Back to Journals » Neuropsychiatric Disease and Treatment » Volume 11
Ranitidine reduced levodopa-induced dyskinesia by remodeling neurochemical changes in hemiparkinsonian model of rats
Authors Shi H, Yang X, Zhao H, Zhang S, Zu J, Zhang W, Shen X, Cui G, Hua F, Yan C
Received 2 January 2015
Accepted for publication 11 March 2015
Published 27 May 2015 Volume 2015:11 Pages 1331—1337
DOI https://doi.org/10.2147/NDT.S80174
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Wai Kwong Tang
Hongjuan Shi,1,2,* Xinxin Yang,2,3,* Hui Zhao,4 Shenyang Zhang,2 Jie Zu,2 Wei Zhang,2 Xia Shen,2 Guiyun Cui,2,3 Fang Hua,2,3 Chuanzhu Yan1
1Department of Neurology, Qilu Hospital of Shangdong University, Jinan, 2Department of Neurology, Affiliated Hospital of Xuzhou Medical College, 3Institute of Neurological Diseases of Xuzhou Medical College, 4Department of Neurology, Xuzhou Central Hospital, Xuzhou, People’s Republic of China
*These authors contributed equally to this work
Background: Levodopa (L-dopa) remains the best drug in the treatment of Parkinson’s disease (PD). Unfortunately, long-term L-dopa caused motor complications, one of which is L-dopa-induced dyskinesia (LID). The precise mechanisms of LID are not fully understood. We have previously reported that ranitidine could reduce LID by inhibiting the activity of protein kinase A pathway in a rat model of PD. It is demonstrated that neurotransmitters such as γ-aminobutyric-acid (GABA) and glutamate (Glu) are also involved in the expression of LID. But whether ranitidine could reduce LID by remodeling the neurochemical changes is unknown.
Methods: In the present study, we produced PD rats by injection of 6-hydroxydopamine. Then PD rats were treated with vehicle, L-dopa (6 mg/kg, plus benserazide 12 mg/kg, intraperitoneal [ip]) or L-dopa (6 mg/kg, plus benserazide 12 mg/kg, ip) plus ranitidine (10 mg/kg, oral). Abnormal voluntary movements were adopted to measure the antidyskinetic effect of ranitidine in PD rats. Rotarod tests were used to observe whether ranitidine treatment affects the antiparkinsonian effect of L-dopa. In vivo microdialysis was used to measure nigral GABA and striatal Glu in PD rats.
Results: We found that ranitidine pretreatment reduced abnormal voluntary movements in L-dopa-primed PD rats without affecting the antiparkinsonian effect of L-dopa. In parallel with behavioral improvement, ranitidine pretreatment reduced protein kinase A activity and suppressed the surge of nigral GABA and striatal Glu.
Conclusion: These data indicated that ranitidine could reduce LID by modeling neurochemical changes induced by L-dopa, suggesting a novel mechanism of ranitidine in the treatment of LID.
Keywords: ranitidine, Parkinson’s disease, levodopa-induced dyskinesia, PKA, γ-aminobutyric-acid, glutamate
Introduction
Parkinson’s disease (PD) is a kind of neurodegenerative disease, which is characterized by loss of dopaminergic neurons in the substantia nigra.1 Levodopa (L-dopa) remains the best drug in the treatment of PD.2 However, long-term L-dopa treatment caused many complications, one of which is dyskinesia.3–5 Until now, the pathogenesis of L-dopa-induced dyskinesia (LID) is not fully understood. Numerous studies indicated that pulsatile dopamine (DA) stimulation plays an important role in LID,6–8 which changed the plasticity of striatal neurons.9 Unregulated DA release from DA and non-DA neurons causes upregulation and abnormal trafficking of D1 receptors in striatal neurons along with abnormal downstream signaling responses.10 In addition to striatum, substantia nigra also plays an important role in the expression of LID.11 It is reported that elevated extracellular DA levels were observed in the substantia nigra of L-dopa-primed PD rats.11 Pulsatile DA stimulation not only changes the firing pattern of neurons but also changes the related neurochemical alternations.12 It is demonstrated that increased levels of nigral γ-aminobutyric-acid (GABA) were observed in LID rats,12 which reflects a hyperactivity in the direct striatonigral pathway. This hyperactivity plays a crucial role in LID. In addition to GABA, glutamate (Glu) also has a close relationship with LID.13–15 Indeed, upon DA depletion and subsequent L-dopa or D1R agonist treatment, augmentation of striatal Glu levels and increased expression of striatal Glu receptors have been postulated to result in dyskinetic behaviors.
