")
Back to Journals » Cancer Management and Research » Volume 10
Rasip1 is a RUNX1 target gene and promotes migration of NSCLC cells
Authors Chen Y, Zhang L , Liu L, Sun S , Zhao X, Wang Y, Zhang Y, Du J, Gu L
Received 19 March 2018
Accepted for publication 22 June 2018
Published 12 October 2018 Volume 2018:10 Pages 4537—4552
DOI https://doi.org/10.2147/CMAR.S168438
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Antonella D'Anneo
Yan Chen,1 Lin Zhang,1 Lei Liu,1 Shixiu Sun,2 Xuyang Zhao,1 Yueyuan Wang,2 Yujie Zhang,2,3 Jun Du,2,3 Luo Gu1–3
1Department of Biochemistry and Molecular Biology, Nanjing Medical University, Nanjing, Jiangsu, People’s Republic of China; 2Department of Physiology, Nanjing Medical University, Nanjing, Jiangsu, People’s Republic of China; 3Cancer Center, Nanjing Medical University, Nanjing, Jiangsu, People’s Republic of China
Background: Runt-related transcription factor 1 (RUNX1), an essential regulator of hematopoiesis, is overexpressed in patients with nonsmall-cell lung cancer (NSCLC) and is correlated with enhanced metastatic ability. Ras-interacting protein 1 (Rasip1), a potential oncogene, is required for blood vessel formation, and recently, it has been shown that Rasip1 is widely expressed in NSCLC patients. We noticed that Rasip1 promoter contains several potential RUNX1-binding sequences. However, the relationship between Rasip1 and RUNX1 in NSCLC is still unknown. In this study, the potential function of RUNX1 involving in Rasip1 expression and the potential role of Rasip1 in lung cancer cells were investigated.
Materials and methods: Rasip1 and RUNX1 expressions were analyzed by quantitative reverse transcription polymerase chain reaction (qRT-PCR) and Western blotting in NSCLC cells lines. A549 and H1299 cells were transfected with plasmids or interfering RNA (siRNA) to upregulate or downregulate the expression of Rasip1 and RUNX1. Cell motility was assessed by transwell and wound-healing assay. Location of Rasip1 and RUNX1 was detected via immunofluorescence. Meanwhile, chromatin immunoprecipitation was done using an anti-RUNX1 antibody. Rasip1 promoter was constructed, and cells were lysed for the analysis of luciferase activity.
Results: In this study, we showed that ectopic expression or knockdown of RUNX1 resulted in a significant increase or reduction in Rasip1 expression, respectively. RUNX1 bound directly to a specific DNA sequence within Rasip1 promoter and modulated its transcription. Furthermore, silencing of Rasip1 inhibited the migration of RUNX1-overexpressing NSCLC cells through inactivation of Rac1 pathway. Moreover, we found that Rasip1 was expressed ubiquitously in NSCLC cells lines and enhanced cell migration. In addition, EGFR signaling was involved both in the expression and the subcellular localization of Rasip1.
Conclusion: Our data indicated that Rasip1 is regulated in part by the transcription factor RUNX1 and might be developed as a therapeutic target for NSCLC.
Keywords: Rasip1, RUNX1, EGF, migration, nonsmall-cell lung cancer
Introduction
Nonsmall-cell lung cancer (NSCLC) accounts for ~80% of all lung cancer cases, and its metastasis is a major cause of high mortality.1 The EGFR tyrosine kinase inhibitors (TKIs) are an effective treatment for NSCLC patients with EGFR-activating mutations.2 However, EGFR T790M and K-Ras mutations result in resistance to TKIs, which limit the effectiveness of chemotherapy.3 Therapies targeting Ras downstream effectors have been adopted in NSCLC patients.4,5 Thus, identification of novel Ras effectors as promising NSCLC therapeutic targets is needed.
Ras-interacting protein 1 (Rasip1), an emerging Ras effector, has been identified as the endothelial-restricted protein that contains a Ras-associating (RA) domain and a dilute (DIL) domain.6 A previous study indicated that Rasip1 is essential for vascular development and angiogenesis. Depletion of Rasip1 in mouse islet endothelial cells (MS1) inhibited angiogenesis and cell motility.7 Loss of Rasip1 in human umbilical vein endothelial cells impaired cell–cell attachment and increased basal permeability.8 In addition, elimination of Rasip1 in endothelial cell (ECs) reduced cell polarity, which was necessary for Rap1-induced cell spreading and endothelial barrier.9,10 Rasip1 mediated Cdc42 and Rac1 signaling during vascular tubulogenesis.9 Activated Rac1 is known to induce the lamellipodia formation or membrane ruffles, which plays a critical role for tumor metastasis.11,12 Rasip1 has been found in the human lung tissue and NSCLC patients. However, the role of Rasip1 in NSCLC pathogenesis remains unknown.
Runt-related transcription factor 1 (RUNX1, also known as AML1), a member of the RUNX family, contains a conserved Runt domain that binds to core-binding factor subunit-β (CBFβ) and specific DNA sequences (5′-TGTGGTT-3′). RUNX1 is required for normal hematopoietic development, and the function of RUNX1 in leukemia is well established.13 It is now clear that RUNX1 plays an essential and paradoxical role in cancer development and progression. In hematopoietic diseases, RUNX1 mutations often lead to accelerated tumor development,14 whereas some level of wild-type (WT) RUNX1 activity is still necessary to promote the leukemogenic cell growth and survival.15 RUNX1 has been identified as a downregulated gene in metastasis-prone solid tumors, acting as a tumor suppressor.16 However, the expression of RUNX1 is upregulated in patients with epithelial cancers and promotes tumor growth and metastasis.17–19 In lung adenocarcinoma, RUNX1 is one of the significantly overexpressed genes and could be regarded as a biomarker for cancer diagnosis.17,20 Unfortunately, the RUNX1 target gene(s) in lung cancer is still unclear. To our interest, several potential RUNX1-binding sequences were found ubiquitously within Rasip1 promoter. We are inspired to hypothesize that RUNX1 may act as a transcription factor of Rasip1.
In this study, we found that Rasip1 could enhance Rac1 activity and ERK phosphorylation, thereby promoting RUNX1-mediated migration in NSCLC cells lines. RUNX1 bound directly to Rasip1 promoter and enhanced the expression of Rasip1. In addition, EGF could induce the plasma membrane translocation of Rasip1 and affect Rasip1 expression. These findings revealed RUNX1 as a transcriptional regulator of Rasip1 and uncovered a critical role of Rasip1 in NSCLC metastasis.
Materials and methods
Cell lines and cell culture
Human BEAS-2B, MRC-5, H1299, A549, SPC-A-1, PC9 and NCI-H292 cell lines were purchased from the Cell Biology Institute of Chinese Academy of Sciences (Shanghai, China) and cultured in DMEM (Biological Industries, Beit-Haemek, Israel) with 10% fetal bovine serum (FBS; Thermo Fisher Scientific, Waltham, MA, USA). The cells were serum-starved overnight, and then treated with EGF (R&D Systems, Inc., Minneapolis, MN, USA) and U0126 (Promega Corporation, Fitchburg, WI, USA) or CBFβ-Runx1 Inhibitor II Ro5-3335 (EMD Millipore, Billerica, MA, USA) for various time periods as indicated before harvest.
Plasmids and siRNAs
Full-length cDNA of human Rasip1 was amplified from pDONRTM223-Rasip1 plasmid (Youbio, Hunan, China) using the following sequences: 5′-CGGAATTCCGGCCACCATGCTGTCTGGTGAACG-3′ (sense) and 5′-GGGGTACCCCAGGAGACGTGGCCACG-3′ (antisense). EcoRI, KpnI restriction sites are underlined. The PCR products were cloned into the pEGFP-N1 vector (Clontech, Palo Alto, CA, USA). The primers for human RUNX1 are as follows: 5′-CCGGAATTCATGCGTATCCCCGTAG-3′ (sense) and 5′-CCGCTCGAGGTAGGGCCTCCACACG-3′ (antisense). EcoRI, XhoI restriction sites are underlined. The PCR products were subcloned into the pCMV-C-HA vector (Beyotime, Nantong, China). A549 and H1299 cells were transfected with plasmids or siRNAs (GenePharma, Shanghai, China) using Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA). The siRNA sequences targeting Rasip1 are as follows: siRasip1-#1, 5′-GCAGCCUCUGUCAAGUCUUTT-3′; siRasip1-#2, 5′-CCAGGACUUUGUGGUGUAUTT-3′; and siRasip1-#3, 5′-GCGAACCUUGAAAUGUCACTT-3′. The siRNA sequences targeting RUNX1 are as follows: siRUNX1-#1, 5′-CCAGGUUGCAAGAUUUAAUTT-3′; siRUNX1-#2, 5′-UCGAAGUGGAAGAGGGAAATT-3′; and siRUNX1-#3, 5′-GGCAGAAACUAGAUGAUCATT-3′. Those sequences targeting RUNX2 are as follows: siRUNX2-#1, 5′-GGUCCUAUGACCAGUCUUATT-3′; siRUNX2-#2, 5′-CUCUGCACCAAGUCCUUUUTT-3′; and siRUNX2-#3, 5′-CCAGCCACCUUUACUUACATT-3′. Those sequences targeting RUNX3 are as follows: siRUNX3-#1, 5′-UGACGAGAACUACUCCGCUTT-3′; siRUNX3-#2, 5′-CCUUCAAGGUGGUGGCAUUTT-3′; and siRUNX3-#3, 5′-CCCUGACCAUCACUGUGUUTT-3′.
