Back to Journals » Drug Design, Development and Therapy » Volume 10
Rapid disintegrating tablets of simvastatin dispersions in polyoxyethylene–polypropylene block copolymer for maximized disintegration and dissolution
Authors Balata GF, Zidan AS, Abourehab MA, Essa EA
Received 10 June 2016
Accepted for publication 26 July 2016
Published 3 October 2016 Volume 2016:10 Pages 3211—3223
DOI https://doi.org/10.2147/DDDT.S114724
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Gehan F Balata,1,2 Ahmad S Zidan,2 Mohamad AS Abourehab,1,3 Ebtessam A Essa4
1Department of Pharmaceutics, Faculty of Pharmacy, Umm Al-Qura University, Makkah, Saudi Arabia; 2Department of Pharmaceutics, Faculty of Pharmacy, Zagazig University, Zagazig, 3Department of Pharmaceutics, Faculty of Pharmacy, El-Minia University, El-Minia, 4Department of Pharmaceutics, Faculty of Pharmacy, Tanta University, Tanta, Egypt
Abstract: The objective of this research was to improve the dissolution of simvastatin and to incorporate it in rapid disintegrating tablets (RDTs) with an optimized disintegration and dissolution characteristics. Polyoxyethylene–polypropylene block copolymer (poloxamer 188) was employed as a hydrophilic carrier to prepare simvastatin solid dispersions (SDs). Fourier transform infrared spectroscopy, differential scanning calorimetry (DSC) and X-ray diffractometry were employed to understand the interaction between the drug and the carrier in the solid state. The results obtained from Fourier transform infrared spectroscopy showed absence of any chemical interaction between the drug and poloxamer. The results of differential scanning calorimetry and X-ray diffractometry confirmed the conversion of simvastatin to distorted crystalline state. The SD of 1:2 w/w drug to carrier ratio showed the highest dissolution; hence, it was incorporated in RDT formulations using a 32 full factorial design and response surface methodology. The initial assessments of RDTs demonstrated an acceptable flow, hardness, and friability to indicate good mechanical strength. The interaction and Pareto charts indicated that percentage of croscarmellose sodium incorporated was the most important factor affecting the disintegration time and dissolution parameter followed by the hardness value and their interaction effect. Compression force showed a superior influence to increase RDT’s porosity and to fasten disintegration rather than swelling action by croscarmellose sodium. On the other hand, croscarmellose sodium was most important for the initial simvastatin release. The results suggest the potential use of poloxamer 188-based SD in RDT for the oral delivery of poor water-soluble antihyperlipidemic drug, simvastatin.
Keywords: simvastatin, poloxamer 188, croscarmellose sodium, full factorial design, dissolution
Introduction
Rapid disintegrating tablets (RDTs) have been used in drug manufacturing field for many nonprescription and/or prescription drugs to estimate about 500 RDT-based new drug application- and abbreviated new drug application-approved medications. According to the statistics of the pharmaceutical market over the past decade, the RDT market share can be estimated to approach $13 billion in 2015.1 RDTs as a dosage form are suitable for children, elderly, and patients with dysphagia. However, the RDT formulations have expanded to most patient populations.2 Different techniques have been proposed in the literature to manufacture RDTs with more preference to freeze drying. Generally, the process selected to manufacture RDTs could significantly alter the resultant RDTs’ critical characteristics of the disintegration pattern and the tablets’ hardness.3 Freeze drying process can produce RDTs with very short disintegration time due to the highly porous matrix formed; however, very low mechanical strengths of the tablets also result that could limit packaging, handling, and further distribution.4 On the other hand, simple direct compression of powder beds incorporating an optimized mixture of superdisintegrants in swellable matrix can produce RDTs with better hardness values, optimized porosities, disintegration rate, and eventually lower production costs.5 Ideally, RDTs should possess rapid disintegration pattern, acceptable taste, and no grittiness feeling in mouth.6 It is also worth noting that European Pharmacopoeia identified RDTs as those tablets that disintegrate in <3 minutes before swallowing. On the other hand, United States Food and Drug Administration defined them with disintegration time of ≤30 seconds.2
Dietary and lifestyle changes in recent decades have resulted in many chronic diseases such as dyslipidemia. Dyslipidemia constitutes a serious problem all over the world as it is considered the primary predisposing factors for many heart illnesses resulting in mortality and medical costs.7,8 Literature reported emergence of atherosclerosis, cardiac infarction, and ischemic heart disease that develop early in life.9,10 The occurrence of fibrous thrombosis increases from 10% to 70% from young patients to adults.9 Daniels and Greer reported that there is a strong correlation between the rise in triglycerides and cholesterol, hypertension and obesity, and the incidence of cardiovascular diseases.11 Statins are one of the most commonly prescribed medications for decreasing deaths due to cardiac ischemia in hyperlipidemic patients.12 Daniels and Greer also reported that statins contribute to reduce the elevated cholesterol levels by ~60% according to the dose used.11 Statins are considered primary medications of choice for treatment of dyslipidemia. All statins are available in conventional tablet formulations with very limited availability as disintegrating oral tablet to suit other patients’ populations.13 As one of the most widely prescribed statins, simvastatin is demonstrated to significantly reduce the levels of all types of blood lipids including triglycerides, low-density lipoproteins, apolipoproteins, very-low-density lipoproteins, and cholesterol. However, the increase in high-density lipoproteins and apolipoproteins was not significant after 10 months of treatment.14 Simvastatin is offered in both conventional and extended-release tablets that cannot be divided, crushed, or chewed by patients. Moreover, it is also not available in a liquid form. With the increased demand of statins for pediatric population, other formulation approaches with flexible dosing capacity and administration potential are then required. Simvastatin exists as crystalline powdered substance with very low solubility in water and gastric fluids.15 Various approaches have been proposed in the literature to improve the solubility of simvastatin including nanocrystals, co-solvency, recrystallization, and self-emulsification.16,17 None of these methods have been applied to propose simvastatin dispersions in hydrophilic RDTs. Consequently, the aim of the current investigation was to prepare RDTs of simvastatin using its solid dispersion (SD) with a hydrophilic carrier while optimizing disintegration and porosity of RDTs. The hydrophilic carrier investigated in this study was poloxamer 188. Poloxamers are “polyoxyethylene–polypropylene block copolymer nonionic surfactants that are mostly employed to improve the wettability, solubilization and bioavailability of various hydrophobic medications using several methodologies such as melting agglomeration, solvent evaporation, and co-melting.”18,19 Poloxamer 188 has low melting point (~56°C–57°C) and is well known for its oral safety. Melting–solvent evaporation co-technique was employed to prepare different SD formulations at different simvastatin to poloxamer 188 weight ratios. Attenuated total reflectance–Fourier transform infrared spectroscopy (ATR-FTIR) and X-ray diffraction (XRD) as well as differential scanning calorimetric (DSC) analyses were used to understand the chemical and physical compatibilities between simvastatin and poloxamer 188. The SD formulation with the highest solubility and dissolution was then incorporated in RDT matrix tablets. The influences of changing RDT hardness value and superdisintegrant concentration on the time to achieve complete disintegration and simvastatin dissolution were studied. A two-factored three-level factorial design was employed to optimize the RDT formulation for maximum simvastatin dissolution, shorter disintegration, and highest water wicking action.
Methods
Materials
Simvastatin was purchased from Artemis Biotech Ltd (Jeedimetla, Hyderabad, India). Poloxamer 188 was obtained from Sigma-Aldrich Biochemie GmbH (Hamburg, Germany). Methanol, acetonitrile, and magnesium stearate were obtained from VWR scientific Inc (Minneapolis, MN, USA). Avicel PH101 and Croscarmellose Sodium known as Ac-Di-Sol® were obtained from FMC Corp. (Newark, DE, USA). Mannitol was purchased from Biesterfeld Siemsgluss Int. GmbH (Fort Lauderdale, FL, USA). All chemicals were of either high pressure liquid chromatography (HPLC) or analytical grade. They were used as received.