It is demonstrated that histamine system is involved in the expression of LID.16 A recent study has showed that famotidine could reduce LID in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned macaque model of PD.17 We also have reported that ranitidine could reduce LID by downregulating protein kinase A (PKA) pathway in a rat model of PD.16 However, whether ranitidine could reduce LID by remodeling the neurochemical changes is still unknown. Thus, this study was performed to verify this hypothesis.
Methods
Experimental design
As shown in Figure 1, PD rats were induced by 6-hydroxydopamine (6-OHDA) lesions. Three weeks later, apomorphine-induced rotations were used to select valid PD rats. Then valid PD rats were randomly divided into three groups (n=8 in each group) and received vehicle, L-dopa (6 mg/kg, plus benserazide 12 mg/kg, intraperitoneal [ip]) or L-dopa (6 mg/kg, plus benserazide 12 mg/kg, ip) plus ranitidine (10 mg/kg, oral) for 21 days. The dose of ranitidine was chosen according to our previous report.16 Ranitidine was given to rats 5 minutes before L-dopa treatment. As control, another group of sham-operated rats (n=8) received vehicle treatment. On test days, rotarod test (days 10 and 20 of L-dopa/benserazide treatment) and abnormal in voluntary movements (AIMs) (days 0, 5, 9, 12, 17, 19, and 21 of L-dopa/benserazide treatment) were measured by an observer who was blind to the experiment. On day 20 of L-dopa treatment, probes were implanted in the DA-lesioned SNr and ipsilateral striatum of dyskinetic rats. Microdialysis was performed on day 21 of L-dopa treatment and lasted for 3 days. All rats were sacrificed at the end of experiment on day 23 of L-dopa treatment. Striatal PKA levels were determined by enzyme-linked immunosorbent assay (ELISA). Striatal Glu and nigral GABA levels were measured by high-performance liquid chromatography analysis.
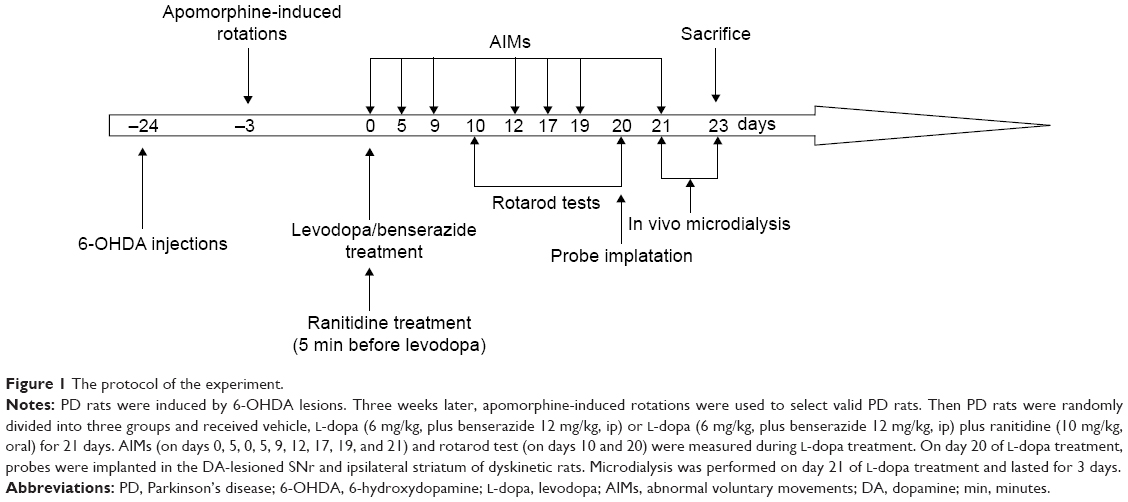 | Figure 1 The protocol of the experiment. |
Animals
Fifty-six adult rats (Sprague-Dawley, males, 180–220 g) were used in this study. All protocols involving the animals were approved by the Institutional Review Board of the Affiliated Hospital of Xuzhou medical college and were performed according to the guidelines of the National Institutes of Health for the care and use of laboratory animals (NIH publication No 80-23). All experimental procedures were conducted in conformity with institutional guidelines for the care and use of laboratory animals in Xuzhou Medical College, Xuzhou, China, and conformed to the National Institutes of Health for the care and use of laboratory animals (NIH publication No 80-23).