Western blotting and quantitative reverse transcription polymerase chain reaction (qRT-PCR)
Western blotting was performed as previously described.21 The cells were lysed in RIPA buffer containing phenylmethane sulfonyl fluoride (PMSF) and protease inhibitor cocktail. The cellular lysates were detected by Western blotting with antibodies against Rasip1, RUNX1, RUNX3 (Abcam, San Francisco, USA), ERK, phospho-ERK, HA, phospho-RUNX1 (Cell Signaling Technology, Danvers, MA, USA), Rac1 (EMD Millipore), Rasip1, RUNX2 (Protein Tech Group, Wuhan, China) and GAPDH (Thermo Fisher Scientific). Total RNA was extracted by TRIzol reagent (Thermo Fisher Scientific). The reverse transcriptase reaction was performed using a HiScript II Q RT SuperMix (Vazyme, Nanjing, China). The qRT-PCR was conducted using the ABI StepOne™ real-time PCR system (Thermo Fisher Scientific) with AceQ qPCR SYBR Green Master Mix (Vazyme). Primers for Rasip1, RUNX1, RUNX2, RUNX3 and GAPDH are listed in Table 1.
 | Table 1 Primers for Rasip1, RUNX1, RUNX2, RUNX3 and GAPDH Abbreviation: Rasip1, Ras-interacting protein 1. |
Immunofluorescence microscopy
Twelve hours after serum starvation, cells were treated with or without 50 ng/mL EGF for 30 minutes. Cells were fixed in 4% paraformaldehyde for 20 minutes and then permeabilized with 0.1% TritonX-100 before blocking in 1% BSA for 1 hour. Cells were incubated with anti-Rasip1 (Abgent, San Diego, CA, USA) or anti-HA (Cell Signaling Technology) antibodies overnight at 4°C. After several washes, cells were then incubated with Alexa-coupled secondary antibody (Thermo Fisher Scientific) and FITC-phalloidin (Sigma-Aldrich Co., St Louis, MO, USA). DAPI (Southern Biotech, Birmingham, AL, USA) was added and incubated with cells before observation. Images were acquired with an Olympus BX51 microscope.
Rac1 activation assay
Activation of Rac1 was detected according to the method previously described.22 Briefly, GST-PAK-CRIB fusion proteins were expressed in BL21 bacteria and captured on MagneGST Glutathione Particles (Promega Corporation). Equal concentrations of total cellular protein were incubated with fusion proteins at 4°C for 60 minutes to pull down GTP-Rac1. After incubation, beads were washed and resuspended in 2× SDS sample buffer and boiled. Rac1 activation was analyzed by Western blotting with anti-Rac1 antibody.
Chromatin immunoprecipitation (ChIP) assay
The ChIP assay was performed using the ChIP Assay Kit (Upstate; EMD Millipore), as previously described.23 Nuclear DNAs were sonicated to an average length of about <1,000 bp and incubated with RUNX1 antibody or normal rabbit IgG (Abcam). The DNA bound to the antibody was extracted by using TIANamp Genomic DNA Kit (Tiangen, Beijing, China) and confirmed by PCR. PCR products were analyzed on a 1% agarose gel. Primers used for the Rasip1 promoter are as follows: 5′-ACTTGTGTTCTGGATCTATGGGCCT-3′ (sense); 5′-AGTACAGGGGTGAGAAGATCACA-3′ (anti-sense).
Luciferase report assay
The 2.1 kb human Rasip1 promoter fragment was amplified by using PCR Phanta® Super-Fidelity DNA Polymerase (Vazyme) and cloned into pGL3 Basic vector (Promega Corporation). The four short fragments of the Rasip1 promoter containing different numbers of RUNX family binding sites were cloned using a DNA fragment and subcloned into pGL3 Basic vectors. KpnI and HindIII restriction sites were underlined. The primers are listed in Table 2. Cells growing in 24-well plates were transfected with Rasip1 promoter reporter plasmid (full length or short fragments) and RUNX1 expression plasmid (wide-type or mutation) by using lipofectamine 2000 for 48 hours. The plasmid pRL-SV40 (Renilla luciferase) was used as an internal control to normalize for transfection efficiency. Cells were lysed, and luminescence was measured on a GloMax 20/20 luminometer (Promega Corporation) using the Dual-Luciferase Assay Kit (Promega Corporation).
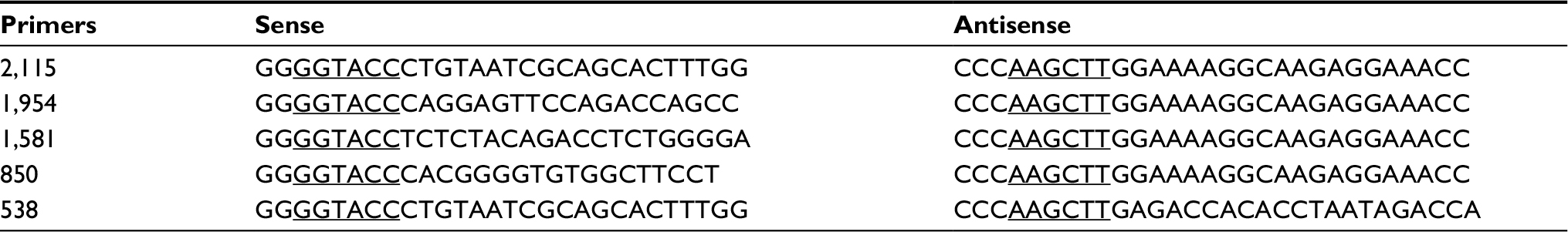 | Table 2 Primers for five sections of the Rasip1 promoter Abbreviation: Rasip1, Ras-interacting protein 1. |
Mutagenesis
The pCMV-C-HA vector containing RUNX1 and the fragments of Rasip1 promoter were used as templates. Mutations were generated by site-directed mutagenesis using the Fast Mutagenesis Kit V2 (Vazyme). All mutagenic primers are listed in Table 3.
 | Table 3 Primers for R174Q, S249A, Rasip1 promoter mutants 1–4 Abbreviation: Rasip1, Ras-interacting protein 1. |
Wound-healing assay
Twenty-four hours after transient transfection, wound-healing assay was carried out. Briefly, the cell monolayer was grown to confluence and scratched with a sterile P200 pipette tip. The cell monolayers were rinsed with PBS to remove nonadherent cells, and new medium was added. Images were collected immediately by an inverted microscope (Carl Zeiss Meditec AG, Jena, Germany). Cells were also visualized after 12 hours of incubation at 37°C for comparison of wound healing.
Transwell assay
After 24 hours of transfection, cells were isolated and suspended in DMEM without FBS. 4×104 cells were added in the upper chamber of the Transwell (EMD Millipore). The lower chamber was filled with 600 mL of DMEM with 10% FBS. The cells were allowed to migrate for 12 hours to reach the lower surface of the membrane. Then, the cells were fixed in 4% formaldehyde and stained with 0.1% crystal violet. Nonmigrated cells were scraped off using a cotton swab, and migrated cells were counted under an inverted microscope (TS100; Nikon Corporation, Tokyo, Japan).
Statistical analysis
All experiments were repeated at least three times. All data are presented as mean ± SD. Statistical analysis was performed using the Prism 5.0 software (GraphPad Software, Inc., La Jolla, CA, USA). Differences between two groups were analyzed by Student’s t-test. One-way ANOVA was used to compare the differences among multiple groups. P<0.05 was considered statistically significant.
Results
Rasip1 is involved in migration of NSCLC cells
Rasip1 mRNA was widely expressed in human and mouse tissues, whereas the protein was found only in lung tissue.24 We searched the Cosmic database and found that Rasip1 mRNA expression was increased in many cancers including NSCLC. Although Rasip1 protein has been detected in A549 cell, which is well known to harbor mutation in K-Ras,25 its protein expression profile in NSCLC cells lines has not been clarified. Therefore, we examined the expression of Rasip1 in a panel of NSCLC cells lines and found that Rasip1 mRNA and protein were expressed ubiquitously, particularly in A549 and NCI-H292 cells (Figure 1A). We also noted that the expression of Rasip1 in H1299 was lowest among the NSCLC cells lines. Therefore, we chose A549 and H1299 cell lines for functional study. To investigate the effect of Rasip1 on migration activity in NSCLC cells lines, the plasmid encoding Rasip1 tagged with GFP (Rasip1-GFP) was used to transfect the cells, and the migration rate of cells was detected. The cells were transfected with control vector and Rasip1-GFP plasmid, respectively. Western blotting was performed to assess the transfection efficiency (Figure 1B), and the overexpression of Rasip1 enhanced the migration ability of A549 and H1299 cells (Figures 1C, D and S1C). Immunofluorescence was also performed to check the transfection efficiency (Figure S1A). Then, interfering RNA (siRNA) was used to knockdown Rasip1 expression in A549 and H1299 cells. siRasip1 efficiently abolished the expression of Rasip1 (Figures 1E and S1B), and silencing Rasip1 dramatically impaired the migration ability of cancer cell (Figures 1F, G and S1D). In addition, we found that Rasip1 had no obvious effects on proliferation in NSCLC cells lines (data not shown). Previous study has shown that the levels of activated Rac1 and phosphorylated ERK (p-ERK) were markedly decreased in siRasip1-depleted ECs.9 To identify whether Rac1 and ERK were involved in Rasip1-mediated lung cancer migration, A549 and H1299 cells were transfected with siRasip1 and Rasip1-GFP plasmid, respectively. Deletion of Rasip1 in NSCLC cells lines reduced Rac1 activity and ERK phosphorylation, while overexpression of Rasip1 enhanced Rac1 activity and increased p-ERK level (Figure 1H). Taken together, these findings suggested that Rasip1 could mediate the migration of NSCLC cells by activating Rac1 and ERK phosphorylation.
