Back to Journals » OncoTargets and Therapy » Volume 13
Rationale for Lung Adenocarcinoma Prevention and Drug Development Based on Molecular Biology During Carcinogenesis
Authors Zhang H, Guo L, Chen J
Received 5 February 2020
Accepted for publication 14 March 2020
Published 14 April 2020 Volume 2020:13 Pages 3085—3091
DOI https://doi.org/10.2147/OTT.S248436
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Takuya Aoki
Hongming Zhang,1,* Liting Guo,2,* Jibei Chen1
1Department of Respiratory Medicine, Yancheng Third People’s Hospital, Affiliated Yancheng Hospital of Southeast University Medical College, Yancheng, Jiangsu Province, People’s Republic of China; 2Department of Oncology, Ruijin Hospital,affiliated with Shanghai Jiao Tong University School of Medicine, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jibei Chen
Department of Respiratory Medicine, Yancheng Third People’s Hospital, Affiliated Yancheng Hospital of Southeast University Medical College, Jiangsu Province Tel +86 515 8160 6123
Email [email protected]
Abstract: Lung adenocarcinoma (LUAD) is the most common and aggressive subtype of lung cancer with the greatest heterogeneity and aggression. Inspite of recent years’ achievements in understanding the pathogenesis of this disease, as well as the development of new therapeutic approaches, our knowledge on crucial early molecular events during its development is still rudimentary. Recent classification and grading of LUAD has postulated that LUAD does not arise spontaneously, but through a stepwise process from lung adenomatous premalignancy atypical adenomatous hyperplasia to adenocarcinoma in situ, minimally invasive adenocarcinoma, and eventually frankly invasive predominant adenocarcinoma. In this review, we discuss the molecular processes that drive the evolutionary process that results in the formation of LUAD. We also describe how to handle lung premalignancy in clinical settings based on the most recent advances in genomic biology and our own understanding of lung cancer prevention.
Keywords: lung adenocarcinoma, molecular biology, pathogenesis, cancer prevention
Introduction
Lung cancer is the predominant cause of cancer deaths worldwide.1,2 Most patients with lung cancer present with advanced-stage disease when diagnosed, and even in patients with early-stage resectable or locally advanced disease receiving definitive chemo- or radiation therapy, up to 90% of will still inevitably have disease recurrence, with a 5-year survival rate <60%.2,3 Non-small-cell lung cancer (NSCLC) represents 80%–85% of lung cancers, and can be subdivided into adenocarcinoma, squamous-cell carcinoma, and large-cell carcinoma.4 Among all NSCLC subtypes, lung adenocarcinoma (LUAD) is the most heterogeneous and aggressive and has a very high tumor-mutation burden associated with EGFR, KRAS, BRAF, ERBB2, TP53, ALK, STK11, and TTE1 mutations.5–8 Despite advances in chemotherapy, radiation therapy, and targeted therapy in the last decade, prevention and early detection and treatment of lung cancer is still challenging, due to limited awareness of molecular mechanisms mediating early lung carcinogenesis and also the very late diagnosis of the majority of them.9
The intriguing development of next-generation sequencing in recent years has defined a large number of driver mutations of cancer, leading to malignancy-therapy advances.10 The establishment of the Pre-Cancer Genome Atlas, a project for characterization of molecular evolutions from premalignant lesions to invasive carcinoma, as well as corresponding changes in the tumor microenvironment (TME) in 2016, has also helped to provide us the insight to consider another direction for cancer treatment, ie, cancer prevention.11 A complex and stepwise process from the only known type of preneoplasia, atypical adenomatous hyperplasia (AAH), to a more pronounced cellular atypia adenocarcinoma in situ (AIS) to microinvasive lesion minimally invasive adenocarcinoma (MIA), and finally to LUAD involving several genetic and epigenetic alterations has been postulated based on a variety of pathological, molecular, and clinical studies.6,12–14 In this review, we set out an organizing framework for understanding molecular biology in LUAD premalignancy, in order to set out a theoretical background for cancer prevention–drug development. In addition, we highlight possible intervention approaches that might prevent progression of premalignant lesion to LUAD.
