Back to Journals » The Application of Clinical Genetics » Volume 15
RASopathy Cohort of Patients Enrolled in a Brazilian Reference Center for Rare Diseases: A Novel Familial LZTR1 Variant and Recurrent Mutations
Authors Chaves Rabelo N ![]() , Gomes ME, de Oliveira Moraes I, Cantagalli Pfisterer J, Loss de Morais G, Antunes D
, Gomes ME, de Oliveira Moraes I, Cantagalli Pfisterer J, Loss de Morais G, Antunes D ![]() , Caffarena ER, Llerena Jr J, Gonzalez S
, Caffarena ER, Llerena Jr J, Gonzalez S
Received 7 May 2022
Accepted for publication 3 September 2022
Published 21 October 2022 Volume 2022:15 Pages 153—170
DOI https://doi.org/10.2147/TACG.S372761
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Natana Chaves Rabelo,1– 3 Maria Eduarda Gomes,1– 3 Isabelle de Oliveira Moraes,1– 3 Juliana Cantagalli Pfisterer,1– 3 Guilherme Loss de Morais,4 Deborah Antunes,5 Ernesto Raúl Caffarena,6 Juan Llerena Jr,1,2,7,8 Sayonara Gonzalez1– 3
1Centro de Genética Médica IFF/Fiocruz, Rio de Janeiro, RJ, Brazil; 2Centro de Referência para Doenças Raras IFF/Fiocruz, Rio de Janeiro, RJ, Brazil; 3Laboratório de Medicina Genômica IFF/Fiocruz, Rio de Janeiro, RJ, Brazil; 4Laboratório Nacional de Computação Científica, Petrópolis, RJ, Brazil; 5Laboratório de Genômica Funcional e Bioinformática, Instituto Oswaldo Cruz/Fiocruz, Rio de Janeiro, RJ, Brazil; 6Grupo de Biofísica Computacional e Modelagem Molecular, Programa de Computação Científica, Fundação Oswaldo Cruz/Fiocruz, Rio de Janeiro, RJ, Brazil; 7Faculdade de Medicina de Petrópolis, FASE, Petrópolis, RJ, Brazil; 8INAGEMP, Rio de Janeiro, RJ, Brazil
Correspondence: Juan Llerena Jr, Email [email protected]
Purpose: Noonan syndrome and related disorders are genetic conditions affecting 1:1000– 2000 individuals. Variants causing hyperactivation of the RAS/MAPK pathway lead to phenotypic overlap between syndromes, in addition to an increased risk of pediatric tumors. DNA sequencing methods have been optimized to provide a molecular diagnosis for clinical and genetic heterogeneity conditions. This work aimed to investigate the genetic basis in RASopathy patients through Next Generation Sequencing in a Reference Center for Rare Diseases (IFF/Fiocruz) and implement the precision medicine at a public health institute in Brazil.
Patients and Methods: This study comprises 26 cases with clinical suspicion of RASopathies. Sanger sequencing was used to screen variants in exons usually affected in the PTPN11 and HRAS genes for cases with clinical features of Noonan and Costello syndrome, respectively. Posteriorly, negative and new cases with clinical suspicion of RASopathy were analyzed by clinical or whole-exome sequencing.
Results: Molecular analysis revealed recurrent variants and a novel LZTR1 missense variant: 24 unrelated individuals with pathogenic variants [PTPN11(11), NF1(2), SOS1(2), SHOC2(2), HRAS(1), BRAF(1), LZTR (1), RAF1(1), KRAS(1), RIT1(1), a patient with co-occurrence of PTPN11 and NF1 mutations (1)]; familial cases carrying a known pathogenic variant in PTPN11 (mother-two children), and a previously undescribed paternally inherited variant in LZTR1. The comparative modeling analysis of the novel LZTR1 variant p.Pro225Leu showed local and global changes in the secondary and tertiary structures, showing a decrease of about 1% in the β-sheet content. Furthermore, evolutionary conservation indicated that Pro225 is in a highly conserved region, as observed for known dominant pathogenic variants in this protein.
Conclusion: Bringing precision medicine through NGS towards congenital syndromes promotes a better understanding of complex clinical and/or undiagnosed cases. The National Policy for Rare Diseases in Brazil emphasizes the importance of incorporating and optimizing diagnostic methodologies in the Unified Brazilian Health System (SUS). Therefore, this work is an important step for the NGS inclusion in diagnostic genetic routine in the public health system.
Keywords: RASopathy, RAS/MAPK pathway, genetic heterogeneity, cancer risk, DNA sequencing
Introduction
The RASopathies constitute one of the largest groups of genetic conditions, affecting approximately 1 in 1000–2000 individuals with molecular and clinical heterogeneity presentation. Noonan syndrome (NS; OMIM: 163950) is the most common disorder in this group, mainly characterized by short stature, facial features (including low-set and posteriorly rotated ears, downslanting palpebral fissures, epicanthal folds, ptosis, hypertelorism, and others), congenital heart defects and developmental delay of variable degree. Other manifestations can include broad or webbed neck, pectus carinatum or pectus excavatum, cryptorchidism in males, coagulation defects and pediatric cancer predisposition.1,2
Phenotypic variations of NS include the Noonan syndrome with multiple lentigines (NSML; formerly known as LEOPARD syndrome; OMIM # 151100) and Noonan-like syndrome with loose anagen hair 1 and 2 (NSLH1; OMIM # 607721 and NSLH2; OMIM # 617506). Furthermore, phenotypic overlap is observed in other syndromes that also comprise the RASopathy group, like the Cardiofaciocutaneous syndrome (CFCS; OMIM # 115150); Costello syndrome (CS; OMIM # 218040); and Neurofibromatosis type 1 (NF1; OMIM # 162200).
RASopathies are caused by dysregulation of RAS/mitogen-activated protein kinase (MAPK) cell signaling pathway, essential to cell growth, survival, and proliferation.3,4 Many genes responsible for encoding proteins involved in this pathway are associated with RASopathies (Figure 1). In NS, PTPN11 harbors most variants (50%), followed by SOS1 (10–13%); RAF1 (5%); RIT1 (5%); KRAS (<5%); LZTR1 (~8%); SOS2 (~4%); BRAF (<2%); MAP2K1 (<2%); NRAS (<1%); and CBL (<1%).5 Other genes have been proposed to be associated with NS, such as RRAS2, RASA2, and A2ML1, but further investigations are needed.6,7 The related disorders can share mutations in the same genes, what leads to the phenotypic overlap among affected individuals.
