crossing the BBB. Two recently isolated porcine AAVs were demonstrated to have some tropism for the CNS.98 A third approach has been to artificially manipulate the AAV capsid to produce novel AAV vectors that cross the BBB more efficiently than AAV9.99 This approach was demonstrated in a murine model of MPSVII. Importantly, MPSVII is a difficult target for AAV9-based gene therapy because the disease results in an accumulation of sialic acid within the CNS; sialic acid generally inhibits the AAV9 transduction of cells. In this model, the capsid-modified AAV9 vectors were able to overcome these issues, enter the CNS, and correct the cognitive deficits and storage lesions.99
Ex vivo gene therapy for neuropathic LSDs
Another strategy to bypass the BBB is the use of HSCs or other cells naturally capable of crossing the BBB on their own. These cells can be harvested from the patient, modified with RV or LV ex vivo, and then returned to the patient, where they can theoretically cross the BBB and provide enzymes to neighboring cells within the CNS via cross-correction. This approach has been explored in MLD models.103 MLD is a neuropathic LSD caused by a deficiency in arylsulfatase A (ARSA). Patients with <1% ARSA activity present at 1–2 years of age with progressive muscle weakness, vision loss, convulsions, and dementia, and they typically die by the age of 5 years.15,16 There is currently no therapy approved for this fatal disorder.
Experiments in a MLD mouse model have demonstrated that HSC gene therapy can succeed in correcting lysosomal storage and neurologic damage in affected mice.100 Interestingly, this model also highlighted the importance of gene therapy for LSDs, as only HSCs that received gene therapy were able to prevent neurologic deficits.101 This is likely because genetically modified HSCs produce up 15 times the normal amount of the required LSD protein, and therefore were able to secrete more enzyme per cell for cross-correction.102 This work has been translated into a Phase I/II clinical trial for MLD where three presymptomatic MLD patients were treated with HSC gene therapy.103 Autologous HSCs were harvested from the patients, transduced ex vivo with a LV expressing ARSA, and the transduced cells were infused into the patients that had also undergone preconditioning of their bone marrow via myeloablation with busulfan. The reinfused cells engrafted within the patients, maintaining a high-level of detectable ARSA activity within a wide variety of cells and the cerebrospinal fluid. The therapy forestalled the onset of cognitive deficits and lessened the motor deficits in all three patients, as compared to untreated older siblings with MLD and historical controls.103 Importantly, no adverse outcomes were associated with the administration of the vector, and no antibodies developed against the vector or the transgene. The safety and efficacy of this trial demonstrated the potential of ex vivo LV gene therapy for the treatment of LSDs.
In addition to LVs, nonviral methods for modifying cells ex vivo are in development, and the use of these methods for the treatment of LSDs has recently been patented.104 These methods include the potential use of zinc-finger nucleases,105 transcriptional activator-like effector nucleases,106 and systems using clustered regularly interspaced short palindromic repeat-CRISPR-associated protein 9 (CRISPR-Cas9)107 in attempts to insert potentially therapeutic transgenes into a locus of choice. These can also potentially be used in combination with integrase-deficient LVs to allow LVs to achieve targeted transgene insertion.108 Their potential applications in LSDs has been recently reviewed, and readers are directed there for more details.109
Trans-BBB enzyme for neuropathic LSDs
Another approach to supply the brain with the deficient enzyme is to modify the gene coding the enzyme so that the enzyme produced by gene therapy in the periphery is able to cross the BBB itself. For example the lysosomal enzyme ARSA was fused to several of these targeting motifs, including the Tat domain from HIV, the angiopep peptide, and the receptor-binding domains from human apolipoproteins B and E. The biodistribution of these fusion proteins was altered by the presence of the additional ligand motifs, demonstrating the potential for trans-BBB enzyme therapy.110 Recent work has also shown that lysosomal enzymes modified with ligand motifs that allow them to bind the human insulin receptor are better able to cross the BBB and distribute throughout the brain in rhesus monkeys.111 This approach has also been utilized in gene therapy for MPSIIIA mice.71 A liver-directed AAV8-based gene therapy vector produced a LSD protein that had been modified to include the secretion signal from iduronate-2-sulfatase to enhance secretion and the BBB-binding domain (BBBBD) from apolipoprotein B to cross the BBB. The BBBBD from apolipoprotein B is recognized by low-density lipoprotein receptors and it allows the enzyme to be trafficked into the brain. A single intravenous injection of the AAV2/8 carrying the modified SGSH gene was better able to foster the detection of SGSH activity throughout the periphery and the CNS of the animals treated with this version of the SGSH gene.71 MPSIIIA mice that received this treatment also showed decreased brain pathology and recovered normal behavior. This novel approach combines the advantages of liver-directed gene therapy with the ability to improve targeting of LSD enzymes into the CNS.
