Back to Journals » Infection and Drug Resistance » Volume 19
Rapid Identification of Drug-Resistant Tuberculosis Using Nanopore Targeted Next-Generation Sequencing from Sputum and Culture Isolates: Accuracy and Limitations
Authors Dokrungkoon T, Chumponsuk T ![]() , Suwannakarn K, Sripichai O, Ngamskulrungroj P
, Suwannakarn K, Sripichai O, Ngamskulrungroj P ![]()
Received 19 October 2025
Accepted for publication 22 January 2026
Published 3 February 2026 Volume 2026:19 571825
DOI https://doi.org/10.2147/IDR.S571825
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Hazrat Bilal
Thanadon Dokrungkoon,1 Tawatchai Chumponsuk,1 Kamol Suwannakarn,1 Orapan Sripichai,2 Popchai Ngamskulrungroj1
1Department of Microbiology, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand; 2National Institute of Health, Department of Medical Sciences, Ministry of Public Health, Nonthaburi, Thailand
Correspondence: Popchai Ngamskulrungroj, Department of Microbiology, Faculty of Medicine Siriraj Hospital, Mahidol University, 2 Wanglang Road, Siriraj, Bangkok Noi, Bangkok, 10700, Thailand, Tel +66 2 419 7053, Fax +66 2 418 4148, Email [email protected]
Purpose: Drug-resistant tuberculosis (DR-TB) complicates treatment and requires diagnostic approaches capable of comprehensive resistance profiling of Mycobacterium tuberculosis (MTB). This study evaluated the diagnostic performance of the Oxford Nanopore Technologies (ONT) custom TB-DR sequencing assay, a targeted Next-Generation Sequencing (tNGS) using Nanopore sequencing technology, in sputum and culture isolates. The assay targets resistance-associated variants across 24 genes covering 13 anti-tuberculosis drugs and integrates the hsp65 gene and direct repeat (DR) region for species identification and lineage determination.
Methods: DNA was extracted from 88 clinical samples, comprising 30 uncultured sputum specimens (10 MTB-positive, 10 non-tuberculous mycobacteria, and 10 mycobacteria-negative controls) to evaluate species identification, and 58 MTB culture isolates. The culture isolates represented diverse phenotypic resistance profiles, including mono-drug resistant, multidrug-resistant, and pre-extensively drug-resistant strains. tNGS profiles were compared with pDST to evaluate diagnostic performance for drug-resistance profiling, including sensitivity, specificity, and test agreement.
Results: Profiling success in sputum samples was dependent on mycobacterial load. Among MTB-positive sputum specimens, 6 (60%) produced results, including 2 complete resistance profiles and 4 partial profiles limited to species identification; the remaining specimens failed due to low mycobacterial load (smear-negative and high Ct values). All NTM samples were correctly identified, and all mycobacteria-negative controls tested negative. In contrast, 57 of 58 (98.3%) culture isolates yielded complete resistance profiles. Compared with pDST, sensitivity and specificity exceeded 90% for most drugs (except streptomycin, 85.7% sensitivity), with very strong agreement (κ > 0.8).
Conclusion: The ONT custom TB-DR sequencing assay provides comprehensive resistance profiling with high concordance to pDST in samples yielding complete sequencing profiles and enables species and lineage identification with a shorter analytical turnaround time compared with phenotypic testing. Performance in sputum specimens was influenced by mycobacterial load. Further studies involving larger and more diverse cohorts are needed to validate clinical applicability.
Keywords: Mycobacterium tuberculosis, Nanopore sequencing, tNGS, drug-resistant tuberculosis, rapid diagnostics
Introduction
Tuberculosis (TB), caused by Mycobacterium tuberculosis (MTB), persists as a formidable global health threat. Data from the Global Tuberculosis Report 2025 confirm that TB remains the leading cause of death from a single infectious agent, following the COVID-19 pandemic period, and presents a heavy burden worldwide.1 Despite extensive global efforts, the incidence of drug-resistant tuberculosis (DR-TB) remains a critical concern.1 In this context, accurate and timely drug susceptibility testing (DST) is critical, not only for guiding effective treatment regimens but also for curbing the transmission of resistant strains.1
A major challenge in TB diagnostics is the gap between speed and comprehensiveness. While phenotypic drug susceptibility testing (pDST) remains the gold standard, it is labor-intensive and delayed by the requirement for mycobacterial culture. Rapid molecular assays like Xpert MTB/RIF, line probe assays (LPA), and MTB real-time PCR panels have revolutionized turnaround times.2,3 However, these molecular methods are primarily limited to detecting common resistance-associated variants (RAVs) for specific drugs, often resulting in incomplete resistance profiles.4 Whole genome sequencing (WGS) offers a more comprehensive approach, providing detailed genomic data for resistance profiling and phylogenetic analysis. However, WGS is typically performed on cultured MTB isolates and requires biosafety level 3 facilities, delaying treatment decisions by weeks. While WGS enrichment from uncultured sputum is a promising development,5 the high costs and advanced bioinformatics expertise required to make it inaccessible to many low- and middle-income countries.6 However, bypassing culture to perform sequencing directly on sputum introduces a practical trade-off in diagnostic performance. Sequencing performed on cultured isolates has been shown to provide reliable drug-resistance profiling, with good concordance to pDST. In contrast, studies of direct sputum sequencing indicate that not all specimens yield sufficient data for complete resistance prediction, with performance influenced by the amount of mycobacterial DNA present in the specimen.7,8
To bridge this gap, targeted next-generation sequencing (tNGS) has emerged as a practical alternative recommended by the WHO for patients requiring comprehensive DST with shorter turnaround times.9 By amplifying and sequencing only specific drug-resistance regions, tNGS offers a balance between the depth of WGS and the sensitivity required for direct specimen analysis. Accordingly, the WHO Consolidated Guidelines on Tuberculosis (2025) have endorsed this class of diagnostics, including platforms such as the Deeplex® Myc-TB assay (GenoScreen) and TBSeq (ShengTing Medical Technology Co).10 These amplicon-based approaches allow for simultaneous comprehensive profiling without the high cost and infrastructure demands of WGS.11
The ONT custom TB-DR sequencing assay (Oxford Nanopore Technologies) is a tNGS platform utilizing Nanopore sequencing technology, specifically designed for TB diagnostics. The assay detects RAVs in 24 target genes of MTB linked to 13 anti-TB drugs: rifampicin (rpoB), isoniazid (katG, inhA, fabG1), pyrazinamide (pncA), ethambutol (embB, embA), streptomycin (rrs, gidB, rpsL), kanamycin (eis, rrs), amikacin (rrs), capreomycin (tlyA, rrs), fluoroquinolones (gyrA, gyrB), ethionamide (ethA, inhA, fabG1), linezolid (rplC, rrl), bedaquiline and clofazimine (rv0678), and delamanid (fgd1, ddn, atpE, fbiA, fbiB, fbiC). It also includes the hsp65 gene for differentiating MTB from non-tuberculous mycobacteria (NTM) and the direct repeat (DR) locus for spoligotyping, enabling genotyping of the Mycobacterium tuberculosis complex (MTBC) to track strains and infer lineages. Notably, the platform can process up to 22 samples per flow cell, along with positive and negative controls, making it suitable for moderate-throughput settings. Therefore, this study aimed to validate this assay on both uncultured sputum and culture isolates to assess its diagnostic performance, including sensitivity, specificity, accuracy, and agreement with pDST as the gold standard.