 |
Figure 1 Schematic representation of the canonical RAS/MAPK pathway and RASopathy-associated genes. This pathway is activated by growth factors binding to receptors tyrosine kinase (RTK) creating binding sites for the adaptor protein GRB2 and promoting relocation of SOS to the plasma membrane. The SHP2 (codified by PTPN11 gene) participates in the RAS activation by dephosphorylating several inhibitory phosphotyrosines. RAS switches from a guanosine diphosphate (GDP)-bound inactive form and the guanosine triphosphate (GTP)-bound active form. In the active state, the phosphorylation cascade RAF-MEK-ERK is initialized. The scaffold protein SHOC2 mediates the activation of the PP1C phosphatase, which dephosphorylates an inhibitory residue on RAF. Thus, the protein kinases MEK1 and MEK2 are activate and phosphorylate to activate the serine-/threonine-specific protein kinases - ERK, the RAS downstream effector pathway. NF1 and SPRED1 proteins are negative regulators of the RAS/MAPK pathway. The hyperactive of this pathway is commonly associated with RASopathies and the proteins in color ovals indicate the associations with diseases represented by corresponding rectangles, showed in the left side. Double color ovals effect indicates genes associated with more than one syndrome. In yellow, blue and rose, mutations on RAS/RAF activators causing NS and NS-like syndromes, and double color yellow-rose indicates association with both NS and NSML. In brown and green, mutations responsible for CS and CFCS, respectively, affect the RAS/MAPK backbone. In purple and pink, mutations in NF1 and SPRED1 negative regulators proteins cause neurofibromatosis type 1 and Legius.*Genes that encoding these proteins are still under investigation to determine their links with RASopathy. |
Different molecular strategies have been used in the diagnosis of these heterogeneous syndromes. DNA analysis by Sanger sequencing can be suitable in specific conditions, such as in the Costello syndrome, wherein the HRAS is the only gene currently known to be associated.8,9 Molecular screening for NS can also be performed by Sanger sequencing of a few hotspots located in exons 3 and 8 on the PTPN11.10,11 However, from a practical standpoint, these approaches are no longer effective and become more costly.12
Next-generation sequencing (NGS) is a highly informative and robust technique that simultaneously provides data from several genomic regions, incrementing the chances of identifying pathogenic variants related to a particular clinical condition. The technique also makes it possible to explore novel candidate genes that could not have been otherwise.12–14 Whole or clinical exome sequencing (WES or CES) has been mainly used in the diagnostic routine in the field of clinical genetics, contributing to the diagnosis of different clinical and molecular conditions.15–17
Herein we present our results of the molecular investigation in a cohort of Noonan syndrome or related disorders. The project was a pilot study intended to implement NGS approach in a Reference Center for Rare Diseases – Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira/Fiocruz (RSFD/IFF/Fiocruz), as part of the Brazilian National Unified Health System (SUS).
Materials and Methods
Clinical Evaluations
This study was approved by the Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira/Fiocruz Human Research Ethics Committee (approval number: 2.071.943) and a written informed consent was obtained from patients and/or parents for publication of the clinical details and clinical images, in compliance with the Declaration of Helsinki. From 2017 to 2021, twenty-six patients, comprising unrelated patients and two familial cases with clinical features of Noonan/Noonan-like syndrome, were selected for the molecular investigation.
The clinical features of patients are shown in Table 1.
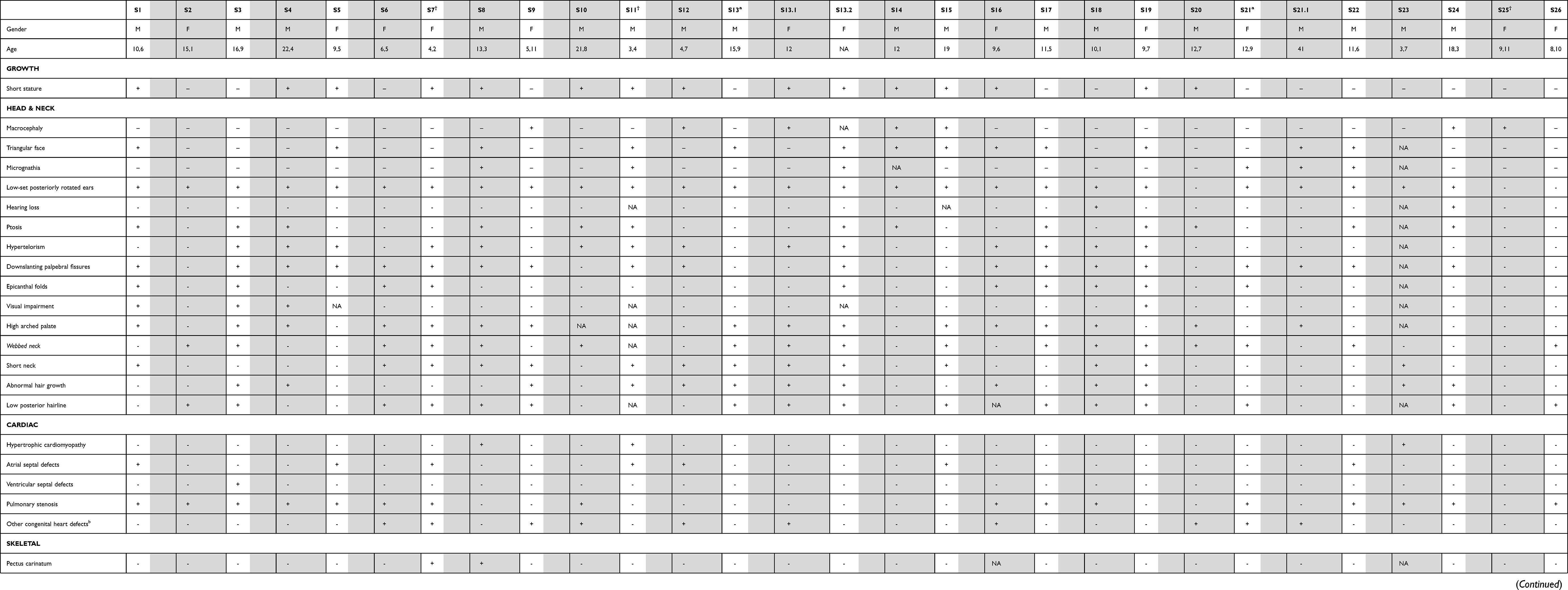 |
Table 1 Clinical Features of Patients |
DNA Samples
Genomic DNA (gDNA) was isolated from the peripheral blood of the probands and their parents/siblings (if available) using the PureLink Genomic DNA Kit (Invitrogen, California, USA), according to the manufacturer’s instructions.
DNA Analysis by Sanger Sequencing
The molecular screening was initially performed in six NS suspected cases [S1-S6], by Sanger sequencing18 of exons 3 and 8 of the PTPN11 gene; and in three CS suspected cases [S7-S9], in which exons 2–6 of the HRAS gene were sequenced, according to the prevalence of pathogenic variants in these regions for the suspected syndromes.8,10 Oligonucleotide sequences and PCR conditions are available upon request.
Fragments were sequenced in the automated DNA sequencer ABI 3730 (Applied Biosystems) using BigDye Sequencing Buffer (Applied Biosystems). Sequence data were analyzed using BioEdit Software version 7.2 (Ibis Biosciences).
DNA Analysis by Next-Generation Sequencing (NGS)
As described below, the next-generation sequencing was performed through clinical or whole-exome sequencing. Sequencing was performed in paired-end mode using the NextSeq 500 platform (Illumina). All pathogenic and candidate variants found by NGS were validated by Sanger sequencing.