Intranasal delivery
There have also been attempts to introduce viral vectors to the brain via intranasal delivery. Early work in rats demonstrated that adenovirus could enter the CNS and transduce the β-galactosidase gene into the rat brain following intranasal delivery.112 This delivery system was also used with AAV8 to introduce a physiologically relevant levels of α-l-iduronidase (IDUA) into a mouse model of MPSI (IDUA–/– mice).113 Due to the substantial anatomical differences between the nasal cavity of rodents and humans,114,115 this approach likely needs to be evaluated in primates to confirm its potential for high levels of efficacy in human populations.
Current limitations in gene therapy for LSDs
Despite the many recent achievements in the gene therapy for LSDs, there remain several obstacles to overcome. A major obstacle that many gene therapy vectors face is pre-existing immunity against the vector itself, which can be a major limitation when translating these approaches to the actual human population, especially with use of AAV.116,117 Although great promise exists for the use of AAV-based therapies, most of the human population also have pre-existing humoral immunity to AAV serotypes, with 72% of the population having antibodies against AAV2, 67% against AAV1, 47% against AAV9, 46% against AAV6, 40% against AAV5, and 38% against AAV8.116 Furthermore, under certain conditions, even low titers of anti-AAV antibodies have been shown to completely block AAV gene therapy attempts.42,118,119 Additionally, anti-AAV capsid antibodies have been shown to cross-react unpredictably across AAV serotypes, so antibodies against one AAV serotype may prevent any other AAV from functioning as a vector.116,120 Finally, in certain circumstances, preformed neutralizing AAV antibodies have been shown to partially decrease transgene expression, even when injected into immune-privileged sites, such as the brain121 or eye.122 Current clinical trials circumvent this issue by carefully excluding research participants harboring pre-existing anti-AAV antibodies; however, this can exclude 72%–33% of the patient population from treatment.116,123 Groups have proposed using empty capsids as decoys for antibodies (thereby decreasing the ratio of antibody to target) with variable success.124,125
Another limitation in the development and translation of gene therapy for LSDs is the animal models. While the animal models for LSDs are among some of the best for any disease (being true homologues), interspecies differences have still proven relevant when translating from one species to another. In addition to the limitations mentioned earlier, differences in the immune responses between animal models have demonstrated that immune responses to gene therapy vectors can vary between species. For instance, when neonatal MPSI dogs received retroviral gene therapy, they did not mount an immune response to the protein expressed by the transduced transgene (α-l-iduronidase),126 despite the ability of adult MPSI dogs to develop a potent antibody response to the same protein directly.127,128 However, when a very similar vector was administered to neonatal MPSI cats, the cats developed a very strong T-cell-mediated response against the protein encoded by the vector that rapidly eliminated transfected cells.62 The critical importance of these differences was illustrated by the clinical Phase I/II study of an AAV2-based vector expressing factor IX. In preclinical trials, hemophilia B dogs showed no evidence of potent immune responses to the vector;129,130 however, human trial participants experienced potent T-cell responses that eliminated AAV-transfected cells.45,50 The differences in the immune responses between species are an inherent limitation as to the use of animal models; this is a caveat that must be remembered when translating any therapy from an animal model to human subjects.
Expert opinion
Of the numerous human genetic diseases, LSDs likely provide the highest chance for the clinical success of gene therapy strategies given contemporary understandings of the limitations of gene therapy approaches. Currently, gene therapy clinical trials are underway for Fabry disease, Gaucher disease, MLD, MPSII, MPSIIIA, MPSIIIB, INCL, and Pompe disease.131 In addition, preclinical research has demonstrated special advantages that gene therapy might have for use in the treatment of LSDs. The ability of liver-directed gene therapy to produce robust amounts of LSD enzymes systemically, as well as to potentially induce tolerance, is highly promising, especially for the subset of patients who develop potent immune responses to ERT.8 In addition, many of the neuropathic LSDs currently have no treatments, and the recent clinical trials demonstrate that gene therapy may provide a safe and potentially therapeutic option for these diseases. Taken together, these exciting advances show that gene therapy for LSDs is overcoming previous limitations and moving forward on several fronts. As these therapies are translated into clinical practice, the role of administering clinical gene therapy may reside firmly in the venue of the metabolic geneticist or other clinician that specializes in the treatment of LSDs.
Disclosure
The authors report no conflicts of interest in this work.