6-OHDA lesions
All animals received injections of a total of 8 μg of 6-OHDA (dissolved in 4 μL of 0.9% physiological saline containing 0.02% ascorbic acid; Sigma-Aldrich Co., St Louis, MO, USA). The coordinates were calculated using the rat brain atlas18 as follows: anterior–posterior (AP): −4.4 mm, medial–lateral (ML): −1.2 mm, and dorsal–ventral (DV): −7.8 mm. The tooth bar was set to −2.4 mm. Three weeks after injections, the rats that exhibited a stable apomorphine-induced rotational asymmetry of at least seven full turns per minute away from the lesioned side were selected for the next experiment.
Behavioral test
Rotarod tests were performed after 2 hours of L-dopa treatment; the speed of rotating cylinder was stepwise increased from 3 rpm to 30 rpm until animals fell off the rod as previously described.19 When the animal falls off the rotating drum, individual sensors at the bottom of each separate compartment automatically record the amount of time spent on the cylinder.
Rats were monitored for AIMs using a procedure as we have reported previously.20 On test days, rats were individually placed in plastic trays 5 minutes before the drug treatments. Following injections, each rat was assessed for exhibition of axial, limb, orolingual, and locomotor movements. At 20-minute intervals (ie, 20 minutes, 40 minutes, 60 minutes, 80 minutes, etc), AIMs were rated for 60 seconds for each rat for a total of 120 minutes, during which a severity score of 0–4 was assigned for each AIM’s category. For each AIM’s category, the scores were summed for each time point.
Striatal PKA analysis
Striatal supernatants of rats were measured for PKA concentrations by a sandwich ELISA procedure according to the manufacturer’s instructions (Kexin, Shanghai, People’s Republic of China). Optical density was determined by a enzyme standard instrument (WD-2102A; Liuyi, Beijing, People’s Republic of China).
In vivo microdialysis
Two microdialysis probes were implanted in the DA-lesioned SNr (AP −5.3, ML −2.2, VD −8.6) and ipsilateral striatum (AP +0.6, ML −3.5, DV −6.0) of dyskinetic rats as previously described.11 The microdialysis probes in the substantia nigra and striatum were implanted in the same rat. Forty-eight hours after surgery, probes were perfused with a modified Ringer solution (CaCl2 1.2 mM, KCl 2.7 mM, NaCl 148 mM, MgCl2 0.85 mM) at a 3 μL/min flow rate. Microdialyzate samples were collected every 15 minutes. Three baseline samples were collected before drug treatment.
Endogenous Glu and GABA analysis
Striatal Glu and nigral GABA levels were measured by a high-performance liquid chromatography/fluorimetric detection system as described previously.14 Thirty microliters of o-phthaldialdehyde/mercaptoethanol reagent was added to 30 μL aliquots of sample. Then 50 μL of the mixture was separated in a 5-C18 ChromSep analytical column (Chrompack, Middelburg, the Netherlands). The flow rate of the mobile phase perfused at 0.48 mL/min (0.1 M sodium acetate, 10% methanol, and 2.2% tetrahydrofuran, pH 6.5).
