Back to Journals » Neuropsychiatric Disease and Treatment » Volume 15
Rare compound heterozygous missense SPATA7 variations and risk of schizophrenia; whole-exome sequencing in a consanguineous family with affected siblings, follow-up sequencing and a case-control study
Authors Igeta H, Watanabe Y ![]() , Morikawa R, Ikeda M, Otsuka I, Hoya S, Koizumi M, Egawa J, Hishimoto A, Iwata N
, Morikawa R, Ikeda M, Otsuka I, Hoya S, Koizumi M, Egawa J, Hishimoto A, Iwata N ![]() , Someya T
, Someya T
Received 8 June 2019
Accepted for publication 23 July 2019
Published 19 August 2019 Volume 2019:15 Pages 2353—2363
DOI https://doi.org/10.2147/NDT.S218773
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Hirofumi Igeta,1 Yuichiro Watanabe,1 Ryo Morikawa,1 Masashi Ikeda,2 Ikuo Otsuka,3 Satoshi Hoya,1 Masataka Koizumi,1 Jun Egawa,1 Akitoyo Hishimoto,3 Nakao Iwata,2 Toshiyuki Someya1
1Department of Psychiatry, Niigata University Graduate School of Medical and Dental Sciences, Niigata, Japan; 2Department of Psychiatry, Fujita Health University School of Medicine, Toyoake, Aichi, Japan; 3Department of Psychiatry, Kobe University Graduate School of Medicine, Kobe, Hyogo, Japan
Correspondence: Yuichiro Watanabe
Department of Psychiatry, Niigata University Graduate School of Medical and Dental Sciences, 757 Asahimachidori-ichibancho, Chuo-ku, Niigata 951-8510, Japan
Tel +81 25 227 2213
Fax +81 25 227 0777
Email [email protected]
Purpose: Whole-exome sequencing (WES) of multiplex families is a promising strategy for identifying causative variations for common diseases. To identify rare recessive risk variations for schizophrenia, we performed a WES study in a consanguineous family with affected siblings. We then performed follow-up sequencing of SPATA7 in schizophrenia-affected families. In addition, we performed a case-control study to investigate association between SPATA7 variations and schizophrenia.
Patients and methods: WES was performed on two affected siblings and their unaffected parents, who were second cousins, of a multiplex schizophrenia family. Subsequently, we sequenced the coding region of SPATA7, a potential risk gene identified by the WES analysis, in 142 affected offspring from 137 families for whom parental DNA samples were available. We further tested rare recessive SPATA7 variations, identified by WES and sequencing, for associations with schizophrenia in 2,756 patients and 2,646 controls.
Results: Our WES analysis identified rare compound heterozygous missense SPATA7 variations, p.Asp134Gly and p.Ile332Thr, in both affected siblings. Sequencing SPATA7 coding regions from 137 families identified no rare recessive variations in affected offspring. In the case-control study, we did not detect the rare compound heterozygous SPATA7 missense variations in patients or controls.
Conclusion: Our data does not support the role of the rare compound heterozygous SPATA7 missense variations p.Asp134Gly and p.Ile332Thr in conferring a substantial risk of schizophrenia.
Keywords: Japanese, multiplex schizophrenia family, next-generation sequencing, recessive variations
Introduction
Schizophrenia is a complex disorder with heritability of approximately 80%.1 Understanding the genetic architecture of schizophrenia has progressed steadily.2–4 Genome-wide association studies (GWASs) have discovered common loci associated with schizophrenia.5–7 Intriguingly, association of the major histocompatibility complex locus with schizophrenia involves structurally distinct alleles of C4 that affect the expression of C4A and C4B in the brain.8 However, the heritability of schizophrenia is not fully explained by common variations, suggesting that rare variations also contribute to schizophrenia liability.9 Indeed, rare copy number variations are associated with schizophrenia.10–12 Whole-exome sequencing (WES) studies have demonstrated that rare sequence variations play a substantial role in the genetic etiology of schizophrenia.13–15 Of note, SETD1A was identified as a risk gene for schizophrenia with a large effect.16,17
WES and whole-genome sequencing (WGS) of multiplex families is a promising strategy for identifying causative variations for common diseases.18,19 The number of WES and WGS studies that have examined multiplex schizophrenia families is still limited, but they have detected highly penetrant variations in GRM5,20 UNC13B,21 SHANK2 and SMARCA1,22 RELN,23 TAAR1,24 RBM12,25 CSPG4,26 PTPRA,27 ITGΒ4,28 TIMP2,29 and TENM4.30
Two recent studies suggested that a combined strategy including identity-by-descent (IBD) mapping and WES may be useful in identifying rare risk variations for schizophrenia inherited from common ancestors31,32 Harold et al performed IBD mapping using Irish schizophrenia GWAS data and identified potential risk haplotypes.31 Subsequently, they conducted WES and identified PCNT p.Gly1452Arg as a potential risk haplotype, although this missense variation was not associated with schizophrenia in replication samples. In the other study, Salvoro et al performed IBD mapping and WES in multiplex families with schizophrenia, bipolar disorder, and schizoaffective disorder from Chioggia, Italy.32 Among potential risk haplotypes, they found significant enrichment of non-synonymous variations of genes involved in extracellular matrix biology and axon guidance processes.