References
Futerman AH, van Meer G. The cell biology of lysosomal storage disorders. Nat Rev Mol Cell Biol. 2004;5(7):554–565. | |
Staretz-Chacham O, Lang TC, LaMarca ME, Krasnewich D, Sidransky E. Lysosomal storage disorders in the newborn. Pediatrics. 2009;123(4):1191–1207. | |
Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281(3):249–254. | |
Platt FM, Boland B, van der Spoel AC. The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J Cell Biol. 2012;199(5):723–734. | |
Fratantoni JC, Hall CW, Neufeld EF. Hurler and Hunter syndromes: mutual correction of the defect in cultured fibroblasts. Science. 1968; 162(3853):570–572. | |
Eng CM, Banikazemi M, Gordon RE, et al. A phase 1/2 clinical trial of enzyme replacement in fabry disease: pharmacokinetic, substrate clearance, and safety studies. Am J Hum Genet. 2001;68(3):711–722. | |
Amalfitano A, Bengur AR, Morse RP, et al. Recombinant human acid alpha-glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial. Genet Med. 2001;3(2):132–138. | |
Muenzer J, Bodamer O, Burton B, Clarke L, Frenking GS et al. The role of enzyme replacement therapy in severe Hunter syndrome—an expert panel consensus. Eur J Pediatr. 2012;171(1):181–188. | |
Sands MS. A Hitchhiker’s Guide to the Blood–brain Barrier: In Trans Delivery of a Therapeutic Enzyme. Mol Ther. 2014;22(3):483–484. | |
Kishnani PS, Goldenberg PC, DeArmey SL, et al. Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants. Mol Genet Metab. 2010;99(1):26–33. | |
Banugaria SG, Patel TT, Mackey J, et al. Persistence of high sustained antibodies to enzyme replacement therapy despite extensive immunomodulatory therapy in an infant with Pompe disease: need for agents to target antibody-secreting plasma cells. Mol Genet Metab. 2012;105(4):677–680. | |
Bradbury AM, Gurda BL, Casal ML, Ponder KP, Vite CH, Haskins ME. A review of gene therapy in canine and feline models of lysosomal storage disorders. Hum Gene Ther Clin Dev. 2015;26(1):27–37. | |
Haskins ME, Giger U, Patterson DF. Chapter 6. Animal models of lysosomal storage diseases: their development and clinical relevance. In: Mehta A, Beck M, Sunder-Plassmann G, editors. Fabry Disease: Perspectives from 5 Years of FOS. Oxford, UK: Oxford PharmaGenesis; 2006. Available from: http://www.ncbi.nlm.nih.gov/books/NBK11578/. Accessed February 11, 2015. | |
Umapathysivam K, Hopwood JJ, Meikle PJ. Correlation of acid alpha-glucosidase and glycogen content in skin fibroblasts with age of onset in Pompe disease. Clin Chim Acta. 2005;361(1–2):191–198. | |
Leinekugel P, Michel S, Conzelmann E, Sandhoff K. Quantitative correlation between the residual activity of beta-hexosaminidase A and arylsulfatase A and the severity of the resulting lysosomal storage disease. Hum Genet. 1992;88(5):513–523. | |
Polten A, Fluharty AL, Fluharty CB, Kappler J, von Figura K, Gieselmann V. Molecular basis of different forms of metachromatic leukodystrophy. N Engl J Med. 1991;324(1):18–22. | |
Mechtler TP, Stary S, Metz TF, et al. Neonatal screening for lysosomal storage disorders: feasibility and incidence from a nationwide study in Austria. Lancet. 2012;379(9813):335–341. | |
Spada M, Pagliardini S, Yasuda M, et al. High incidence of later-onset fabry disease revealed by newborn screening. Am J Hum Genet. 2006;79(1):31–40. | |
Brunetti-Pierri N, Ng T, Iannitti D, Cioffi W, Stapleton G, Law M, et al. Transgene expression up to 7 years in nonhuman primates following hepatic transduction with helper-dependent adenoviral vectors. Hum Gene Ther. 2013;24(8):761–765. | |
Sabatino DE, Lange AM, Altynova ES, Sarkar R, Zhou S, et al. Efficacy and safety of long-term Pprophylaxis in severe hemophilia A dogs following liver gene therapy using AAV vectors. Mol Ther. 2011;19(3):442–449. | |
Kim EY, Hong YB, Lai Z, Cho YH, Brady RO, Jung SC. Long-term expression of the human glucocerebrosidase gene in vivo after transplantation of bone-marrow-derived cells transformed with a lentivirus vector. J Gene Med. 2005;7(7):878–887. | |
Miyake N, Miyake K, Asakawa N, Yamamoto M, Shimada T. Long-term correction of biochemical and neurological abnormalities in MLD mice model by neonatal systemic injection of an AAV serotype 9 vector. Gene Ther. 2014;21(4):427–433. | |