Statistical analysis
The data are expressed as the mean ± standard error of the mean. Behavioral data were analyzed by one-way repeated-measures analysis of variance (ANOVA) followed by the Newman–Keuls post hoc test (AIMs) and one-way ANOVA followed by the Neuman–Keuls post hoc test (rotarod performances). Others were analyzed by one-way ANOVA followed by the Newman–Keuls post hoc test. Statistical significance was set at P<0.05.
Results
Ranitidine pretreatment did not alter L-dopa efficacy in PD rats
The rotarod test was used to measure the effect of ranitidine treatment on L-dopa efficacy. As shown in Figure 2A, the results showed that ranitidine pretreatment did not change the benefit of L-dopa treatment in PD rats.
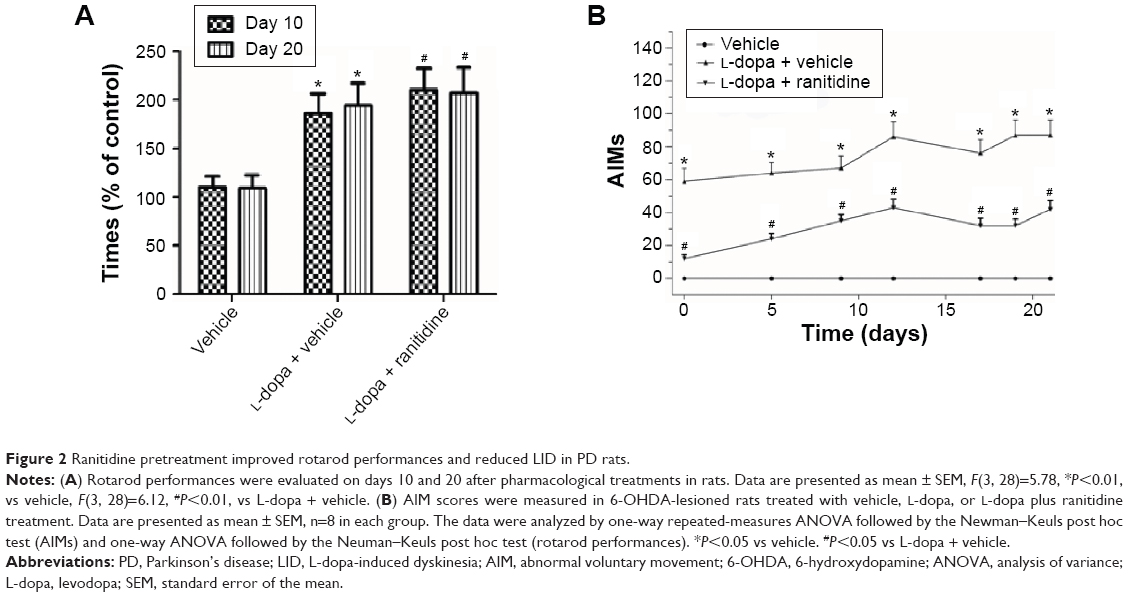 | Figure 2 Ranitidine pretreatment improved rotarod performances and reduced LID in PD rats. |
Ranitidine pretreatment reduced LID in L-dopa-primed PD rats
As shown in Figure 2B, AIM scores were increased after repeated L-dopa administration, which suggested PD rats developed full dyskinesia. However, ranitidine pretreatment prevented the emergence of LID as evidenced by decreased AIM scores.
Ranitidine reduced PKA levels in dyskinetic rats
As shown in Figure 3, after 6-OHDA lesions, PKA levels were significantly increased in rats; the following L-dopa treatment induced further increase of PKA in PD rats. However, ranitidine treatment suppressed the increase of PKA in L-dopa-primed PD rats.