Pathological and Molecular Alterations During Lung Carcinogenesis
Our lungs are continuously exposed to the outside environment, and thus harbor a complex network protecting the host from tissue damage and infection.15,16 The multistage stepwise fashion of tumor development has been demonstrated in various anatomical organs.17,18 Therefore, a paradigm referred to as “field cancerization” has been created for areas of histologically normal-appearing tissue that exhibit molecular abnormalities during early tumorigenesis.17,19 Besides cigarette smoking, environmental exposure, such as air pollution and workplace exposure to asbestos, diesel exhaust, or certain chemicals, also creates a field of injury in airway epithelial cells: dysregulated repair by progenitor cells forming a clonal group of indefinitely self-renewing daughter cells in the initial phase, and proliferation and expansion of premalignant cells resulting from genetic and epigenetic alterations gradually displacing the normal epithelium.20–24
AAH and AIS: Field Cancerization
Lung preneoplastic-lesion AAH and preinvasive-lesion AIS present cellular atypia characteristic of field cancerization.17,20,25,26 As the only known type of preneoplastic lesion, AAH represents the initial step in LUAD pathogenesis.13,14,27–29 It is a small, atypical proliferation (usually 0.5 cm or less) of type II pneumocytes along preexisting alveolar walls most commonly discovered as an incidental histological finding in 5%–20% of lung cancer specimens after resection.12,27,30,31 Telomere attrition has been demonstrated to occur as an initiating event in AAH progression to LUAD.32 An intriguing finding is that shared mutations of two classical genes, KRAS and EGFR, of AAH and LUAD in the same individual have also been detected,33–37 though Sakamoto et al38 showed that harboring a KRAS gene mutation might not ensure AAH’s further progress to LUAD. Other molecular aberrations identified in AAH include FGFR3,39 BRAF,40 TP53,29,41 STK11,42 and ERBB2 (HER2) mutations,43 upregulation of cyclin D1,44 survivin,45 paxillin,46 and Ki67,47 suppression of TBX2,48 and loss of heterozygosity in chromosomes 3p, 9p, 9q, 16p, 17p and 17q.29,49–52 Epigenetic modifications including DNA methylation of CDKN2A-Ex2 and PTPRN2 have also been reported in AAH.25,53 Genes bearing somatic mutations or epigenetic modifications often encode tumor-associated antigensable to elicit immunoresponse; therefore, the immune system is capable of recognizing AAH. T-effector and cytotoxic cell infiltration and upregulation of immunocheckpoint PDL1 and CTLA4 in AAH compared to normal lung tissue suggest that the T cells might have already been activated in the AAH stage.40,54 Activation of protumor (TH2; CCR2, CTLA4) and reduction in antitumor (TH1; IL12A, GZMB, TBX21) immunofunctioning-associated gene sets observed in the development from normal lung to AAH further confirm the involvement of aberrant immunopathways in AAH.55
Growth and progression of AAH with a more pronounced cellular atypia in morphology evolve into AIS, a small localized (≤3 cm) adenocarcinoma with restricted growth of neoplastic cells along preexisting alveolar structures.56 AIS is also regarded as a preinvasive form of LUAD according to recent classification and grading.28 Progression from AAH to AIS occurs over an extended period, with different alterations detected. A recent study by Izumchenko et al57 indicated that only a few gene mutations were shared between AAH and AIS, and that the most frequent base-pair substitutions were C–T transitions in AAH, but G–A in MIA. Tanaka et al58 showed that BI1 is expressed in AIS, but not in AAH. Chung et al59 combined AAH and AIS as preinvasive lesions and investigated their multistep progression to adenocarcinoma. Methylation of HOXA1, TMEFF2, and RARB was frequently observed in preinvasive lesions, suggesting epigenetic alterations in different genes involved in different stages.
Invasive Lesions: MIA and Adenocarcinoma
Malignant cells become invasive once they leave the epidermis.60–62 These invasive lesions generally harbor driver mutations, including EGFR, KRAS, ALK, ERBB2, BRAF, AKT1, PIK3CA, MAP2K1, and MET mutations;63,64 however, few of these have been reported in precursor lesions.33–43 A subset of invasive adenomatous lesions appears to arise spontaneously in the absence of any precursor lesions for only around 15% of LUAD-harboring precursor lesions,65 though it may also be possible that the precursors are no longer detectable at diagnosis.
Substantive changes occur during the evolution to invasive lesions from MIA and adenocarcinoma from preinvasive lesions. In contrast to AAH and AIS, microinvasion is present in MIA, small, solitary, and discrete microinvasive lesions no larger than 3 cm with a predominantly lepidic pattern and invasion <5 mm in any one focus, the majority of which are nonmucinous.6,14 Currently, genomic studies into MIA have been limited compared to preneoplastic lesions, though unsurprisingly a higher frequency of KRAS, EGFR, TP53, and NF1 mutations has been found in MIA than AAH and AIS.57,66 A recent study by Qian et al66 also demonstrated an increasing frequency in mutations of KRAS, TP53, and NF1 in MIA than its earlier stage — AIS.