Here, we performed a three-stage study to identify rare recessive variations that play a substantial role in conferring schizophrenia risk. First, we undertook a WES study in a multiplex family with two siblings with schizophrenia whose unaffected parents were second cousins. Second, we sequenced the coding region of SPATA7, a potential risk gene identified by the WES study, in 142 affected offspring from 137 families for whom parental DNA samples were available. Third, we conducted a case-control study to examine association of rare recessive SPATA7 variations, identified by WES and sequencing, with schizophrenia in 2,756 patients and 2,646 controls.
Materials and methods
Participants
This study was conducted in accordance with the Declaration of Helsinki and was approved by the Ethics Committee of each participating institute. All participants gave written informed consent, and all were of Japanese descent.
We included two siblings with schizophrenia (#4 and #5) and their unaffected parents (#1 and #2) in a WES study (Figure 1). In this family, the female proband (#4) and her younger sister (#5) were diagnosed with schizophrenia. Their older sister (#3) was suspected of having postpartum depression. Their younger brother (#6) died one day after a Caesarean section delivery. Their younger brother (#7) was not diagnosed with any psychiatric disorder. Their unaffected father (#1) and mother (#2) were second cousins. Diagnoses of each family member were made using Diagnostic and Statistical Manual of Mental Disorders, 4th Edition (DSM-IV) criteria, as previously described.33
 |
Figure 1 Pedigree of a consanguineous family with two schizophrenia siblings. The female proband (#4), indicated by an arrow, and her younger sister (#5) were diagnosed with schizophrenia, indicated by black shaded symbols. Their older sister (#3) was suspected of having postpartum depression, indicated by a gray shaded symbol. Their parents (#1 and #2) and younger brothers (#6 and #7) were not diagnosed with any psychiatric disorder, indicated by unshaded symbols. Their younger brother (#6) died one day after a Caesarean section delivery, indicated by a diagonal line through the symbol. Their parents (#1 and #2) were second cousins, indicated by a double line between individuals. Squares and circles represent males and females, respectively. Crosses represent individuals from whom genomic DNA samples were available. |
For sequencing SPATA7 coding regions, we included 142 affected offspring (79 men and 63 women; mean age, 29.4±9.0 years) from 137 families for whom parental DNA samples were available for genotyping. These affected offspring were diagnosed with schizophrenia according to DSM-IV or DSM-5 criteria and were not included in the case-control study.
The case-control study population comprised 2,756 patients with schizophrenia and 2,646 controls, who were recruited from Fujita Health University,6 Kobe University,34 and Niigata University35 (Table 1). The patients were diagnosed according to DSM-IV or DSM-5 criteria. Controls had no personal or family history (first-degree relatives) of psychiatric disorders.
 |
Table 1 Characteristics of case-control study participants |
Wes
From the family, we obtained genomic DNA samples from the proband (#4), her affected younger sister (#5), and their unaffected father (#1) and mother (#2; Figure 1). WES was performed at Takara Bio Inc. (Shiga, Japan), using the HiSeq2500 system (Illumina, San Diego, CA, USA). We prepared exome libraries using the SureSelect Human All Exon V6 Kit (Agilent, Santa Clara, CA, USA). WES data were processed using GeneData Expressionist for Genomic Profiling v9.1.4a (Genedata, Basel, Switzerland). Adaptor sequences and low-quality reads were removed from raw sequence reads using Trimmomatic v0.1.9 (http://www.usadellab.org/cms/?page=trimmomatic).36 Cleaned sequence reads were mapped against the reference human genome (UCSC hg19) using the Burrows–Wheeler Aligner-MEM v0.7.12 (http://bio-bwa.sourceforge.net/).37 Variations were annotated using SnpEff v3.6c (http://snpeff.sourceforge.net/)38 and VCFtools v0.1.9 (https://vcftools.github.io/index.html).39 We calculated the coefficient of relationship from the WES data for each pair of individuals using peddy (https://github.com/brentp/peddy).40
To prioritize variations, we applied the following filtering steps (Table 2). First, we included variations on autosomes. Second, we included variations covered by ≥10 reads. Third, we included “HIGH” or “MODERATE” Effect_Impact variations predicted using SnpEff v3.6c. Fourth, we included recessive homozygous and compound heterozygous variations identified in both affected siblings. Fifth, we included rare variations with mutant allele frequency <0.01 in the Japanese Multi Omics Reference Panel (jMorp) 3.5KJPNv2 (https://jmorp.megabank.tohoku.ac.jp/201808/),41 the Human Genetic Variation Database (HGVD) v1.42 (http://www.genome.med.kyoto-u.ac.jp/SnpDB/),42 the BioBank Japan Whole-Genome Sequencing (BBJWGS) database (http://jenger.riken.jp/),43 Japanese data from the 1000 Genomes Project (1KGP) phase 3 (https://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/),44 and East Asian data from the Genome Aggregation Database (gnomAD) v2.1 (non-neuro) (http://gnomad.broadinstitute.org/).45