Chromatographic analysis for the determination of simvastatin
The quantitation of simvastatin was done according to an adopted and validated chromatographic-based HPLC analysis.20 Agilent HPLC (Agilent Technologies, Santa Clara, CA, USA) with a diode detector set at 238 nm. Simvastatin elution was achieved at 32°C by injecting 10 μL injection volume onto Luna (2) RP-18 (250×4.6 mm, 5 μm packing) column (Phenomenex, Torrance, CA, USA). The mobile phase was composed of acetonitrile and phosphate buffer (pH 6.8; 0.01 M) at 4:6 (v/v) and was pumped isocratically at a flow rate of 1.2 mL/min.
Phase solubility study
An excess amount of simvastatin was placed in scintillation vials having various levels (0.001–0.012 M) of a poloxamer 188 solution in ethanol/water (10/90 V/V) mixture (5.0 mL). The vials were shaken at 25°C until equilibrium on water bath shaker (Julabo Inc., Allentown, PA, USA). The samples were centrifuged at 14,000 rpm for 1 hour. The resulting supernatants were filtered through a PTFE membrane filter with 0.45 μm pore size and analyzed for simvastatin contents by the aforementioned HPLC chromatographic method.
Preparation of SDs
Dispersions of simvastatin were formulated by the melting followed by solvent evaporation method. Different SD formulations were prepared at four drug-to-carrier weight ratios as shown in Table 1. Simvastatin and the specified amount of poloxamer 188 were dissolved in 2 mL of isopropyl alcohol with heating at the melting temperature of poloxamer 188 (60°C). The resultant solutions were allowed to cool overnight at room temperature followed by 24 hours vacuum drying to completely evaporate the alcohol, and thus, solid masses were formed. The obtained powders were then triturated in the mortar followed by sieving (40 mesh). These processed SD powders were kept in a desiccator for further analyses. The physical mixtures (PMs) of simvastatin and poloxamer 188 at the corresponding weight ratios were formulated by tumbling-mixing (Glen Mills Inc., Clifton, NJ, USA) the sieved fractions of simvastatin and the carrier for 25 minutes.
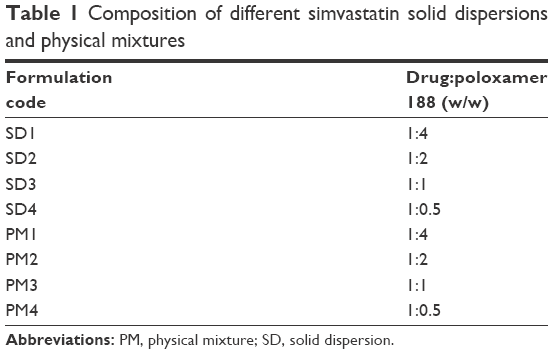 | Table 1 Composition of different simvastatin solid dispersions and physical mixtures |
Characterization of SDs
In vitro simvastatin dissolution characteristics from its SDs and PMs were compared with those of the raw drug using a dialysis bag diffusion technique. Specified weights of the formulations equivalent to 5 mg simvastatin were filled into Float-A-Lyzer cellulose ester dialysis tubes (1 mL, molecular weight cut-off of 10 kDa, Spectrum Laboratories, Los Angeles, CA, USA) and floated in a receiver media of 200 mL of phosphate buffer (pH 7.4). The media was kept under a stirring rate of 120 rpm at 37°C±0.5°C. One milliliter samples were withdrawn after 10, 20, 30, 45, 60, and 90 minutes for simvastatin analysis. The withdrawn volumes were compensated with fresh volume of phosphate buffer (pH 7.4). Samples were diluted appropriately and their simvastatin contents were measured using the aforementioned chromatographic method. Each dissolution experiment was done in triplicate. Dissolution efficiency parameters (DE10 and DE60) were determined as the percentage of the area under dissolution curves at the given times to the area of the rectangle describing 100% dissolution within the same period.21
FTIR experiments were performed to characterize any possible interaction that might exist in the SD of simvastatin and the carrier. FTIR spectra over the range 4,000–500 cm−1 were collected using a Perkin Elmer ATR-FTIR spectrometer (Waltham, MA, USA). DSC analysis was done to understand the thermal behavior of raw drug, carrier, and their SDs and PMs. Thermograms were collected using Perkin-Elmer differential calorimeter. Approximately 3–5 mg samples were weighted in a standard aluminum pan and hermetically sealed using hydraulic press. The samples were then exposed to a heating rate of 5°C/min up to 300°C using dry nitrogen as carrier gas. The powder XRD patterns of the same samples were performed using a XRD X’Pert 1 X-ray Diffractometer (Amstelplein 2, Philips, Amsterdam, the Netherlands). The data were obtained over a diffraction angle of 18°–80°.
Preparation of RDTs
The SDs and various excipient powders were directly compressed into RDTs. Croscarmellose sodium was employed at the specified percentage as superdisintegrant. Mg-stearate was employed at a concentration of 1% as lubricant. Microcrystalline cellulose (Avicel PH101) and mannitol at a loading ratio of 1:4 w/w were employed as fillers to produce RDTs of 200 mg weight equivalent to 5 mg simvastatin. This low percentage of microcrystalline cellulose was added to mannitol for its capillary and swelling action and its synergistic actions with Ac-Di-Sol.22 The powders were blended for 5 minutes followed by sieving (40 mesh) and then compressed into flat-faced RDTs using a single punch press and 10 mm die and punches.
Evaluation of the prepared RDTs
The mean hardness of ten tablets from each formulation was measured by a TBH 325 Erweka hardness tester (Erweka GmbH, Heusenstamm, Germany). RDT weights were assessed by determining the variability within individual weights of ten tablets from each batch. Diameters and thicknesses of RDTs were determined by digital caliber. The friability of RDTs was determined by calculating the percentage weight loss of 30 RDTs after shaking in Erweka friabilator at 25 rpm for 5 minutes. The disintegration time of RDTs was determined by measuring the average time needed for complete disintegration of six RDTs in Erweka USP disintegration tester using 900 mL double distilled water as the disintegration medium at 37°C±0.5°C. Since, the mechanism of disintegration can be described by the determination of water absorption ratio, one tablet from each formulation was weighted (Wa) and positioned over the tissue paper in petri dish containing 10 mL of methylene blue solution. The time needed to completely wet the RDT surface was determined as the wetting time. Water absorption (wicking) ratio was then calculated by determining the percentage increase in RDT weight by the wetting process. Simvastatin dissolution from RDTs was determined in 500 mL of phosphate buffer (pH 7.4) using apparatus II of USP dissolution instrument at 100 rpm and 37°C±0.5°C. Approximately 1 mL aliquots were withdrawn after 5, 10, 15, 20, and 30 minutes for simvastatin quantitation. Each experiment was performed in triplicate.