Whole Exome Sequencing (WES)
WES was performed in three individuals negative by Sanger sequencing [S3, S4 e S7], one trio-case [S12] (which was previously negative for a commercial Noonan NGS panel); and a proband of a familial Noonan case [S13]. The genomic DNA library was prepared and captured using Nextera Rapid Capture Expanded Exome (Illumina). Sequencing was performed using the NextSeq 500 MID OUTPUT v2 kit 300 cycles (Illumina).
Clinical Exome Sequencing (CES)
CES was performed in three negative cases previously sequenced by Sanger [S5, S6, S8] and 15 other non-related cases [S10, S11, S14-S26]. In addition, DNA libraries were prepared using the TruSight One Expanded Sequencing Panel kit (Illumina), which were analyzed approximately 6700 target genes (list of genes available in: https://www.illumina.com/products/by-type/clinical-research-products/trusight-one.html). Sequencing was performed using the NextSeq 500/550 HIGH OUTPUT v2 kit 300 cycles.
NGS Data Analysis
Whole exome data analysis was performed by in house pipeline while the CES data analysis was performed by Vastation app,19 as shown below (Figure 2):
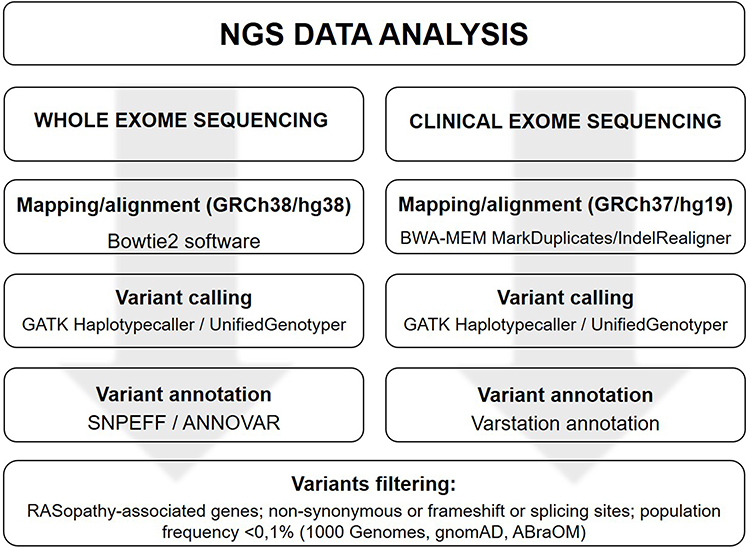 |
Figure 2 NGS data analysis. |
Comparative Modeling and Structural Analysis of LZTR1 Kelch Domain
We have performed comparative modeling for the LZTR1 variant through Swiss-Model server,20 using input parameters as default. The high-resolution structure of the Kelch Protein from Thlaspi Arvense (PDB ID: 5A10)21 was used as a template to create the three-dimensional structures of the LZTR1 Kelch domain (residues 50–425) wild-type and mutant. The long insertion (residues 328–381) was not modeled.
The resulting structures were refined using the GalaxyRefine22 service of GalaxyWEB server.23 Initial and optimized models were evaluated using the Swiss-Model server’s structure assessment tool. Models’ secondary structure composition was calculated using 2struc server24, and topology diagrams were drawn using Pro-origami25 and Inkscape (http://www.inkscape.org). Finally, figures corresponding to the sequence alignment and three-dimensional structures were generated through ALINE26 software and UCSF ChimeraX software.27
The ConSurf server28 was used to estimate the evolutionary conservation of amino acid positions in the LZTR1 protein based on the phylogenetic relations between homologous sequences.
Results
This study describes the molecular analysis of 26 cases with clinical suspicion of RASopathies.
Clinical features are presented in Table 1 and Figure 3.
 |
Figure 3 Clinical features of patients. (A) Patients with Noonan syndrome: (I) 14 individuals with variants in PTPN11 gene, three of them from the same family - S14, S14.1, S14.2; (II) Patient with variant in SOS1, the *Indicates that the image of the other case with variant in this gene (S23:T266K) is not available; (III) Patient with variant in RIT1; (IV) Patient with variant in KRAS; (V) Patient with variant in RAF1; (VI) Three individuals with variants in LZTR1 gene, two of them from the same family - S21, S21.1. (B) Patients with Noonan-like syndrome and other RASopathies: (I) Patient with Noonan syndrome-like with loose anagen hair 1 and a variant in SHOC2, the *Indicates that the image of the other case with variant in this gene (S11:S12G) is not available; (II) Patient with Cardiofaciocutaneous syndrome and a variant in BRAF, (III) Patient with Costello syndrome and a variant in HRAS; (IV) Patients with variants in NF1 gene, who present neurofibromatosis type 1 and Noonan-neurofibromatosis syndrome; (V) Patient with co-occurrence of PTPN11 and NF1 mutations presenting phenotype of Noonan syndrome and neurofibromatosis type 1. |
Recurrent Variants in Genes of the RAS/MAPK Pathway
The molecular analysis (Sanger, CES or WES) identified heterozygous pathogenic and likely pathogenic variants in the RASopathy-genes in 24 unrelated cases (PTPN11 [12], NF1 [2], SOS1 [2], SHOC2 [2], HRAS [1], BRAF [1], LZTR1 [1], RAF1 [1], KRAS [1], RIT1 [1], and a patient with co-occurrence of PTPN11 and NF1 mutations [1]). A familial case carrying a mutation in PTPN11 was also identified.
The molecular results are shown in Table 2.
 |
Table 2 Molecular Results of Cases with Noonan Syndrome and Related Disorders |
A Novel Variant in the LZTR1 Gene
A familial case carrying a novel missense variant in the LZTR1 gene was identified: p.Pro225Leu (S21 and S21.1). This variant has not been reported previously in the literature or described in the public genomic databases (gnomAD, aBraOM). This is a non-conservative amino acid substitution and in silico prediction classified this variant as probably damaging (MutationTaster, PolyPhen-2, SIFT, and PROVEAN).
In addition, multiple alignments of 100 vertebrate species and measurements of evolutionary conservation using phastCons and phyloP (through UCSC Genome Browser - https://genome.ucsc.edu/) indicated the nucleotide conservation score of 7.9 and the amino acid affected as highly conserved across species (97/100) (Multiz/autoMZ). When we performed conservation analysis through the Consurf Server, it was observed that Pro225 amino acid is in a highly conserved region, as well as other monoallelic variants already described (Figure 4A). On the other hand, most of the variants inherited as a recessive trait already described are in less conserved regions of the protein (Figure 4B).
 |
Figure 4 Evolutionary conservation of LZTR1 protein for amino acid regions associated with Noonan syndrome. (A) Monoallelic variants location previously described in literature (M91, Y119, H287, Y193, R284, S247). The novel variant (P225) is indicated by the red arrow. (B) Biallelic variants location previously described in literature (H121, I205, R107, Q217, R294). Notes: The degree of which amino acid position evolutionarily conserved is presented by the stronger shades of green indicating less conservation, and stronger shades of purple indicating greater conservation. |
The missense variant p.Pro225Leu is located in the kelch domain of the LZTR1 protein. A symmetric structure of six blades characterizes this domain, and each of them is composed by four antiparallel β-strands, as previously described.29,30 Several loops connect the β-strands within each Kelch motif, joining them to the adjacent motif. Mutant and wild-type LZTR1 structural model comparison showed differences in the loop that connects strand D (blade III) with strand A (blade IV) and changes in blade III in the local secondary structure of the Kelch domain (Figure 5A). Additionally, structure changes in most of the blades can be seen in the tertiary model (Figure 5B). Global changes promoted a decrease of approximately 1% in the β-sheet content (34.04% wide-type versus 32.97% mutant).