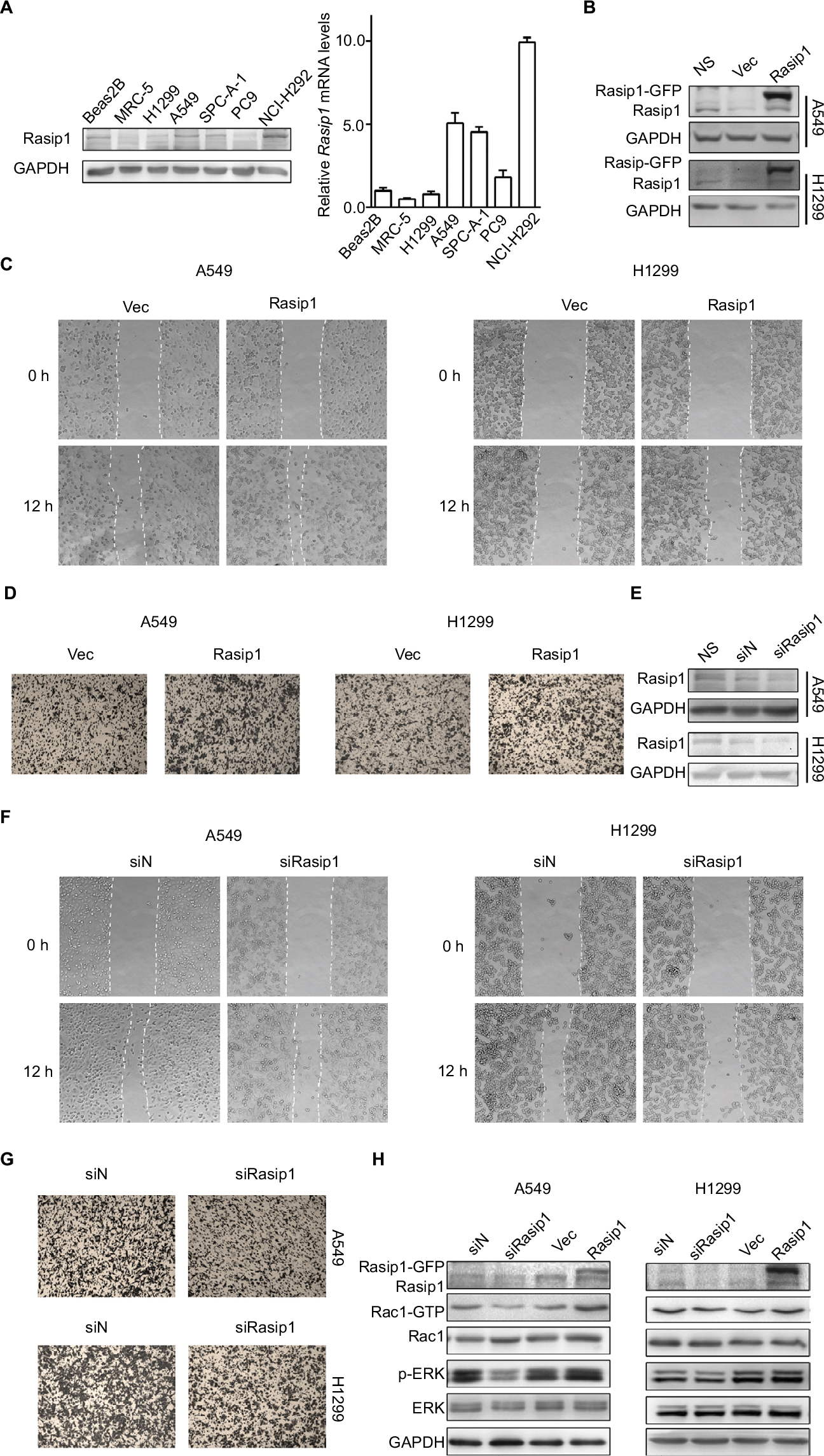 | Figure 1 Rasip1 enhances migration of NSCLC cells. Notes: (A) The endogenous expression levels of Rasip1 in NSCLC cells lines were analyzed by Western blotting and qRT-PCR. Rasip1 mRNAs are normalized to gene expression levels in a human bronchial epithelial cell line (BEAS-2B). MRC-5, human fetal lung fibroblasts, is also a normal lung cell line. (B) The pEGFP-N1 vector (control) and Rasip1-GFP plasmid were overexpressed in H1299 and A549 cells, respectively. Forty-eight hours after transfection, Western blotting was performed to detect the expression of Rasip1 and GAPDH. (C) After 24 hours of transfection, wound-healing assay was performed to evaluate the migration of the cells. The images were taken at the 0 and 12th hour by an inverted microscope. (D) After 24 hours of transfection, cell motility was measured by transwell assay. Cells were counted at time point 12 hours. (E) A549 and H1299 cells were transfected with negative siRNA (control) or Rasip1 siRNAs. Forty-eight hours after transfection, knockdown efficiencies were assessed by Western blotting. (F) At 24th hour after Rasip1 knockdown, wound-healing assay was done to evaluate migration ability. (G) At 24th hour after deletion of Rasip1, transwell assay was performed to evaluate cell motility. (H) A549 cells and H1299 cells were transfected with negative siRNA, Rasip1 siRNAs, pEGFP-N1 vector and Rasip1-GFP plasmid, respectively. After 48 hours of transfection, lysates were detected by Western blotting with antibodies against Rasip1, Rac1, p-ERK, ERK and GAPDH. The GTP-bound and total Rac1 were detected with anti-Rac1 antibody. All the experiments were repeated at least three times. Abbreviations: h, hours; NSCLC, nonsmall-cell lung cancer; qRT-PCR, quantitative reverse transcription polymerase chain reaction; Rasip1, Ras-interacting protein 1; p-ERK, phosphorylated ERK. |
RUNX1 is required for the expression of Rasip1
To explore molecular mechanisms involved in regulation of Rasip1 expression, we analyzed the Rasip1 promoter by the JASPAR CORE database and noted that RUNX may be a putative transcription factor. By using the Evolutionarily Conserved Region Browser, we found that 2.0 kb of the Rasip1 promoter region was highly conserved among various species including human, mouse, rat and zebrafish. We aligned multiple Rasip1 promoter regions and identified many potential RUNX-binding sites (Figure 2A). In mammals, there are three members of the RUNX family, including RUNX1, RUNX2 and RUNX3. To determine which member of the RUNX family regulated the expression of Rasip1, we examined their mRNA and protein expressions in NSCLC cells lines. The expression of RUNX1 was significantly higher when compared with RUNX2 and RUNX3 (Figure 2B and C). Knockdown of RUNX1 in NSCLC cells lines remarkably reduced Rasip1 expression compared with siRUNX2 and siRUNX3 (Figure 2D and E). These data suggested that RUNX1 was essential for Rasip1 expression. The structure of RUNX1 common point mutations and the protein phosphorylation sites are indicated by the bars and asterisk (Figure 2F). To rule out potential nonspecific effects of RUNX1, two RUNX1 mutant (R174Q and S249A) plasmids were constructed. The R174Q mutant could still dimerize with CBFβ, but reduce DNA binding ability and transcription function. The S249A mutant failed to be phosphorylated, and its transcriptional activity was impaired. In A549 and H1299 cells, overexpression of RUNX1 resulted in a significant increase in Rasip1 expression, at both mRNA (Figure 2G) and protein levels (Figure 2H). However, overexpression of the R174Q and S249A plasmids in NSCLC cells lines had no significant effect on Rasip1 expression, at the mRNA and protein levels. These results confirmed that RUNX1 could be a key transcription factor of Rasip1.
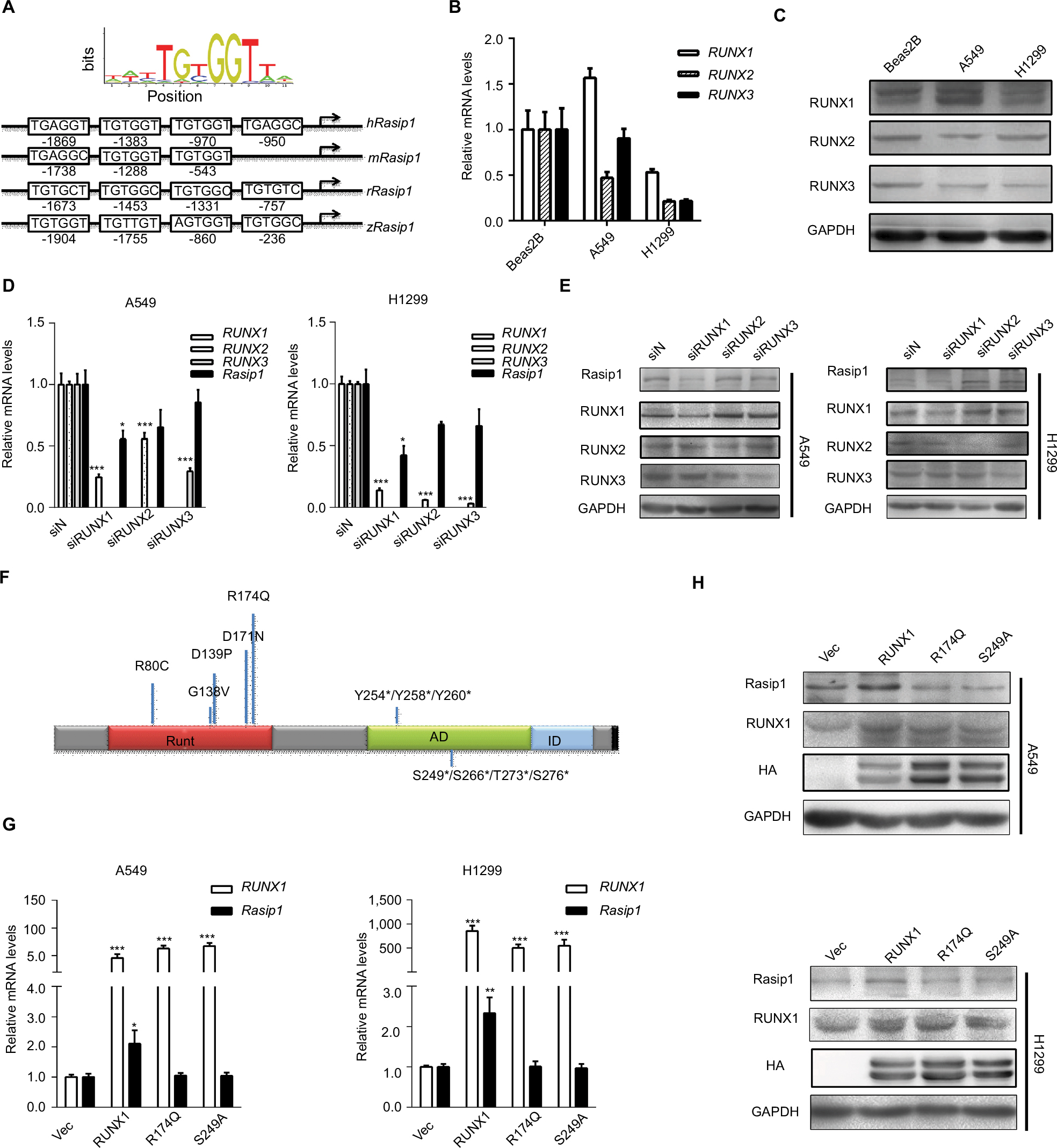 | Figure 2 RUNX1 promotes Rasip1 expression. Notes: (A) Schematic depiction of the sequence alignment of RUNX binding site from various species within the Rasip1 promoter region. (B) The endogenous expression levels of RUNX1, RUNX2 or RUNX3 mRNA were measured by qRT-PCR in A549 cells and H1299 cells. Data are normalized to gene expression levels in BEAS-2B cell. (C) The RUNX1, RUNX2 or RUNX3 protein was measured by Western blotting in BEAS-2B, A549 and H1299 cells. (D) A549 cells and H1299 cells were transfected with negative siRNA, siRUNX1, siRUNX2 and siRUNX3, respectively. After 24 hours of transfection, qRT-PCR was performed to detect the expression of Rasip1 mRNA. (E) After 48 hours of transfection, expression of Rasip1 was detected by Western blotting. (F) The structure of RUNX1 with somatic mutations at the RD and the protein phosphorylation sites. Numbers indicate positions of amino acid residues of RUNX1 from the N terminus. Runt (the DNA- and CBFB-binding domain), AD (the activation domain) and ID (the inhibitory domain) are indicated. The bars above in each diagram represent the relative frequency of RUNX1 point mutations in cancer, and the phosphorylation residues were indicated by the asterisk. (G) A549 and H1299 cells were transfected with RUNX1-HA, R174Q-HA, S249A-HA plasmid and pCMV-C-HA vector (control), respectively. RUNX1 and Rasip1 mRNA expression were analyzed by qRT-PCR. (H) Western blotting was performed by using anti-RUNX1 and anti-Rasip1 antibodies. All the experiments were repeated at least three times. Abbreviations: h, hours; qRT-PCR, quantitative reverse transcription polymerase chain reaction; Rasip1, Ras-interacting protein 1; RUNX1, Runt-related transcription factor 1; RD, Runt domain. |