Experimental design
A two-factored three-level full-factorial design was employed to understand the influences of croscarmellose sodium concentration in RDT (X1), and the compression force (X2) on RDT’s disintegration time, simvastatin dissolution after 15 and 30 minutes and water wicking ratio (Table 2). The following polynomial prediction equation was used to build up the regression models:
Yi = A + y1X1 + y2X2 + y3X1X2 + y4X11 + y5X22 + E |
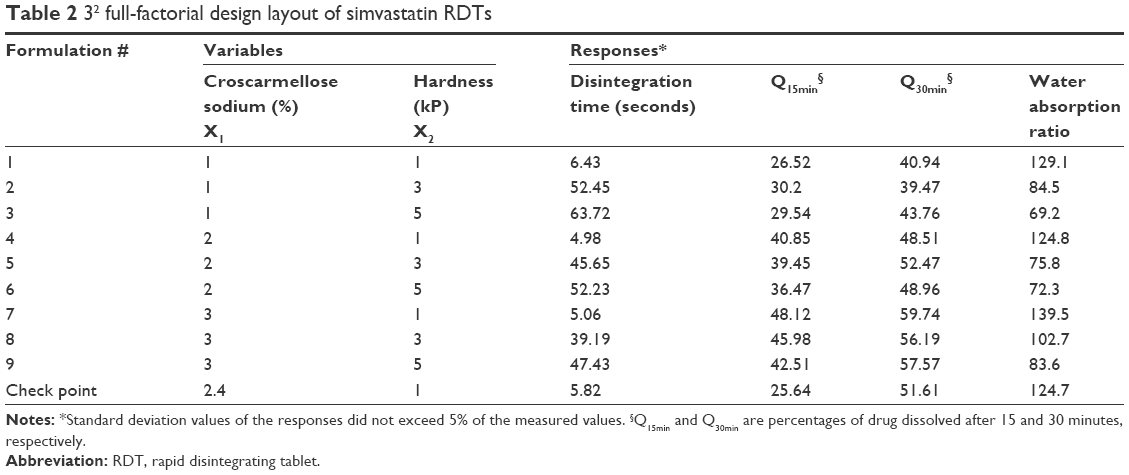 | Table 2 32 full-factorial design layout of simvastatin RDTs |
where Yi is the response to be predicted. A is the intercept of regression model. y1 and y2 are coefficients of changing the variables linearly. y3 is the coefficient for collinear interaction of the two variables. y4 and y5 are the polynomial coefficients of variables. E is the model universal error that combines the individual error constant for each variable into a universal error constant of the combined error.
Results and discussion
The solubility data of simvastatin at various percentages of poloxamer 188 are illustrated in Figure 1. The solubility of raw simvastatin in 10% v/v hydroalcoholic medium was 17.86 μg/mL which is in good agreement with solubility values reported by other researchers in the literature.23 The obtained data demonstrated that drug solubility was linearly enhanced by increasing poloxamer 188 concentrations to suggest an AL phase solubility diagram.24
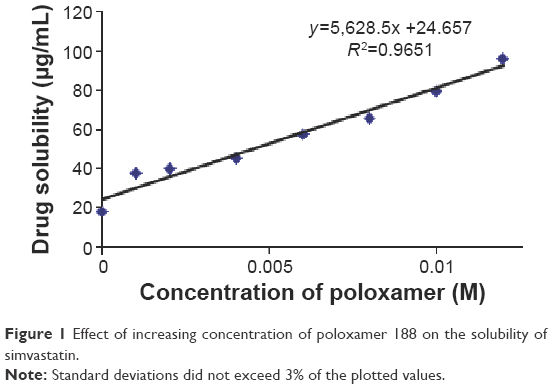 | Figure 1 Effect of increasing concentration of poloxamer 188 on the solubility of simvastatin. |
Characterization of simvastatin SDs
The dissolution profiles and parameters of raw simvastatin and its SDs and PMs are represented in Figure 2. Raw simvastatin exhibited poor dissolution properties with DE10 and DE60 of ~5.6% and 16.9%, respectively. This result might be attributed to poor solubility and wetting properties of simvastatin.15 The existence of poloxamer 188 improved the dissolution rate of simvastatin from its PM and SD samples. In addition, SDs showed higher dissolution parameters than the corresponding PMs. The surface activity of poloxamer (hydrophilic/lipophilic balance =29) might facilitate wetting of drug particles and its further conversion from the crystalline to molecular and colloidal dispersion state within the hydrophilic matrix of poloxamer.23 Further scanning electron microscope, DSC, XRD, and FTIR studies were performed to support this hypothesis. The drug:carrier ratio demonstrated a significant enhancement in simvastatin dissolution up to simvastatin:poloxamer ratio of 1:2. The further increase in poloxamer percentage resulted in a decrease in drug dissolution due to the increase in viscosity in the microdomain of drug particles.
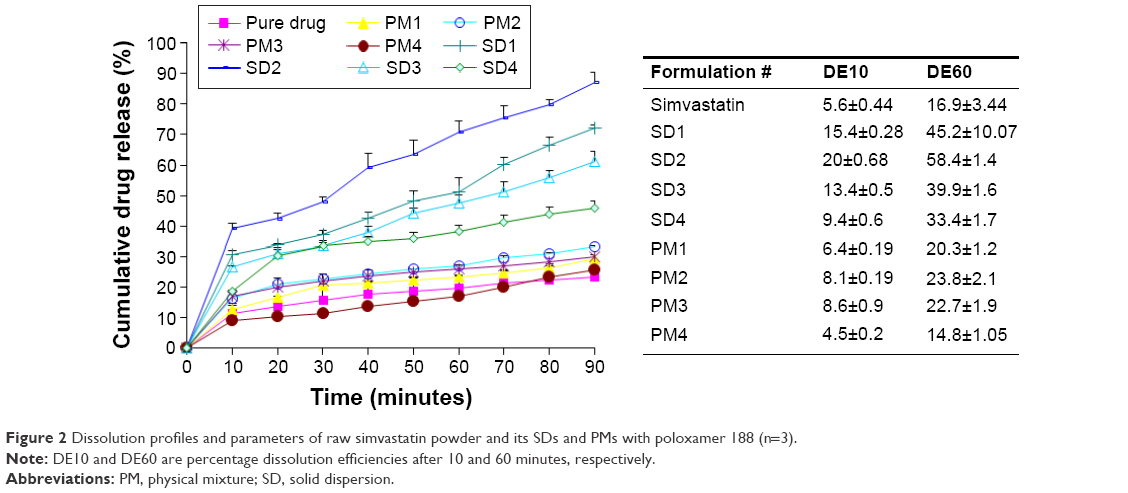 | Figure 2 Dissolution profiles and parameters of raw simvastatin powder and its SDs and PMs with poloxamer 188 (n=3). |
FTIR spectra of simvastatin, poloxamer 188, and their PMs and SDs are shown in Figure 3. Raw simvastatin showed major peaks at wave-numbers 3,551 cm−1 due to OH stretch; 3,009, 2,961, and 2,884 cm−1 due to both CH stretch; and 1,698 cm−1 due to stretching band of carbonyl group of the ester and/or lactone. All simvastatin-specific peaks were shown in the spectra of its SDs and PMs to demonstrate no chemical interaction between simvastatin and poloxamer 188 in their mixtures. DSC thermograms and XRD diffractograms of the same FTIR samples are depicted in Figure 4A and B. Thermogram of powdered raw drug showed a melting thermal transition at 142.15°C and a latent heat of fusion (ΔH) of −36.6 mJ indicating its crystalline