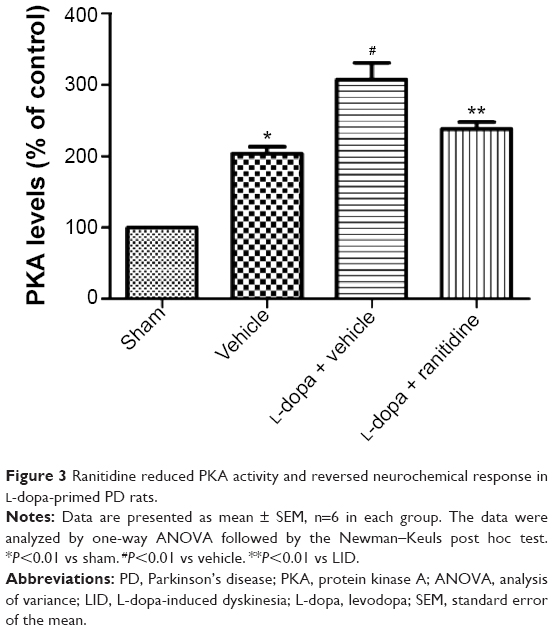 | Figure 3 Ranitidine reduced PKA activity and reversed neurochemical response in L-dopa-primed PD rats. |
Ranitidine pretreatment reduced nigral GABA levels in dyskinetic rats
Microdialysis was used to investigate whether the antidyskinetic action of ranitidine was accompanied by changes in the nigral output. We found that long-term L-dopa treatment induced increased nigral GABA levels in PD rats (Figure 4A), which is in line with previous studies.12 Conversely, ranitidine treatment prevented this increase while ameliorating LID in L-dopa-primed PD rats.
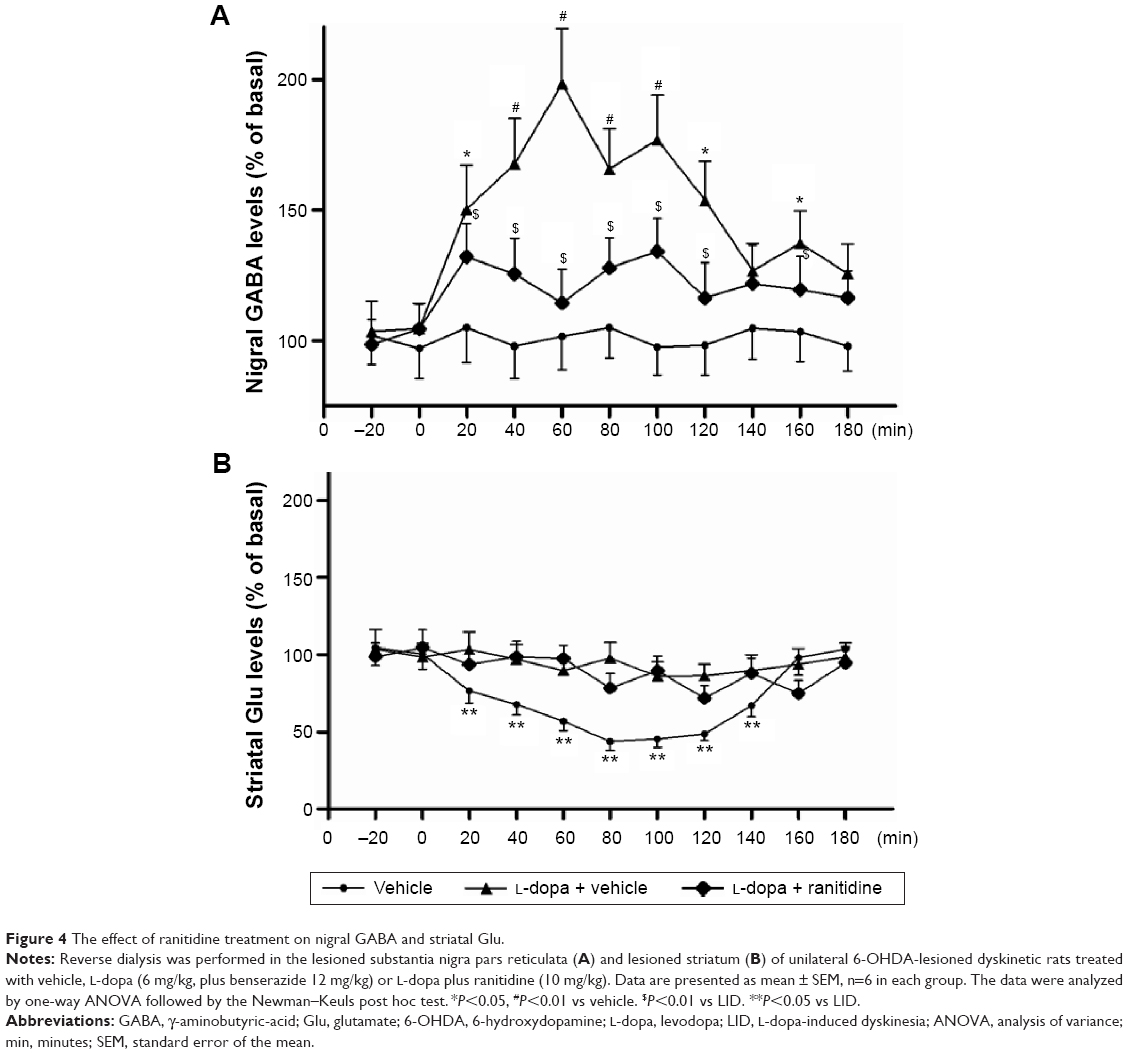 | Figure 4 The effect of ranitidine treatment on nigral GABA and striatal Glu. |
Ranitidine pretreatment prevented the reduction of striatal Glu in dyskinetic rats
The extracellular Glu levels were measured in sham-operated rats and 6-OHDA-lesioned rats treated with vehicle, L-dopa or L-dopa plus ranitidine. As shown in Figure 4B, there was an obvious efflux of Glu in L-dopa-primed 6-OHDA rats. Conversely, ranitidine pretreatment depressed the surge of Glu in PD rats.
Discussion
LID is one of main complications during PD treatment. A previous study showed that famotidine, a kind of histamine 2 receptor antagonist, could reduce LID in patients with PD. In the present study, we found that ranitidine could reduce LID without affecting antiparkinsonian effect of L-dopa, which is in line with our previous study.16 In parallel with behavioral reversal, ranitidine treatment reduced PKA activity and remodeled neurochemical change caused by long-term L-dopa treatment as evidenced by reducing the rise of nigral GABA and striatal Glu.