 |
Table 2 Filtering steps applied to variations identified by WES |
To validate prioritized variations, we performed Sanger sequencing using a 3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA, USA), as previously described.46
Sequencing the SPATA7 coding region
The SPATA7 coding region (RefSeq accession number, NM_018418) was sequenced in 142 affected offspring from 137 families. In 32 offspring, we screened for rare recessive SPATA7 variations using our published35,47 and unpublished WES data. In the remaining 110 offspring, we performed Sanger sequencing. Primer sequences for amplification are listed in Table S1.48
Case-control study
We performed an association study of rare recessive SPATA7 variations, prioritized via WES and sequencing, with schizophrenia in 2,756 patients and 2,646 controls. We genotyped p.Asp134Gly and p.Ile332Thr in our case-control samples, using the TaqMan 5′-exonuclease assay (Thermo Fisher Scientific, Waltham, MA, USA; Table S2), as previously described.33
In silico analysis
We performed in silico analysis to predict the functional effects of SPATA7 variations identified via WES and resequencing using Polymorphism Phenotyping v2 (PolyPhen-2; http://genetics.bwh.harvard.edu/pph2/),49 Protein Variation Effect Analyzer v1.1 (PROVEN; http://provean.jcvi.org/genome_submit_2.php?species=human),50 and Combined Annotation Dependent Depletion (CADD; http://cadd.gs.washington.edu/home) scores.51
Results
The mean read depth varied from 48.0× to 67.6×, and 97.1–98.1% of the target regions were covered by 10 or more reads (Table S3). We identified a total of 213,038 variations via WES (Table 2). The coefficient of relationship observed for the parents (#1 and #2) was 0.038, which was similar to 0.031, the value expected for second cousins (Table S4). The coefficients of relationship observed for the other pairs of individuals ranged from 0.430 to 0.522, which were similar to 0.5, the value expected for parent-offspring or siblings. After the filtering steps (Table 2), we prioritized rare compound heterozygous missense variations in SPATA7 (Table 3). One was previously unidentified: an A to G transition (g.88892604A>G) at codon 134 resulting in an aspartic acid to glycine substitution (p.Asp134Gly). The other, a T to C transition (g.88895774T>C) at codon 332 resulting in an isoleucine to threonine substitution (p.Ile332Thr), had been previously reported (rs534658921). Unaffected father (#1) and mother (#2) transmitted the mutant p.Ile332Thr and p.Asp134Gly alleles, respectively, to both affected siblings (#4 and #5). In silico analysis predicted these variations to be “benign” and “neutral” using PolyPhen-2 and PROVEN, respectively (Table 3). CADD scores for p.Asp134Gly and p.Ile332Thr were 3.243 and 8.805, respectively, indicating that these variations were not deleterious.
 |
Table 3 Rare compound heterozygous missense SPATA7 variations prioritized by WES |
Sequencing SPATA7 coding regions identified eight variations in 142 affected offspring (Table S5). However, there were no rare recessive variations. In the case-control study, p.Asp134Gly was not found in 2,732 patients or 2,627 controls, while heterozygous p.Ile332Thr was observed in five patients and one control (Table 4). In these individuals, we did not detect other rare variations by sequencing SPATA7 coding regions. The frequency of mutant alleles (0.0002) of p.Ile332Thr in our control group was similar to that in large databases including jMorp (0.0001), HGVD (0.0004), and gnomAD (0.0005; Table 3).
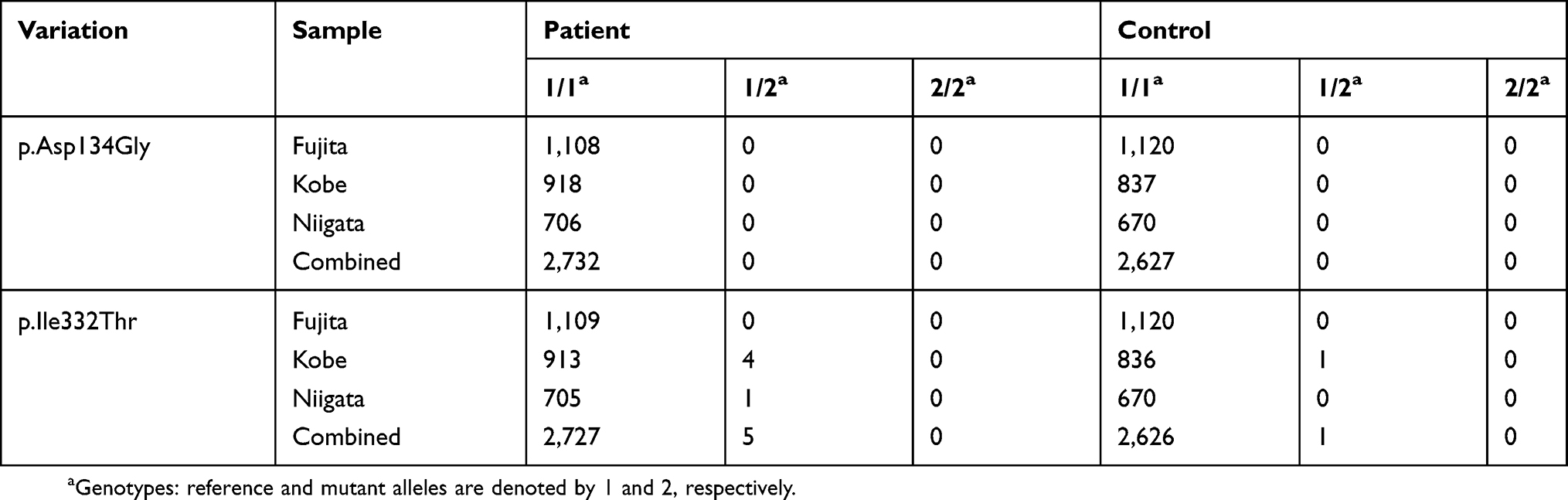 |
Table 4 Genotyping of two missense SPATA7 variations in the case-control study |
Discussion
In the first-stage of this study, we did not identify rare recessive homozygous variations, but rare compound heterozygous missense SPATA7 variations, p.Asp134Gly and p.Ile332Thr, via WES in a family with two affected siblings whose unaffected parents were second cousins. Even in a consanguineous pedigree, a disease trait may be caused by compound heterozygous variations.52 For example, rare compound heterozygous missense AACS variations were identified in a consanguineous Pakistani family with autosomal recessive intellectual disability.53 In the second-stage of our study, sequencing SPATA7 coding regions did not detect rare recessive variations in 142 affected offspring for whom parental DNA samples were available for genotyping. In the third-stage of the study, we did not provide statistical evidence for the associations of SPATA7 p.Asp134Gly and p.Ile332Thr with schizophrenia in 2,756 patients and 2,646 controls.