nature.25 Raw poloxamer 188 powder also showed a melting endotherm at 49.19°C with ΔH value of −110.08 mJ. The thermograms of all PMs showed the same endotherms corresponding to both drug and carrier melting transitions with shifting of drug peak to a lower melting transition and reduced intensity. This would indicate a distorted crystallinity of simvastatin in its PM with poloxamer 188.26 All the thermograms of SDs exhibited only one endotherm at 55°C corresponding to the carrier except SD4 that showed both endotherms. The absence of simvastatin melting transition might be explained by the presence of distorted crystallinity of simvastatin in its dispersion with poloxamer 188 or dissolving drug crystals within a matrix of poloxamer 188.27 The XRD diffractogram of the raw drug demonstrated its crystallinity as shown by its various sharp peaks at 2θ of 28.0°, 22.4°, 19.0°, 17.8°, 16.9°,14.8°, and 10.8°. Poloxamer 188 is crystalline as well and its diffractogram showed two characteristic peaks at 19° and 23.8°. All PMs and SDs exhibited the presence of the characteristic peaks of both drug and carrier with reduced intensity. This observation agreed with those observed by DSC analysis to confirm a reduction in drug crystallinity.28
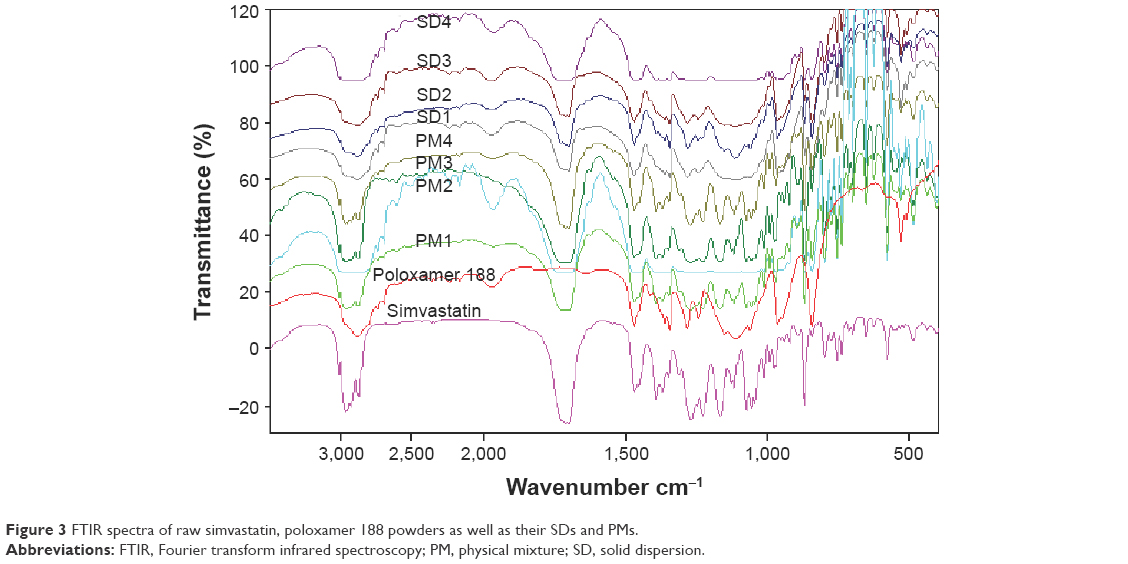 | Figure 3 FTIR spectra of raw simvastatin, poloxamer 188 powders as well as their SDs and PMs. |
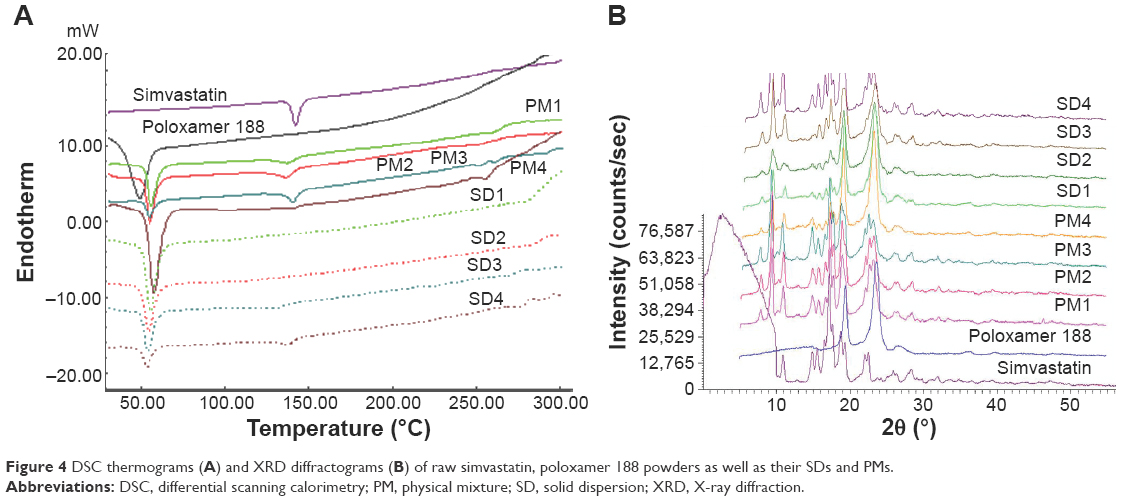 | Figure 4 DSC thermograms (A) and XRD diffractograms (B) of raw simvastatin, poloxamer 188 powders as well as their SDs and PMs. |
Characterization of simvastatin RDTs
Having this SD formulation, simvastatin was proposed in a hydrophilic SD of poloxamer 188 appropriate for RDT manufacturing. The preformulation of this water insoluble drug into a fast dissolving yet free flowable hydrophilic powder to be compressed into tablets is critical as a flexible dosing for pediatric and geriatric patients. To statistically understand the effects of various variables on the quality specifications of RDTs, a two-factored three-leveled factorial design of experiments was utilized. Based on our preliminary testing, the level of the disintegrant added (X1) and hardness values of the RDTs (X2) were chosen as experimental factors. On the other hand, RDTS’ disintegration performance, drug dissolution in 15 and 30 minutes (Q15min and Q30min) and water wicking percentage were the responses investigated (Yi).
Table 3 demonstrates that all RDT formulations exhibited uniform weight from 218.5 to 222.9 mg with variabilities not exceeding 0.9 mg. The diameters of RDTs were fluctuating between 9.9 and 10.8 mm with variabilities not exceeding 0.04 mm. The thickness values of RDTs ranged between 2.1 and 2.3 mm, with variabilities not exceeding 0.02 mm. Simvastatin assay was also performed for all RDT formulations to show that its values ranged between 98.6% and 102.1%, with variabilities not exceeding 1.8% to agree with the pharmacopeial specifications for assay testing. RDTs’ friability demonstrated that all RDTs resisted chapping with no signs of cracking or broken parts upon shaking in the friabilator. Furthermore, the weight difference of RDTs (<1%) during friability testing was still complying with the pharmacopeial specifications to pass the friability testing. This would demonstrate that these RDTs were resistant to cracking and can withstand the operations of packaging, handling, and distribution. The hardness testing of these RDTs was also performed. The obtained hardness values of all batches were in good agreement with the specified compression forces in the design with correlation coefficient of 0.8765.