Seregin SS, Amalfitano A. Gene therapy for lysosomal storage diseases: progress, challenges and future prospects. Curr Pharm Des. 2011;17(24):2558–2574. | |
Kamata Y, Tanabe A, Kanaji A, Kosuga M, Fukuhara Y, et al. Long-term normalization in the central nervous system, ocular manifestations, and skeletal deformities by a single systemic adenovirus injection into neonatal mice with mucopolysaccharidosis VII. Gene Ther. 2003;10(5):406–414. | |
Ginn SL, Alexander IE, Edelstein ML, Abedi MR, Wixon J. Gene therapy clinical trials worldwide to 2012 – an update. J Gene Med. 2013;15(2):65–77. | |
Appledorn DM, Patial S, McBride A, et al. Adenovirus vector-induced innate inflammatory mediators, MAPK signaling, as well as adaptive immune responses are dependent upon both TLR2 and TLR9 in vivo. J Immunol. 2008;181(3):2134–2144. | |
Tian J, Xu Z, Smith JS, Hofherr SE, Barry MA, Byrnes AP. Adenovirus activates complement by distinctly different mechanisms in vitro and in vivo: indirect complement activation by virions in vivo. J Virol. 2009; 83(11):5648–5658. | |
Muruve DA, Pétrilli V, Zaiss AK, et al. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature. 2008;452(7183):103–107. | |
Seregin SS, Appledorn DM, McBride AJ, et al. Transient pretreatment with glucocorticoid ablates innate toxicity of systemically delivered adenoviral vectors without reducing efficacy. Mol Ther. 2009;17(4):685–696. | |
Oka K, Mullins CE, Kushwaha RS, Leen AM, Chan L. Gene therapy for rhesus monkeys heterozygous for LDL receptor deficiency by balloon catheter hepatic delivery of helper-dependent adenoviral vector. Gene Ther. 2015;22(1):87–95. | |
Brunetti-Pierri N, Ng T, Iannitti D, et al. Transgene expression up to 7 years in nonhuman primates following hepatic transduction with helper-dependent adenoviral vectors. Hum Gene Ther. 2013;24(8):761–765. | |
Brunetti-Pierri N, Ng P. Helper-dependent adenoviral vectors for liver-directed gene therapy. Hum Mol Genet. 2011;20(R1):R7–R13. | |
Palmer DJ, Ng P. Helper-dependent adenoviral vectors for gene therapy. Hum Gene Ther. 2005;16(1):1–16. | |
Cerullo V, Seiler MP, Mane V, et al. Toll-like receptor 9 triggers an innate immune response to helper-dependent adenoviral vectors. Mol Ther. 2007;15(2):378–385. | |
Zhao H, Hemmi H, Akira S, Cheng SH, Scheule RK, Yew NS. Contribution of Toll-like receptor 9 signaling to the acute inflammatory response to nonviral vectors. Mol Ther. 2004;9(2):241–248. | |
Keriel A, René C, Galer C, Zabner J, Kremer EJ. Canine adenovirus vectors for lung-directed gene transfer: efficacy, immune response, and duration of transgene expression using helper-dependent vectors. J Virol. 2006;80(3):1487–1496. | |
Soudais C, Laplace-Builhe C, Kissa K, Kremer EJ. Preferential transduction of neurons by canine adenovirus vectors and their efficient retrograde transport in vivo. FASEB J. 2001;15(12):2283–2285. | |
Ariza L, Giménez-Llort L, Cubizolle A, et al. Central nervous system delivery of helper-dependent canine adenovirus corrects neuropathology and behavior in mucopolysaccharidosis type VII mice. Hum Gene Ther. 2014;25(3):199–211. | |
Aiuti A, Biasco L, Scaramuzza S, et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013;341(6148):1233151. | |
Hacein-Bey-Abina S, Von Kalle C, Schmidt M, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302(5644):415–419. | |
McCormack MP, Rabbitts TH. Activation of the T-cell oncogene LMO2 after gene therapy for X-linked severe combined immunodeficiency. N Engl J Med. 2004;350(9):913–922. | |
Schambach A. Toward a safer integration profile of MLV-based retroviral vectors. Mol Ther. 2014;22(8):1405–1406. | |
Zhou S, Mody D, DeRavin SS, et al. A self-inactivating lentiviral vector for SCID-X1 gene therapy that does not activate LMO2 expression in human T cells. Blood. 2010;116(6):900–908. | |
Zincarelli C, Soltys S, Rengo G, Rabinowitz JE. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol Ther. 2008;16(6):1073–1080. | |
Mingozzi F, Maus MV, Hui DJ, et al. CD8(+) T-cell responses to adeno-associated virus capsid in humans. Nat Med. 2007;13(4):419–422. | |
Zhu J, Huang X, Yang Y. The TLR9-MyD88 pathway is critical for adaptive immune responses to adeno-associated virus gene therapy vectors in mice. J Clin Invest. 2009;119(8):2388–2398. | |