 |
Figure 5 Models of Kelch motifs of LZTR1 showing conformational changes caused by missense mutation p.P225L: (A) The secondary structure showing the P225 residue in the blade III of the Kelch motifs of LZTR1 wild type and (B) mutant – L225; (C) the tertiary structure of the wild-type and (D) mutant domain, showing within each panel the backbone (left) and solvent-exposed surface (right) showing mutant residue colored in red if surface-exposed. Notes: These Kelch motifs are organized to form a largely symmetric six-bladed β-propeller structure, and each motif constitutes a twisted β-sheet blade (motifs are numbered I to VI, starting from the N-terminus) composed by four antiparallel β-strands. |
Discussion
Molecular and clinical heterogeneity in RASopathies has been extensively documented over the years, constituting one of the largest groups of neurodevelopmental diseases.31 The NGS technique has contributed to expanding our knowledge about molecular physiopathology and the genotype-phenotype correlations.6,31–33
In the current study, we performed DNA sequencing using one or more molecular approaches (WES, CES, and/or Sanger) in a cohort of 26 Brazilian patients with clinical findings of Noonan syndrome or related disorders. Pathogenic or likely pathogenic variants were identified in 10 of the ~22 RASopathy-associated genes to date: PTPN11 [12]; NF1 [3]; SHOC2 [2]; SOS1 [2]; LZTR1 [2]; RIT1 [1]; RAF1 [1]; KRAS [1]; BRAF [1]; HRAS [1].
The clinical and genetic heterogeneity of the RASopathies syndromes per se indicates the need for a more comprehensive technique for molecular diagnosis; however, except for specific clinical phenotypes orientating a molecular target as a first-tier in the diagnostic flowchart.2,6,34,35 In such cases, Sanger sequencing can be used, especially when genes carry hotspots for the suspected phenotypes. By this mean, we screened six unrelated cases with NS phenotype for exons 3 and 8 in the PTPN11 gene. Although these exons are considered to carry the most frequent variants found in this syndrome,10,11 in the present study, only two out of six cases (S1 and S2) presented a missense pathogenic variant in exon 8 (c.923 A>G; p.Asn308Ser). Thereafter, the other four patients were sequenced by WES or CES, being identified pathogenic variants in exon 12 and 13 of the PTPN11 gene (S3 and S5, respectively); and in two other genes, SOS1 (S6) and BRAF (S4). In the latter patient, clinical reassignment for Cardiofaciocutaneous syndrome 1 was possible due to the p.Gln257Arg variant.
Sanger sequencing was also performed for a group of patients whose Costello syndrome phenotypes were considered (S7-S9). The only case presenting a pathogenic variant in HRAS was the S9, with the most common variant in CS (Gly12Ser), responsible for approximately 80% of the cases.8 Germline pathogenic variants leading to amino acid substitutions of the glycine residue at positions 12 or 13 are typical in CS,36,37 indicating this region as a mutational hotspot. However, due to the phenotypic overlap among Costello, Noonan, and CFC syndromes, Sanger sequencing may not always be successful as the first tier of choice. For S7 and S8 cases, the NGS had a decisive role in identifying mutations in different genes (PTPN11 and RAF1, respectively), which led to a clinical reassignment to NS.10,38
Among the patients carrying pathogenic variants in PTPN11 in the current study (n = 13), exon 8 was the most involved, while exon 3 did not show a higher prevalence than exon 12 and 13. In the other studies involving large Brazilian and non-Brazilian cohorts of RASopathies, exons 3 and 8 of the PTPN11 gene harbored the majority of mutations in NS cases (Figure 6).10,11 This difference probably occurred due to the smaller number of individuals in our study compared to the others. On the other hand, exon 13 seems to be more frequently affected in Brazilian patients, as observed in previously reported cases11 and in the present study. Thus, if Sanger sequencing is the approach of choice as the first tier for PTPN11 screening mutations, we recommend the inclusion of exon 13, in addition to exons 3 and 8, in the Brazilians with suspicion of NS.
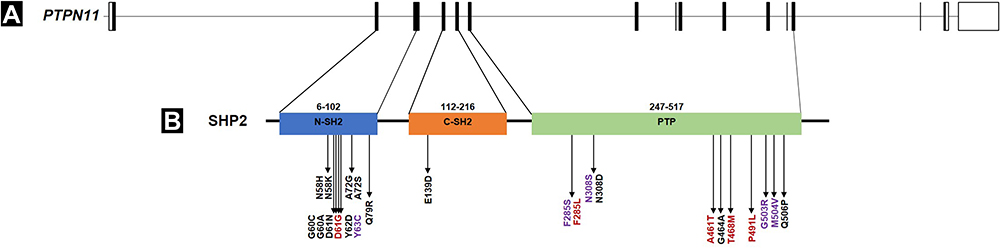 |
Figure 6 Scheme of the organization of PTPN11 gene, the SHP2 protein domains and distribution of residues usually affected by missense mutations associated with Noonan syndrome in Brazilian patients. (A) Exons (rectangles) and introns (bridging gaps/lines) boundaries of PTPN11 gene (NM_002834.5). Those exons that are translated as peptides are depicted in black, while those that are not, are left blank. (B) Presentation of SHP2 protein and corresponding functional domains: the two SH2 (Src Homology 2) domains (blue and Orange boxes) and the PTP (protein tyrosine phosphatase) domain (green box). The corresponding start and end position are showed above each domain (Uniprot: Q06124) while the distribution of the mutations in Brazilian patients are listed underneath. The mutations are colored differently: black for variants found in another Brazilian cohort,11 red for variants found in the present study, and purple for variants found in both studies. |
The phenotypic variability in RASopathies is remarkable, ranging from mild to severe features. Clinical manifestations can be highly variable, as observed in cases S7 and S17, with the same PTPN11 mutation (F285S). It is worth mentioning that in the case S7, in addition to presenting this pathogenic variant in PTPN11, she has also presented a frameshift variant in A2ML1 (Ser482fs), which was earlier considered to be modulating the clinical severity. The A2ML1 gene encodes the secreted protease inhibitor a-2-macroglobulin (A2M)-like-1, and missense variants have already been associated with the NS phenotype.7 Since then, this gene has been investigated along with the other genes associated with NS. However, a recent study identified some of the same A2ML1 variants previously reported in unaffected relatives of probands. The authors also described the co-occurrence of variants in A2ML1 and other RAS/MAPK pathway genes in affected patients, which does not cause a more severe phenotype.39 Furthermore, the frameshift variant Ser482fs was recently submitted to ClinVar (ID: 241884) as a benign variant, which suggests the severity of the phenotype in S7 patient was not due to this second variant. Unfortunately, she died during this study due to cardiac surgical complications.