RUNX1 acts as a transcription factor of Rasip1
To elucidate the molecular mechanism by which RUNX1 regulates expression of Rasip1, a 2.1 kb Rasip1 promoter was constructed and its transcriptional activity was detected. The 2.1 kb Rasip1 promoter activity increased ~50% in RUNX1-overexpressed cells (Figure 3A). Moreover, the effects of RUNX1 on Rasip1 promoter occupancy were tested by ChIP assay. Notably, an increased occupancy of RUNX1 in the region of Rasip1 promoter was observed in EGF-treated cells (Figure 3B); these findings were consistent with the role of EGF in regulating RUNX1 transcriptional activity.26 To define the key sequences in the Rasip1 promoter required for transcription, a series of truncated promoters that contained putative RUNX1-binding sequences and the plasmid-encoding RUNX1 tagged with HA (RUNX1-HA) were constructed. 1,954 bp and 1,581 bp fragments had increased transcriptional activity (Figure 3C). There were three putative RUNX1-binding sequences in 1,954 bp fragment. To further identify the more important RUNX1-binding sites in the fragment, four mutation promoter constructs were generated (Figure 3D). The promoter activity dropped sharply to 30% when the -1,383 bp and -950 bp sites were both mutated (Figure 3E). Taken together, these observations indicated that RUNX1 could bind directly to Rasip1 promoter to regulate its transcription.
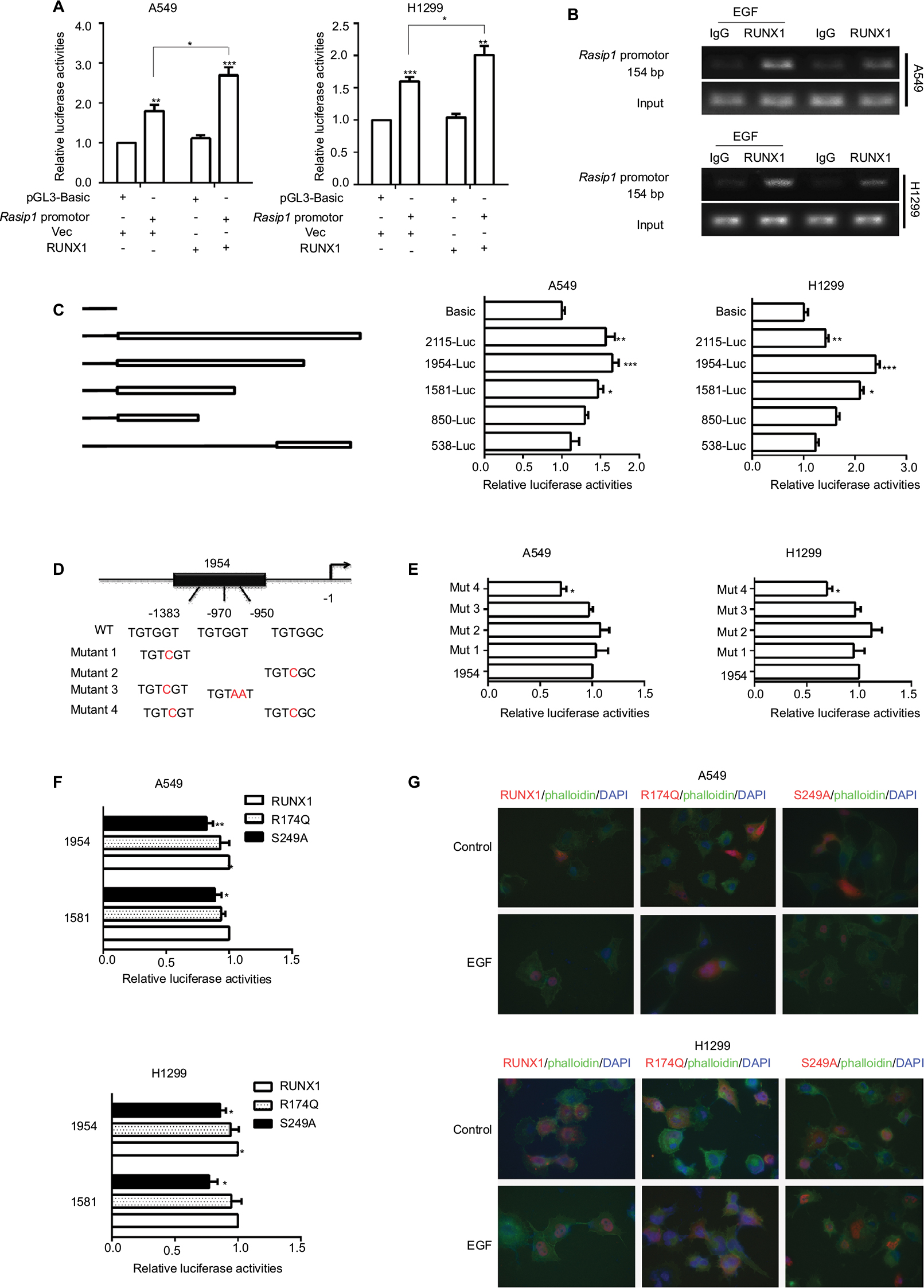 | Figure 3 RUNX1 binds to Rasip1 promoter and increases Rasip1 transcription. Notes: (A) A549 and H1299 cells were cotransfected with the pGL3-Basic (control) or Rasip1-Luc reporter and RUNX1-HA plasmid or pCMV-C-HA vector, respectively. Forty-eight hours later, cell extracts were analyzed for luciferase activity. (B) A549 and H1299 cells that were either treated or untreated with EGF (50 ng/mL) for 2 hours. ChIP was performed to analyze the RUNX1 occupancy within the Rasip1 promoter. Normal rabbit IgG was used as negative controls. PCR amplification was performed using primers corresponding to the Rasip1 promoter sequence. (C) Different alignments of the Rasip1-Luc reporter were illustrated to the right. A549 and H1299 cells were transfected with the Rasip1-Luc reporter and RUNX1-HA plasmid for 48 hours. Cells were lysed for the analysis of luciferase activity. (D) Schema of the 1,954 bp fragment with positioning of the putative RUNX1-binding sequences. (E) The putative RUNX1-binding site in 1,954 bp-Luc reporter was mutated by site-directed mutagenesis. H1299 and A549 cells were cotransfected with the mutagenized promoter constructs and RUNX1-HA plasmid for 48 hours and then analyzed for luciferase activity. The 1,954-Luc (WT) reporter was used as the control. (F) A549 and H1299 cells were cotransfected with 1,954 bp/1,581 bp-Luc reporter and the WT RUNX1-HA or mutants (R174Q-HA and S249A-HA) plasmid for 48 hours. Then, the cells were lysed to analyze luciferase activity. (G) A549 and H1299 cells were transfected with the RUNX1-HA, R174Q-HA or S249A-HA plasmid, respectively, and stimulated with EGF for 1 hour. Then, photos of the cells were taken and analyzed for the cellular localization of RUNX1. RUNX1/R174Q/S249A-HA, red; phalloidin, green and nucleus (DAPI), blue. All the experiments were repeated at least three times. Abbreviations: Rasip1, Ras-interacting protein 1; RUNX1, Runt-related transcription factor 1; WT, wild type. |
To determine the molecular basis of the differences between RUNX1 and its mutants (R174Q and S249A) in regulating Rasip1 expression, three truncated Rasip1 promoters (1,954 bp and 1,581 bp) with high transcriptional activity were cotransfected with the RUNX1-HA or its mutant plasmids in NSCLC cells lines. The S249A mutant did not have a remarkable effect on the transcriptional activities of truncated promoters when compared with WT RUNX1. In contrast to a previous study,27 the R174Q mutant still could enhance the transcriptional activity of three truncated promoters (Figure 3F). To explore how the R174Q mutant impairs its transcription function, immunofluorescence assay was performed. After EGF stimulation, the S249A mutant could be translocated from the cytoplasm to the nucleus, similar to WT RUNX1, whereas the R174Q mutant was still partially localized in the cytoplasm of transfected cells (Figure 3G). These results indicated that the R174Q disrupted its nuclear localization and consequently lost its transcription activity.