There is no converging evidence that rare recessive variations play an important role in the genetic etiology of schizophrenia. In a WES study of seven Italian schizophrenia patients with a high number of large runs of homozygosity (ROH), Giacopuzzi et al identified 119 low frequency, homozygous, recessive, non-synonymous and splice-cite variations in 107 genes within ROH regions.54 These genes significantly overlapped with the composite set of 1,796 genes of a Swedish case-control sample.14 Using WES data of the Swedish case-control sample, Magri et al found that rare homozygous variations in genes of the gamma-aminobutyric acid system were more frequent in patients (6/4,225) compared with controls (0/5,834).55 However, Ruderfer et al observed no significant difference in rare recessive gene-disrupting variations between Swedish patients (229 of 2,477) and controls (233 of 2,481).56 WES of 604 Bulgarian parent-affected offspring trios did not find an increased burden of rare recessive non-synonymous variations.57 To draw any conclusion on the effect of rare recessive variations on schizophrenia, further studies should be performed using sufficiently large sample sizes.
SPATA7 encodes spermatogenesis-associated protein 7 (SPATA7). Spata7 was identified in rat testis, and SPATA7 was isolated by screening a human testis library.58 SPATA7 mRNA levels are high in retina, brain and testis.59 Recessive loss-of-function SPATA7 variations cause Laber congenital amaurosis and juvenile retinitis pigmentosa.48,59,60 In mouse retina, SPATA7 plays a critical role in the proper localization of proteins at the distal connecting cilium.61,62 However, the functions of SPATA7 in the brain remain unclear. In silico analysis predicted the SPATA7 variations, p.Asp134Gly and p.Ile332Thr, to be not damaging. Nevertheless, functional analyses are required to assess the functional implications of these variations. Earlier WES studies also reported no significant association between rare SPATA7 variations and schizophrenia14 and no de novo SPATA7 variations in schizophrenia.13 SPATA7 expression in the dorsolateral prefrontal cortex was not altered in schizophrenia patients.63 There were no available data regarding the methylation of SPATA7 in the three postmortem brain studies that are registered in the schizophrenia database (SZDB) v2 (http://www.szdb.org/index.html).64 Taken together, these findings do not support the role of SPATA7 in the development of schizophrenia.
There are some limitations to our study. First, our WES study had no power to statistically analyze the results and assess their significance. Therefore, we performed follow-up sequencing of SPATA7 and a case-control study. However, we did not confirm the findings from the WES study. Second, we prioritized rare recessive variations because we hypothesized that these variations play a substantial role in conferring risk for schizophrenia in the consanguineous family with affected siblings. However, the inclusion of compound heterozygous variations may partially contradict the original study design. When we included low frequency, homozygous, recessive variations with mutant allele frequency <0.05, we identified two missense variations: HEBP2 p.Arg140Gln (rs3734303) and UPK2 p.Arg152Cy (rs137900462). Even when we genotyped rs3734303 and rs137900462 in our case-control samples, we found no significant associations between these two low frequency missense variations and schizophrenia (Table S6). It is possible that we overlooked the role of other kinds of variations, eg de novo variations13,17 and copy number variations.10–12 In our family, we identified no rare de novo variations or large homozygous deletions shared by two affected siblings. Third, genomic DNA samples from three siblings (#3, #6 and #7) were not available, and thus we were unable to assess whether they had rare compound heterozygous missense SPATA7 variations (p.Asp134Gly and p.Ile332Thr). Therefore, it was difficult to distinguish whether the variations that were prioritized in the family were potential risk variations or were coincidentally shared by two affected siblings. Fourth, our results did not exclude the possibility that common variations are implicated in schizophrenia vulnerability in consanguineous families. Interestingly, a WGS study of eight families with monozygotic twin pairs discordant for schizophrenia revealed that polygenic risk scores were higher in probands than in unaffected parents.65 Because we performed WES but not WGS, we were unable to calculate the polygenic risk scores.