Materials and Methods
Study Population and Samples
This study was conducted retrospectively using leftover DNA extracted from a total of 88 clinical samples stored between 2022 and 2024 at the routine Mycobacterium Laboratory, Department of Microbiology, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand. The archived samples were obtained from patients presenting with symptoms of pulmonary tuberculosis at Siriraj Hospital, a tertiary referral center.
We strategically selected archived samples to validate the performance of the assay across different clinical contexts. The study population was structured to address two specific objectives. First, to evaluate diagnostic accuracy using direct clinical specimens, the cohort included 30 DNA samples extracted from uncultured sputum, stratified into 10 confirmed M. tuberculosis (MTB), 10 non-tuberculous mycobacteria (NTM), and 10 mycobacteria-negative samples to strictly assess specificity of the assay. Second, to evaluate the accuracy of drug resistance prediction, the remaining 58 DNA samples derived from culture isolates were selected to maximize the diversity of phenotypic resistance profiles. This enriched cohort included 10 isoniazid-resistant (IR-TB), 11 rifampicin-resistant (RR-TB), 16 multidrug-resistant TB (MDR-TB), 4 pre-extensively drug-resistant TB (pre-XDR), and 17 pan-susceptible isolates, aiming to challenge the assay against both first- and second-line anti-TB drugs. Drug susceptibility profiles for the 58 MTB isolates were determined using standard phenotypic DST methods (MGIT 960 and agar proportion), which served as the gold standard for evaluating the genotypic predictions of the tNGS assay.
All samples underwent routine diagnostic identification serving as the reference standard, including acid-fast bacillus (AFB) staining, liquid medium culture, Löwenstein-Jensen (LJ) culture, and Anyplex™ MTB/NTM Real-time Detection (Seegene Inc., Republic of Korea). Specifically, NTM were identified using the Anyplex™ MTB/NTM assay, while the mycobacteria-negative group consisted of clinical sputum specimens confirmed negative for both MTB and NTM by real-time PCR (true negative samples).
Specimen Processing and DNA Extraction
Sputum Samples
Single uncultured sputum samples were collected, typically in volumes of 4–5 mL (or the maximum available volume from a single collection). To decontaminate the samples, an equal volume of NALC-NaOH solution (2% w/v NALC, 1% v/v NaOH) was added to the sputum in a 50 mL tube. The mixture was shaken for 5–20 seconds to liquefy the sputum and ensure thorough contact with the decontaminating solution. The tubes were then allowed to stand for 15 minutes, followed by adding Phosphate Buffer Saline (PBS) pH 6.8 or distilled water to a final volume of 45 mL. The tubes were capped tightly and inverted several times to mix. Subsequently, the samples were centrifuged at 3000g for 15 minutes. The supernatant was discarded, and 500 µL of PBS was added to the pellet. The mixture was resuspended by pipetting up and down. For culture purposes, 250 µL of the suspension was spread on Löwenstein–Jensen (LJ) medium. The remaining 250 µL was used for genomic DNA extraction using the automated magLEAD® 12gC system (Precision System Science Co., Ltd., Japan) in accordance with the manufacturer’s instructions12 for further analysis.
Culture Isolates
For mycobacterial colonies grown on LJ medium, DNA extraction was performed using the thermolysis method.13 Using a sterile inoculation loop, 2–3 loopfuls of the colonies were transferred to a 2 mL tube containing 0.5 mL of distilled water. The suspension was then heated at 100°C for 3 minutes. After heating, the samples were centrifuged at 13,000g for 5 minutes at 4°C. The supernatant (300 µL) was transferred to a new 1.5 mL tube, and DNA concentration was measured using a Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific, USA). The extracted DNA was stored at −20°C until subsequent analysis.
Anyplex™ MTB/NTM Real-Time Detection Assay
The Anyplex™ MTB/NTM Real-time Detection assays (Seegene Inc., Republic of Korea) were conducted according to the manufacturer’s instructions, as described previously.14 Available results from the routine Mycobacterium Laboratory, Department of Microbiology, Faculty of Medicine Siriraj Hospital were retrieved for analysis. Of the 15 samples with quantitative data, 10 were from sputum samples identified as MTB-positive, and 5 were from sputum samples classified in the NTM group. The Cycle threshold (Ct) values obtained from these assays were used to assess the impact of mycobacterial load on tNGS performance in sputum samples.
Phenotypic Drug Susceptibility Testing by Agar Proportion and MGIT
Drug susceptibility testing (DST) for all culture-positive isolates was conducted using the MGIT 960 system (Becton Dickinson Diagnostic System, Sparks, MD, USA) at the routine Mycobacterium Laboratory, Department of Microbiology, Faculty of Medicine Siriraj Hospital. The results were retrieved from the laboratory for analysis in this study. The assays utilized critical concentrations recommended by the World Health Organization (WHO): 0.1 mg/L for isoniazid (INH), 1 mg/L for rifampicin (RIF), 5 mg/L for ethambutol (EMB), and 2 mg/L for streptomycin (STR), using BD BACTEC™ MGIT™ 960 SIRE kits. Additionally, pyrazinamide (PZA) susceptibility was tested using the BD BACTEC™ MGIT™ PZA kit with a critical concentration of 100 mg/L.15 For the remaining drugs, DST was conducted using the agar proportion method on Middlebrook 7H10 agar. The critical concentration applied was 2 mg/L for ofloxacin (OFX). For the injectable agents, the critical concentrations were 4 mg/L for amikacin (AMK), 5 mg/L for kanamycin (KAN), and 5 mg/L for ethionamide (ETH). These concentrations were based on WHO and CLSI guidelines, ensuring accurate identification of drug resistance across all tested isolates.15,16
Library Generation for Targeted Next-Generation Sequencing
The amplification, purification, and quantification of the ONT custom TB-DR sequencing assay amplicons were performed in accordance with the Oxford Nanopore Tuberculosis Drug Resistance test’s user manual. In brief, 5 μL of DNA extract from clinical samples underwent PCR amplification using the ONT custom TB-DR sequencing assay, including primer sets designed and developed by Nanopore (Oxford Nanopore Technologies, UK). Libraries were then prepared using the Rapid Barcoding Kit SQK-RBK110.96 (Oxford Nanopore Technologies, UK), following the manufacturer’s instructions.17 Post-barcoding, the samples were pooled and purified using AMPure XP Beads (Beckman Coulter, USA) in accordance with the manufacturer’s protocols.17 The quantification of the purified libraries was performed using the Qubit dsDNA BR Assay Kit (Thermo Fisher Scientific, USA) on a Qubit fluorometer (Thermo Fisher Scientific, USA), according to the manufacturer’s instructions. For progression to the sequencing step, the library concentration had to reach a minimum of 50 ng/μL.18
Oxford Nanopore Targeted Next-Generation Sequencing (tNGS)
The sequencing adapter was attached to the qualified library in accordance with the manufacturer’s instructions prior to initiating the sequencing step. The MinION flow cell (Oxford Nanopore Technologies, UK) was primed with the designated priming buffers as per the manufacturer’s guidelines.17 Subsequently, 75 μL of the library mixture, consisting of a batch of 22 pooled samples and 2 controls, was loaded onto the flow cell following the prescribed protocols.17 The sequencing configuration was strictly set as per the Oxford Nanopore Tuberculosis Drug Resistance test’s user manual, using the high-accuracy base-calling mode in the MinKNOW software. Sequencing was conducted using the MinION Mk1B Oxford Nanopore sequencer (Oxford Nanopore Technologies, UK), which was connected to a high-performance computer. Sequencing aimed for an approximate depth of 100X to accurately detect genetic resistance markers in the specified regions.