Once the invasive area of MIA extends >5 mm in diameter or meets invasion criteria, the lesion becomes an invasive predominant adenocarcinoma.6,14 The mutational landscape of early-stage LUAD has been investigated by multiple studies: EGFR, TP53, KRAS, STK11, and NF1 are substantially mutated in LUAD, and driver mutations in EGFR, BRAF, MET, and TP53 are almost always clonal, with acquired mutations directly linked to patient prognosis.8,13,67 It has also been established that mutations acquired during progression are linked to patient prognosis.13 With acquisition of more mutations, the immune system also becomes more complicated in early-stage LUAD. An example is that inactivation of STK11 will result in accumulation of immunosuppressive neutrophils and reduced PDL1 expression, as well as fewer tumor-infiltrating lymphocytes.68
Rationale for LUAD Prevention: A Clinical Insight
In clinical practice, preinvasive and early invasive LUAD lesions can be detected by computed tomography or during histopathological studies on surgically resected specimens, and present as pulmonary nodules with ground-glass opacity (GGO): circumscribed hazy lesions with preservation of bronchial and vascular margins.9,69 Prediction of GGO nodules is difficult, as a considerable proportion will disappear spontaneously; however, approximately 10% will progress to invasive cancer.70–72 Therefore, management of incidentally detected GGO nodules is recommended as a follow-up for a minimum 3–4 years.73,74 According to the American College of Chest Physicians, increased size or solid-component development, pure GGO nodules >10 mm with confirmed persistence, mixed (GGO >50%) GGO nodules >8 mm with confirmed persistence, or mixed GGO nodules >15 mm without follow-up should be considered for surgery.75,76 However, patients may still relapse following resection: according to a report from Cho et al77 5.1% (five of 97) of patients with GGO nodules experienced recurrence after resection, while Nakao et al reported that 8% (four of 50) of patients with GGO nodules recurred after limited section.78 For patients with early-stage NSCLC following standard treatment surgery, as many as 40% of patients with stage I and 66% of stage II NSCLC are still found to relapse and finally die within 5 years.79–82 In addition, surgical resection may not be feasible for patients carrying multiple potentially aggressive transformation nodules.83 Therefore, drug development for lung cancer prevention and interception becomes our next-step consideration. Gene mutations can lead to activation of specific oncogenic pathways, leading to the occurrence of cancer. Therefore, targeting key molecular events will help in lung cancer interception (Figure 1). Molecular changes also have major effects on the TME, and thus secretion of specific inhibitory cytokines or production of chemokines and other factors related to immunosuppression. A typical example is that early KRAS-mutated pancreatic neoplastic cells can secrete VEGF, GM-CSF, and cytokines to recruit Tregs, myeloid-derived suppressor cells, adipocytes, neutrophils, macrophage, and chemokines, leading to a progressively immunosuppressive TME that contributes to immunoescape.84,85 In NSCLC, EGFR and STK11 mutations are more likely to have low levels of PDL1 expression and mutational burden, thus lacking benefit in immunoheckpoint blockadetherapy.86–90 As to the positivity of EGFRT790M, Haratani et al91 showed that as a result of higher PDL1 expression level in EGFRT790M-negative patients, they are more likely to benefit from nivolumab after EGFR TKI treatment. A recent study by Hastings et al92 suggested that EGFR-mutant tumors have generally low response to immunocheckpoint inhibitors, but outcomes varied by allele, eg, lung tumors with EGFRΔ19 alterations harbored a lower tumor-mutation burden compared with EGFRL858R lung tumors. In contrast, TP53 and KRAS mutations and loss of PTEN and STK11 were observed to increase with PDL1 expression and mutational burden, as well as activated T-effector and IFNγ signature in LUAD, and thus remarkable clinical benefit to PD1 inhibitors are found in patients with TP53 and KRAS mutations.7,68,93,94 Therefore, TME interception could also be a potential interception method.
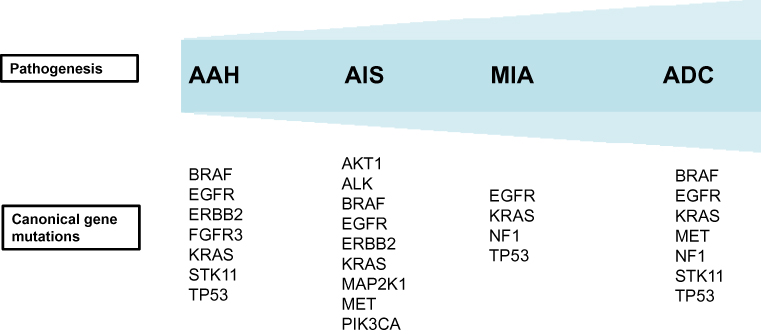 |
Figure 1 Summary of genetic alterations in multistep progression of lung adenocarcinoma. Abbreviations: AAH, atypical adenomatous hyperplasia; AIS, aAdenocarcinoma in situ; MIA, minimally invasive adenocarcinoma; ADC, adenocarcinoma. |
Conclusion
As the most common subtype of lung cancer, LUAD has gained great attention from oncologists. In this review, based on the most recent International Association for the Study of Lung Cancer–American Thoracic Society–European Respiratory Society International Multidisciplinary Classification of Lung Adenocarcinoma, we elaborated a conceptual framework to reveal the stepwise genomic evolution of LUAD from preinvasive AAH and AIS lesions to invasive MIA lesions and adenocarcinoma. We also proposed a scheme for LUAD prevention based on our clinical perspective.