The precise mechanisms of LID are not fully understood. Previous studies showed that PKA pathway plays an important role in the expression of LID.9,11,20,21 Inhibiting PKA pathway was helpful in improving LID in PD rats. Besides, it is reported that histamine via H2 receptors indirectly increases the firing frequency of projection neurons in the substantia nigra pars reticulate so as to the overactive basal ganglia output in PD.22 Therefore, H2 antagonists could potentially act synergistically with L-dopa to reduce overactivity of the substantia nigra pars reticulata.23 Previous studies have also indicated that H2 antagonists have the ability to modulate striatal release of acetylcholine. In addition, histamine H2 receptor stimulation decreases acetylcholine in the striatum and then reduces DA D1-mediated activation, leading to increased activity in the direct striatal output pathway. These data showed that histamine H2 receptor may be involved in emergency of LID. Numerous studies have shown that long-term L-dopa treatment induced LID in animal models of PD.24,25 In the present study, we found that PD rats developed full development of LID as evidenced by increased AIM scores. But ranitidine pretreatment reduced AIM scores in L-dopa-primed PD rats. Rotarod test was often used to measure the efficacy of L-dopa treatment in PD rats. In the present study, we found that L-dopa treatment improved rotarod performance in PD rats, which suggested that ranitidine pretreatment did not affect the benefit of L-dopa treatment.
Recent studies showed that striatal Glu was involved in the development of LID.26 Glu itself could not induce LID, but it may serve to modulate dyskinetic behaviors as suggested by a study reporting that direct intrastriatal administrations of Glu antagonists reduced LID.27 In the present study, we found that 6-OHDA lesions induced a surge of striatal Glu in PD rats primed with L-dopa, which was in line with a previous study.13 Interestingly, ranitidine pretreatment prevented the efflux of striatal Glu while attenuating LID in L-dopa-primed PD rats.