tNGS Bioinformatic Analysis
Following the sequencing run, raw fastq files from each sample were processed for species identification, drug resistance prediction, and lineage analysis using the EPI2ME software (https://epi2me.nanoporetech.com) and its associated pipeline (wf-tb-amr-v2.0.0-alpha.4), which are provided free of charge by the ONT company. In brief, MTB and NTM were identified using the hsp65 gene. Sequencing reads from MTB were aligned to the NC_000962.3 reference genome using minimap2.19 The base composition of predefined variants was determined with bcftools20 for variant calling. Indels were called using clair3.21 Variants were phased with whatshap software.22 Lineage classification was inferred from spoligotyping by mapping reads to the direct repeat locus. The results were compiled and reported in the EPI2ME software. tNGS profile completeness classification was based on amplicon coverage, as implemented in the wf-tb-amr workflow executed within the EPI2ME software: a complete profile required coverage of 15 out of 24 drug-resistance genes, the hsp65 gene, and the internal control, all with a median depth >20x. Partial profiles met only hsp65 coverage, while failed profiles lacked sufficient coverage across all targets. Drug-resistance profiling was specific to MTB, as this platform is not designed for NTM resistance detection.
To verify the accuracy and reliability of the ONT pipeline in identifying and predicting drug-resistant TB, results were cross-validated using TB-profiler23 v6.2.1 (CLI version with the --platform nanopore parameter), applied directly to the targeted sequencing products to generate resistance profiles derived strictly from the drug-resistance gene amplicons, and a manual variant calling pipeline incorporating freebayes24 v1.3.6 for variant calling and snpEff25 v5.2 for annotation.
Statistical Analysis
Statistical analyses were conducted using RStudio (v2024.04.2 build 764) with the epiR package (v2.0.75). Genotypic resistance data for each drug were compared with the corresponding phenotypic drug susceptibility testing (pDST) results across all samples. Diagnostic performance indicators, including sensitivity, specificity, positive predictive value (PPV), negative predictive value (NPV), and accuracy, were computed using the epi.tests function. The agreement between genotypic and phenotypic resistance profiles was assessed using Cohen’s Kappa statistic, calculated via the epi.kappa function. The strength of agreement was interpreted based on the Kappa coefficient (κ), following the guidelines set by Landis and Koch:26 κ values below 0.00 indicate poor agreement; 0.00–0.20, slight agreement; 0.21–0.40, fair agreement; 0.41–0.60, moderate agreement; 0.61–0.80, strong agreement; and 0.81–1.00, very strong agreement.
Results
tNGS Performance on Sputum Samples
A total of 30 DNA samples extracted directly from uncultured sputum were analyzed. Among the 10 confirmed M. tuberculosis (MTB) samples, species identification was successful in 6 samples (60%), all of which were correctly identified as MTB. Two of these (20%) produced complete profiles (Lineage 2-Beijing and Lineage 1 EAI2-Nonthaburi) and were correctly identified as fully susceptible to all anti-tuberculosis agents. The other four samples (40%) yielded partial profiles sufficient only for species identification, providing insufficient data for drug resistance profiling, while the remaining 4 samples (40%) failed to generate sufficient data for analysis.
Sequencing success was strongly associated with mycobacterial load. Complete profiles (20%) were obtained exclusively from samples with high bacterial loads (AFB grades 2+ and 1+; Ct values 26.81–27.76) and achieved a median sequencing depth >100x. In contrast, samples yielding partial profiles (40%), including one scanty AFB sample, were associated with lower loads (Ct >31) and variable depth coverage (27x–570x). The failed profiles (40%) generally had very low bacterial loads (AFB-negative; Ct >35).
Regarding assay specificity, 20 control samples (10 NTM and 10 mycobacteria-negative) were evaluated. The assay correctly identified NTM in 3 samples (specifically M. iranicum and M. abscessus). However, similar to the low-load MTB samples, 7 NTM samples with high Ct values failed to yield species-level identification. Importantly, no false-positive MTB detection was observed in the 10 mycobacteria-negative samples, confirming 100% specificity for MTB detection. Detailed results for the control samples are provided in Supplementary Table S1.
tNGS Performance on Culture Isolates
Of the 58 culture isolates sequenced, 57 (98.3%) were successfully profiled. One sample failed to profile due to internal control failure, indicating that the PCR failed and no reads were generated, even after repeat attempts. All 57 successfully profiled samples provided “complete profiles” (100%), with a mean depth >200x for most drug-resistance genes and 200x–500x for hsp65. Lineage analysis using EPI2ME (wf-tb-amr) software from the Nanopore tNGS platform found the majority to be Lineage 2-Beijing (78.0%, n=45). Lineage 1 EAI accounted for 13.6% (n=7), Lineage 4 Euro-American made up 5.1% (n=3), and one sample belonged to Manu-ancestor, while one sample failed lineage analysis (Table 1).