RUNX1 promotes lung cancer cell migration in a Rasip1-dependent manner
RUNX1 regulates a list of target genes and is linked to metastasis of lung adenocarcinoma.20,28 It has been reported that knockdown of RUNX1 in H1299 cell inhibited cell invasion.29 To investigate whether Rasip1 was involved in RUNX1-mediated cell migration, RUNX1-HA plasmid and siRasip1 were cotransfected into NSCLC cells lines. RUNX1 remarkably increased the migration ability of NSCLC cells, which was reduced by depletion of Rasip1 (Figures 4A and S2A). Similar results were obtained using the transwell assay (Figures 4B and S2B). To determine the functional impact of Rasip1 on RUNX1-mediated cell migration, we examined the downstream molecules. Overexpression of RUNX1 in A549 and H1299 cells also increased the Rac1 activity, whereas siRasip1 treatment attenuated these changes (Figure 4C). Collectively, these data suggested that Rasip1 might be a downstream mediator of RUNX1 in NSCLC cells lines.
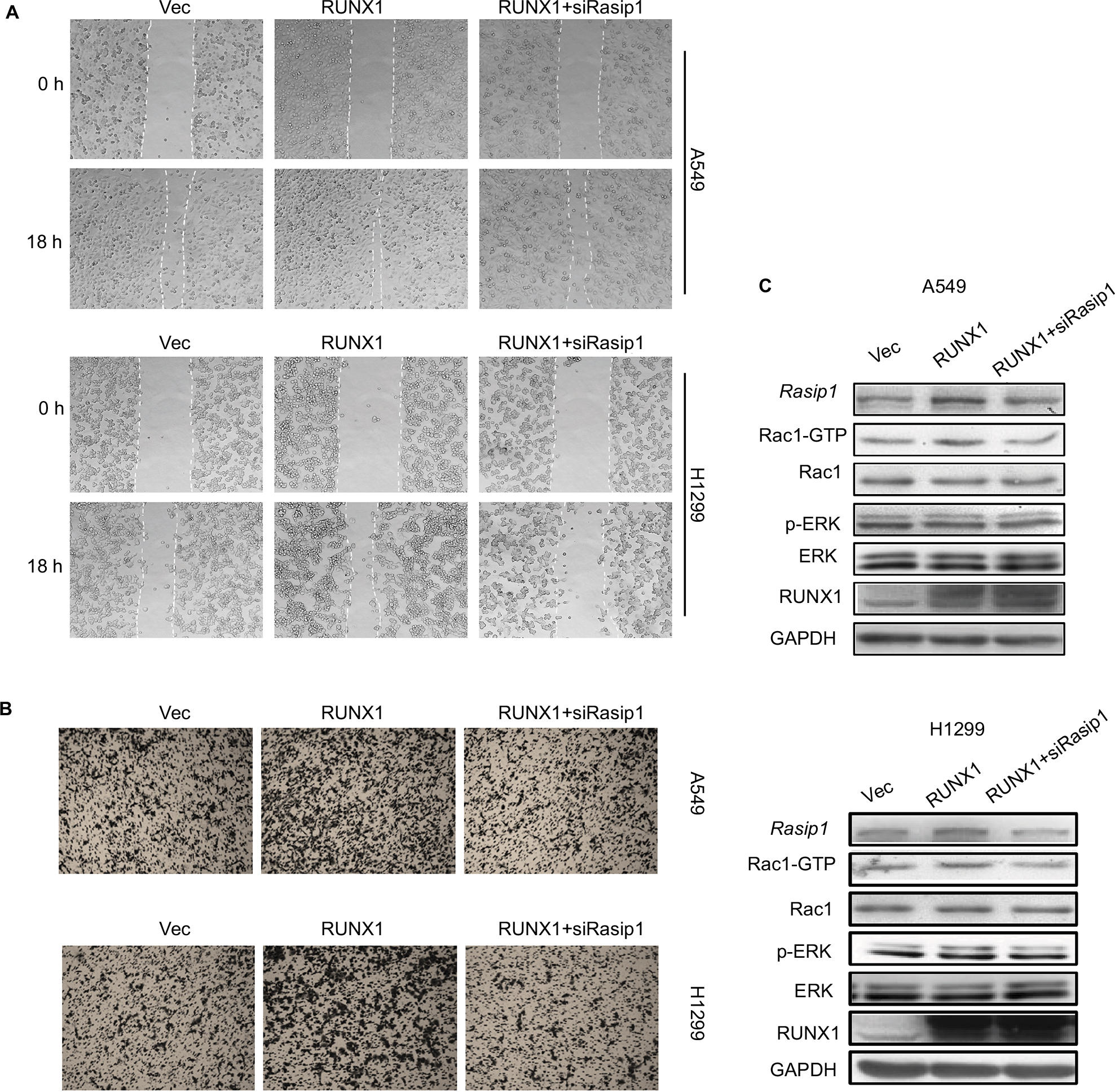 | Figure 4 Deletion of Rasip1 blocks RUNX1-mediated migration. Notes: (A) pCMV-C-HA vector or RUNX1-HA plasmid and negative siRNAs or Rasip1 siRNAs were cotransfected into H1299 and A549 cells, respectively. Wound-healing assay was performed, and images were taken at 0 and 18th hour by an inverted microscope. The empty vector was used as the control. (B) After 24 hours of transfection, transwell assay was performed, and cells were counted at the 12th hour after plating. (C) After 48 hours of transfection, cells were lysed and the lysates were detected by Western blotting for the Rasip1, Rac1, p-ERK, ERK, RUNX1 and GAPDH. The Rac1-GTP was pulled down by GST-PAK-CRIB fusion proteins. All the experiments were repeated at least three times. Abbreviations: Rasip1, Ras-interacting protein 1; RUNX1, Runt-related transcription factor 1; p-ERK, phosphorylated ERK. |
EGF stimulates Rasip1 expression and induces the plasma membrane translocation of Rasip1
EGFR is commonly overexpressed and mutated in NSCLC patients, resulting in an increased cell proliferation, survival and metastasis.2 RUNX1 was phosphorylated by ERK at S249 and S266 residues, and its transcriptional activity was enhanced.26 However, phosphorylated RUNX1 (p-RUNX1) was susceptible to degradation by the ubiquitin-proteasome pathway.13 To explore whether the expression of Rasip1 is influenced by EGF stimulation, we treated NSCLC cells lines with EGF, and a time course of Rasip1 expression was detected. EGF stimulation led to a rapid increase in the Rasip1 expression. Rasip1 mRNA increased to the first peak after 2 hours of EGF treatment and was followed by a decrease in its initial level after 4 hours (data not shown). ERK inhibitor (U0126) and RUNX1 inhibitor (Ro5-3335) completely suppressed EGF-induced Rasip1 mRNA expression (Figure 5A). Subsequently, we found that Rasip1 protein also increased after 2 hours of EGF stimulation (Figure 5B). In accordance with the previous studies, EGF induced the phosphorylation of RUNX1 at Ser249. Considering that the phosphorylation of RUNX1 promoted its degradation in protein level, we analyzed expression of total RUNX1 and found that it decreased after EGF stimulation. As we have mentioned earlier, Rasip1 could increase the phosphorylation of ERK. Together, these results suggested that this complexity feedback may be a mechanism for keeping Rasip1 overexpression in NSCLC cells lines.