The advent of sequencing technologies has allowed us to understand pathogenesis during LUAD development; however our knowledge of its detailed pathogenesis remains somewhat superficial. Firstly, the current treatment method for pulmonary nodules with GGO in the clinic is observation and a wait-and-see approach once they are detected by computed tomography scan. Associations between antigen expression and the mechanism of cellular transformation are unknown. In addition, how long GGO nodules should be followed up, appropriate timing for surgical resection, and the extent of resection are other questions.95 Secondly, many risk factors contribute to the development of LUAD, and recognizing these risk factors can be a potential consideration for prevention. As we all know, human papillomavirus vaccination has already been successfully used in cervical cancer. However, many gene-modified autologous vaccines for LUAD have already been tested, but not with the effects we expected.10,96 Thirdly, alterations in the TME during carcinogenesis, especially immuneffector processes mediating cancer elimination, equilibrium, and escape, need to be elucidated. Potential interception of LUAD development would be enhancing the immune system’s recognition and elimination or weakening immuno escape of the abnormal preneoplasias, ie, immunoprevention. Therefore, identifying molecular mechanisms and antigen expression in premalignancy responsible for immunoediting becomes crucial. Last but not least, beyond what we have summarized, other challenges associated need to be addressed, includingmaintenance of the lung environment, key driving events, and personalized cancer intervention.
Overall, this review serves to enrich our knowledge of molecular aspects ofLUAD pathogenesis. A limitation of this review is that we did not explicitly address the concrete genomic evolution during early lung adenomatous progression due to current superficial understanding, but we believe that our viewpoints will help to propel precancer research in the next few years and eventually expand cancer prevention to a greater subset of patients.
Disclosure
The authors have declared that no competing interest exists in this work.
References
1. Tan W-L, Jain A, Takano A, et al. Novel therapeutic targets on the horizon for lung cancer. Lancet Oncol. 2016;17(8):e347–e62. doi:10.1016/S1470-2045(16)30123-1
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34. doi:10.3322/caac.21551
3. Barnett R. Lung cancer. The Lancet. 2017;390(10098):928. doi:10.1016/S0140-6736(17)32243-2
4. Reck M, Rabe KF. Precision diagnosis and treatment for advanced non-small-cell lung cancer. N Engl J Med. 2017;377(9):849–861. doi:10.1056/NEJMra1703413
5. Ding L, Getz G, Wheeler DA, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455(7216):1069–1075. doi:10.1038/nature07423
6. Travis WD, Brambilla E, Noguchi M, et al. International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society International Multidisciplinary Classification of Lung Adenocarcinoma. J Thor Oncol. 2011;6(2):244–285. doi:10.1097/JTO.0b013e318206a221
7. Rizvi NA, Hellmann MD, Snyder A, et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science (New York, NY). 2015;348(6230):124–128. doi:10.1126/science.aaa1348
8. Devarakonda S, Morgensztern D, Govindan R. Genomic alterations in lung adenocarcinoma. Lancet Oncol. 2015;16(7):e342–e51. doi:10.1016/S1470-2045(15)00077-7
9. Mulshine JL, D’Amico TA. Issues with implementing a high-quality lung cancer screening program. CA Cancer J Clin. 2014;64(5):352–363. doi:10.3322/caac.21239
10. Spira A, Disis ML, Schiller JT, et al. Leveraging premalignant biology for immune-based cancer prevention. Proc Natl Acad Sci U S A. 2016;113(39):10750–10758. doi:10.1073/pnas.1608077113
11. Campbell JD, Mazzilli SA, Reid ME, et al. The case for a Pre-Cancer Genome Atlas (PCGA). Cancer Prev Res. 2016;9(2):119–124. doi:10.1158/1940-6207.CAPR-16-0024
12. Maeshima AM, Tochigi N, Yoshida A, Asamura H, Tsuta K, Tsuda H. Clinicopathologic analysis of multiple (five or more) atypical adenomatous hyperplasias (AAHs) of the lung: evidence for the AAH-adenocarcinoma sequence. J Thor Oncol. 2010;5(4):466–471. doi:10.1097/JTO.0b013e3181ce3b73
13. Noguchi M. Stepwise progression of pulmonary adenocarcinoma—clinical and molecular implications. Cancer Metastasis Rev. 2010;29(1):15–21. doi:10.1007/s10555-010-9210-y
14. Aoyagi Y, Yokose T, Minami Y, et al. Accumulation of losses of heterozygosity and multistep carcinogenesis in pulmonary adenocarcinoma. Cancer Res. 2001;61(21):7950–7954.
15. Hunninghake GW, Gadek J, Kawanami O, Ferrans V, Crystal R. Inflammatory and immune processes in the human lung in health and disease: evaluation by bronchoalveolar lavage. Am J Pathol. 1979;97(1):149.