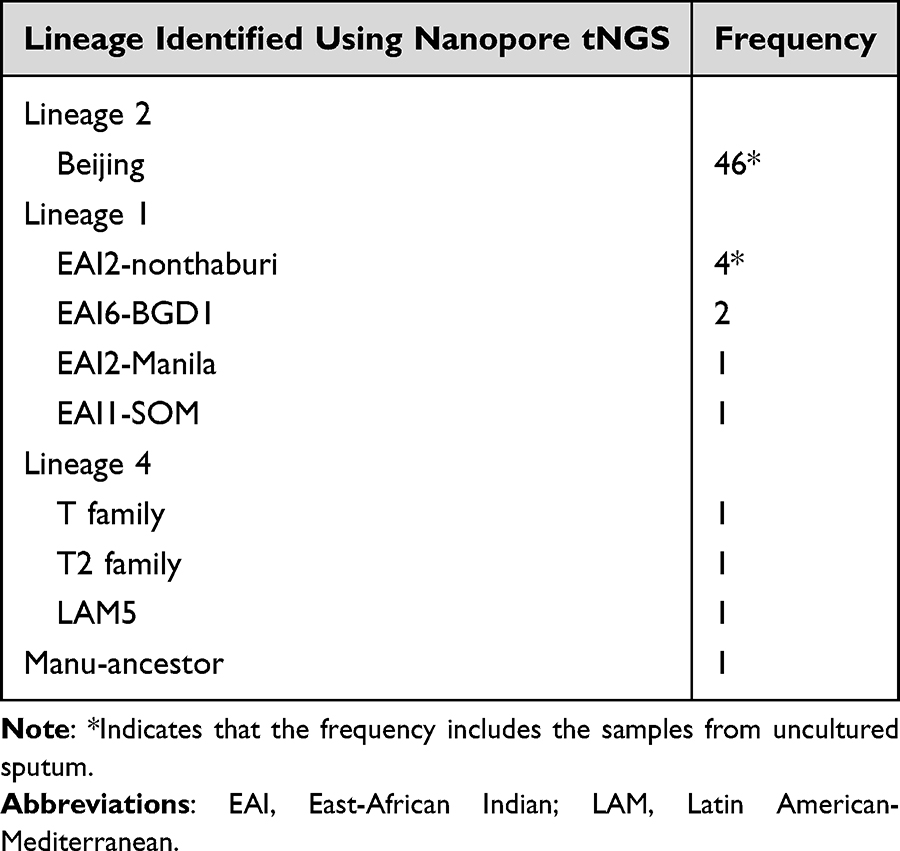 |
Table 1 Lineage Identification of Clinical Samples Using Nanopore tNGS |
Compared to sputum samples, the culture isolates showed significantly higher success rates and more consistent depth coverage. While sputum samples displayed variability due to differences in MTB DNA quantity (as indicated by AFB grades and Ct values), all successfully profiled culture isolates reached complete profiles with high mean depth. This indicates that DNA extracted from culture isolates yielded consistently higher quality and quantity of MTB DNA, enabling more reliable profiling across drug-resistance genes and lineage determination.
The Accuracy of Nanopore tNGS Assay Compared with pDST
The accuracy of the nanopore tNGS platform was assessed by comparing its performance with pDST as the gold standard. A total of 59 samples (57 culture isolates and 2 sputum samples) were included in the analysis. However, pDST for pyrazinamide, streptomycin, and second-line drugs was performed only upon clinical indication; therefore, concordance analyses for these drugs were based on isolates with available paired phenotypic and genotypic data. Detailed phenotypic and genotypic profiles of these isolates are provided in Supplementary Table S2 and Supplementary Figure S1. The results showed high concordance between nanopore tNGS and pDST across both sample types (over 85%). Diagnostic metrics, including sensitivity, specificity, PPV, and NPV, were calculated for each drug assessed (Table 2), with genotypic resistance profiles demonstrating concordance with pDST across both sample types.
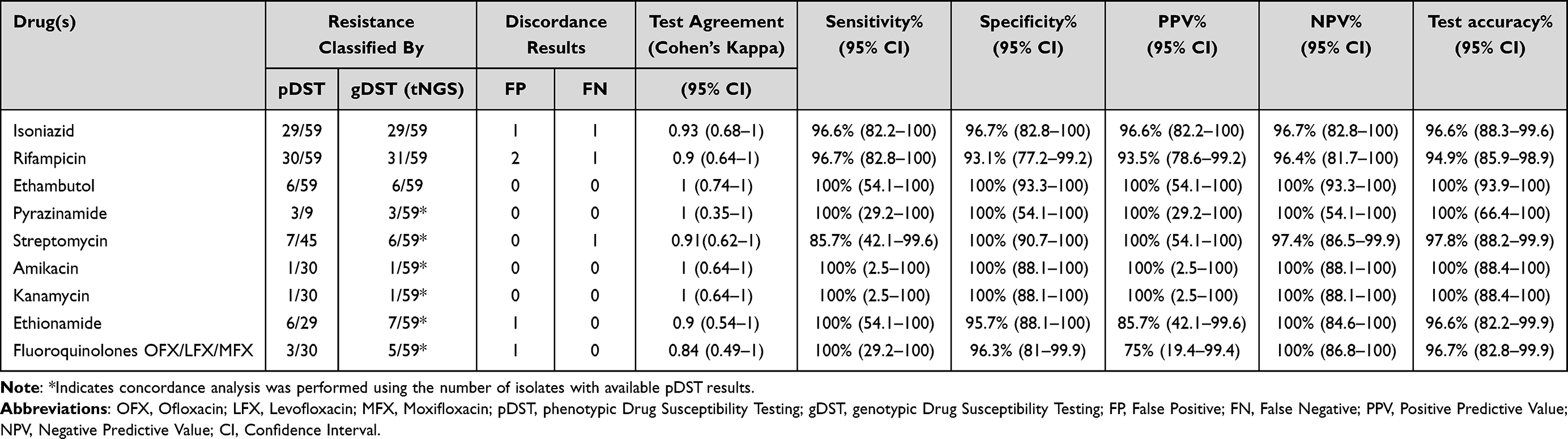 |
Table 2 Diagnostic Indicators of Nanopore tNGS Compared with pDST of Clinical Samples |
First-line drugs, including isoniazid (INH), rifampicin (RIF), ethambutol (EMB), and pyrazinamide (PZA), all showed high sensitivity and specificity (>90%). Specifically, INH had 96.6% sensitivity and 96.7% specificity, RIF had 96.7% sensitivity and 93.1% specificity. While both EMB and PZA exhibited 100% sensitivity and specificity. Streptomycin (SM) showed 85.7% sensitivity and 100% specificity. For second-line injectable drugs (SLIs), amikacin (AMK) and kanamycin (KAN) both showed 100% sensitivity and specificity. Ethionamide (ETH) exhibited 100% sensitivity and 95.7% specificity, while fluoroquinolones (OFX/LFX/MFX) had 100% sensitivity and 96.3% specificity. (Table 2). Overall, sensitivity, specificity, PPV, NPV, and accuracy were consistently high across the drugs analyzed. The concordance results were evaluated using Cohen’s kappa coefficients (κ) to assess the level of agreement between tNGS and pDST. For first-line drugs, Cohen’s kappa coefficients were 0.93 for INH, 0.9 for RIF, 1 for EMB, 0.91 for SM, and 1 for PZA, all indicating very strong agreement. In SLIs, kappa coefficients were 0.9 for ETH, 0.84 for FQs, and 1 for AMK and KAN, also indicating very strong agreement (Table 2).
Despite the high concordance between tNGS and pDST, discordant results were observed, including five false-positive and three false-negative calls. These discrepancies primarily involved RIF and INH, with single discordant cases observed for SM, ETH, and FQs. Notably, while perfect agreement (κ = 1) was achieved for EMB, PZA, AMK, and KAN, these findings should be interpreted with caution due to the limited number of resistant isolates, particularly for the second-line injectables, where only a single resistant isolate was detected.