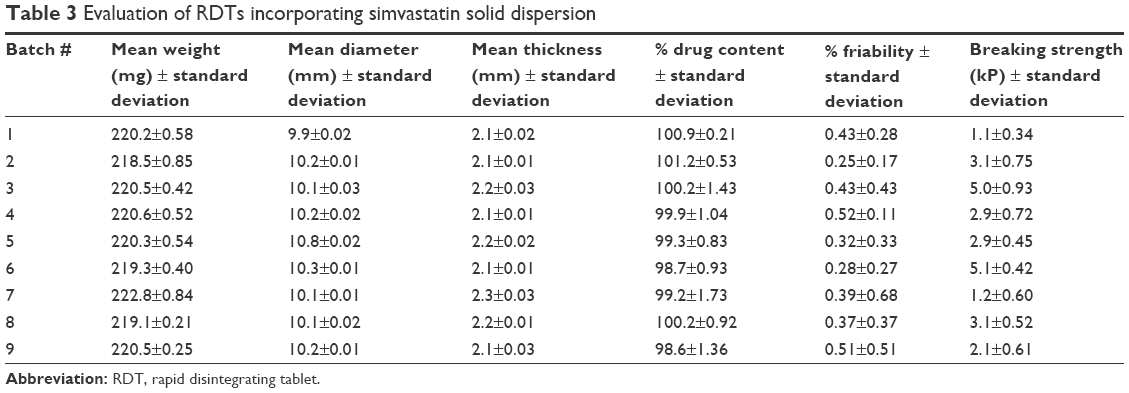 | Table 3 Evaluation of RDTs incorporating simvastatin solid dispersion |
The effects of disintegrant and hardness values and their various combinations on disintegration, dissolution, and water absorption of RDTs are summarized in Table 2. The nine RDT formulations exhibited significant differences among the values of the dependent variables by changing the levels of the investigated factors. The time for complete disintegration and water wicking percentage changed from 4.98 and 69.2 seconds to 63.72 and 139.5 seconds as the breaking strengths of RDTs changed from 1 kP (Formulation 7) up to 5 kP (Formulation 3), respectively. Those RDT formulations were prepared with the highest and lowest croscarmellose sodium percentages. The higher added percentage of the superdisintegrant and lower compression force resulted in faster RDT disintegration and maximum water wicking.29 RDT swelling, disintegration, and then erosion would be the encountered processes to describe simvastatin release.30 The percentages of simvastatin released from the prepared RDTs after 15 and 30 minutes approached 48.12% and 59.74% from batch 7 which was prepared with 3% w/w croscarmellose sodium and compressed with the least compression force. This fast simvastatin dissolution could also be governed by dissolution-enhancing action of the poloxamer 188 within the RDT matrix. Table 4 shows multiple regression analysis for the individual and polynomial variables, and their colinearities on the investigated responses. The regression models showed prediction efficiencies of 94%, 95%, 98% and 92% for the disintegration time, Q15min, Q30min, and water wicking percentage, respectively. The individual terms of each independent variable as well as the polynomial term of RDTs’ breaking strength values influenced RDT disintegration process and water wicking behavior significantly (P<0.05). Croscarmellose sodium percentage showed a significant positive effect on Q15min. On the other hand, it was adversely affected by the hardness of RDTs (X2), the collinear action of the superdisintegrant and the breaking strength of RDTs (X1X2), and the polynomial action of breaking strength of RDTs (X22). Q30min was not significantly affected by all the investigated factors with the exception of the level of the superdisintegrant that showed a significant action with a direct proportionation.
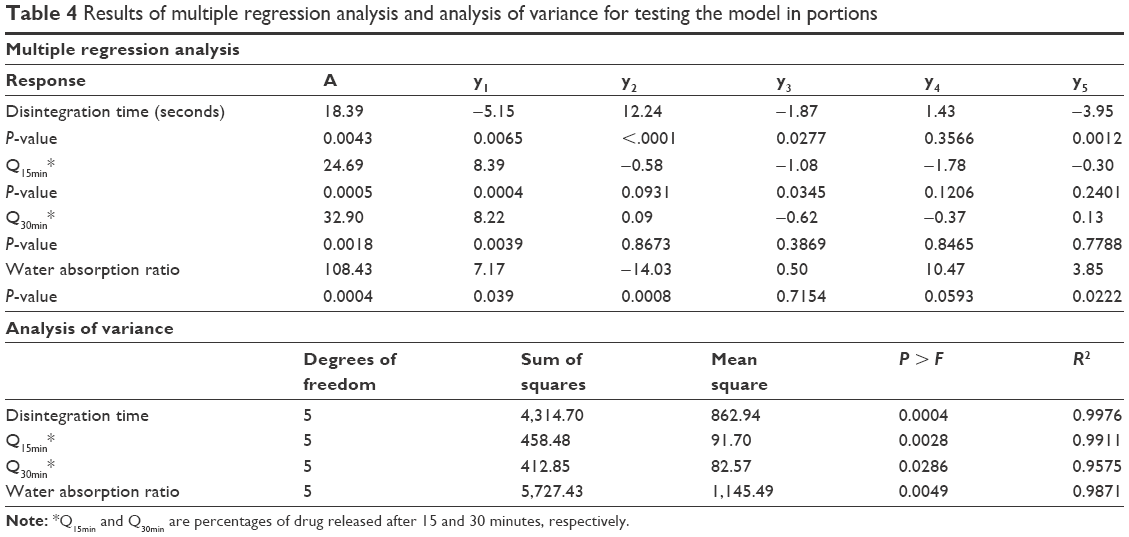 | Table 4 Results of multiple regression analysis and analysis of variance for testing the model in portions |
Response surface and contour plots
The three dimensional surface diagrams overlaid (Figure 5) with two dimensional contour diagrams were employed to understand the polynomial correlations of the investigated dependent factors at the highest and lowest values of the independent variables (Figure 5). At low values of X2, increasing croscarmellose sodium percentages was nonsignificant to enhance the disintegration of RDTs or to maximize the water wicking action. Contrarywise, compressing the powders into RDTs under high compression forces with increasing X1 from 1% to 3% produced shortening in disintegration time from 63.72 to 41.43 seconds and an increase in water absorption from 70% to 96%, respectively (Figure 5). At the extreme values of the compression force, adding more superdisintegrant resulted in higher simvastatin release rates. Although the exact mechanisms of croscarmellose sodium action within the RDT matrices have not been fully unveiled and are still under investigation by many researchers, water wicking, swelling of the hydrophilic components, relaxation after stress by plastic components, repelling forces among particles with similar electrostatic charges, or work by wicking action could be considered individually or combined as advocated mechanisms for RDTS’ disintegration.31,32 At the extreme percentages of the superdisintegrant, increasing the compression force resulted in longer time for complete disintegration, decline in water wicking action, and shallow simvastatin release rates (Figure 5). Compression force would affect water wicking by influencing the tablet porosity.33 At low compression forces, relaxation after stress by plastic components could be overawed by resulting porous matrix properties. At moderate value of the compression force, a highest superdisintegrant action might prevail. At high compression forces, water wicking might be reduced significantly by the low porosity with more contribution of deformation action by the superdisintegrant.34 Consequently, the effects of hardness of RDTs on disintegration, water wicking, and subsequent drug dissolution are greatly modulated by the disintegration mechanism.