The current work identified pathogenic variants in less common genes associated with NS (RAF1-S8, KRAS-S10, and RIT1-S22). Likewise, an increase in heart defects was also previously associated with variants in the RAF1 and RIT1 genes, as well as is described for variants in PTPN11.40,41 The S8 case presented hypertrophic cardiomyopathy and the S22, pulmonary stenosis, consistent with the literature.
Another atypical case with a severe phenotype (S24) presented with co-occurrence of PTPN11 and NF1 pathogenic heterozygous variants. The patient presented an overlapping of LEOPARD and neurofibromatosis type 1 characteristics without Lisch nodules and neurofibromas. The co-occurrence of pathogenic variants in these RASopathies genes are less frequent but have already been described involving PTPN11 and SOS1,42 PTPN11 e SHOC2,43 NF1 e KRAS,44 and PTPN11 and NF1 genes.45–47 Cardiac and neurological manifestations were more severe in patients presenting mutations in both PTPN11 and NF1 genes than observed in cases with only one of these genes mutated,47 similar to our S24 case.
The phenotypic combination of NS and neurofibromatosis 1 has also been reported in patients with a single variant in the NF1 gene,48–51 as found in our patient (S26) with both phenotypes but showing only one pathogenic variant in this gene (H1366fs). In our study, the NGS analysis indicating only one pathogenic variant in the NF1 gene reinforces the effectiveness of this approach, whereas, in previous work,51 the analysis of the PTPN11 and NF1 genes was performed by Sanger to conclude that there was not a second variant.
In addition to the known pathogenic variants identified in this study, a novel missense variant in the LZTR1 gene was found in a familial case S21 (P225L) segregating with the NS phenotype.
Since when the LZTR1 gene (Leucine zipper-like transcriptional regulator 1) was associated with NS phenotype,6 several studies have reported different pathogenic variants, showing dominant and recessive inheritance patterns.30,52–54 This gene codes for a protein that belongs to the superfamily of BTB/POZ (broad complex, tramtrack, and bric-A-brac/poxvirus and zinc finger). LZTR1 has an N-terminal domain composed of six Kelch motifs, followed by two BTB domains. The Kelch and BTB domains mediate protein–protein interactions, and LZTR1 acts as a substrate-specific adapter of a BCR (BTB-CUL3-RBX1) E3 ubiquitin-protein ligase complex that mediates Ras ubiquitination (HRAS, NRAS, and KRAS). Therefore, this protein is a negative regulator of the RAS-MAPK signaling pathway, controlling, and decreasing RAS levels.30,55,56
A three-dimensional model previously proposed of the LZTR1 Kelch domain demonstrated that the Kelch motifs form an essentially symmetric six-bladed β-propeller structure, where each motif constitutes a twisted β-sheet blade composed of four antiparallel β-strands. These strands stabilize the substrate-binding domain, and the monoallelic variants previously described affected the loops connecting the β-strands.30 Their results suggested that the Kelch domain-substrates interaction would be affected by monoallelic variants, which could change RAS ubiquitination.
We evaluated the potential impact of the new P225L variant in the LZTR1 structure through the in silico modeling based on the same homology model previously proposed.30 As noted for the other monoallelic variants, the P225L affects one of the β-strands loops, specifically the loop connecting the “a” and “d” strands. These results indicated that the variant promotes structural changes in the bi- and three-dimensional models in LZTR1-mutant.
To date, every LZTR1 pathogenic dominant variant described resides between codons 119 and 287, while the variants identified in recessive inheritance are located throughout the protein, including intronic regions (Figure 7).53 Additionally, in silico analysis of evolutionary conservation demonstrated the maximum conservation degree to position P225, where we identified the novel variant. It is worthy to mention that all other dominant NS variants previously described are located at sequences of maximum conservation degree. Conversely, the location of the recessive phenotype variants recently associated with NS53,54 has a low degree of conservation.
 |
Figure 7 Scheme of the domain organization of LZTR1 gene, LZTR1 protein domains and distribution of residues affected by missense mutations associated with autosomal dominant Noonan syndrome. (A) Exons (rectangles) and introns (bridging gaps/lines) boundaries of LZTR1 gene (NM_006767.4). Those exons that are translated as peptides are depicted in black, while those that are not, are left blank. (B) Presentation of LZTR1 protein and corresponding functional domains: the six K (Kelch) domains (Orange boxes) and the two BTB (Broad-Complex, Tramtrack and Bric a brac) domains (green boxes). The corresponding start and end position are showed above each domain (Uniprot: Q8N653), note that the mixed color gradient indicates overlapping of K6 and BTB1 domains (443–450). The distribution of the mutations associated with autosomal dominant Noonan syndrome is listed underneath and colored differently: black for variants previously reported in the literature, red for variants detected only in the present study, and purple for variant previously reported in the literature and also found in this study. |
Phenotypic variability has also been observed among NS patients carrying LZTR1 mutations. Indeed, the other dominant variant found in the current study (Gly248Arg-S20) and previously described6,52 was associated with the clinical presentation of individuals with some distinct characteristics. Additionally, as shown in a series of familial cases, clinical characteristics were quite variable, intra- and interfamily.57 Corroborating with such observation, in our study, the father (S21.1) and daughter (S21) carrying the novel LZTR1 variant showed distinct phenotypes. The daughter showed a typical NS phenotype, including pterygium colli, pectus excavatum, shield thorax, fetal pads, and congenital cardiopathy; and the father with a very mild clinical presentation showing down-slanting palpebral fissures, cubitus valgus, fetal pads, pectus excavatum and a pulmonary cardiac murmur with a systolic click (Table 1).
A significant clinical implication considering the RASopathy cases is the occurrence of cancer, particularly in Noonan and Costello syndromes.4,58
The overall risk of malignancy to NS was calculated for childhood cancer, and a standardized incidence ratio of 8.1 was established.58 The overall cancer risk by the age of 20 years old is estimated to be 4%.59 Because the cancer risk falls below 5%, routine cancer surveillance has not been recommended by the American Association for Cancer Research. However, for those with PTPN11 or KRAS variants known to be associated with the myeloproliferative disorder or JMML, physical examination every three to six months from time of diagnosis to age five years can be important.60
On the other hand, cancer risk in patients with CS was 42.4 times higher (rhabdomyosarcoma).58 Cases of acute lymphoblastic leukemia (ALL) have been reported in individuals with CFCS,61–63 in addition to hepatoblastoma,64 non-Hodgkin lymphoma65 and large B-cell lymphoma.63
No patients developed cancer to date in our cohort, even those who presented a PTPN11 mutation. The mean age in our 12 patients carrying a mutation in this gene was 11.9 years, whereas, in the other cohorts studied the average age of cancer onset was 3.6 years.58 The referral of a child in early life to a clinical geneticist with a diagnosis and follow-up care for cancer risk could improve the survival of individuals. This is a crucial issue to be considered, especially to predict if there is a bias for NS mortality rates in Brazil due to underdiagnosed cases in public health services.