In addition to Glu, GABA is also proved to be involved in the expression of LID.14,27 It is reported that peak-dose dyskinesia involves activation of the striatonigral GABA pathway, and increase of GABA release in SNr was observed after L-dopa challenge.12 Ranitidine could reduce L-dopa-mediated Glu efflux, suggesting that ranitidine may act presynaptically on glutamatergic corticostriatal neurons. But whether ranitidine could affect postsynaptic mechanism such as GABA release to substantia nigra is not known. In the present study, we found that nigral GABA levels were increased after L-dopa priming in PD rats. However, ranitidine pretreatment reduced LID while suppressing the surge of nigral GABA. These data indicated that ranitidine could reduce LID via affecting presynaptic and postsynaptic mechanisms. Moreover, we assumed that inhibition of PKA activity was helpful in reducing Glu and GABA.
Taken together, in this study, we found that ranitidine reduced LID without affecting the antiparkinsonian effect of L-dopa. Ranitidine pretreatment reduced PKA activity and reversed neurochemical changes caused by long-term L-dopa treatment in PD rats, suggesting a novel mechanism of ranitidine in the treatment of LID.
Acknowledgments
The study was supported by the National Natural Science Foundation of China ( 81301077, 81271344, and 81271268), the Natural Science Foundation of Jiangsu Higher Education Institutions (13KJB320026), Jiangsu Province Health Department-funded project (H200926), Jiangsu Specially-Appointed Professor Program to Dr Hua, and the social development of science and technology plan (XM13B058) for Dr Zhao.
Disclosure
The authors have no conflicts of interest to declare.
References
Schulz-Schaeffer WJ. The synaptic pathology of α-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol. 2010;120:131–143. | ||
Connolly BS, Lang AE. Pharmacological treatment of Parkinson disease: a review. JAMA. 2014;311:1670–1683. | ||
Cerasa A, Fasano A, Morgante F, Koch G, Quattrone A. Maladaptive plasticity in levodopa-induced dyskinesias and tardive dyskinesias: old and new insights on the effects of dopamine receptor pharmacology. Front Neurol. 2014;5:49. | ||
Rodnitzky RL, Narayanan NS. Amantadine’s role in the treatment of levodopa-induced dyskinesia. Neurology. 2014;82:288–289. | ||
Picconi B, Calabresi P. Targeting metabotropic glutamate receptors as a new strategy against levodopa-induced dyskinesia in Parkinson’s disease? Mov Disord. 2014;29:715–719. | ||
Bezard E, Tronci E, Pioli EY, et al. Study of the antidyskinetic effect of eltoprazine in animal models of levodopa-induced dyskinesia. Mov Disord. 2013;28:1088–1096. | ||
Heiman M, Heilbut A, Francardo V, et al. Molecular adaptations of striatal spiny projection neurons during levodopa-induced dyskinesia. Proc Natl Acad Sci U S A. 2014;111:4578–4583. | ||
Manson A, Stirpe P, Schrag A. Levodopa-induced-dyskinesias clinical features, incidence, risk factors, management and impact on quality of life. J Parkinsons Dis. 2012;2:189–198. | ||
Cerovic M, Bagetta V, Pendolino V, et al. Derangement of Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1) and extracellular signal-regulated kinase (ERK) dependent striatal plasticity in L-dopa-induced dyskinesia. Biol Psychiatry. 2015;77(2):106–115. | ||
Bishop C, George JA, Buchta W, et al. Serotonin transporter inhibition attenuates L-dopa-induced dyskinesia without compromising L-DOPA efficacy in hemi-parkinsonian rats. Eur J Neurosci. 2012;36:2839–2848. | ||