Mingozzi F, Meulenberg JJ, Hui DJ, et al. AAV-1-mediated gene transfer to skeletal muscle in humans results in dose-dependent activation of capsid-specific T cells. Blood. 2009;114(10):2077–2086. | |
Vandenberghe LH, Wang L, Somanathan S, et al. Heparin binding directs activation of T cells against adeno-associated virus serotype 2 capsid. Nat Med. 2006;12(8):967–971. | |
Wang L, Dobrzynski E, Schlachterman A, Cao O, Herzog RW. Systemic protein delivery by muscle-gene transfer is limited by a local immune response. Blood. 2005;105(11):4226–4234. | |
Manno CS, Pierce GF, Arruda VR, et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med. 2006;12(3):342–347. | |
McPhee SW, Janson CG, Li C, et al. Immune responses to AAV in a phase I study for Canavan disease. J Gene Med. 2006;8(5):577–588. | |
Nathwani AC, Tuddenham EG, Rangarajan S, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. 2011;365(25):2357–2365. | |
Donsante A, Miller DG, Li Y, Vogler C, Brunt EM, et al. AAV vector integration sites in mouse hepatocellular carcinoma. Science. 2007;317(5837):477. | |
Chandler RJ, LaFave MC, Varshney GK, Trivedi NS, Carrillo-Carrasco N, et al. Vector design influences hepatic genotoxicity after adeno-associated virus gene therapy. J Clin Invest. 2015;125(2):870–880. | |
Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27(1):59–65. | |
Fu H, Dirosario J, Killedar S, Zaraspe K, McCarty DM. Correction of neurological disease of mucopolysaccharidosis IIIB in adult mice by rAAV9 trans-blood-brain barrier gene delivery. Mol Ther. 2011;19(6):1025–1033. | |
Ruzo A, Marcó S, García M, et al. Correction of pathological accumulation of glycosaminoglycans in central nervous system and peripheral tissues of MPSIIIA mice through systemic AAV9 gene transfer. Hum Gene Ther. 2012;23(12):1237–1246. | |
Cecchini S, Virag T, Kotin RM. Reproducible high yields of recombinant adeno-associated virus produced using invertebrate cells in 0.02- to 200-liter cultures. Hum Gene Ther. 2011;22(8):1021–1030. | |
Ylä-Herttuala S. Endgame: glybera finally recommended for approval as the first gene therapy drug in the European Union. Mol Ther. 2012;20(10):1831–1832. | |
Tripathy SK, Black HB, Goldwasser E, Leiden JM. Immune responses to transgene-encoded proteins limit the stability of gene expression after injection of replication-defective adenovirus vectors. Nat Med. 1996;2(5):545–550. | |
Limberis MP, Bell CL, Heath J, Wilson JM. Activation of transgene-specific T cells following lentivirus-mediated gene delivery to mouse lung. Mol Ther. 2010;18(1):143–150. | |
Ponder KP, Wang B, Wang P, et al. Mucopolysaccharidosis I cats mount a cytotoxic T lymphocyte response after neonatal gene therapy that can be blocked with CTLA4-Ig. Mol Ther. 2006;14(1):5–13. | |
Ding EY, Hodges BL, Hu H, et al. Long-term efficacy after [E1-, polymerase-] adenovirus-mediated transfer of human acid-alpha-glucosidase gene into glycogen storage disease type II knockout mice. Hum Gene Ther. 2001;12(8):955–965. | |
McVie-Wylie AJ, Ding EY, Lawson T, et al. Multiple muscles in the AMD quail can be “cross-corrected” of pathologic glycogen accumulation after intravenous injection of an [E1-, polymerase-] adenovirus vector encoding human acid-alpha-glucosidase. J Gene Med. 2003;5(5):399–406. | |
Xu F, Ding E, Liao SX, et al. Improved efficacy of gene therapy approaches for Pompe disease using a new, immune-deficient GSD-II mouse model. Gene Ther. 2004;11(21):1590–1598. | |
Franco LM, Sun B, Yang X, et al. Evasion of immune responses to introduced human acid alpha-glucosidase by liver-restricted expression in glycogen storage disease type II. Mol Ther. 2005;12(5):876–884. | |
Weeratna RD, Wu T, Efler SM, Zhang L, Davis HL. Designing gene therapy vectors: avoiding immune responses by using tissue-specific promoters. Gene Ther. 2001;8(24):1872–1878. | |
LoDuca PA, Hoffman BE, Herzog RW. Hepatic gene transfer as a means of tolerance induction to transgene products. Curr Gene Ther. 2009; 9(2):104–114. | |
Zhang P, Sun B, Osada T, et al. Immunodominant liver-specific expression suppresses transgene-directed immune responses in murine pompe disease. Hum Gene Ther. 2012;23(5):460–472. | |