16. Mizgerd JP. Respiratory infection and the impact of pulmonary immunity on lung health and disease. Am J Respir Crit Care Med. 2012;186(9):824–829. doi:10.1164/rccm.201206-1063PP
17. Curtius K, Wright NA, Graham TA. An evolutionary perspective on field cancerization. Nat Rev Cancer. 2018;18(1):19–32. doi:10.1038/nrc.2017.102
18. Sahai E, Swanton C. Evolution and cancer medicine - transformative insights. Nat Rev Clin Oncol. 2017;14(12):709–710. doi:10.1038/nrclinonc.2017.121
19. Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. 1953;6(5):963–968. doi:10.1002/1097-0142(195309)6:5<963::AID-CNCR2820060515>3.0.CO;2-Q
20. Gomperts BN, Walser TC, Spira A, Dubinett SM. Enriching the molecular definition of the airway “field of cancerization”: establishing new paradigms for the patient at risk for lung cancer. Cancer Prev Res. 2013;6(1):4–7. doi:10.1158/1940-6207.CAPR-12-0470
21. Sellers TA, Weaver TW, Phillips B, Altmann M, Rich SS. Environmental factors can confound identification of a major gene effect: results from a segregation analysis of a simulated population of lung cancer families. Genet Epidemiol. 1998;15(3):251–262. doi:10.1002/(ISSN)1098-2272
22. Sellers TA, Bailey-Wilson JE, Elston RC, et al. Evidence for mendelian inheritance in the pathogenesis of lung cancer. J Natl Cancer Inst. 1990;82(15):1272–1279. doi:10.1093/jnci/82.15.1272
23. Thorgeirsson TE, Geller F, Sulem P, et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature. 2008;452(7187):638–642. doi:10.1038/nature06846
24. Kantrowitz J, Sinjab A, Xu L, et al. Genome-wide gene expression changes in the normal-appearing airway during the evolution of smoking-associated lung adenocarcinoma. Cancer Prev Res. 2018;11(4). doi:10.1158/1940-6207.CAPR-17-0295.
25. Kadara H, Scheet P, Wistuba II, Spira AE. Early events in the molecular pathogenesis of lung cancer. Cancer Prev Res. 2016;9(7):518–527. doi:10.1158/1940-6207.CAPR-15-0400
26. Klebe S, Henderson D. Facts and fiction: premalignant lesions of lung tissues. Pathol J RCPA. 2013;45(3):305–315.
27. Kitagawa H, Goto A, Niki T, Hironaka M, Nakajima J, Fukayama M. Lung adenocarcinoma associated with atypical adenomatous hyperplasia. A clinicopathological study with special reference to smoking and cancer multiplicity. Pathol Int. 2003;53(12):823–827. doi:10.1046/j.1440-1827.2003.01570.x
28. Weichert W, Warth A. Early lung cancer with lepidic pattern: adenocarcinoma in situ, minimally invasive adenocarcinoma, and lepidic predominant adenocarcinoma. Curr Opin Pulm Med. 2014;20(4):309–316. doi:10.1097/MCP.0000000000000065
29. Westra WH. Early glandular neoplasia of the lung. Respir Res. 2000;1(3):8. doi:10.1186/rr28
30. Seki M, Akasaka Y. Multiple lung adenocarcinomas and AAH treated by surgical resection. Lung Cancer. 2007;55(2):237–240. doi:10.1016/j.lungcan.2006.10.007
31. Min JH, Lee HY, Lee KS, et al. Stepwise evolution from a focal pure pulmonary ground-glass opacity nodule into an invasive lung adenocarcinoma: an observation for more than 10 years. Lung Cancer. 2010;69(1):123–126. doi:10.1016/j.lungcan.2010.04.022
32. Lantuejoul S, Raynaud C, Salameire D, et al. Telomere maintenance and DNA damage responses during lung carcinogenesis. Clin Cancer Res. 2010;16(11):2979–2988. doi:10.1158/1078-0432.CCR-10-0142
33. Ikeda K, Nomori H, Ohba Y, et al. Epidermal growth factor receptor mutations in multicentric lung adenocarcinomas and atypical adenomatous hyperplasias. J Thor Oncol. 2008;3(5):467–471. doi:10.1097/JTO.0b013e31816b4b14
34. Westra WH, Baas IO, Hruban RH, et al. K-ras oncogene activation in atypical alveolar hyperplasias of the human lung. Cancer Res. 1996;56(9):2224–2228.
35. Yoshida Y, Shibata T, Kokubu A, et al. Mutations of the epidermal growth factor receptor gene in atypical adenomatous hyperplasia and bronchioloalveolar carcinoma of the lung. Lung Cancer. 2005;50(1):1–8. doi:10.1016/j.lungcan.2005.04.012
36. Kitamura H, Kameda Y, Ito T, Hayashi H. Atypical adenomatous hyperplasia of the lung: implications for the pathogenesis of peripheral lung adenocarcinoma. Am J Clin Pathol. 1999;111(5):610–622. doi:10.1093/ajcp/111.5.610
37. Tang X, Shigematsu H, Bekele BN, et al. EGFR tyrosine kinase domain mutations are detected in histologically normal respiratory epithelium in lung cancer patients. Cancer Res. 2005;65(17):7568–7572. doi:10.1158/0008-5472.CAN-05-1705
38. Sakamoto H, Shimizu J, Horio Y, et al. Disproportionate representation of KRAS gene mutation in atypical adenomatous hyperplasia, but even distribution of EGFR gene mutation from preinvasive to invasive adenocarcinomas. J Pathol. 2007;212(3):287–294. doi:10.1002/(ISSN)1096-9896
39. Xu X, Li N, Zhao R, Zhu L, Shao J, Zhang J. Targeted next-generation sequencing for analyzing the genetic alterations in atypical adenomatous hyperplasia and adenocarcinoma in situ. J Cancer Res Clin Oncol. 2017;143(12):2447–2453. doi:10.1007/s00432-017-2500-9
40. Sivakumar S, Lucas FAS, McDowell TL, et al. Genomic landscape of atypical adenomatous hyperplasia reveals divergent modes to lung adenocarcinoma. Cancer Res. 2017;77(22):6119–6130. doi:10.1158/0008-5472.CAN-17-1605
41. Kohno H, Hiroshima K, Toyozaki T, Fujisawa T, Ohwada H. p53 mutation and allelic loss of chromosome 3p, 9p of preneoplastic lesions in patients with nonsmall cell lung carcinoma. Cancer. 1999;85(2):341–347.