Lindgren HS, Andersson DR, Lagerkvist S, Nissbrandt H, Cenci MA. L-dopa-induced dopamine efflux in the striatum and the substantia nigra in a rat model of Parkinson’s disease: temporal and quantitative relationship to the expression of dyskinesia. J Neurochem. 2010;112:1465–1476. | ||
Papathanou M, van der Laan R, Jenner P, Rose S, McCreary AC. Levodopa infusion does not decrease the onset of abnormal involuntary movements in parkinsonian rats. Mov Disord. 2013;28(8): 1072–1079. | ||
Dupre KB, Ostock CY, Eskow Jaunarajs KL, et al. Local modulation of striatal glutamate efflux by serotonin 1A receptor stimulation in dyskinetic, hemiparkinsonian rats. Exp Neurol. 2011;229:288–299. | ||
Mela F, Marti M, Bido S, Cenci MA, Morari M. In vivo evidence for a differential contribution of striatal and nigral D1 and D2 receptors to L-dopa induced dyskinesia and the accompanying surge of nigral amino acid levels. Neurobiol Dis. 2012;45:573–582. | ||
Morgese MG, Cassano T, Gaetani S, et al. Neurochemical changes in the striatum of dyskinetic rats after administration of the cannabinoid agonist WIN55,212-2. Neurochem Int. 2009;54:56–64. | ||
Cui G, Yang X, Wang X, et al. Ranitidine reduced levodopa-induced dyskinesia in a rat model of Parkinson’s disease. Neuropsychiatr Dis Treat. 2014;10:39–46. | ||
Johnston TH, van der Meij A, Brotchie JM, Fox SH. Effect of histamine H2 receptor antagonism on levodopa-induced dyskinesia in the MPTP-macaque model of Parkinson’s disease. Mov Disord. 2010;25:1379–1390. | ||
Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. San Diego, CA, USA: Academic Press; 2007. | ||
Liu M, Choi DY, Hunter RL, et al. Trichloroethylene induces dopaminergic neurodegeneration in Fisher 344 rats. J Neurochem. 2010;112:773–783. | ||
Yang X, Chen Y, Hong X, et al. Levodopa/benserazide microspheres reduced levodopa-induced dyskinesia by downregulating phosphorylated GluR1 expression in 6-OHDA-lesioned rats. Drug Des Devel Ther. 2012;6:341–347. | ||
Azkona G, Marcilla I, López de Maturana R, et al. Sustained increase of PKA activity in the postcommissural putamen of dyskinetic monkeys. Mol Neurobiol. 2014;50:1131–1141. | ||
Zhou FW, Xu JJ, Zhao Y, LeDoux MS, Zhou FM. Opposite functions of histamine H1 and H2 receptors and H3 receptor in substantia nigra pars reticulata. J Neurophysiol. 2006;96:1581–1591. | ||
Sittig N, Davidowa H. Histamine reduces firing and bursting of anterior and intralaminar thalamic neurons and activates striatal cells in anesthetized rats. Behav Brain Res. 2001;124:137–143. | ||
Pinna A, Bonaventura J, Farré D, et al. L-dopa disrupts adenosine A(2A)-cannabinoid CB(1)-dopamine D(2) receptor heteromer cross-talk in the striatum of hemiparkinsonian rats: biochemical and behavioral studies. Exp Neurol. 2014;253:180–191. | ||
Tronci E, Lisci C, Stancampiano R, et al. 5-Hydroxy-tryptophan for the treatment of L-dopa-induced dyskinesia in the rat Parkinson’s disease model. Neurobiol Dis. 2013;60:108–114. | ||
Nevalainen N, Lundblad M, Gerhardt GA, Strömberg I. Striatal glutamate release in L-dopa-induced dyskinetic animals. PLoS One. 2013;8:e55706. | ||
Rangel-Barajas C, Silva I, Lopéz-Santiago LM, Aceves J, Erlij D, Florán B. L-dopa-induced dyskinesia in hemiparkinsonian rats is associated with up-regulation of adenylyl cyclase type V/VI and increased GABA release in the substantia nigra reticulata. Neurobiol Dis. 2011;41:51–61. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.