Hinderer C, Bell P, Gurda BL, et al. Liver-directed gene therapy corrects cardiovascular lesions in feline mucopolysaccharidosis type I. Proc Natl Acad Sci U S A. 2014;111(41):14894–14899. | |
Sorrentino NC, D’Orsi L, Sambri I, et al. A highly secreted sulphamidase engineered to cross the blood-brain barrier corrects brain lesions of mice with mucopolysaccharidoses type IIIA. EMBO Mol Med. 2013;5(5):675–690. | |
Kiang A, Hartman ZC, Liao S, et al. Fully deleted adenovirus persistently expressing GAA accomplishes long-term skeletal muscle glycogen correction in tolerant and nontolerant GSD-II mice. Mol Ther. 2006;13(1):127–134. | |
Koeberl DD, Sun B, Bird A, Chen YT, Oka K, Chan L. Efficacy of helper-dependent adenovirus vector-mediated gene therapy in murine glycogen storage disease type Ia. Mol Ther. 2007;15(7):1253–1258. | |
Ziegler RJ, Lonning SM, Armentano D, et al. AAV2 vector harboring a liver-restricted promoter facilitates sustained expression of therapeutic levels of alpha-galactosidase A and the induction of immune tolerance in Fabry mice. Mol Ther. 2004;9(2):231–240. | |
Ziegler RJ, Cherry M, Barbon CM, et al. Correction of the biochemical and functional deficits in fabry mice following AAV8-mediated hepatic expression of alpha-galactosidase A. Mol Ther. 2007;15(3):492–500. | |
Descamps D, Benihoud K. Two key challenges for effective adenovirus-mediated liver gene therapy: innate immune responses and hepatocyte-specific transduction. Curr Gene Ther. 2009;9(2):115–127. | |
Nunes FA, Furth EE, Wilson JM, Raper SE. Gene transfer into the liver of nonhuman primates with E1-deleted recombinant adenoviral vectors: safety of readministration. Hum Gene Ther. 1999;10(15):2515–2526. | |
Morral N, O’Neal WK, Rice K, et al. Lethal toxicity, severe endothelial injury, and a threshold effect with high doses of an adenoviral vector in baboons. Hum Gene Ther. 2002;13(1):143–154. | |
Brunetti-Pierri N, Stapleton GE, Law M, et al. Efficient, long-term hepatic gene transfer using clinically relevant HDAd doses by balloon occlusion catheter delivery in nonhuman primates. Mol Ther. 2009; 17(2):327–333. | |
Han SO, Li S, Brooks ED, et al. Enhanced efficacy from gene therapy in Pompe disease using coreceptor blockade. Hum Gene Ther. 2015;26(1):26–35. | |
Fontanellas A, Hervás-Stubbs S, Mauleón I, et al. Intensive pharmacological immunosuppression allows for repetitive liver gene transfer with recombinant adenovirus in nonhuman primates. Mol Ther. 2010;18(4):754–765. | |
Smith TA, White BD, Gardner JM, Kaleko M, McClelland A. Transient immunosuppression permits successful repetitive intravenous administration of an adenovirus vector. Gene Ther. 1996;3(6):496–502. | |
Jiang H, Couto LB, Patarroyo-White S, et al. Effects of transient immunosuppression on adenoassociated, virus-mediated, liver-directed gene transfer in rhesus macaques and implications for human gene therapy. Blood. 2006;108(10):3321–3328. | |
Tardieu M, Zérah M, Husson B, et al. Intracerebral administration of adeno-associated viral vector serotype rh.10 carrying human SGSH and SUMF1 cDNAs in children with mucopolysaccharidosis type IIIA disease: results of a phase I/II trial. Hum Gene Ther. 2014;25(6):506–516. | |
Pardridge WM. Drug and gene targeting to the brain with molecular Trojan horses. Nat Rev Drug Discov. 2002;1(2):131–139. | |
Abbott NJ, Rönnbäck L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7(1):41–53. | |
Hawkins-Salsbury JA, Cooper JD, Sands MS. Pathogenesis and therapies for infantile neuronal ceroid lipofuscinosis (infantile CLN1 disease). Biochim Biophys Acta. 2013;1832(11):1906–1909. | |
Valstar MJ, Neijs S, Bruggenwirth HT, et al. Mucopolysaccharidosis type IIIA: clinical spectrum and genotype-phenotype correlations. Ann Neurol. 2010;68(6):876–887. | |
Passini MA, Dodge JC, Bu J, et al. Intracranial delivery of CLN2 reduces brain pathology in a mouse model of classical late infantile neuronal ceroid lipofuscinosis. J Neurosci. 2006;26(5):1334–1342. | |
Hackett NR, Redmond DE, Sondhi D, et al. Safety of direct administration of AAV2(CU)hCLN2, a candidate treatment for the central nervous system manifestations of late infantile neuronal ceroid lipofuscinosis, to the brain of rats and nonhuman primates. Hum Gene Ther. 2005;16(12):1484–1503. | |