42. Makowski L, Hayes D. Role of LKB1 in lung cancer development. Br J Cancer. 2008;99(5):683. doi:10.1038/sj.bjc.6604515
43. Awaya H, Takeshima Y, Furonaka O, Kohno N, Inai K. Gene amplification and protein expression of EGFR and HER2 by chromogenic in situ hybridisation and immunohistochemistry in atypical adenomatous hyperplasia and adenocarcinoma of the lung. J Clin Pathol. 2005;58(10):1076–1080. doi:10.1136/jcp.2004.025585
44. Kurasono Y, Ito T, Kameda Y, Nakamura N, Kitamura H. Expression of cyclin D1, retinoblastoma gene protein, and p16 MTS1 protein in atypical adenomatous hyperplasia and adenocarcinoma of the lung. Virchows Archiv. 1998;432(3):207–215. doi:10.1007/s004280050157
45. Nakanishi K, Kawai T, Kumaki F, Hiroi S, Mukai M, Ikeda E. Survivin expression in atypical adenomatous hyperplasia of the lung. Am J Clin Pathol. 2003;120(5):712–719. doi:10.1309/GWTN2JTAN6K73YDE
46. Mackinnon AC, Tretiakova M, Henderson L, et al. Paxillin expression and amplification in early lung lesions of high-risk patients, lung adenocarcinoma and metastatic disease. J Clin Pathol. 2010;2010.075853(jcp).
47. Koga T, Hashimoto S, Sugio K, et al. Lung adenocarcinoma with bronchioloalveolar carcinoma component is frequently associated with foci of high-grade atypical adenomatous hyperplasia. Am J Clin Pathol. 2002;117(3):464–470. doi:10.1309/CHXA-3MH0-B7FD-JGUL
48. Khalil AA, Sivakumar S, Lucas FAS, et al. TBX2 subfamily suppression in lung cancer pathogenesis: a high-potential marker for early detection. Oncotarget. 2017;8(40):68230–68241. doi:10.18632/oncotarget.19938
49. Takamochi K, Ogura T, Suzuki K, et al. Loss of heterozygosity on chromosomes 9q and 16p in atypical adenomatous hyperplasia concomitant with adenocarcinoma of the lung. Am J Pathol. 2001;159(5):1941–1948. doi:10.1016/S0002-9440(10)63041-6
50. Suzuki K, Ogura T, Yokose T, et al. Loss of heterozygosity in the tuberous sclerosis gene associated regions in adenocarcinoma of the lung accompanied by multiple atypical adenomatous hyperplasia. Int J Cancer. 1998;79(4):384–389. doi:10.1002/(ISSN)1097-0215
51. Kitaguchi S, Takeshima Y, Nishisaka T, Inai K. Proliferative activity, p53 expression and loss of heterozygosity on 3p, 9p and 17p in atypical adenomatous hyperplasia of the lung. Hiroshima J Med Sci. 1998;47(1):17–26.
52. Jakubek Y, Lang W, Vattathil S, et al. Genomic landscape established by allelic imbalance in the cancerization field of a normal appearing airway. Cancer Res. 2016;76(13):3676–3683. doi:10.1158/0008-5472.CAN-15-3064
53. Selamat SA, Galler JS, Joshi AD, et al. DNA methylation changes in atypical adenomatous hyperplasia, adenocarcinoma in situ, and lung adenocarcinoma. PLoS One. 2011;6(6):e21443. doi:10.1371/journal.pone.0021443
54. Grimes BS. The Immune Contexture and Genomic Landscape of Lung Adenomatous Premalignancy. Los Angeles: University of California; 2017.
55. Sivakumar S, San Lucas F, McDowell T, et al. Genomic landscape of atypical adenomatous hyperplasia and their progression to lung adenocarcinomas. J Thor Oncol. 2017;12(8):S1546. doi:10.1016/j.jtho.2017.06.061
56. Borczuk AC. Assessment of invasion in lung adenocarcinoma classification, including adenocarcinoma in situ and minimally invasive adenocarcinoma. Modern Pathol. 2012;25(S1).
57. Izumchenko E, Chang X, Brait M, et al. Targeted sequencing reveals clonal genetic changes in the progression of early lung neoplasms and paired circulating DNA. Nat Commun. 2015;6(1):8258. doi:10.1038/ncomms9258
58. Tanaka R, Ishiyama T, Uchihara T, et al. Expression of the Bax inhibitor‐1 gene in pulmonary adenocarcinoma. Cancer. 2006;106(3):648–653.