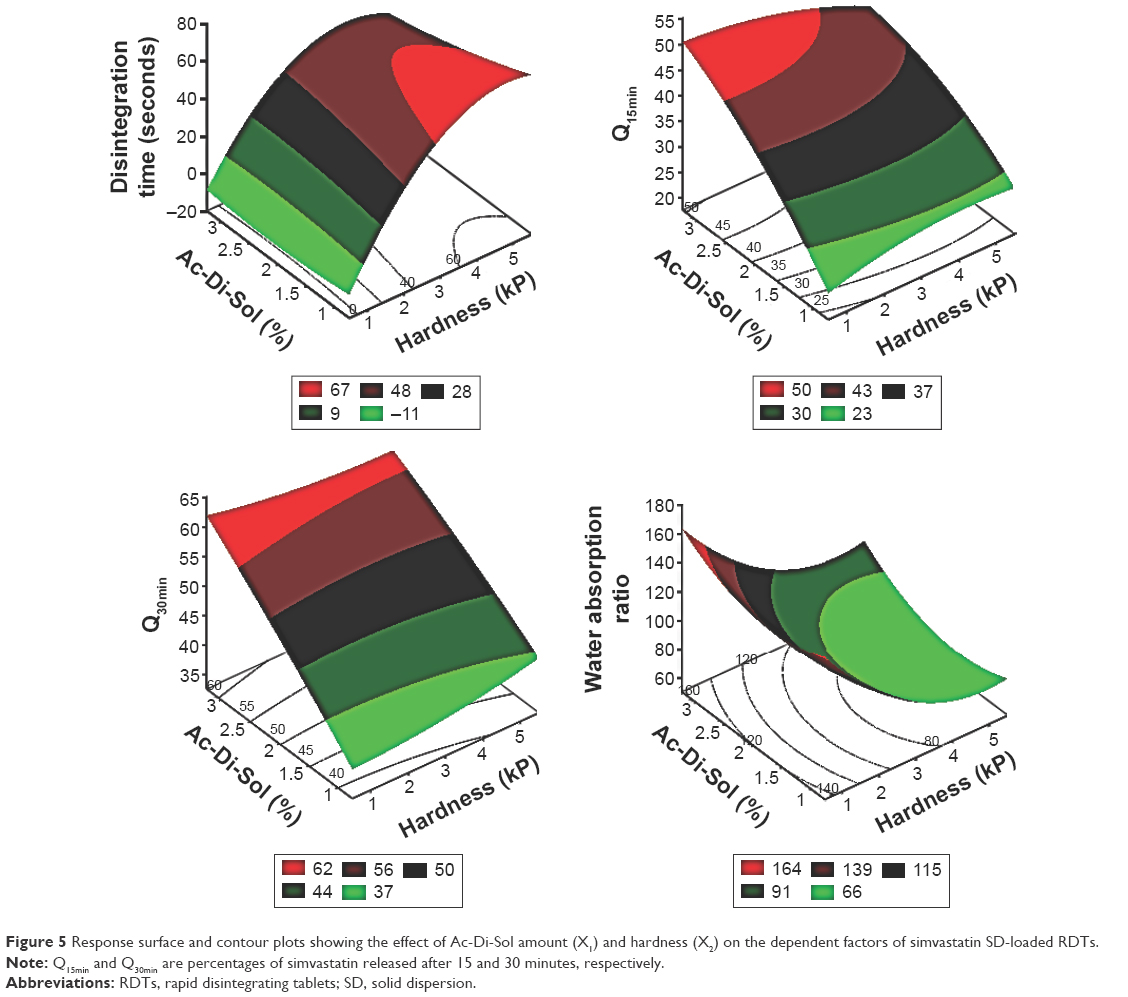 | Figure 5 Response surface and contour plots showing the effect of Ac-Di-Sol amount (X1) and hardness (X2) on the dependent factors of simvastatin SD-loaded RDTs. |
Analysis of variance
Regression of the experimental values of the responses versus the predicted values demonstrated linear relationships with correlation coefficients (R2) >0.95 (Figure 6 and Table 4). For all the measured responses, regression of the obtained residual values versus the prediction values of the model showed random scattering about the zero value. No missing terms, outliers, or effective points were observed to indicate the normal distribution among the recoded data (Figure 6). Moreover, the recorded Cook’s distances for each response were significantly different than the conforming threshold value to indicate no deviation within each data set. The results of analysis of variance showed sum of squares of values 4,314.7, 458.4, 412.8, and 5,727.4 with mean squares of 862.9, 91.7, 82.5, and 1,145.4 for the disintegration time, Q15min, Q30min, and water absorption ratio, respectively. R2 values of 0.9934, 0.9943, 0.9856, and 0.9890 for disintegration time, Q15min, Q30min, and water absorption ratio, respectively, were also obtained to indicate an acceptable fit with at least 95% of covariance explained by the models. This was confirmed by Prob > F values of 0.0004, 0.0028, 0.0286, and 0.0049 for RDTs’ disintegration time, Q15min, Q30min, and water absorption ratio, respectively. Therefore, these results would demonstrate the precision of the developed models to estimate these RDT characteristics.
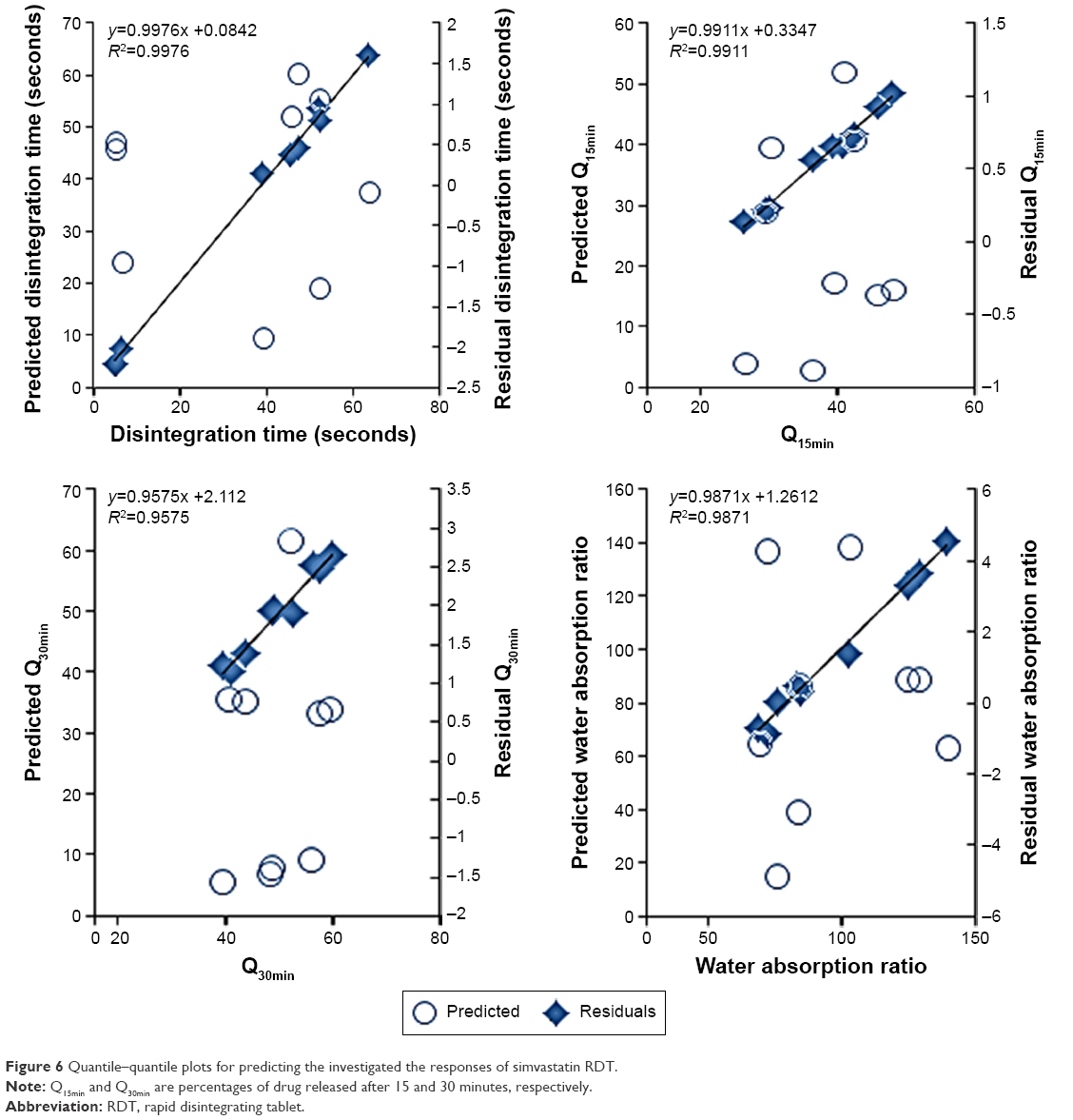 | Figure 6 Quantile–quantile plots for predicting the investigated the responses of simvastatin RDT. |
Ranking and interactions among variables
The collinear relationships between croscarmellose sodium percentages and compression forces were also investigated as a tool to represent the design space of RDTs (Figure 7). An interaction means the abolished effect of one variable at a certain value of the other variable. In these plots, the effects’ parallel lines demonstrate no interaction; however, the intersecting lines indicate that these factors interact instantaneously at certain values.35 Figure 7 demonstrates no colinearities between X1 and X2 at their extreme values on the disintegration time. However, changing X1 level from its lowest to highest percentage while compressing the RDTs under low compression force did not show the same disintegration rate. Hence, it could be revealed by the superior influence of compaction force to enhance RDTs’ porous structure and to facilitate RDT disintegration instead of only the swelling action by croscarmellose sodium. Similarly, the influences of X2 on simvastatin dissolved after 15 and 30 minutes abolished extreme percentages of the superdisintegrant, respectively. This would indicate the superior action of croscarmellose sodium than RDTs’ porosity for the initial simvastatin release. On the other hand, more croscarmellose sodium percentage should be used for complete disintegration and dissolution of simvastatin. The ranking of the individual and polynomial factors and their interactions for their effects of the responses are also presented on Pareto charts (Figure 7). The variables could be ranked as X2 > X22 > X1 > X1X2 > X12 for their effects on the disintegration of RDTs. However, X2 > X22 > X1 > X12 > X1X2 were their ranking for the effect on water wicking percentage. The level of the superdisintegrant was the primary variable to influence simvastatin release rate and then the compression force. Most of the other variables and their collinear and quadratic effects showed minimal influences on Q15min, Q30min (Figure 7).