Among patients with neurofibromatosis type 1, about 2–3% develop plexiform tumors and/or other types of tumors such as schwannomas and neurofibromas.66 Case S22 presented two facial schwannomas aggravated by Moyamoya disease (MMD). Unfortunately, a stroke caused her death. It is not uncommon the MMD and NF1 co-occurrence;67–69 and, the risk of this vasculopathy is greater in patients with variants in NF1 than in the general population.70
Finally, Brazil has a unified health system (SUS) that manages most of the patients with rare diseases. Furthermore, establishing a National Policy for Rare Diseases in the country (MS PORTARIA N° 3.123, 2016 DECEMBER, 28)71 emphasizes the importance of robust genetic techniques incorporation and optimization towards a precise molecular diagnosis. In this way, the NGS approach has been successfully introduced in our clinical practice.
Conclusion
We identified a novel familial variant in LZTR1 gene associated with Noonan syndrome phenotype and we demonstrated protein structural changes by in silico modeling of LZTR1-mutant. Additionally, the P225L is located in a known conserved region that harbors the other monoallelic pathogenic variants, which reinforces the possible pathogenicity of this variant. The diagnostic molecular precision towards congenital syndromes promotes a better understanding of complex clinical cases; and unravels genetic profile within heterogeneous clinical and genetic pathways, such as the RASopathy group. The genetic counseling and prenatal diagnosis opportunities have become envisaged for a broader number of patients in the Brazilian SUS, increasing demand for faster and more accurate diagnostics. The proper clinical prognosis and therapeutic opportunities are challenged by such precision medicine, providing an important contribution in this area.
Acknowledgments
We acknowledge all physicians who provided patient samples and data. We wish to thank all patients and families who participated in this study. The authors are grateful to the Genomic Platforms of FIOCRUZ (Rede de Plataformas Tecnológicas Fiocruz). We acknowledge the Darcy Fontoura de Almeida Computational Genomics Unit (LNCC/MCTI) and the Bioinformatics Laboratory (LNCC/MCTI) for carrying out the whole exome sequencing and bioinformatic analysis.
Funding
This study was funded in part by the Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira – IFF/Fiocruz and Genomic Platforms of FIOCRUZ (Rede de Plataformas Tecnológicas Fiocruz); CAPES/INCT – MCTI/CNPq/CAPES/FAPs 16/2014 – 88887.599057/2021-00; CAPES/DS – 88882.443088/2019-01; CAPES 3384/2013 23038.010041/2013-13; and FAPERJ (E-26/202.903/2015 - Cientista do Nosso Estado). Conselho Nacional de Desenvolvimento Científico e Tecnológico - 465549/2014-4; Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - 88887.136366/2017-00; Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul - 17/2551-0000521-0.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Rauen KA. The RASopathies. Annu Rev Genomics Hum Genet. 2013;14:355–369. doi:10.1146/annurev-genom-091212-153523
2. Tidyman WE, Rauen KA. Pathogenetics of the RASopathies. Hum Mol Genet. 2016;25:123–132. doi:10.1093/hmg/ddw191
3. Yoon S, Seger R. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors. 2006;24(1):21–44. doi:10.1080/02699050500284218
4. Hernández-Martín A, Torrelo A. Rasopathies: developmental disorders that predispose to cancer and skin manifestations. Actas Dermosifiliogr. 2011;102:402–416. doi:10.1016/j.ad.2011.02.010
5. Roberts AE. Noonan syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al. editors. GeneReviews® [Internet]. Seattle: University of Washington, Seattle; 2001:1993–2022. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1124/.
6. Chen PC, Yin J, Yu HW, et al. Next-generation sequencing identifies rare variants associated with Noonan syndrome. Proc Natl Acad Sci USA. 2014;111:11473–11478. doi:10.1073/pnas.1324128111
7. Vissers LE, Bonetti M, Overman JP, et al. Heterozygous germline mutations in A2ML1 are associated with a disorder clinically related to noonan syndrome. Eur J Hum Genet. 2015;23:317–324. doi:10.1038/ejhg.2014.115
8. Gripp KW, Lin AE. Costello syndrome: a Ras/mitogen activated protein kinase pathway syndrome (rasopathy) resulting from HRAS germline mutations. Genet Med. 2012;14(3):285–292. doi:10.1038/gim.0b013e31822dd91f
9. Gripp KW, Rauen KA. Costello syndrome. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews® [Internet]. Seattle: University of Washington, Seattle; 2006:1993–2022.
10. Tartaglia M, Kalidas K, Shaw A, et al. PTPN11 mutations in noonan syndrome: molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity. Am J Hum Genet. 2002;70:1555–1563. doi:10.1086/340847
11. Bertola DR, Castro MAA, Yamamoto GL, et al. Phenotype-genotype analysis of 242 individuals with RASopathies: 18-year experience of a tertiary center in Brazil. Am J Med Genet C Semin Med Genet. 2020;184(4):896–911. doi:10.1002/ajmg.c.31851
12. Lapunzina P, López RO, Rodríguez-Laguna L, et al. Impact of NGS in the medical sciences: genetic syndromes with an increased risk of developing cancer as an example of the use of new technologies. Genet Mol Biol. 2014;37(1 SUPPL):241–249. doi:10.1590/S1415-47572014000200010
13. von Bubnoff A. Next-generation sequencing: the race is on. Cell. 2008;132:721–723. doi:10.1016/j.cell.2008.02.028
14. Choi M, Scholl UI, Ji W, et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci USA. 2009;106:19096–19101. doi:10.1073/pnas.0910672106
15. Lee H, Deignan JL, Dorrani N, et al. Clinical exome sequencing for genetic identification of rare mendelian disorders. JAMA. 2014;312(18):1880–1887. doi:10.1001/jama.2014.14604
16. Matthijs G, Souche E, Alders M, et al.; European Society of Human Genetics. Guidelines for diagnostic next-generation sequencing. Eur J Hum Genet. 2016;24(1):2–5. doi:10.1038/ejhg.2015.226
17. Ramos-Fuentes F, González-Meneses A, Ars E, et al. Genetic diagnosis of rare diseases: past and present. Adv Ther. 2020;37(2):29–37. doi:10.1007/s12325-019-01176-1
18. Sanger F, Nicklen S, Coulson R. DNA sequencing with chain-terminating. Biochemistry. 1977;74:5463–7546.
19. Faria ACO, Caraciolo MP, Minillo RM, et al. Varstation: a complete and efficient tool to support NGS data analysis. bioRxiv. 2019:833582. doi:10.1101/833582
20. Waterhouse A, Martino B, Stefan B, et al. Swiss-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296–W303. doi:10.1093/nar/gky427
21. Gumz F, Joern K, Daniela E, et al. The crystal structure of the thiocyanate-forming protein from Thlaspi arvense, a kelch protein involved in glucosinolate breakdown. Plant Mol Biol. 2015;89(1–2):67–81. doi:10.1007/s11103-015-0351-9
22. Heo L, Park H, Seok C. GalaxyRefine: protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013;41:W384–W388. doi:10.1093/nar/gkt458
23. Ko J, Park H, Heo L, et al. GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res. 2012;40(W1):W294–W297. doi:10.1093/nar/gks493
24. Klose DP, Wallace BA, Janes RW. 2Struc: the secondary structure server. Bioinformatics. 2010;26(20):2624–2625. doi:10.1093/bioinformatics/btq480
25. Stivala A, Wybrow M, Wirth A, et al. Automatic generation of protein structure cartoons with pro-origami. Bioinformatics. 2011;27(23):3315–3316. doi:10.1093/bioinformatics/btr575
26. Bond CS, Schüttelkopf AW. ALINE: a WYSIWYG protein-sequence alignment editor for publication-quality alignments. Acta Crystallogr Sect D Struct Biol. 2009;D65:510–512.