59. Chung JH, Lee HJ, Kim BH, Cho NY, Kang GH. DNA methylation profile during multistage progression of pulmonary adenocarcinomas. Virchows Archiv. 2011;459(2):201–211. doi:10.1007/s00428-011-1079-9
60. Chiosea S, Jelezcova E, Chandran U, et al. Overexpression of Dicer in precursor lesions of lung adenocarcinoma. Cancer Res. 2007;67(5):2345–2350. doi:10.1158/0008-5472.CAN-06-3533
61. Chapman A, Kerr K. The association between atypical adenomatous hyperplasia and primary lung cancer. Br J Cancer. 2000;83(5):632. doi:10.1054/bjoc.2000.1317
62. Noguchi M, Shimosato Y. The development and progression of adenocarcinoma of the lung. Lung Cancer. 1994;131–142.
63. Kris M, Johnson B, Kwiatkowski D, et al. Identification of driver mutations in tumor specimens from 1000 patients with lung adenocarcinoma: the NCI’s Lung Cancer Mutation Consortium (LCMC). J Clin Oncol. 2011;29(18_suppl):CRA7506–CRA. doi:10.1200/jco.2011.29.15_suppl.cra7506
64. Pao W, Girard N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011;12(2):175–180. doi:10.1016/S1470-2045(10)70087-5
65. Chung J-H, Choe G, Jheon S, et al. Epidermal growth factor receptor mutation and pathologic-radiologic correlation between multiple lung nodules with ground-glass opacity differentiates multicentric origin from intrapulmonary spread. J Thor Oncol. 2009;4(12):1490–1495. doi:10.1097/JTO.0b013e3181bc9731
66. Qian J, Zhao S, Zou Y, et al. Genomic underpinnings of tumor behavior in in situ and early lung adenocarcinoma. Am J Respir Crit Care Med. 2019.
67. Jamal-Hanjani M, Wilson GA, McGranahan N, et al. Tracking the evolution of non-small-cell lung cancer. N Engl J Med. 2017;376(22):2109–2121. doi:10.1056/NEJMoa1616288
68. Koyama S, Akbay EA, Li YY, et al. STK11/LKB1 deficiency promotes neutrophil recruitment and proinflammatory cytokine production to suppress T-cell activity in the lung tumor microenvironment. Cancer Res. 2016;76(5):999–1008. doi:10.1158/0008-5472.CAN-15-1439
69. Naidich DP, Bankier AA, MacMahon H, et al. Recommendations for the management of subsolid pulmonary nodules detected at CT: a statement from the Fleischner society. Radiology. 2013;266(1):304–317. doi:10.1148/radiol.12120628
70. Gulati CM, Schreiner AM, Libby DM, Port JL, Altorki NK, Gelbman BD. Outcomes of unresected ground-glass nodules with cytology suspicious for adenocarcinoma. J Thor Oncol. 2014;9(5):685–691. doi:10.1097/JTO.0000000000000143
71. Voltaggio L, Cimino-Mathews A, Bishop JA, et al. Current concepts in the diagnosis and pathobiology of intraepithelial neoplasia: a review by organ system. CA Cancer J Clin. 2016;66(5):408–436. doi:10.3322/caac.21350
72. McWilliams A, Tammemagi MC, Mayo JR, et al. Probability of cancer in pulmonary nodules detected on first screening CT. N Engl J Med. 2013;369(10):910–919. doi:10.1056/NEJMoa1214726
73. Lu W, Cham MD, Qi L, et al. The impact of chemotherapy on persistent ground-glass nodules in patients with lung adenocarcinoma. J Thorac Dis. 2017;9(11):4743–4749. doi:10.21037/jtd
74. Kobayashi Y, Mitsudomi T. Management of ground-glass opacities: should all pulmonary lesions with ground-glass opacity be surgically resected? Transl Lung Cancer Res. 2013;2(5):354–363. doi:10.3978/j.issn.2218-6751.2013.09.03
75. Gould MK, Donington J, Lynch WR, et al. Evaluation of individuals with pulmonary nodules: when is it lung cancer? Diagnosis and management of lung cancer, 3rd ed: american College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2013;143(5 Suppl):e93S–e120S. doi:10.1378/chest.12-2351
76. Kobayashi Y, Mitsudomi T, Sakao Y, Yatabe Y. Genetic features of pulmonary adenocarcinoma presenting with ground-glass nodules: the differences between nodules with and without growth. Ann Oncol. 2015;26(1):156–161. doi:10.1093/annonc/mdu505
77. Cho JH, Choi YS, Kim J, Kim HK, Zo JI, Shim YM. Long-term outcomes of wedge resection for pulmonary ground-glass opacity nodules. Ann Thorac Surg. 2015;99(1):218–222. doi:10.1016/j.athoracsur.2014.07.068
78. Nakao M, Yoshida J, Goto K, et al. Long-term outcomes of 50 cases of limited-resection trial for pulmonary ground-glass opacity nodules. J Thor Oncol. 2012;7(10):1563–1566. doi:10.1097/JTO.0b013e3182l641b5c