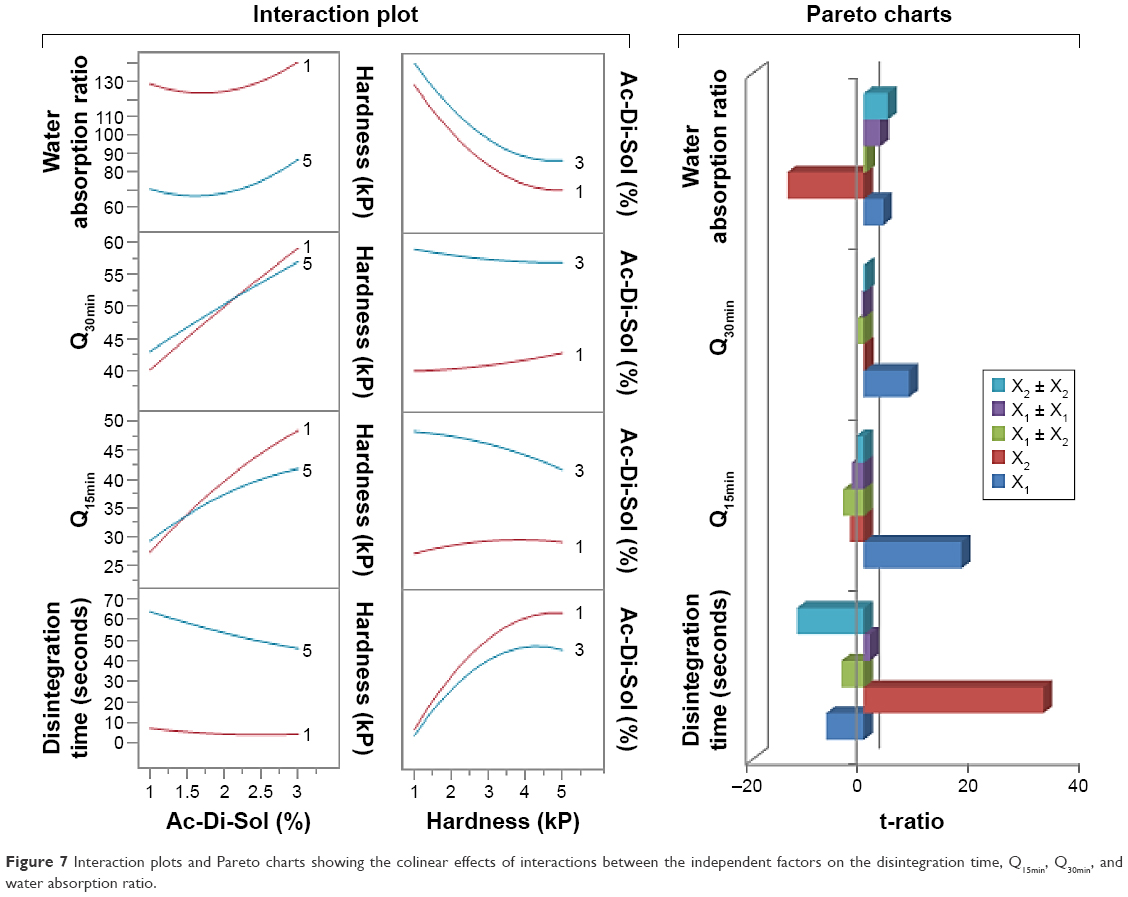 | Figure 7 Interaction plots and Pareto charts showing the colinear effects of interactions between the independent factors on the disintegration time, Q15min, Q30min, and water absorption ratio. |
Optimization of RDTs
A constraint for disintegration time of RDTs was used on the basis of the recommendations of the US Food and Drug Administration for RDTs to be completely disintegrated in <30 seconds.2 For water absorption ratio, Q15min and Q30min, maximized functions were used. An individualized function was employed to optimize each response yielded different percentages for Ac-Di-Sol and different compression forces as well. Consequently, a generalized desirability function was advocated to normalize the individualized functions into single-objective function.36,37 Applying the generalized desirability function to the model resulted in predicted disintegration time, Q15min, Q30min, and the water absorption ratio of 5.12 seconds, 45.23%, 61.78%, and 144.05, respectively, at the highest percentage of croscarmellose sodium of 3% and lowest compression force of 1 kP. Fresh RDT batch was then actually prepared to validate the optimization function with these conditions and yielded disintegration time, Q15min, Q30min, and water absorption ratio of 5.5 seconds, 47%, 62%, and 145.7, respectively. These measured responses were more or less similar to the actual values of the responses with minimal standardized residuals to confirm the robustness of the model within this design space to predict these responses.
Conclusion
Poloxamer 188 was employed to improve the wettability of simvastatin and hence its solubilization in aqueous media. The ratio of drug to poloxamer was critical for the improvement of dissolution with highest dissolution occurring at 1:2 ratio. This SD was further incorporated into a RDT matrix by direct compression. Two-factored three-leveled experimental design along with the response surface methodology was employed to optimize the RDTs for the shortest disintegration time and highest simvastatin dissolution. The level of the superdisintegrant was the primary variable to control disintegrations of RDTs and simvastatin dissolution characteristics. Compression force showed a superior influence to increase porosity of RDTs and to fasten disintegration rather than swelling action by croscarmellose sodium. On the other hand, croscarmellose sodium was most important for the initial simvastatin release. Hence, proper selection of the superdisintegrant and the applied compression force is most critical to optimize simvastatin RDTs.
Acknowledgment
The authors are grateful for the financial support from the Pharmaceutical Sciences Research Centre, Umm Al-Qura University, Makkah, KSA (Project No 4331013).
Disclosure
The authors report no conflicts of interest in this work.