Distribution of Resistance-Associated Variants (RAVs)
The distribution of mutations associated with drug resistance in the clinical samples was summarized in Table 3, providing an overview of the most common resistance-associated variants across the genes analyzed. The most frequently detected variants were the katG S315T, rpoB S450L, rpoB H445D, and embB M306V, including embB G406D. Additionally, the inhA promoter mutations, such as −777C>T (−15C>T for fabG1) and −154G>A, were observed. Pyrazinamide resistance was commonly associated with pncA mutations, including −11A>C, observed. Streptomycin resistance was predominantly associated with rpsL K43R.
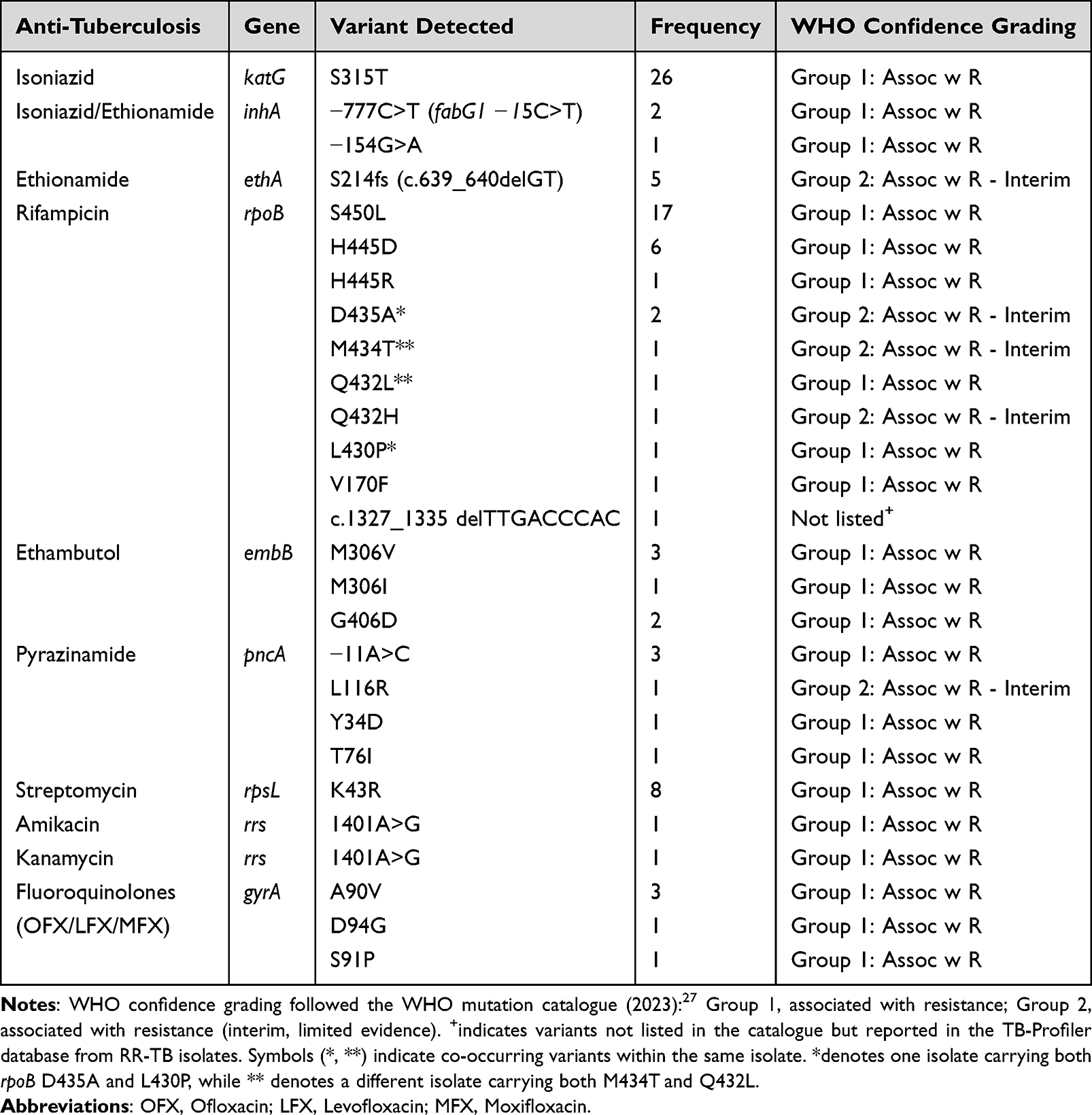 |
Table 3 The Distribution of Resistance-Associated Variants (RAVs) Identified in Clinical Samples Using Nanopore tNGS |
In this study, we identified a deletion mutation in rpoB (c.1327_1335delTTGACCCAC) linked to rifampicin resistance, along with other rpoB variants (Table 3), which illustrates the diversity of mutations contributing to drug resistance. Moreover, multiple rpoB mutations Q432L, D435A, M434T, and L430P were found co-harboring in specific isolates, as indicated by symbols (* and **), suggesting complex resistance mechanisms. Mutations in second-line drug resistance genes were also observed, including notable mutations linked to fluoroquinolone and ethionamide resistance. Fluoroquinolone resistance was linked to gyrA mutations, including A90V and D94G. Mutations in the ethA gene, specifically the S214fs frameshift (c.639_640delGT) mutation, were associated with ethionamide resistance (Table 3).
Discussion
Conventional molecular diagnostics such as Xpert MTB/RIF, LPAs, and MTB real-time PCR panels have substantially improved TB detection28–30 but remain limited by their narrow targets, often failing to provide complete profiles for second-line or repurposed drugs.4,31 While WGS offers the detailed genomic analysis missing from these assays,32,33 its routine application is hindered by the need for high-quality DNA from culture, which reintroduces diagnostic delays and demands specialized infrastructure.5,23,34 In this context, tNGS has emerged as a pragmatic alternative, offering broader resistance coverage than conventional molecular assays while reducing turnaround time, particularly when a comprehensive drug resistance profile is required.2,35
In this study, we evaluated the accuracy of nanopore-based tNGS using the ONT custom TB-DR sequencing assay for predicting drug resistance directly from uncultured sputum and culture isolates. The tNGS approach demonstrated over 90% test accuracy for most first-line and second-line drugs. Specifically, accuracy exceeded 90% for first-line drugs such as INH, RIF, EMB, and PZA, consistent with previous findings.36–39 For second-line drugs, the assay achieved 100% accuracy for AMK and KAN and 96.7% for FQs. The high accuracy observed for SLIs was based on a single positive resistance case, emphasizing the need for larger-scale studies with more resistant cases to validate these findings comprehensively. Additionally, Cohen’s kappa coefficient (κ) was within the range of very strong agreement (0.8–1) across all tested drugs, reflecting the concordance between tNGS and pDST in detecting drug-resistant TB.