Conclusion
Our data provide no evidence for the contribution of the rare compound heterozygous SPATA7 missense variations p.Asp134Gly and p.Ile332Thr to the risk of schizophrenia.
Acknowledgment
The authors thank the patients, their families, and the healthy volunteers for their participation. We would also like to thank Ms Yamazaki, Ms Aizawa and Ms Nagashima for excellent technical assistance. This work was supported by Grants-in-Aid for Scientific Research (16K19754 to HI) from the Japan Society for the Promotion of Science, by a grant from the Niigata Medical Association (to HI), and by a grant from SENSHIN Medical Research Foundation (to YW). We thank Jeremy Allen, PhD, and Sydney Koke, MFA, from Edanz Group for editing a draft of this manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Owen MJ, Sawa A, Mortensen PB. Schizophrenia. Lancet. 2016;388(10039):86–97. doi:10.1016/S0140-6736(15)01121-6
2. Giegling I, Hosak L, Mössner R, et al. Genetics of schizophrenia: a consensus paper of the WFSBP Task Force on Genetics. World J Biol Psychiatry. 2017;18(7):492–505. doi:10.1080/15622975.2016.1268715
3. Kanazawa T, Bousman CA, Liu C, Everall IP. Schizophrenia genetics in the genome-wide era: a review of Japanese studies. NPJ Schizophr. 2017;3(1):27.
4. Yue W, Yu X, Zhang D. Progress in genome-wide association studies of schizophrenia in Han Chinese populations. NPJ Schizophr. 2017;3(1):24.
5. Li Z, Chen J, Yu H, et al. Genome-wide association analysis identifies 30 new susceptibility loci for schizophrenia. Nat Genet. 2017;49(11):1576–1583.
6. Ikeda M, Takahashi A, Kamatani Y, et al. Genome-wide association study detected novel susceptibility genes for schizophrenia and shared trans-populations/diseases genetic effect. Schizophr Bull. 2019;45(4):824–834.
7. Pardiñas AF, Holmans P, Pocklington AJ, et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat Genet. 2018;50(3):381–389.
8. Sekar A, Bialas AR, de Rivera H, et al. Schizophrenia risk from complex variation of complement component 4. Nature. 2016;530(7589):177–183.
9. Sullivan PF, Agrawal A, Bulik CM, et al. Psychiatric genomics: an update and an agenda. Am J Psychiatry. 2018;175(1):15–27.
10. Li Z, Chen J, Xu Y, et al. Genome-wide analysis of the role of copy number variation in schizophrenia risk in Chinese. Biol Psychiatry. 2016;80(4):331–337. doi:10.1016/j.biopsych.2015.11.012
11. Marshall CR, Howrigan DP, Merico D, et al. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat Genet. 2017;49(1):27–35. doi:10.1038/ng.3725
12. Kushima I, Aleksic B, Nakatochi M, et al. High-resolution copy number variation analysis of schizophrenia in Japan. Mol Psychiatry. 2017;22(3):430–440. doi:10.1038/mp.2016.88
13. Fromer M, Pocklington AJ, Kavanagh DH, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506(7487):179–184. doi:10.1038/nature12929
14. Purcell SM, Moran JL, Fromer M, et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014;506(7487):185–190. doi:10.1038/nature12975
15. Genovese G, Fromer M, Stahl EA, et al. Increased burden of ultra-rare protein-altering variants among 4,877 individuals with schizophrenia. Nat Neurosci. 2016;19(11):1433–1441. doi:10.1038/nn.4402
16. Singh T, Kurki MI, Curtis D, et al. Rare loss-of-function variants in SETD1A are associated with schizophrenia and developmental disorders. Nat Neurosci. 2016;19(4):571–577. doi:10.1038/nn.4267
17. Takata A, Ionita-Laza I, Gogos JA, Xu B, Karayiorgou M. De novo synonymous mutations in regulatory elements contribute to the genetic etiology of autism and schizophrenia. Neuron. 2016;89(5):940–947. doi:10.1016/j.neuron.2016.02.024
18. Cirulli ET, Goldstein DB. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat Rev Genet. 2010;11(6):415–425. doi:10.1038/nrg2779
19. Glahn DC, Nimgaonkar VL, Raventós H, et al. Rediscovering the value of families for psychiatric genetics research. Mol Psychiatry. 2019;24(4):523–535.