27. Goddard TD, Huang CC, Meng EC, et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 2018;27:14–25. doi:10.1002/pro.3235
28. Ashkenazy H, Abadi S, Martz E, et al. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016;44(W1):W344–W350. doi:10.1093/nar/gkw408
29. Li X, Zhang D, Hannink M, et al. Crystal structure of the Kelch domain of human Keap1. J Biol Chem. 2004;279(52):54750–54758. doi:10.1074/jbc.M410073200
30. Motta M, Fidan M, Bellacchio E, et al. Dominant Noonan syndrome-causing LZTR1 mutations specifically affect the Kelch domain substrate-recognition surface and enhance RAS-MAPK signaling. Hum Mol Genet. 2019;28(6):1007–1022. doi:10.1093/hmg/ddy412
31. Tajan M, Paccoud R, Branka S, et al. The RASopathy family: consequences of germline activation of the RAS/MAPK pathway. Endocr Rev. 2018;39(5):676–700. doi:10.1210/er.2017-00232
32. Yamamoto GL, Aguena M, Gos M, et al. Rare variants in SOS2 and LZTR1 are associated with Noonan syndrome. J Med Genet. 2015;52(6):413–421. doi:10.1136/jmedgenet-2015-103018
33. Aoki Y, Niihori T, Inoue S, et al. Recent advances in RASopathies. J Hum Genet. 2016;61(1):33–39. doi:10.1038/jhg.2015.114
34. Lee BH, Yoo H-W. Noonan syndrome and RASopathies: clinical features, diagnosis and management. J Genet Med. 2019;16(1):1–9. doi:10.5734/JGM.2019.16.1.1
35. Lepri FR, Scavelli R, Digilio MC, et al. Diagnosis of noonan syndrome and related disorders using target next generation sequencing. BMC Med Genet. 2014;15:14. doi:10.1186/1471-2350-15-14
36. Sol-Church K, Gripp KW. The molecular basis of Costello syndrome. In: Zenker M, editor. Noonan Syndrome and Related Disorders. Monographs in Human Genetics. Basel, Switzerland: Karger; 2009:94–103.
37. Giannoulatou E, McVean G, Taylor IB, et al. Contributions of intrinsic mutation rate and selfish selection to levels of de novo HRAS mutations in the paternal germline. Proc Natl Acad Sci USA. 2013;110:20152–20157. doi:10.1073/pnas.1311381110
38. Razzaque MA, Nishizawa T, Komoike Y, et al. Germline gain-of-function mutations in RAF1 cause Noonan syndrome. Nat Genet. 2007;39(8):1013–1017. doi:10.1038/ng2078
39. Brinkmann J, Lissewski C, Pinna V, et al. The clinical significance of A2ML1 variants in Noonan syndrome has to be reconsidered. Eur J Hum Genet. 2021;29(3):524–527. doi:10.1038/s41431-020-00743-3
40. Pandit B, Sarkozy A, Pennacchio LA, et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet. 2007;39(8):1007–1012. doi:10.1038/ng2073
41. El Bouchikhi I, Belhassan K, Moufid FZ, et al. Noonan syndrome-causing genes: molecular update and an assessment of the mutation rate. Int J Pediatr Adolesc Med. 2016;3(4):133–142. doi:10.1016/j.ijpam.2016.06.003
42. Brasil AS, Malaquias AC, Wanderley LT, et al. Co-occurring PTPN11 and SOS1 gene mutations in Noonan syndrome: does this predict a more severe phenotype? Arq Bras Endocrinol Metabol. 2010;54(8):717–722. doi:10.1590/S0004-27302010000800009
43. Ekvall S, Hagenäs L, Allanson J, et al. Co-occurring SHOC2 and PTPN11 mutations in a patient with severe/complex Noonan syndrome-like phenotype. Am J Med Genet A. 2011;155A(6):1217–1224. doi:10.1002/ajmg.a.33987
44. Baquedano LI, Izquierdo ÁS, Oliván Del Cacho MJ. Rasopathies case report: concurrence of two pathogenic variations de novo in NF1 and KRAS genes in a patient. BMC Pediatr. 2019;19(1):92–95. doi:10.1186/s12887-019-1463-1
45. Bertola DR, Pereira AC, Passetti F, et al. Neurofibromatosis-Noonan syndrome: molecular evidence of the concurrence of both disorders in a patient. Am J Med Genet A. 2005;136(3):242–245. doi:10.1002/ajmg.a.30813
46. Thiel C, Wilken M, Zenker M, et al. Independent NF1 and PTPN11 mutations in a family with neurofibromatosis-Noonan syndrome. Am J Med Genet A. 2009;149A(6):1263–1267. doi:10.1002/ajmg.a.32837
47. Prada CE, Zarate YA, Hagenbuch S, et al. Lethal presentation of neurofibromatosis and Noonan syndrome. Am J Med Genet A. 2011;155A(6):1360–1366. doi:10.1002/ajmg.a.33996
48. De Luca A, Bottillo I, Sarkozy A, et al. NF1 gene mutations represent the major molecular event underlying neurofibromatosis-Noonan syndrome. Am J Hum Genet. 2005;77(6):1092–1101. doi:10.1086/498454
49. Nyström AM, Ekvall S, Allanson J, et al. Noonan syndrome and neurofibromatosis type I in a family with a novel mutation in NF1. Clin Genet. 2009;76(6):524–534. doi:10.1111/j.1399-0004.2009.01233.x
50. Ekvall S, Sjörs K, Jonzon A, et al. Novel association of neurofibromatosis type 1-causing mutations in families with neurofibromatosis-Noonan syndrome. Am J Med Genet A. 2014;164A(3):579–587. doi:10.1002/ajmg.a.36313
51. Baralle D, Mattocks C, Kalidas K, et al. Different mutations in the NF1 gene are associated with Neurofibromatosis-Noonan syndrome (NFNS). Am J Med Genet A. 2003;119A(1):1–8. doi:10.1002/ajmg.a.20023
52. Umeki I, Niihori T, Abe T, et al. Delineation of LZTR1 mutation-positive patients with Noonan syndrome and identification of LZTR1 binding to RAF1-PPP1CB complexes. Hum Genet. 2019;138(1):21–35. doi:10.1007/s00439-018-1951-7
53. Johnston JJ, van der Smagt JJ, Rosenfeld JA, et al. Autosomal recessive Noonan syndrome associated with biallelic LZTR1 variants. Genet Med. 2018;20(10):1175–1185. doi:10.1038/gim.2017.249
54. Pagnamenta AT, Kaisaki PJ, Bennett F, et al. Delineation of dominant and recessive forms of LZTR1-associated Noonan syndrome. Clin Genet. 2019;95(6):693–703. doi:10.1111/cge.13533
55. Steklov M, Pandolfi S, Baietti MF, et al. Mutations in LZTR1 drive human disease by dysregulating RAS ubiquitination. Science. 2018;362(6419):1177–1182. doi:10.1126/science.aap7607
56. Bigenzahn JW, Collu GM, Kartnig F, et al. LZTR1 is a regulator of RAS ubiquitination and signaling. Science. 2018;362(6419):1171–1177. doi:10.1126/science.aap8210
57. Chinton J, Huckstadt V, Mucciolo M, et al. Providing more evidence on LZTR1 variants in Noonan syndrome patients. Am J Med Genet A. 2020;182(2):409–414. doi:10.1002/ajmg.a.61445
58. Kratz CP, Franke L, Peters H, et al. Cancer spectrum and frequency among children with noonan, costello, and cardio-facio-cutaneous syndromes. Br J Cancer. 2015;112:1392–1397. doi:10.1038/bjc.2015.75
59. Lodi M, Boccuto L, Carai A, et al. Low-grade gliomas in patients with noonan syndrome: case-based review of the literature. Diagnostics. 2020;10(8):582. doi:10.3390/diagnostics10080582
60. Villani A, Greer MC, Kalish JM, et al. Recommendations for cancer surveillance in individuals with RASopathies and other rare genetic conditions with increased cancer risk. Clin Cancer Res. 2017;23(12):e83–e90. doi:10.1158/1078-0432.CCR-17-0631
61. Niihori T, Aoki Y, Narumi Y, et al. Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat Gen. 2006;38:2005–2007. doi:10.1038/ng1749
62. Makita Y, Narumi Y, Yoshida M, et al. Leukemia in Cardio-facio-cutaneous (CFC) syndrome: a patient with a germline mutation in BRAF proto-oncogene. J Pediatr Hematol Oncol. 2007;29(5):287–290. doi:10.1097/MPH.0b013e3180547136
63. Rauen KA, Tidyman WE, Estep AL, et al. Molecular and functional analysis of a novel MEK2 mutation in cardio-facio-cutaneous syndrome: transmission through four generations. Am J Med Genet A. 2010;152A:807–814.