Regarding the discrepancies observed in some resistance profiles, primarily in isoniazid and rifampicin, including false-negative and false-positive cases. False negatives were primarily attributed to the amplicon-based design of the ONT custom TB-DR sequencing assay, which could miss resistance-conferring mutations located outside the targeted regions. Detailed analysis of amplicon coverage using the Integrative Genomics Viewer (IGV) confirmed that the assay focuses on hotspot regions of genes where resistance mutations are most commonly found. However, rare mutations outside these targeted areas were occasionally undetected, contributing to discrepancies. For instance, one false-negative case involved a rpoB deletion (c.1327_1335delTTGACCCAC), which was not identified by the assay analyzing software (EPI2ME) but was detected through cross-validation with TB-profiler and manual variant calling. This discrepancy highlights the platform’s reliance on the WHO DR-TB mutation catalogue,27 which lacks this specific mutation. In contrast, the TB-profiler database includes additional mutations, demonstrating the critical role of database selection in the accuracy of resistance prediction.
False positives were also identified, such as the rpoB L452P mutation, which did not result in phenotypic resistance. This disputed variant has been associated with low-level rifampin resistance but may not consistently manifest phenotypically, as reported by Miotto et al.40 Variants like L452P can result in minimal inhibitory concentrations (MICs) below the phenotypic resistance threshold, leading to classification as susceptible despite clinical associations with treatment failures. These findings suggest that integrating genotypic data from the ONT custom TB-DR sequencing assay with pDST results could provide a more accurate understanding of resistance, particularly for disputed mutations.
Our analysis identified several clinically relevant mutations associated with resistance to first- and second-line drugs. The katG S315T mutation was the most frequently observed, confirming its established role in high-level INH resistance.41,42 Mutations in the inhA promoter, such as −777C>T (also known as fabG1 −15C>T or inhA −15C>T), and −154G>A were also detected, supporting their roles in low-level INH resistance and potential ethionamide cross-resistance.43 These findings align with prior reports showing high frequencies of katG S315T and inhA promoter mutations in resistant strains.44 Additionally, an ETH-resistant variant (ethA S214fs, c.639_640delGT), which disrupts ethionamide activation, was detected, further highlighting the utility of the assay in identifying diverse resistance mechanisms.45,46
Mutations in the rpoB gene, such as the well-known S450L, were frequently observed and are strongly associated with high-level RIF resistance.47 The rpoB L430P mutation, a disputed variant, was also identified. Interestingly, L430P was detected alongside D435A, a mutation associated with high-level RIF resistance, suggesting that the presence of D435A may reinforce the phenotypic resistance linked to L430P.40 Other significant mutations included embB M306V, associated with EMB resistance,48 and pncA mutations, such as −11A>C and L116R, linked to PZA resistance.49,50 The rrs 1401A>G mutation, a marker for high-level resistance to AMK and KAN, was also identified, consistent with previous findings.51 FQ resistance was commonly associated with gyrA mutations A90V and D94G, which are well-established markers for this drug class.52
The assay includes primers targeting resistance genes for drugs in the BPaL regimen53 (eg, rplC and rrl for linezolid,54 rv0678 for bedaquiline and clofazimine,55,56 and fgd1, ddn, atpE, fbiA, fbiB, and fbiC for delamanid and pretomanid).57 However, the absence of phenotypically resistant isolates for these drugs limited our ability to fully evaluate the assay’s accuracy for these agents. Further validation with resistant reference samples is needed to strengthen its clinical application. Additionally, the high prevalence of Beijing lineage isolates (approximately 80%) observed in this study suggests lineage-specific resistance profiles, consistent with prior findings.58
Sequencing depth was a key limitation; specifically, complete resistance profiles were obtained in only 2 of 10 (20%) MTB-positive sputum samples, strictly associated with high mycobacterial loads. Samples with higher Ct values (>35) and negative AFB grades often failed to produce complete profiles, reinforcing the importance of sample quality for reliable sequencing. This finding aligns with previous studies indicating a relationship between Ct values and sequencing success.11 Enhancing preprocessing steps, such as mycobacterial DNA enrichment or host DNA depletion, could improve sequencing performance for sputum samples.4 Although a major advantage of tNGS is the potential to bypass culture requiring BSL-3 facilities, our results show that culture isolates remain superior to sputum for sequencing quality. DNA from culture isolates consistently yielded complete resistance profiles; in contrast, direct sputum sequencing was inconsistent and dependent on mycobacterial load. Speed is a clear benefit of tNGS, but culture remains an essential backup for samples with low bacterial load to ensure accuracy.
The absence of WGS for verification presents another limitation, as resistance mutations outside the targeted regions may have been missed. Specifically, the accuracy of lineage classification requires further validation; for instance, the detection of the Manu-ancestor lineage, which is rarely reported in Thailand,59 may represent either true identification or misclassification by the analysis pipeline. Furthermore, we recognize that the overall sample size was relatively small (n=88), and the diversity of resistance profiles was constrained by the low prevalence of certain strains in our setting. Additionally, incorporating a broader range of resistant isolates, particularly for second-line and repurposed drugs like bedaquiline, delamanid, and clofazimine, in future studies will help to comprehensively evaluate the platform’s utility in MDR-TB treatment planning.
Conclusion and Perspectives
The ONT custom TB-DR sequencing assay provides a comprehensive approach to drug-resistance profiling and shows high concordance with phenotypic drug susceptibility testing for first-line drugs in samples yielding complete sequencing profiles, while also supporting species identification and lineage determination. The sequencing workflow offers a shorter turnaround time during the analytical phase compared with conventional phenotypic testing, particularly for comprehensive resistance profiling; however, when culture is required, the overall diagnostic timeline remains constrained by culture incubation. Reduced sequencing depth in sputum samples with low mycobacterial load highlights the importance of specimen quality and identifies areas for further technical optimization, including DNA enrichment strategies. Future studies involving larger and more diverse cohorts, particularly those including isolates resistant to newer anti-tuberculosis drugs, are required to better define diagnostic performance and clinical applicability.
Data Sharing Statement
The sequencing data generated in this study are available in the NCBI Sequence Read Archive (SRA) repository under the BioProject accession number PRJNA1196685.
Ethics Approval and Consent to Participate
This study was approved by the Siriraj Institutional Review Board (SI-IRB), Mahidol University, Bangkok, Thailand (COA No. SI 583/2024), in accordance with the ethical standards of the Declaration of Helsinki and applicable national regulations. A waiver of informed consent was granted by the SI-IRB, as the study used leftover DNA samples and corresponding laboratory data from routine diagnostic procedures without any direct patient contact or identifiable information.
Acknowledgments
We thank the Mycobacteriology and Mycology Laboratory, Department of Microbiology, Faculty of Medicine Siriraj Hospital, Mahidol University, for providing the DNA samples, Anyplex™ MTB/NTM results, and phenotypic DST results.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the establishment fund of Siriraj Translational Microbial Genomics and Data Center (SiTMiD) provided by the Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand.
Disclosure
The authors report no conflicts of interest in this work.
References
1. World Health Organization. Global Tuberculosis Report 2025. World Health Organization; 2025. Available from: https://iris.who.int/handle/10665/383364.