20. Timms AE, Dorschner MO, Wechsler J, et al. Support for the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia from exome sequencing in multiplex families. JAMA Psychiatry. 2013;70(6):582–590. doi:10.1001/jamapsychiatry.2013.1195
21. Egawa J, Hoya S, Watanabe Y, et al. Rare UNC13B variations and risk of schizophrenia: whole-exome sequencing in a multiplex family and follow-up resequencing and a case-control study. Am J Med Genet B Neuropsychiatr Genet. 2016;171(6):797–805. doi:10.1002/ajmg.b.32444
22. Homann OR, Misura K, Lamas E, et al. Whole-genome sequencing in multiplex families with psychoses reveals mutations in the SHANK2 and SMARCA1 genes segregating with illness. Mol Psychiatry. 2016;21(12):1690–1695. doi:10.1038/mp.2016.24
23. Zhou Z, Hu Z, Zhang L, et al. Identification of RELN variation p.Thr3192Ser in a Chinese family with schizophrenia. Sci Rep. 2016;6:24327.
24. John J, Kukshal P, Bhatia T, et al. Possible role of rare variants in Trace amine associated receptor 1 in schizophrenia. Schizophr Res. 2017;189:190–195. doi:10.1016/j.schres.2017.02.020
25. Steinberg S, Gudmundsdottir S, Sveinbjornsson G, et al. Truncating mutations in RBM12 are associated with psychosis. Nat Genet. 2017;49(8):1251–1254. doi:10.1038/ng.3894
26. de Vrij FM, Bouwkamp CG, Gunhanlar N, et al. Candidate CSPG4 mutations and induced pluripotent stem cell modeling implicate oligodendrocyte progenitor cell dysfunction in familial schizophrenia. Mol Psychiatry. 2019;24(5):757–771. doi:10.1038/s41380-017-0004-2
27. John J, Kukshal P, Sharma A, et al. Rare variants in Protein tyrosine phosphatase, receptor type A (PTPRA) in schizophrenia: evidence from a family based study. Schizophr Res. 2019;206:75–81. doi:10.1016/j.schres.2018.12.012
28. O’Brien NL, Fiorentino A, Curtis D, et al. Rare variant analysis in multiply affected families, association studies and functional analysis suggest a role for the ITGΒ4 gene in schizophrenia and bipolar disorder. Schizophr Res. 2018;199:181–188. doi:10.1016/j.schres.2018.03.001
29. John J, Sharma A, Kukshal P, et al. Rare variants in tissue inhibitor of metalloproteinase 2 as a risk factor for schizophrenia: evidence from familial and cohort analysis. Schizophr Bull. 2019;45(1):256–263. doi:10.1093/schbul/sbx196
30. Xue CB, Xu ZH, Zhu J, et al. Exome sequencing identifies TENM4 as a novel candidate gene for schizophrenia in the SCZD2 locus at 11q14-21. Front Genet. 2019;9:725.
31. Harold D, Connolly S, Riley BP, et al. Population-based identity-by-descent mapping combined with exome sequencing to detect rare risk variants for schizophrenia. Am J Med Genet B Neuropsychiatr Genet. 2019;180(3):223–231. doi:10.1002/ajmg.b.32716
32. Salvoro C, Bortoluzzi S, Coppe A, et al. Rare risk variants identification by identity-by-descent mapping and whole-exome sequencing implicates neuronal development pathways in schizophrenia and bipolar isorder. Mol Neurobiol. 2018;55(9):7366–7376. doi:10.1007/s12035-018-0922-2
33. Watanabe Y, Muratake T, Kaneko N, Nunokawa A, Someya T. No association between the brain-derived neurotrophic factor gene and schizophrenia in a Japanese population. Schizophr Res. 2006;84(1):29–35. doi:10.1016/j.schres.2006.03.011
34. Otsuka I, Watanabe Y, Hishimoto A, et al. Association analysis of the Cadherin13 gene with schizophrenia in the Japanese population. Neuropsychiatr Dis Treat. 2015;11:1381–1393. doi:10.2147/NDT.S84736
35. Hoya S, Watanabe Y, Hishimoto A, et al. Rare FBXO18 variations and risk of schizophrenia: whole-exome sequencing in two parent-affected offspring trios followed by resequencing and case-control studies. Psychiatry Clin Neurosci. 2017;71(8):562–568. doi:10.1111/pcn.12526
36. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi:10.1093/bioinformatics/btu170
37. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi:10.1093/bioinformatics/btp324
38. Cingolani P, Platts A, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012;6(2):80–92. doi:10.4161/fly.19695
39. Danecek P, Auton A, Abecasis G, et al. The variant call format and VCFtools. Bioinformatics. 2011;27(15):2156–2158. doi:10.1093/bioinformatics/btr330
40. Pedersen BS, Quinlan AR. Who’s who? Detecting and resolving sample anomalies in human DNA sequencing studies with peddy. Am J Hum Genet. 2017;100(3):406–413. doi:10.1016/j.ajhg.2017.01.017
41. Tadaka S, Saigusa D, Motoike IN, et al. jMorp: Japanese multi omics reference panel. Nucleic Acids Res. 2018;46(D1):D551–D557.
42. Higasa K, Miyake N, Yoshimura J, et al. Human genetic variation database, a reference database of genetic variations in the Japanese population. J Hum Genet. 2016;61(6):547–553. doi:10.1038/jhg.2016.12
43. Okada Y, Momozawa Y, Sakaue S, et al. Deep whole-genome sequencing reveals recent selection signatures linked to evolution and disease risk of Japanese. Nat Commun. 2018;9(1):1631.