79. Chansky K, Sculier JP, Crowley JJ, Giroux D, Van Meerbeeck J, Goldstraw P. The International association for the study of lung cancer staging project: prognostic factors and pathologic TNM stage in surgically managed non-small cell lung cancer. J Thor Oncol. 2009;4(7):792–801. doi:10.1097/JTO.0b013e3181a7716e
80. Liu S, Wang R, Zhang Y, et al. Precise diagnosis of intraoperative frozen section is an effective method to guide resection strategy for peripheral small-sized lung adenocarcinoma. J Clin Oncol. 2015;34(4):307–313. doi:10.1200/JCO.2015.63.4907
81. Potti A, Mukherjee S, Petersen R, et al. A genomic strategy to refine prognosis in early-stage non–small-cell lung cancer. N Eng J Med. 2006;355(6):570–580. doi:10.1056/NEJMoa060467
82. Ettinger DS, Wood DE, Akerley W, et al. NCCN guidelines insights: non-small cell lung cancer, Version 4.2016. J Natl Compreh Cancer Net. 2016;14(3):255–264. doi:10.6004/jnccn.2016.0031
83. Panse PM, Gruden JF, Viggiano RW, Smith ML, Gotway MB. Multiple ground-glass opacity pulmonary nodules: an unusual thoracic CT appearance of a rare diagnosis. Clin Pulm Med. 2013;20(4):199–201. doi:10.1097/CPM.0b013e3182991986
84. Kaneda MM, Cappello P, Nguyen AV, et al. Macrophage PI3Kγ drives pancreatic ductal adenocarcinoma progression. Cancer Discov. 2016;6(8):870–885. doi:10.1158/2159-8290.CD-15-1346
85. Phan VT, Wu X, Cheng JH, et al. Oncogenic RAS pathway activation promotes resistance to anti-VEGF therapy through G-CSF–induced neutrophil recruitment. Proc Natl Acad Sci. 2013;110(15):6079–6084. doi:10.1073/pnas.1303302110
86. Soo RA, Lim SM, Syn NL, et al. Immune checkpoint inhibitors in epidermal growth factor receptor mutant non-small cell lung cancer: current controversies and future directions. Lung Cancer. 2018;115:12–20. doi:10.1016/j.lungcan.2017.11.009
87. Wang Y, Ellis P. EGFR mutation positive non-small cell lung cancer: can we identify predictors of benefit from immune checkpoint inhibitors. Ann Transl Med. 2017;5(21).
88. Rizvi H, Sanchez-Vega F, La K, et al. Molecular determinants of response to anti-programmed cell death (PD)-1 and Anti-Programmed Death-Ligand (PD-L)-Ligand 1 blockade in patients with non-small-cell lung cancer profiled with targeted next-generation sequencing. J clin oncol. 2018;Jco2017753384.
89. Skoulidis F, Elamin Y, Papadimitrakopoulou V, et al. MA04. 07 impact of major co-mutations on the immune contexture and response of KRAS-mutant lung adenocarcinoma to immunotherapy. J Thor Oncol. 2017;12(1):S361–S2. doi:10.1016/j.jtho.2016.11.402
90. Lee CK, Man J, Lord S, et al. Checkpoint inhibitors in metastatic EGFR-mutated non-small cell lung cancer-a meta-analysis. J Thor Oncol. 2017;12(2):403–407. doi:10.1016/j.jtho.2016.10.007
91. Haratani K, Hayashi H, Tanaka T, et al. Tumor immune microenvironment and nivolumab efficacy in EGFR mutation-positive non-small-cell lung cancer based on T790M status after disease progression during EGFR-TKI treatment. Ann Oncol. 2017;28(7):1532–1539. doi:10.1093/annonc/mdx183
92. Hastings K, Yu HA, Wei W, et al. EGFR mutation subtypes and response to immune checkpoint blockade treatment in non-small-cell lung cancer. Ann Oncol. 2019;30(8):1311–1320. doi:10.1093/annonc/mdz141
93. Dong ZY, Zhong WZ, Zhang XC, et al. Potential predictive value of TP53 and KRAS mutation status for response to PD-1 blockade immunotherapy in lung adenocarcinoma. Clin Cancer Res. 2017;23(12):3012–3024. doi:10.1158/1078-0432.CCR-16-2554
94. Garon EB, Rizvi NA, Hui R, et al. Pembrolizumab for the treatment of non–small-cell lung cancer. N Eng J Med. 2015;372(21):2018–2028. doi:10.1056/NEJMoa1501824
95. Kobayashi Y, Mitsudomi T. Management of ground-glass opacities: should all pulmonary lesions with ground-glass opacity be surgically resected? Transl Lung Cancer Res. 2013;2(5):354.
96. Nemunaitis J, Dillman RO, Schwarzenberger PO, et al. Phase II study of belagenpumatucel-L, a transforming growth factor beta-2 antisense gene-modified allogeneic tumor cell vaccine in non–small-cell lung cancer. J Clin Oncol. 2006;24(29):4721–4730. doi:10.1200/JCO.2005.05.5335
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.