2. Barbosa-Amezcua M, Cuevas-Córdoba B, Fresno C, et al. Rapid identification of drug resistance and phylogeny in M. tuberculosis, directly from sputum samples. Microbiol Spectr. 2022;10(5):e0125222. doi:10.1128/spectrum.01252-22
3. Horsburgh CR, Barry CE, Lange C. Treatment of Tuberculosis. N Engl J Med. 2015;373(22):2149–12. doi:10.1056/NEJMra1413919
4. Jacobson KR, Barnard M, Kleinman MB, et al. Implications of failure to routinely diagnose resistance to second-line drugs in patients with rifampicin-resistant tuberculosis on Xpert MTB/RIF: a multisite observational study. Clin Infect Dis. 2017;64(11):1502–1508. doi:10.1093/cid/cix128
5. Soundararajan L, Kambli P, Priyadarshini S, et al. Whole genome enrichment approach for rapid detection of Mycobacterium tuberculosis and drug resistance-associated mutations from direct sputum sequencing. Tuberculosis. 2020;121:101915. doi:10.1016/j.tube.2020.101915
6. The CRyPTIC Consortium and the 100,000 Genomes Project. Prediction of susceptibility to first-line tuberculosis drugs by DNA sequencing. N Engl J Med. 2018;379(15):1403–1415. doi:10.1056/NEJMoa1800474
7. Votintseva AA, Bradley P, Pankhurst L, et al. Same-day diagnostic and surveillance data for tuberculosis via whole-genome sequencing of direct respiratory samples. J Clin Microbiol. 2017;55(5):1285–1298. doi:10.1128/jcm.02483-16
8. Doyle RM, Burgess C, Williams R, et al. Direct whole-genome sequencing of sputum accurately identifies drug-resistant Mycobacterium tuberculosis faster than MGIT culture sequencing. J Clin Microbiol. 2018;56(8):10–128. doi:10.1128/jcm.00666-18
9. World Health Organization. Use of targeted next-generation sequencing to detect drug-resistant tuberculosis: rapid communication. World Health Organization; 2023. Available from: https://iris.who.int/handle/10665/371687.
10. World Health Organization. WHO Consolidated Guidelines on Tuberculosis: module 3: diagnosis. World Health Organization; 2025. Available from: https://iris.who.int/handle/10665/381003.
11. Murphy SG, Smith C, Lapierre P, et al. Direct detection of drug-resistant Mycobacterium tuberculosis using targeted next generation sequencing. Front Public Health. 2023;11:1206056. doi:10.3389/fpubh.2023.1206056
12. Precision System Science Co. Ltd. MagDEA® Dx SV Instructions for Use (Version 2.3). 2024. Available from: https://www.pss.co.jp/product/manual-pdf/MagDEA_Dx_SV-ifu-en-V2.3.pdf.
13. Koentjoro MP, Donastin A, Prasetyo EN. A simple method of dna extraction of Mycobacterium tuberculosis from sputum cultures for sequencing analysis. Afr J Infect Dis. 2021;15(2 Suppl):19–22. doi:10.21010/ajidv15i2S:2
14. Sawatpanich A, Petsong S, Tumwasorn S, Rotcheewaphan S. Diagnostic performance of the Anyplex MTB/NTM real-time PCR in detection of Mycobacterium tuberculosis complex and nontuberculous mycobacteria from pulmonary and extrapulmonary specimens. Heliyon. 2022;8(12):e11935. doi:10.1016/j.heliyon.2022.e11935
15. World Health Organization. Technical manual for drug susceptibility testing of medicines used in the treatment of tuberculosis. World Health Organization; 2018. Available from: https://iris.who.int/handle/10665/275469.
16. World Health Organization. Technical report on critical concentrations for drug susceptibility testing of medicines used in the treatment of drug-resistant tuberculosis. World Health Organization; 2018. Available from: https://iris.who.int/handle/10665/260470.
17. Oxford Nanopore Technologies. Rapid sequencing gDNA - barcoding (SQK-RBK110.96). 2021. Available from: https://nanoporetech.com/document/rapid-barcoding-kit-96-sqk-rbk110-96.
18. Thermo Fisher Scientific Inc. QubitTM dsDNA BR Assay Kit. 2022. Available from: https://documents.thermofisher.com/TFS-Assets/LSG/manuals/Qubit_dsDNA_BR_Assay_UG.pdf.
19. Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34(18):3094–3100. doi:10.1093/bioinformatics/bty191
20. Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27(21):2987–2993. doi:10.1093/bioinformatics/btr509
21. Zheng Z, Li S, Su J, Leung AWS, Lam TW, Luo R. Symphonizing pileup and full-alignment for deep learning-based long-read variant calling. Nat Comput Sci. 2022;2(12):797–803. doi:10.1038/s43588-022-00387-x
22. Martin M, Patterson M, Garg S, et al. WhatsHap: fast and accurate read-based phasing. bioRxiv. 2016:085050. doi:10.1101/085050
23. Phelan JE, O’Sullivan DM, Machado D, et al. Integrating informatics tools and portable sequencing technology for rapid detection of resistance to anti-tuberculous drugs. Genome Med. 2019;11(1):41. doi:10.1186/s13073-019-0650-x
24. Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. arXiv. 2012. doi:10.48550/arXiv.1207.3907
25. Cingolani P, Platts A, Wang LL, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: sNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6(2):80–92. doi:10.4161/fly.19695
26. Landis JR, Koch GG. The measurement of observer agreement for categorical data. Biometrics. 1977;33(1):159–174.
27. World Health Organization. Catalogue of Mutations in Mycobacterium Tuberculosis Complex and Their Association with Drug Resistance.