Sondhi D, Peterson DA, Giannaris EL, et al. AAV2-mediated CLN2 gene transfer to rodent and non-human primate brain results in long-term TPP-I expression compatible with therapy for LINCL. Gene Ther. 2005;12(22):1618–1632. | |
Worgall S, Sondhi D, Hackett NR, et al. Treatment of late infantile neuronal ceroid lipofuscinosis by CNS administration of a serotype 2 adeno-associated virus expressing CLN2 cDNA. Hum Gene Ther. 2008;19(5):463–474. | |
Zhang H, Yang B, Mu X, et al. Several rAAV vectors efficiently cross the blood-brain barrier and transduce neurons and astrocytes in the neonatal mouse central nervous system. Mol Ther. 2011;19(8):1440–1448. | |
Saunders NR, Joakim Ek C, Dziegielewska KM. The neonatal blood-brain barrier is functionally effective, and immaturity does not explain differential targeting of AAV9. Nat Biotechnol. 2009;27(9):804–805; author reply 805. | |
Gray SJ, Matagne V, Bachaboina L, Yadav S, Ojeda SR, Samulski RJ. Preclinical differences of intravascular AAV9 delivery to neurons and glia: a comparative study of adult mice and nonhuman primates. Mol Ther. 2011;19(6):1058–1069. | |
Murrey DA, Naughton BJ, Duncan FJ, et al. Feasibility and safety of systemic rAAV9-hNAGLU delivery for treating mucopolysaccharidosis IIIB: toxicology, biodistribution, and immunological assessments in primates. Hum Gene Ther Clin Dev. 2014;25(2):72–84. | |
Yang B, Li S, Wang H, et al. Global CNS transduction of adult mice by intravenously delivered rAAVrh.8 and rAAVrh.10 and nonhuman primates by rAAVrh.10. Mol Ther. 2014;22(7):1299–1309. | |
Bello A, Chand A, Aviles J, Soule G, Auricchio A, Kobinger GP. Novel adeno-associated viruses derived from pig tissues transduce most major organs in mice. Sci Rep. 2014;4:6644. | |
Chen YH, Claflin K, Geoghegan JC, Davidson BL. Sialic acid deposition impairs the utility of AAV9, but not peptide-modified AAVs for brain gene therapy in a mouse model of lysosomal storage disease. Mol Ther. 2012;20(7):1393–1399. | |
Biffi A, Capotondo A, Fasano S, et al. Gene therapy of metachromatic leukodystrophy reverses neurological damage and deficits in mice. J Clin Invest. 2006;116(11):3070–3082. | |
Biffi A, De Palma M, Quattrini A, et al. Correction of metachromatic leukodystrophy in the mouse model by transplantation of genetically modified hematopoietic stem cells. J Clin Invest. 2004;113(8):1118–1129. | |
Capotondo A, Cesani M, Pepe S, et al. Safety of arylsulfatase A overexpression for gene therapy of metachromatic leukodystrophy. Hum Gene Ther. 2007;18(9):821–836. | |
Biffi A, Montini E, Lorioli L, et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341(6148):1233158. | |
Rebar EJ, inventor; Sangqmo BioSciences, Inc., assignee. Methods and compositions for the provision of proteins deficient in lysosomal storage diseases. United States patent US 20140112896 A1. 2014 Apr 24. Available from: http://www.google.com/patents/US20140112896. Accessed March 18, 2015. | |
Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet. 2010; 11(9):636–646. | |
Sanjana NE, Cong L, Zhou Y, Cunniff MM, Feng G, Zhang F. A transcription activator-like effector toolbox for genome engineering. Nat Protoc. 2012;7(1):171–192. | |
Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32(4):347–355. | |
Lombardo A, Genovese P, Beausejour CM, et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol. 2007;25(11):1298–1306. | |
de Carvalho TG, da Silveira Matte U, Giugliani R, Baldo G. Genome editing: potential treatment for lysosomal storage diseases. Current Stem Cell Reports. 2015;1(1):9–15. | |
Böckenhoff A, Cramer S, Wölte P, et al. Comparison of five peptide vectors for improved brain delivery of the lysosomal enzyme arylsulfatase A. J Neurosci. 2014;34(9):3122–3129. | |
Boado RJ, Lu JZ, Hui EK, Sumbria RK, Pardridge WM. Pharmacokinetics and brain uptake in the rhesus monkey of a fusion protein of arylsulfatase a and a monoclonal antibody against the human insulin receptor. Biotechnol Bioeng. 2013;110(5):1456–1465. | |
Draghia R, Caillaud C, Manicom R, Pavirani A, Kahn A, Poenaru L. Gene delivery into the central nervous system by nasal instillation in rats. Gene Ther. 1995;2(6):418–423. | |