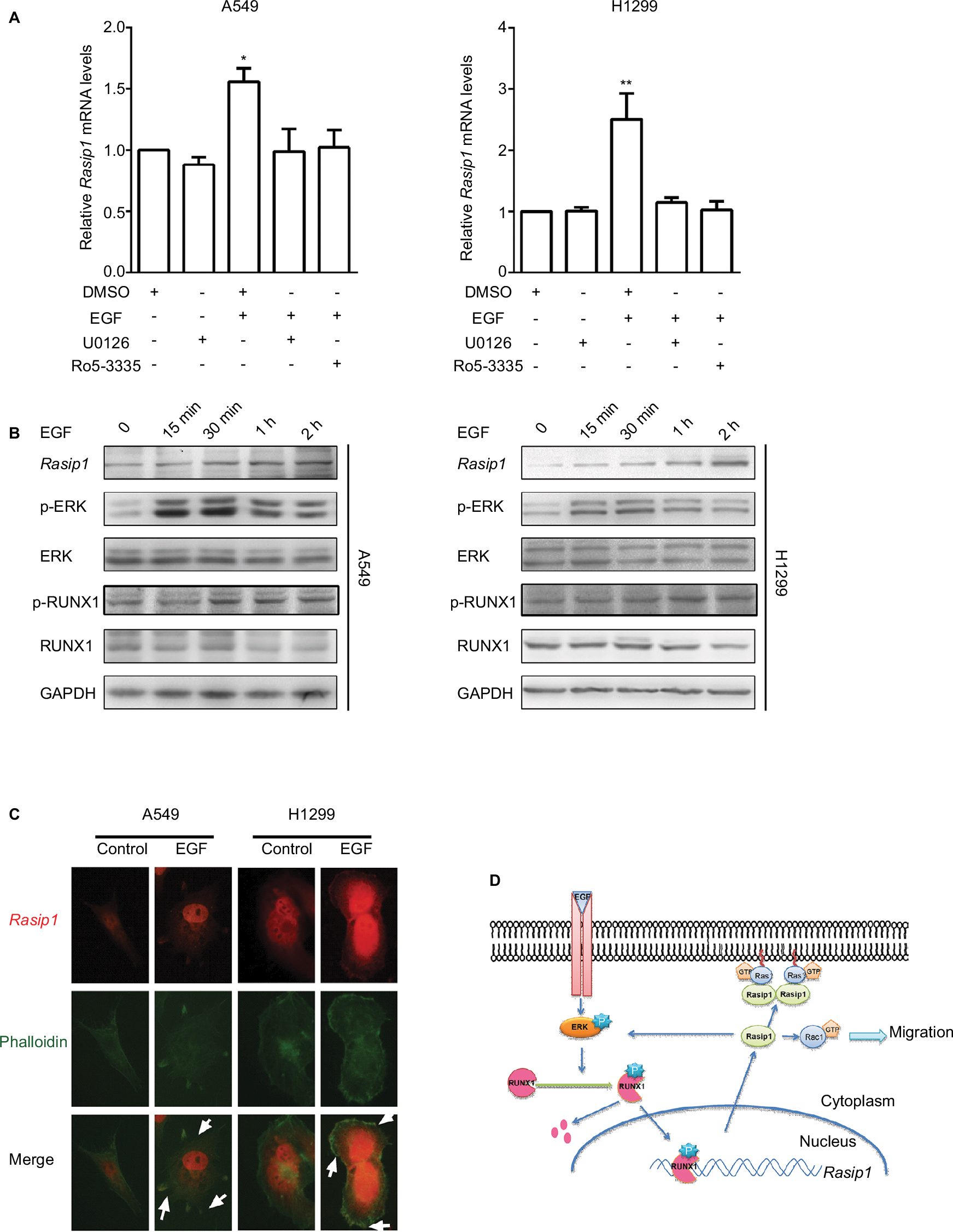 | Figure 5 EGF regulates the expression and subcellular localization of Rasip1. Notes: (A) A549 and H1299 cells were starved and were incubated for 2 hours with either 10 mmol/L U0126 or 25 mmol/L Ro5-3335 prior to EGF treatment (50 ng/mL for 2 hours). Total RNA was extracted, and the Rasip1 mRNA was analyzed by qRT-PCR. (B) Serum-starved cells were stimulated with 50 ng/mL EGF (0, 15 minutes, 30 minutes, 1 hour and 2 hours), and lysates were detected by Western blotting for expression of Rasip1, p-ERK, ERK, p-RUNX1, RUNX1 and GAPDH. (C) A549 cells and H1299 cells cultured on coverslips were starved for 12 hours and treated or untreated with 50 ng/mL EGF for 30 minutes. The cells were fixed and incubated with anti-Rasip1 antibody followed by goat anti-Rabbit Alexa594 secondary antibody. The images were captured by an inverted immunofluorescence microscope. Original colors were Rasip1, red and phalloidin, green. Accumulation of Rasip1 on the plasma membrane was indicated by the arrow. (D) The schematic diagram illustrates the complex feedback of Rasip1 expression and the cellular localization of Rasip1 after EGF treatment. Activation of ERK by EGF stimulation promotes the phosphorylation of RUNX1 at Ser249, increases its transcription activity and, hence, induces its degradation. Phosphorylated RUNX1 binds to a specific DNA sequence within Rasip1 promoter and increases its transcription. In turn, Rasip1 promotes Rac1 and ERK activation, thereby increasing cell migration. Together, this system formed a positive and negative feedback loop to regulate Rasip1 expression. With the extension of EGF treatment, Rasip1 could form a complex with Ras or other protein and be recruited to the plasma membrane. All the experiments were repeated at least three times. *P<0.05 when compared to other groups. **P<0.01 when compared to other groups. Abbreviations: h, hours; min, minutes; Rasip1, Ras-interacting protein 1; RUNX1, Runt-related transcription factor 1; qRT-PCR, quantitative reverse transcription polymerase chain reaction; p-ERK, phosphorylated ERK; p-RUNX1, phosphorylated RUNX1. |
Recently, we have reported that EGF could induce directional migration of NSCLC cells through regulating the localization of RUSC2.30 Previous study has shown that Rasip1 could bind directly to Ras proteins, which are the downstream effectors of EGFR signaling pathway.6 To investigate how Rasip1 is regulated under EGF stimulation, we transfected NSCLC cells lines with Rasip1-GFP plasmid, and the subcellular localization of Rasip1 was determined by immunofluorescence assay. Under resting conditions, Rasip1 was predominantly localized in the cytoplasmic compartment. After EGF stimulation, Rasip1 was recruited to the plasma membrane (Figure 5C). Our findings implied that EGF might activate downstream molecules, to form a complex with Rasip1, and mediate the translocation of Rasip1 to the plasma membrane.
Discussion
Metastasis is a very complex phenomenon, which is responsible for death of NSCLC patients. Targeting metastasis pathways is a potential new therapeutic strategy and has received more and more attention.31 However, the molecular mechanisms underlying metastasis in NSCLC were not fully known. From the database, we noticed that Rasip1, a pan-endothelium marker, was overexpressed in NSCLC patients. However, the expression and function of Rasip1 in NSCLC cells lines are still unknown. In this study, it was identified that Rasip1 was ubiquitously expressed in NSCLC cells lines and that it promoted cell migration through activation of Rac1 and ERK pathways. Rasip1 was colocalized with activated Ras proteins and initially identified around the Golgi apparatus.6 Recent study has highlighted a novel localization of Rasip1; Rap1 activation in EC led to the translocation of Rasip1 to the plasma membrane.32 EGFR is frequently overexpressed and mutated in NSCLC, and its activation initiated intracellular signaling through Ras proteins.33 In this study, we found that Rasip1 could be recruited to the plasma membrane by EGF stimulation. Therefore, we hypothesized that the EGFR pathway spatially controls the cellular localization of Rasip1 and thereby promotes the cell migration. Further studies are needed to focus on the contribution of Rasip1 on the plasma membrane.
The RUNX family is essential for cell survival, proliferation and differentiation. Depletion of RUNX1 in zebrafish abrogates the development of both blood cells and vessels; RUNX2 deficiency in mice arrests osteoblast differentiation; RUNX3 null mice exhibit an asthma-like phenotype, gastric mucosa hyperplasia, neural disorders, as well as abnormalities in CD4 expression.34–36 RUNX genes play essential roles in lung cancer, acting as either tumor suppressors or oncogenes. RUNX2 regulated Snail expression, thereby enhancing lung cancer migration and epithelial-to-mesenchymal transition.37 RUNX3 inactivation by promoter hypermethylation is crucial for the development of lung adenocarcinoma.38 Our results indicated that RUNX1 mRNA was highly expressed in various NSCLC cells lines. Besides hematopoietic diseases, RUNX1 also plays a central role in many solid tumor development and progression. Increased expression of RUNX1 in epithelial cells is required for tumor formation. RUNX1, acting as a skin oncogene, stimulated Stat3 signaling and promoted tumor cell proliferation.39 Elevated RUNX1 expression was associated with poor survival in prostate cancer and triple negative breast cancer.40,41 In serous epithelial ovarian carcinoma, RUNX1 was overexpressed and related to cell proliferation, migration and invasion.19 Depletion of the expression of RUNX1 in H1299 cell inhibited cell invasion. However, different results of RUNX1 gene expression have been reported in lung adenocarcinoma, involving cancer pathogenesis.16,20 Our data confirmed that RUNX1 is involved in the migration of NSCLC cells. Moreover, we identified that Rac1 was activated in RUNX1-mediated cell migration.
As a transcription factor, RUNX1 directly binds to a specific DNA sequence to repress or activate target gene transcription. Identification of target genes of RUNX1 is necessary for understanding the cellular function of this transcriptional factor. Several studies have revealed the target genes of RUNX1 in patients with leukemia, which include MDR-1, GM-CSFR, c-fos, TCRβ enhancer and IL-3.28 RUNX1 directly bonded to the functional sites within PKCβ promoter and regulated its expression, which links RUNX1 to apoptosis in myeloid cells.42 PU.1 has been identified as a target gene of RUNX1 in adult mouse hematopoiesis.43 Downregulation of NR4A3, another RUNX1 target gene, contributed to hematopoiesis deregulation in FPD/AML.44 The target genes of RUNX1 have effect on the cellular programs, including proliferation, differentiation, apoptosis and migration.45 However, little is known about the target genes regulated by RUNX1 in other tissues. In this study, several potential RUNX-binding sites were identified within Rasip1 promoter and silencing of RUNX1 suppressed the Rasip1 expression. In contrast, RUNX1 overexpression significantly enhanced the expression of Rasip1. These findings invoked the possibility that RUNX1 participates in the control of Rasip1 transcription. Indeed, our data indicated that RUNX1 bound directly to Rasip1 promoter. To rule out the transcriptional activity of other RUNX family members, RUNX2 and RUNX3 were knocked down in NSCLC cells lines. The results showed that deletion of RUNX3 had a marginal effect on expression of Rasip1 mRNA, but had no effect on protein expression. However, RUNX2 deletion had no influence on expression of both mRNA and protein of Rasip1. In addition, Rasip1 deletion inhibited RUNX1-mediated migration in NSCLC cells lines. Therefore, it was identified that RUNX1 had the ability to activate Rasip1 transcription in this study.
Given that RUNX1 is a weak transcription factor, its functions are reinforced through posttranslational modifications.46,47 Ser-249/Ser-266 residues of RUNX1 could be phosphorylated by ERK, which activates the interaction of RUNX1 with p300 and increased its transcriptional activity. Our data showed that expression of Rasip1 was increased, and the occupancy of RUNX1 in Rasip1 promoter was enhanced by EGF stimulation. These changes were accompanied by increased RUNX1 phosphorylation after EGF treatment. However, phosphorylated RUNX1 was prone to be degraded by ubiquitin-mediated pathway. We found that the total RUNX1 and Rasip1 expression was reduced by long-term EGF stimulation. As we have noted, Rasip1 could mediate ERK phosphorylation. This multiple feedback loop might lead to upregulation of the expression of Rasip1 in NSCLC cells lines (summarized in Figure 5D).
The R174Q and S249A mutants are well described, which lack transcriptional activity.48,49 Consistent with previous study, the S249A mutant impaired its transcriptional activities. Recent studies demonstrated the presence of R174Q in AML, myelodysplastic syndromes/AML, acute lymphoid leukemia, Familial platelet disorder/AML, as well as breast cancer.50 This study had confirmed that the R174Q plasmid had no significant effect on Rasip1 expression. Interestingly, although Arg174 was reported critical for DNA binding,27 the results of luciferase report assay showed that the R174Q mutant still retained the transcription activity, similar to WT RUNX1. We speculated that another mechanism should contribute to the function of R174Q mutant. As reported, the cellular localization is different between R174Q and WT RUNX1. We found that after EGF stimulation, unlike WT RUNX1, the R174Q mutant was still partially localized in the cytoplasm of transfected cells. Evidence was accumulating that the R174Q mutant might lose its ability to regulate Rasip1 expression through disrupting its nuclear localization. Although RUNX1 was considered to be associated with migration of many cancers, it should be noted that drugs target RUNX1 have not been proven in clinical trials.