44. 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature. 2015;526(7571):68–74. doi:10.1038/nature15393
45. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–291. doi:10.1038/nature19057
46. Nunokawa A, Watanabe Y, Kaneko N, et al. The dopamine D3 receptor (DRD3) gene and risk of schizophrenia: case-control studies and an updated meta-analysis. Schizophr Res. 2010;116(1):61–67. doi:10.1016/j.schres.2009.10.016
47. Watanabe Y, Nunokawa A, Shibuya M, et al. Rare truncating variations and risk of schizophrenia: whole-exome sequencing in three families with affected siblings and a three-stage follow-up study in a Japanese population. Psychiatry Res. 2016;235:13–18.
48. Wang H, Den Hollander AI, Moayedi Y, et al. Mutations in SPATA7 cause Leber congenital amaurosis and juvenile retinitis pigmentosa. Am J Hum Genet. 2009;84(3):380–387.
49. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249.
50. Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012;7(10):e46688.
51. Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47(D1):D886–D894.
52. Ott J, Wang J, Leal SM. Genetic linkage analysis in the age of whole-genome sequencing. Nat Rev Genet. 2015;16(5):275–284.
53. Riazuddin S, Hussain M, Razzaq A, et al. Exome sequencing of Pakistani consanguineous families identifies 30 novel candidate genes for recessive intellectual disability. Mol Psychiatry. 2017;22(11):1604–1614.
54. Giacopuzzi E, Gennarelli M, Minelli A, et al. Exome sequencing in schizophrenic patients with high levels of homozygosity identifies novel and extremely rare mutations in the GABA/glutamatergic pathways. PLoS One. 2017;12(8):e0182778.
55. Magri C, Giacopuzzi E, La Via L, et al. A novel homozygous mutation in GAD1 gene described in a schizophrenic patient impairs activity and dimerization of GAD67 enzyme. Sci Rep. 2018;8(1):15470.
56. Ruderfer DM, Lim ET, Genovese G, et al. No evidence for rare recessive and compound heterozygous disruptive variants in schizophrenia. Eur J Hum Genet. 2015;23(4):555–557.
57. Rees E, Kirov G, Walters JT, et al. Analysis of exome sequence in 604 trios for recessive genotypes in schizophrenia. Transl Psychiatry. 2015;5:e607.
58. Zhang X, Liu H, Zhang Y, et al. A novel gene, RSD-3/HSD-3.1, encodes a meiotic-related protein expressed in rat and human testis. J Mol Med (Berl). 2003;81(6):380–387.
59. Perrault I, Hanein S, Gerard X, et al. Spectrum of SPATA7 mutations in Leber congenital amaurosis and delineation of the associated phenotype. Hum Mutat. 2010;31(3):E1241–1250.
60. Mackay DS, Ocaka LA, Borman AD, et al. Screening of SPATA7 in patients with Leber congenital amaurosis and severe childhood-onset retinal dystrophy reveals disease-causing mutations. Invest Ophthalmol Vis Sci. 2011;52(6):3032–3038.
61. Eblimit A, Nguyen TM, Chen Y, et al. Spata7 is a retinal ciliopathy gene critical for correct RPGRIP1 localization and protein trafficking in the retina. Hum Mol Genet. 2015;24(6):1584–1601.
62. Dharmat R, Eblimit A, Robichaux MA, et al. SPATA7 maintains a novel photoreceptor-specific zone in the distal connecting cilium. J Cell Biol. 2018;217(8):2851–2865.
63. Fromer M, Roussos P, Sieberts SK, et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat Neurosci. 2016;19(11):1442–1453.
64. Wu Y, Yao YG, Luo XJ. SZDB: a database for schizophrenia genetic research. Schizophr Bull. 2017;43(2):459–471.
65. Tang J, Fan Y, Li H, et al. Whole-genome sequencing of monozygotic twins discordant for schizophrenia indicates multiple genetic risk factors for schizophrenia. J Genet Genomics. 2017;44(6):295–306.