Wolf DA, Hanson LR, Aronovich EL, et al. Lysosomal enzyme can bypass the blood-brain barrier and reach the CNS following intranasal administration. Mol Genet Metab. 2012;106(1):131–134. | |
Lochhead JJ, Thorne RG. Intranasal delivery of biologics to the central nervous system. Adv Drug Deliv Rev. 2012;64(7):614–628. | |
Dhuria SV, Hanson LR, Frey WH. Intranasal delivery to the central nervous system: mechanisms and experimental considerations. J Pharm Sci. 2010;99(4):1654–1673. | |
Boutin S, Monteilhet V, Veron P, et al. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum Gene Ther. 2010;21(6):704–712. | |
Hurlbut GD, Ziegler RJ, Nietupski JB, et al. Preexisting immunity and low expression in primates highlight translational challenges for liver-directed AAV8-mediated gene therapy. Mol Ther. 2010;18(11):1983–1994. | |
Scallan CD, Jiang H, Liu T, et al. Human immunoglobulin inhibits liver transduction by AAV vectors at low AAV2 neutralizing titers in SCID mice. Blood. 2006;107(5):1810–1817. | |
Murphy SL, Li H, Zhou S, Schlachterman A, High KA, High K. Prolonged susceptibility to antibody-mediated neutralization for adeno-associated vectors targeted to the liver. Mol Ther. 2008;16(1):138–145. | |
Li C, Diprimio N, Bowles DE, et al. Single amino acid modification of adeno-associated virus capsid changes transduction and humoral immune profiles. J Virol. 2012;86(15):7752–7759. | |
Treleaven CM, Tamsett TJ, Bu J, et al. Gene transfer to the CNS is efficacious in immune-primed mice harboring physiologically relevant titers of anti-AAV antibodies. Mol Ther. 2012;20(9):1713–1723. | |
Kotterman MA, Yin L, Strazzeri JM, Flannery JG, Merigan WH, Schaffer DV. Antibody neutralization poses a barrier to intravitreal adeno-associated viral vector gene delivery to non-human primates. Gene Ther. 2015;22(2):116–126. | |
Thwaite R, Pagès G, Chillón M, Bosch A. AAVrh.10 immunogenicity in mice and humans. Relevance of antibody cross-reactivity in human gene therapy. Gene Ther. 2015;22(2):196–201. | |
Mingozzi F, Anguela XM, Pavani G, et al. Overcoming preexisting humoral immunity to AAV using capsid decoys. Sci Transl Med. 2013; 5(194):194ra92. | |
Monahan PE, Sun J, Gui T, et al. Employing a gain-of-function factor IX variant R338L to advance the efficacy and safety of hemophilia B human gene therapy: preclinical evaluation supporting an ongoing adeno-associated virus clinical trial. Hum Gene Ther. 2015;26(2):69–81. | |
Traas AM, Wang P, Ma X, et al. Correction of clinical manifestations of canine mucopolysaccharidosis I with neonatal retroviral vector gene therapy. Mol Ther. 2007;15(8):1423–1431. | |
Shull RM, Lu X, McEntee MF, Bright RM, Pepper KA, Kohn DB. Myoblast gene therapy in canine mucopolysaccharidosis. I: Abrogation by an immune response to alpha-L-iduronidase. Hum Gene Ther. 1996; 7(13):1595–1603. | |
Lutzko C, Kruth S, Abrams-Ogg AC, et al. Genetically corrected autologous stem cells engraft, but host immune responses limit their utility in canine alpha-L-iduronidase deficiency. Blood. 1999;93(6):1895–1905. | |
Snyder RO, Miao C, Meuse L, et al. Correction of hemophilia B in canine and murine models using recombinant adeno-associated viral vectors. Nat Med. 1999;5(1):64–70. | |
Mount JD, Herzog RW, Tillson DM, et al. Sustained phenotypic correction of hemophilia B dogs with a factor IX null mutation by liver-directed gene therapy. Blood. 2002;99(8):2670–2676. | |
Parenti G, Andria G, Ballabio A. Lysosomal storage diseases: from pathophysiology to therapy. Annu Rev Med. 2015;66:471–486. | |
Ribera A, Haurigot V, Garcia M, et al. Biochemical, histological and functional correction of mucopolysaccharidosis Type IIIB by intra-cerebrospinal fluid gene therapy. Hum Mol Genet. 2015;24(7):2078–2095. | |
Gray SJ, Nagabhushan Kalburgi S, McCown TJ, Jude Samulski R. Global CNS gene delivery and evasion of anti-AAV-neutralizing antibodies by intrathecal AAV administration in non-human primates. Gene Ther. 2013;20(4):450–459. | |
Colle MA, Piguet F, Bertrand L, et al. Efficient intracerebral delivery of AAV5 vector encoding human ARSA in non-human primate. Hum Mol Genet. 2010;19(1):147–158. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.