Conclusion
The results of this study indicated that Rasip1 is a downstream target gene of RUNX1 and is essential for cell migration of NSCLC. Furthermore, we observed that EGF regulated the localization and expression of Rasip1. Thus, our findings pointed out the relationship between RUNX1 and Rasip1 and generated a novel insight into cell migration in NSCLC. Given that Rasip1 also plays a critical role in angiogenesis, targeting Rasip1 may provide a new therapeutic treatment in NSCLC patients.
Acknowledgments
This work was supported by funding from the National Natural Science Foundation of China (No 81372319), a Project Funded by Jiangsu Collaborative Innovation Center for Cancer Personalized Medicine to Luo Gu, the National Natural Science Foundation of China (No 81773107) to Jun Du, the National Natural Science Foundation of China (No 81602561) to Yujie Zhang, the College Graduates Research and Innovation Program of Jiangsu Province (No JX22013334) to Yan Chen and a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Disclosure
The authors report no conflicts of interest in this work.
References
Thatcher N, Chang A, Parikh P, et al. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer). Lancet. 2005;366(9496):1527–1537. | ||
Paez JG, Jänne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497–1500. | ||
van Zandwijk N, Mathy A, Boerrigter L, et al. EGFR and KRAS mutations as criteria for treatment with tyrosine kinase inhibitors: retro- and prospective observations in non-small-cell lung cancer. Ann Oncol. 2007;18(1):99–103. | ||
Takashima A, Faller DV. Targeting the RAS oncogene. Expert Opin Ther Targets. 2013;17(5):507–531. | ||
Keeton AB, Salter EA, Piazza GA. The RAS–effector interaction as a drug target. Cancer Res. 2017;77(2):221–226. | ||
Mitin NY, Ramocki MB, Zullo AJ, der CJ, Konieczny SF, Taparowsky EJ. Identification and characterization of rain, a novel Ras-interacting protein with a unique subcellular localization. J Biol Chem. 2004;279(21):22353–22361. | ||
Xu K, Chong DC, Rankin SA, Zorn AM, Cleaver O. Rasip1 is required for endothelial cell motility, angiogenesis and vessel formation. Dev Biol. 2009;329(2):269–279. | ||
Wilson CW, Parker LH, Hall CJ, et al. Rasip1 regulates vertebrate vascular endothelial junction stability through Epac1-Rap1 signaling. Blood. 2013;122(22):3678–3690. | ||
Xu K, Sacharidou A, Fu S, et al. Blood vessel tubulogenesis requires Rasip1 regulation of GTPase signaling. Dev Cell. 2011;20(4):526–539. | ||
Koo Y, Barry DM, Xu K, et al. Rasip1 is essential to blood vessel stability and angiogenic blood vessel growth. Angiogenesis. 2016;19(2):173–190. | ||
Sanz-Moreno V, Gadea G, Ahn J, et al. Rac activation and inactivation control plasticity of tumor cell movement. Cell. 2008;135(3):510–523. | ||
Parri M, Chiarugi P. Rac and Rho GTPases in cancer cell motility control. Cell Commun Signal. 2010;8(1):23. | ||
Ito Y, Bae S-C, Chuang LSH. The RUNX family: developmental regulators in cancer. Nat Rev Cancer. 2015;15(2):81–95. | ||
Mangan JK, Speck NA. RUNX1 mutations in clonal myeloid disorders: from conventional cytogenetics to next generation sequencing, a story 40 years in the making. Crit Rev Oncog. 2011;16(1–2):77–91. | ||
Goyama S, Schibler J, Cunningham L, et al. Transcription factor RUNX1 promotes survival of acute myeloid leukemia cells. J Clin Invest. 2013;123(9):3876–3888. | ||
Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003;33(1):49–54. | ||
Scheitz CJ, Lee TS, Mcdermitt DJ, Tumbar T. Defining a tissue stem cell-driven Runx1/Stat3 signalling axis in epithelial cancer. EMBO J. 2012;31(21):4124–4139. | ||
Browne G, Taipaleenmäki H, Bishop NM, et al. Runx1 is associated with breast cancer progression in MMTV-PyMT transgenic mice and its depletion in vitro inhibits migration and invasion. J Cell Physiol. 2015;230(10):2522–2532. | ||
Keita M, Bachvarova M, Morin C, et al. The RUNX1 transcription factor is expressed in serous epithelial ovarian carcinoma and contributes to cell proliferation, migration and invasion. Cell Cycle. 2013;12(6):972–986. | ||
Chen R, Khatri P, Mazur PK, et al. A meta-analysis of lung cancer gene expression identifies PTK7 as a survival gene in lung adenocarcinoma. Cancer Res. 2014;74(10):2892–2902. | ||
Zhu Y, Shen T, Liu J, et al. Rab35 is required for Wnt5a/Dvl2-induced Rac1 activation and cell migration in MCF-7 breast cancer cells. Cell Signal. 2013;25(5):1075–1085. | ||
Edlund S, Landström M, Heldin C-H, Aspenström P. Transforming growth factor-β-induced mobilization of actin cytoskeleton requires signaling by small GTPases Cdc42 and RhoA. Mol Biol Cell. 2002;13(3):902–914. | ||
Zhang Y, Du J, Zheng J, et al. EGF-reduced Wnt5a transcription induces epithelial-mesenchymal transition via Arf6-ERK signaling in gastric cancer cells. Oncotarget. 2015;6(9):7244–7261. | ||
Mitin N, Konieczny SF, Taparowsky EJ. RAS and the RAIN/RasIP1 effector. Methods Enzymol. 2006;407:322–335. | ||
Post A, Pannekoek WJ, Ross SH, Verlaan I, Brouwer PM, Bos JL. Rasip1 mediates Rap1 regulation of Rho in endothelial barrier function through ArhGAP29. Proc Natl Acad Sci U S A. 2013;110(28):11427–11432. | ||
Tanaka T, Kurokawa M, Ueki K, et al. The extracellular signal-regulated kinase pathway phosphorylates AML1, an acute myeloid leukemia gene product, and potentially regulates its transactivation ability. Mol Cell Biol. 1996;16(7):3967–3979. | ||
Tahirov TH, Inoue-Bungo T, Morii H, et al. Structural analyses of DNA recognition by the AML1/Runx-1 Runt domain and its allosteric control by CBFbeta. Cell. 2001;104(5):755–767. | ||