Supplementary materials
 |
Table S1 Primer sequences for sequencing SPATA7 coding regions |
 |
Table S2 Probes used for the TaqMan 5′-exonuclease assay |
 |
Table S3 WES quality report summary |
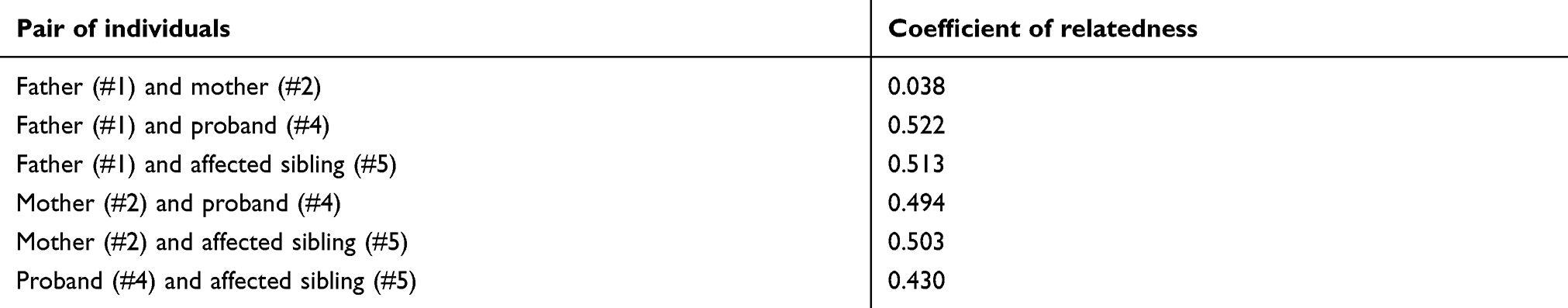 |
Table S4 Coefficient of relatedness from the WES data for each pair of individuals |
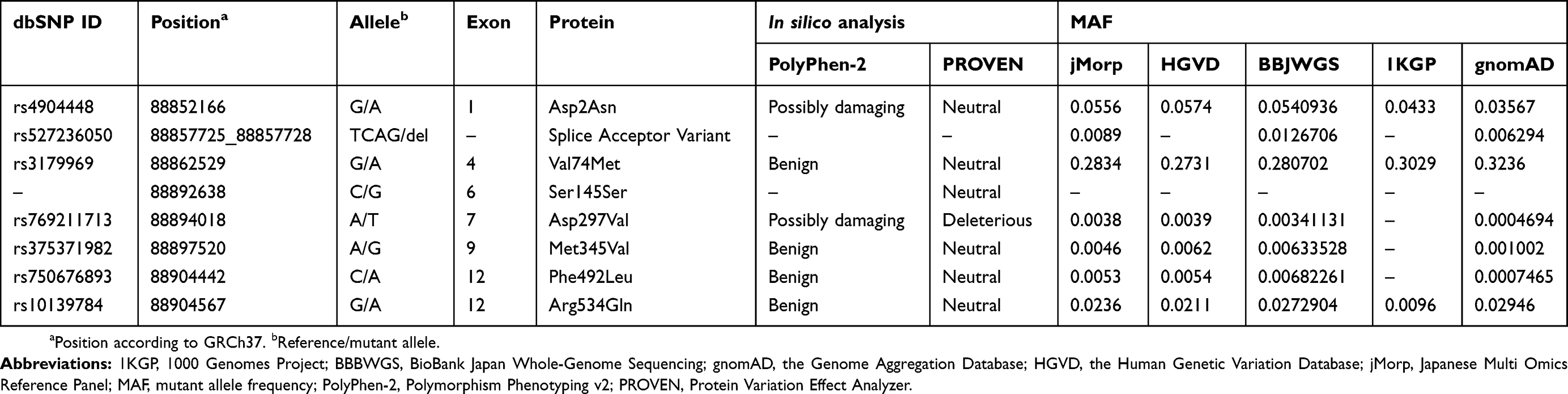 |
Table S5 SPATA7 variations identified by sequencing |
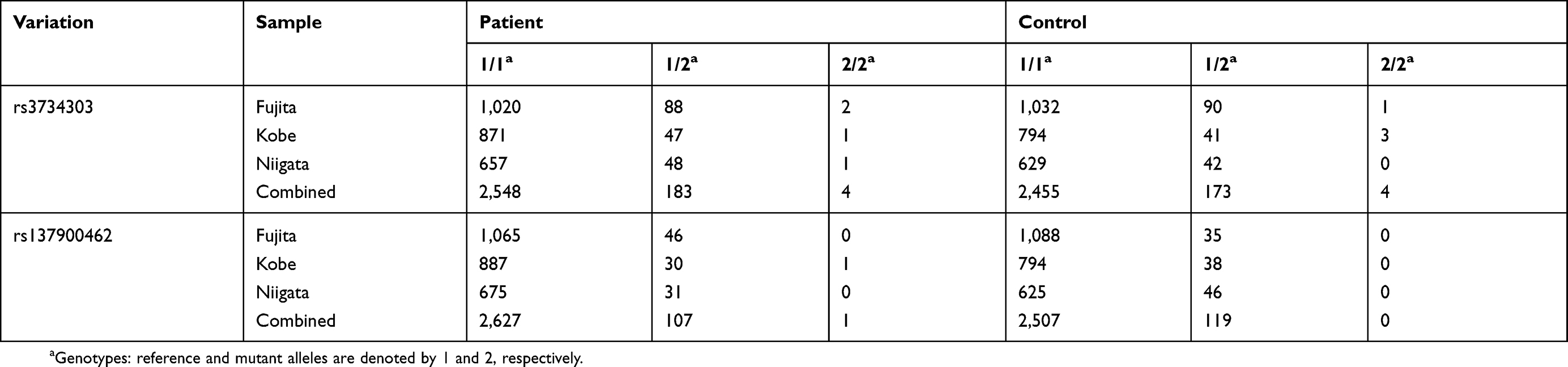 |
Table S6 Genotyping of two uncommon missense variations in the case-control study |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.