References
Hirani JJ, Rathod DA, Vadalia KR. Orally disintegrating tablets: a review. Tropical Journal of Pharmaceutical Research. 2009;8(2):161–172. | ||
Pabari RM, Ramtoola Z. Application of face centred central composite design to optimise compression force and tablet diameter for the formulation of mechanically strong and fast disintegrating orodispersible tablets. Int J Pharm. 2012;430(1–2):18–25. | ||
Fu Y, Yang S, Jeong SH, Kimura S, Park K. Orally fast disintegrating tablets: developments, technologies, taste-masking and clinical studies. Crit Rev Ther Drug Carrier Syst. 2004;21(6):433–476. | ||
Shukla D, Chakraborty S, Singh S, Mishra B. Fabrication and evaluation of taste masked resinate of risperidone and its orally disintegrating tablets. Chem Pharm Bull (Tokyo). 2009;57(4):337–345. | ||
Kawano Y, Ito A, Sasatsu M, Machida Y, Onishi H. [Preparation and evaluation of taste masked orally disintegrating tablets with granules made by the wet granulation method]. Yakugaku Zasshi. 2010;130(12):1737–1742. Japanese. | ||
Hu X, Li Y, Zhang E, et al. Preparation and evaluation of orally disintegrating tablets containing taste-masked microcapsules of berberine hydrochloride. AAPS PharmSciTech. 2013;14(1):29–37. | ||
Smith DG. Epidemiology of dyslipidemia and economic burden on the healthcare system. Am J Manag Care. 2007;13(Suppl 3):S68–S71. | ||
Wietlisbach V, Marques-Vidal P, Kuulasmaa K, Karvanen J, Paccaud F, Project WM. The relation of body mass index and abdominal adiposity with dyslipidemia in 27 general populations of the WHO MONICA Project. Nutr Metab Cardiovasc Dis. 2013;23(5):432–442. | ||
Hakanen M, Lagstrom H, Kaitosaari T, et al. Development of overweight in an atherosclerosis prevention trial starting in early childhood. The STRIP study. Int J Obes (Lond). 2006;30(4):618–626. | ||
Su TC, Liao CC, Chien KL, Hsu SH, Sung FC. An overweight or obese status in childhood predicts subclinical atherosclerosis and prehypertension/hypertension in young adults. J Atheroscler Thromb. 2014;21(11):1170–1182. | ||
Daniels SR, Greer FR; Committee on Nutrition. Lipid screening and cardiovascular health in childhood. Pediatrics. 2008;122(1):198–208. | ||
Belay B, Belamarich PF, Tom-Revzon C. The use of statins in pediatrics: knowledge base, limitations, and future directions. Pediatrics. 2007;119(2):370–380. | ||
Williams D, Feely J. Pharmacokinetic-pharmacodynamic drug interactions with HMG-CoA reductase inhibitors. Clin Pharmacokinet. 2002;41(5):343–370. | ||
Eiland LS, Luttrell PK. Use of statins for dyslipidemia in the pediatric population. J Pediatr Pharmacol Ther. 2010;15(3):160–172. | ||
Kang BK, Lee JS, Chon SK, et al. Development of self-microemulsifying drug delivery systems (SMEDDS) for oral bioavailability enhancement of simvastatin in beagle dogs. Int J Pharm. 2004;274(1–2):65–73. | ||
Jiang T, Han N, Zhao B, Xie Y, Wang S. Enhanced dissolution rate and oral bioavailability of simvastatin nanocrystal prepared by sonoprecipitation. Drug Dev Ind Pharm. 2012;38(10):1230–1239. | ||
Varshosaz J, Tavakoli N, Salamat FA. Enhanced dissolution rate of simvastatin using spherical crystallization technique. Pharm Dev Technol. 2011;16(5):529–535. | ||
Rowe RC, Sheskey PJ, Quinn ME; American Pharmacists Association. Handbook of Pharmaceutical Excipients. London; Chicago; Washington, DC: Pharmaceutical Press; American Pharmacists Association; 2009. | ||
Newa M, Bhandari KH, Li DX, et al. Preparation, characterization and in vivo evaluation of ibuprofen binary solid dispersions with poloxamer 188. Int J Pharm. 2007;343(1–2):228–237. | ||
Guzik L, Mrozik W, Kamysz W. Determination of simvastatin in pharmaceutical dosage forms by optimized and validated method using HPLC/UV. Croat Chem Acta. 2010;83(4):371–377. | ||
Anderson NH, Bauer M, Boussac N, Khan-Malek R, Munden P, Sardaro M. An evaluation of fit factors and dissolution efficiency for the comparison of in vitro dissolution profiles. J Pharm Biomed Anal. 1998;17(4–5):811–822. | ||
Late SG, Yu YY, Banga AK. Effects of disintegration-promoting agent, lubricants and moisture treatment on optimized fast disintegrating tablets. Int J Pharm. 2009;365(1–2):4–11. | ||
Sandeep K, Suresh P, D GG. Effect of non ionic surfactant on the solubility and dissolution of simvastatin. Int Res J Pharm. 2011;2(3):100–102. | ||
Cirri M, Mura P, Rabasco AM, Gines JM, Moyano JR, Gonzalez-Rodriguez ML. Characterization of ibuproxam binary and ternary dispersions with hydrophilic carriers. Drug Dev Ind Pharm. 2004;30(1):65–74. | ||
Affandi MMRMM, Tripathy M, Shah YAA, Majeed ABA. Solubility enhancement of simvastatin by arginine: thermodynamics, solute-solvent interactions, and spectral analysis. Drug Des Dev Ther. 2016;10:959–969. | ||
Bettinetti GP, Sorrenti M, Rossi S, Ferrari F, Mura P, Faucci MT. Assessment of solid-state interactions of naproxen with amorphous cyclodextrin derivatives by DSC. J Pharm Biomed Anal. 2002;30(4):1173–1179. | ||
Sharma A, Jain CP, Tanwar YS. Preparation and characterization of solid dispersions of carvedilol with poloxamer 188. J Chil Chem Soc. 2013;58(1):1553–1557. | ||
Rao M, Mandage Y, Thanki K, Bhise S. Dissolution improvement of simvastatin by surface solid dispersion technology. Dissolut Technol. 2010;17(2):27–34. | ||
Battu SK, Repka MA, Majumdar S, Madhusudan RY. Formulation and evaluation of rapidly disintegrating fenoverine tablets: effect of superdisintegrants. Drug Dev Ind Pharm. 2007;33(11):1225–1232. | ||
Sammour OA, Hammad MA, Zidan AS, Mowafy AG. QbD approach of rapid disintegrating tablets incorporating indomethacin solid dispersion. Pharm Dev Technol. 2011;16(3):219–227. | ||
Fenyvest E, Antal B, Zsadon B, Szejtli J. Cyclodextrin polymer, a new tablet disintegrating agent. Die Pharmazie. 1984;39(7):473–475. | ||
Zhang G, Yang J, Liu H, Zhang J. Sludge ozonation: disintegration, supernatant changes and mechanisms. Bioresour Technol. 2009;100(3):1505–1509. | ||
Pabari R, Ramtoola Z. Effect of a disintegration mechanism on wetting, water absorption, and disintegration time of orodispersible tablets. J Young Pharm. 2012;4(3):157–163. | ||
Hasegawa A, Nakagawa H, Sugimoto I. [The mechanism of disintegration time increase of tablets containing hydroxypropylcellulose by moisture absorption]. Yakugaku Zasshi. 1984;104(5):544–547. Japanese. | ||
Mathialagan T, Viraraghavan T. Biosorption of pentachlorophenol by fungal biomass from aqueous solutions: a factorial design analysis. Environ Technol. 2005;26(5):571–579. | ||
Carlyle WM, Montgomery DC, Runger GC. Optimization problems and methods in quality control and improvement. J Qual Technol. 2000;32(1):1–17. | ||
van Wijk JP, Buirma R, van Tol A, et al. Effects of increasing doses of simvastatin on fasting lipoprotein subfractions, and the effect of high-dose simvastatin on postprandial chylomicron remnant clearance in normotriglyceridemic patients with premature coronary sclerosis. Atherosclerosis. 2005;178(1):147–155. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.