Otto F, Lübbert M, Stock M. Upstream and downstream targets of RUNX proteins. J Cell Biochem. 2003;89(1):9–18. | ||
Wang X, Zhao Y, Qian H, Huang J, Cui F, Mao Z. The miR-101/RUNX1 feedback regulatory loop modulates chemo-sensitivity and invasion in human lung cancer. Int J Clin Exp Med. 2015;8(9):15030–15042. | ||
Duan B, Cui J, Sun S, et al. EGF-stimulated activation of Rab35 regulates RUSC2–GIT2 complex formation to stabilize GIT2 during directional lung cancer cell migration. Cancer Lett. 2016;379(1):70–83. | ||
Steeg PS. Targeting metastasis. Nat Rev Cancer. 2016;16(4):201–218. | ||
Post A, Pannekoek WJ, Ponsioen B, Vliem MJ, Bos JL. Rap1 spatially controls ArhGAP29 to inhibit Rho signaling during endothelial barrier regulation. Mol Cell Biol. 2015;35(14):2495–2502. | ||
Chou Y-T, Lin H-H, Lien Y-C, et al. EGFR promotes lung tumorigenesis by activating miR-7 through a Ras/ERK/Myc pathway that targets the Ets2 transcriptional repressor ERF. Cancer Res. 2010;70(21):8822–8831. | ||
Kalev-Zylinska ML, Horsfield JA, Flores MV, et al. Runx1 is required for zebrafish blood and vessel development and expression of a human RUNX1-CBF2T1 transgene advances a model for studies of leukemogenesis. Development. 2002;129(8):2015–2030. | ||
Komori T, Yagi H, Nomura S, et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89(5):755–764. | ||
Levanon D, Bettoun D, Harris-Cerruti C, et al. The Runx3 transcription factor regulates development and survival of TrkC dorsal root ganglia neurons. EMBO J. 2002;21(13):3454–3463. | ||
Hsu Y-L, Huang M-S, Yang C-J, Hung J-Y, Wu L-Y, Kuo P-L. Lung tumor-associated osteoblast-derived bone morphogenetic protein-2 increased epithelial-to-mesenchymal transition of cancer by Runx2/snail signaling pathway. J Biol Chem. 2011;286(43):37335–37346. | ||
Lee K-S, Lee Y-S, Lee J-M, et al. Runx3 is required for the differentiation of lung epithelial cells and suppression of lung cancer. Oncogene. 2010;29(23):3349–3361. | ||
Hoi CSL, Lee SE, Lu SY, et al. Runx1 directly promotes proliferation of hair follicle stem cells and epithelial tumor formation in mouse skin. Mol Cell Biol. 2010;30(10):2518–2536. | ||
Ferrari N, Mohammed ZM, Nixon C, et al. Expression of RUNX1 correlates with poor patient prognosis in triple negative breast cancer. PLoS One. 2014;9(6):e100759. | ||
Takayama K-I, Suzuki T, Tsutsumi S, et al. RUNX1, an androgen- and EZH2-regulated gene, has differential roles in AR-dependent and -independent prostate cancer. Oncotarget. 2015;6(4):2263–2276. | ||
Hug BA, Ahmed N, Robbins JA, Lazar MA. A chromatin immunoprecipitation screen reveals protein kinase Cβ as a direct RUNX1 target gene. J Biol Chem. 2004;279(2):825–830. | ||
Huang G, Zhang P, Hirai H, et al. PU.1 is a major downstream target of AML1 (RUNX1) in adult mouse hematopoiesis. Nat Genet. 2008;40(1):51–60. | ||
Bluteau D, Gilles L, Hilpert M, et al. Down-regulation of the RUNX1-target gene NR4A3 contributes to hematopoiesis deregulation in familial platelet disorder/acute myelogenous leukemia. Blood. 2011;118(24):6310–6320. | ||
Michaud J, Simpson KM, Escher R, et al. Integrative analysis of RUNX1 downstream pathways and target genes. BMC Genomics. 2008;9(1):363. | ||
Goyama S, Huang G, Kurokawa M, Mulloy JC. Posttranslational modifications of RUNX1 as potential anticancer targets. Oncogene. 2015;34(27):3483–3492. | ||
Wang L, Huang G, Zhao X, et al. Post-translational modifications of Runx1 regulate its activity in the cell. Blood Cells Mol Dis. 2009;43(1):30–34. | ||
Osato M. Point mutations in the RUNX1/AML1 gene: another actor in RUNX leukemia. Oncogene. 2004;23(24):4284–4296. | ||
Zhang Y, Biggs JR, Kraft AS. Phorbol ester treatment of K562 cells regulates the transcriptional activity of AML1c through phosphorylation. J Biol Chem. 2004;279(51):53116–53125. | ||
van Bragt MP, Hu X, Xie Y, Li Z. RUNX 1, a transcription factor mutated in breast cancer, controls the fate of ER-positive mammary luminal cells. Elife. 2004;3:e03881. |
Supplementary materials
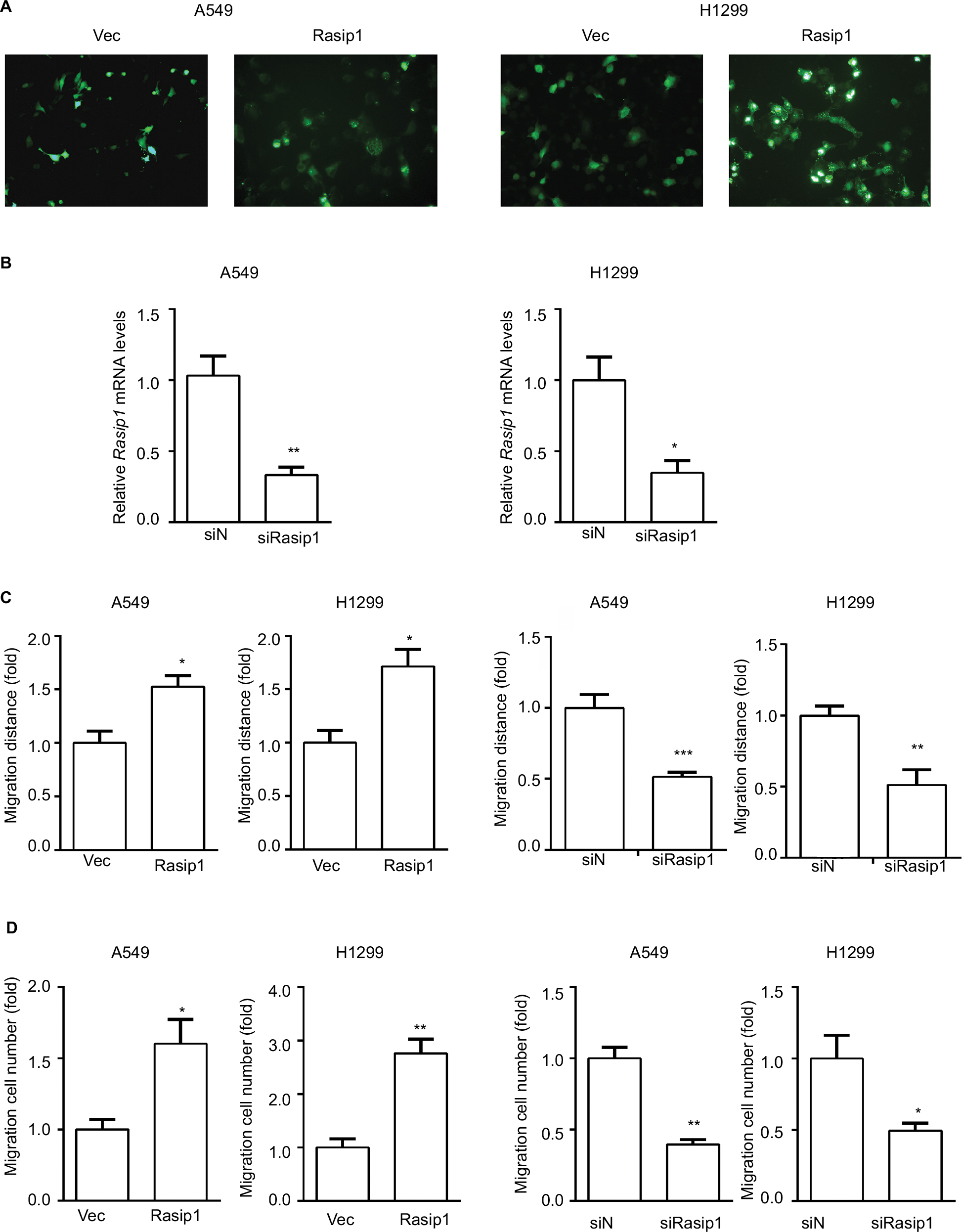 | Figure S1 Rasip1 promotes migration of NSCLC cells. Notes: (A) The pEGFP-N1 vector (control) and Rasip1-GFP plasmid were overexpressed in H1299 and A549 cells, respectively. The images were captured by an inverted immunofluorescence microscope. (B) A549 cells and H1299 cells were transfected with negative siRNA and siRasip1, respectively. After 24 hours of transfection, qRT-PCR was performed to detect the expression of Rasip1 mRNA. (C) Wound-healing assay results were quantified and statistically analyzed. (D) Transwell assay results were quantified and statistically analyzed. Data are presented as mean ± SD, *P<0.05, **P<0.01, ***P<0.005. Abbreviations: NSCLC, nonsmall-cell lung cancer; qRT-PCR, real-time quantitative PCR; Rasip1, Ras-interacting protein 1. |
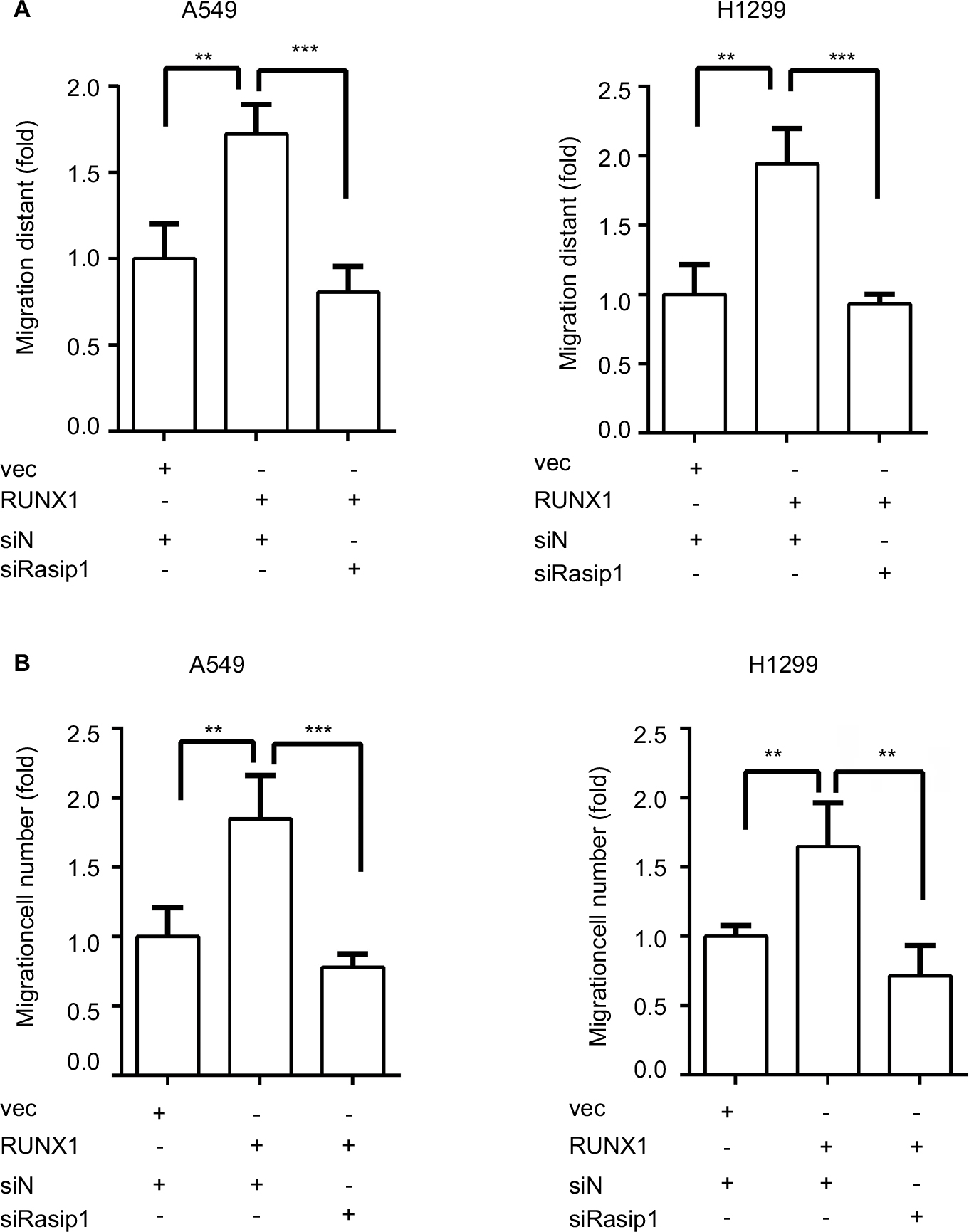 | Figure S2 Rasip1 mediates RUNX1-induced migration. Notes: (A) pCMV-C-HA vector or RUNX1-HA plasmid and negative siRNAs or Rasip1 siRNAs were cotransfected into H1299 and A549 cells, respectively. Wound-healing assay was quantified and statistically analyzed. (B) After 24 hours of transfection, transwell assay was performed and statistically analyzed. Data are presented as mean ± SD, **P<0.01, ***P<0.005. Abbreviations: Rasip1, Ras-interacting protein 1; RUNX1, Runt-related transcription factor 1. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.