28. Boehme CC, Nabeta P, Hillemann D, et al. Rapid molecular detection of tuberculosis and rifampin resistance. N Engl J Med. 2010;363(11):1005–1015. doi:10.1056/NEJMoa0907847
29. Dorman SE, Schumacher SG, Alland D, et al. Xpert MTB/RIF Ultra for detection of Mycobacterium tuberculosis and rifampicin resistance: a prospective multicentre diagnostic accuracy study. Lancet Infect Dis. 2018;18(1):76–84. doi:10.1016/S1473-3099(17)30691-6
30. Nathavitharana RR, Cudahy PGT, Schumacher SG, Steingart KR, Pai M, Denkinger CM. Accuracy of line probe assays for the diagnosis of pulmonary and multidrug-resistant tuberculosis: a systematic review and meta-analysis. Eur Respir J. 2017;49(1). doi:10.1183/13993003.01075-2016
31. Hillemann D, Rüsch-Gerdes S, Richter E. Evaluation of the GenoType MTBDRplus assay for rifampin and isoniazid susceptibility testing of Mycobacterium tuberculosis strains and clinical specimens. J Clin Microbiol. 2007;45(8):2635–2640. doi:10.1128/jcm.00521-07
32. Walker TM, Kohl TA, Omar SV, et al. Whole-genome sequencing for prediction of Mycobacterium tuberculosis drug susceptibility and resistance: a retrospective cohort study. Lancet Infect Dis. 2015;15(10):1193–1202. doi:10.1016/S1473-3099(15)00062-6
33. Pankhurst LJ, Elias CDO, Votintseva AA, et al. Rapid, comprehensive, and affordable mycobacterial diagnosis with whole-genome sequencing: a prospective study. Lancet Respir Med. 2016;4(1):49–58. doi:10.1016/S2213-2600(15)00466-X
34. Brown AC, Bryant JM, Einer-Jensen K, et al. Rapid whole-genome sequencing of Mycobacterium tuberculosis isolates directly from clinical samples. J Clin Microbiol. 2015;53(7):2230–2237. doi:10.1128/jcm.00486-15
35. Kambli P, Ajbani K, Kazi M, et al. Targeted next generation sequencing directly from sputum for comprehensive genetic information on drug resistant Mycobacterium tuberculosis. Tuberculosis. 2021;127:102051. doi:10.1016/j.tube.2021.102051
36. Faksri K, Kaewprasert O, Ong RTH, et al. Comparisons of whole-genome sequencing and phenotypic drug susceptibility testing for Mycobacterium tuberculosis causing MDR-TB and XDR-TB in Thailand. Int J Antimicrob Agents. 2019;54(2):109–116. doi:10.1016/j.ijantimicag.2019.04.004
37. Shea J, Halse TA, Lapierre P, et al. Comprehensive whole-genome sequencing and reporting of drug resistance profiles on clinical cases of Mycobacterium tuberculosis in New York State. J Clin Microbiol. 2017;55(6):1871–1882. doi:10.1128/jcm.00298-17
38. Madrazo-Moya CF, Cancino-Muñoz I, Cuevas-Córdoba B, et al. Whole genomic sequencing as a tool for diagnosis of drug and multidrug-resistance tuberculosis in an endemic region in Mexico. PLoS One. 2019;14(6):e0213046. doi:10.1371/journal.pone.0213046
39. Wu SH, Xiao YX, Hsiao HC, Jou R. Development and assessment of a novel whole-gene-based targeted next-generation sequencing assay for detecting the susceptibility of Mycobacterium tuberculosis to 14 drugs. Microbiol Spectr. 2022;10(6):e02605–22. doi:10.1128/spectrum.02605-22
40. Miotto P, Cabibbe AM, Borroni E, Degano M, Cirillo DM. Role of disputed mutations in the rpoB gene in interpretation of automated liquid MGIT culture results for rifampin susceptibility testing of Mycobacterium tuberculosis. J Clin Microbiol. 2018;56(5). doi:10.1128/jcm.01599-17
41. Zhang Y, Heym B, Allen B, Young D, Cole S. The catalase-peroxidase gene and isoniazid resistance of Mycobacterium tuberculosis. Nature. 1992;358(6387):591–593. doi:10.1038/358591a0
42. Wengenack NL, Uhl JR, St. Amand AL, et al. Recombinant Mycobacterium tuberculosis KatG(S315T) Is a competent catalase-peroxidase with reduced activity toward isoniazid. J Infect Dis. 1997;176(3):722–727. doi:10.1086/514096
43. Rao M, Wollenberg K, Harris M, et al. Lineage classification and antitubercular drug resistance surveillance of Mycobacterium tuberculosis by whole-genome sequencing in Southern India. Microbiol Spectr. 2023;11(5):e04531–22. doi:10.1128/spectrum.04531-22
44. Seifert M, Catanzaro D, Catanzaro A, Rodwell TC. Genetic mutations associated with isoniazid resistance in Mycobacterium tuberculosis: a systematic review. PLoS One. 2015;10(3):e0119628. doi:10.1371/journal.pone.0119628
45. Boonaiam S, Chaiprasert A, Prammananan T, Leechawengwongs M. Genotypic analysis of genes associated with isoniazid and ethionamide resistance in MDR-TB isolates from Thailand. Clin Microbiol Infect. 2010;16(4):396–399. doi:10.1111/j.1469-0691.2009.02838.x
46. Baulard AR, Betts JC, Engohang-Ndong J, et al. Activation of the pro-drug ethionamide is regulated in mycobacteria. J Biol Chem. 2000;275(36):28326–28331. doi:10.1074/jbc.M003744200
47. Ramaswamy S, Musser JM. Molecular genetic basis of antimicrobial agent resistance in Mycobacterium tuberculosis: 1998 update. Tuber Lung Dis. 1998;79(1):3–29. doi:10.1054/tuld.1998.0002
48. Sreevatsan S, Stockbauer KE, Pan X, et al. Ethambutol resistance in Mycobacterium tuberculosis: critical role of embB mutations. Antimicrob Agents Chemother. 1997;41(8):1677. doi:10.1128/aac.41.8.1677
49. Park SK, Lee JY, Chang CL, et al. pncA mutations in clinical Mycobacterium tuberculosis isolates from Korea. BMC Infect Dis. 2001;1:4. doi:10.1186/1471-2334-1-4
50. Lange C, Alghamdi WA, Al-Shaer MH, et al. Perspectives for personalized therapy for patients with multidrug-resistant tuberculosis. J Intern Med. 2018;284(2):163–188. doi:10.1111/joim.12780
51. Maus CE, Plikaytis BB, Shinnick TM. Molecular analysis of cross-resistance to Capreomycin, Kanamycin, Amikacin, and Viomycin in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2005;49(8):3192–3197. doi:10.1128/aac.49.8.3192-3197.2005
52. Li J, Gao X, Luo T, et al. Association of gyrA/B mutations and resistance levels to fluoroquinolones in clinical isolates of Mycobacterium tuberculosis. Emerg Microbes Infect. 2014;3(3):e19. doi:10.1038/emi.2014.21
53. World Health Organization. Key updates to the treatment of drug-resistant tuberculosis: rapid communication, June 2024. World Health Organization; 2024. doi:10.2471/B09123.
54. Gan WC, Ng HF, Ngeow YF. Mechanisms of linezolid resistance in mycobacteria. Pharmaceuticals. 2023;16(6):784. doi:10.3390/ph16060784
55. Andries K, Villellas C, Coeck N, et al. Acquired resistance of Mycobacterium tuberculosis to Bedaquiline. PLoS One. 2014;9(7):e102135. doi:10.1371/journal.pone.0102135
56. Zhang S, Chen J, Cui P, Shi W, Zhang W, Zhang Y. Identification of novel mutations associated with clofazimine resistance in Mycobacterium tuberculosis. J Antimicrob Chemother. 2015;70(9):2507–2510. doi:10.1093/jac/dkv150
57. Liu Y, Shi J, Li L, et al. Spontaneous mutational patterns and novel mutations for delamanid resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2022;66(12):e00531–22. doi:10.1128/aac.00531-22
58. Shanmugam SK, Kumar N, Sembulingam T, et al. Mycobacterium tuberculosis Lineages Associated with mutations and drug resistance in isolates from India. Microbiol Spectr. 2022;10(3):e01594. doi:10.1128/spectrum.01594-21
59. Regmi SM, Chaiprasert A, Coker OO, et al. Draft genome sequence of an extensively drug-resistant Mycobacterium tuberculosis Manu-Ancestor Spoligo-International Type 523 Isolate from Thailand. Genome Announc. 2015;3(1):e01589–14. doi:10.1128/genomeA.01589-14
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.