64. Al-Rahawan MM, Chute DJ, Sol-Church K, et al. Hepatoblastoma and heart transplantation in a patient with cardio-facio-cutaneous syndrome. Am J Med Genet A. 2007;143A:1481–1488.
65. Ohtake A, Aoki Y, Saito Y, et al. Non-Hodgkin lymphoma in a patient with cardiofaciocutaneous syndrome. J Pediatr Hematol Oncol. 2011;33:e342–e346. doi:10.1097/MPH.0b013e3181df5e5b
66. Evans DGR, Salvador H, Chang VY, et al. Cancer and central nervous system tumor surveillance in pediatric neurofibromatosis 1. Clin Cancer Res. 2017;23(12):e46–e53. doi:10.1158/1078-0432.CCR-17-0589
67. Rosser TL, Vezina G, Packer RJ. Cerebrovascular abnormalities in a population of children with neurofibromatosis type 1. Neurology. 2005;64(3):553–555. doi:10.1212/01.WNL.0000150544.00016.69
68. Cairns AG, North KN. Cerebrovascular dysplasia in neurofibromatosis type 1. J Neurol Neurosurg Psychiatry. 2008;79(10):1165–1170. doi:10.1136/jnnp.2007.136457
69. Han C, Yang WZ, Zhang HT, et al. Clinical characteristics and long-term outcomes of moyamoya syndrome associated with neurofibromatosis type 1. J Clin Neurosci. 2015;22(2):286–290. doi:10.1016/j.jocn.2014.05.046
70. Santoro C, Di Rocco F, Kossorotoff M, et al. Moyamoya syndrome in children with neurofibromatosis type 1: Italian-French experience. Am J Med Genet A. 2017;173(6):1521–1530. doi:10.1002/ajmg.a.38212
71. Ministério da Saúde, Gabinete do Ministro. Portaria N° 199, DE 30 De Janeiro De 2014 and Portaria N° 3.123, De 28 De Dezembro De 2016; 2014. Available from: http://bvsms.saude.gov.br/bvs/saudelegis/gm/2014/prt0199_30_01_2014.html.
72. Yoshida R, Nagai T, Hasegawa T, et al. Two novel and one recurrent PTPN11 mutations in LEOPARD syndrome. Am J Med Genet A. 2004;130A(4):432–434. doi:10.1002/ajmg.a.30281
73. Rodriguez-Viciana P, Tetsu O, Tidyman WE, et al. Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science. 2006;311(5765):1287–1290. doi:10.1126/science.1124642
74. Sarkozy A, Conti E, Seripa D, et al. Correlation between PTPN11 gene mutations and congenital heart defects in Noonan and LEOPARD syndromes. J Med Genet. 2003;40(9):704–708. doi:10.1136/jmg.40.9.704
75. Tartaglia M, Pennacchio LA, Zhao C, et al. Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat Genet. 2007;39(1):75–79. doi:10.1038/ng1939
76. Aoki Y, Niihori T, Kawame H, et al. Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat Genet. 2005;37(10):1038–1040. doi:10.1038/ng1641
77. Schubbert S, Zenker M, Rowe SL, et al. Germline KRAS mutations cause Noonan syndrome. Nat Genet. 2006;38(3):331–336. doi:10.1038/ng1748
78. Cordeddu V, Di Schiavi E, Pennacchio LA, et al. Mutation of SHOC2 promotes aberrant protein N-myristoylation and causes Noonan-like syndrome with loose anagen hair. Nat Genet. 2009;41(9):1022–1026. doi:10.1038/ng.425
79. Tartaglia M, Martinelli S, Stella L, et al. Diversity and functional consequences of germline and somatic PTPN11 mutations in human disease. Am J Hum Genet. 2006;78(2):279–290. doi:10.1086/499925
80. Tartaglia M, Mehler EL, Goldberg R, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet. 2001;29(4):465–468. doi:10.1038/ng772
81. Aoki Y, Niihori T, Banjo T, et al. Gain-of-function mutations in RIT1 cause Noonan syndrome, a RAS/MAPK pathway syndrome. Am J Hum Genet. 2013;93(1):173–180. doi:10.1016/j.ajhg.2013.05.021
82. Roberts AE, Araki T, Swanson KD, et al. Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat Genet. 2007;39(1):70–74. doi:10.1038/ng1926
83. Messiaen L, Callens T, De Paepe A, et al. Characterisation of two different nonsense mutations, C6792A and C6792G, causing skipping of exon 37 in the NF1 gene. Hum Genet. 1997;101(1):75–80. doi:10.1007/s004390050590
84. Digilio MC, Conti E, Sarkozy A, et al. Grouping of multiple-lentigines/LEOPARD and Noonan syndromes on the PTPN11 gene. Am J Hum Genet. 2002;71(2):389–394. doi:10.1086/341528
85. Syrbe S, Eberle K, Strenge S, et al. Neurofibromatose Typ 1 und assoziierte Krankheiten bei 27 Kindern und Jugendlichen [Neurofibromatosis type 1 and associated clinical abnormalities in 27 children]. Klin Padiatr. 2007;219(6):326–332. doi:10.1055/s-2007-973086
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.