Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 13
Randomized, double-blind, placebo-controlled trial to assess the efficacy and safety of three doses of co-suspension delivery technology glycopyrronium MDI in Japanese patients with moderate-to-severe COPD
Authors Fukushima Y ![]() , Nakatani Y
, Nakatani Y ![]() , Ide Y, Sekino H, St Rose E
, Ide Y, Sekino H, St Rose E ![]() , Siddiqui S
, Siddiqui S ![]() , Maes A, Reisner C
, Maes A, Reisner C ![]()
Received 8 December 2017
Accepted for publication 12 March 2018
Published 13 April 2018 Volume 2018:13 Pages 1187—1194
DOI https://doi.org/10.2147/COPD.S159246
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Yasushi Fukushima,1 Yuji Nakatani,2 Yumiko Ide,3 Hisakuni Sekino,4 Earl St Rose,5 Shahid Siddiqui,6 Andrea Maes,5 Colin Reisner5,6
1Department of Internal Medicine, Fukuwa Clinic, Tokyo, Japan; 2Department of Internal Medicine, Nakatani Hospital, Hyogo, Japan; 3Department of Internal Medicine, Tokyo Center Clinic, Tokyo, Japan; 4Department of Internal Medicine, Sekino Hospital, Tokyo, Japan; 5Pearl – a member of the AstraZeneca Group, Morristown, NJ, USA; 6AstraZeneca, Gaithersburg, MD, USA
Purpose: Due to the burden of COPD in Japan, new pharmacologic treatments are needed to meet patient requirements. This study assessed the efficacy and safety of glycopyrronium (GP) delivered via metered dose inhaler (MDI) in Japanese patients with moderate-to-severe COPD.
Methods: This Phase IIb, multicenter, randomized, double-blind, 7-day, crossover study compared GP MDI 28.8, 14.4, and 7.2 µg with placebo MDI (all administered as two inhalations, twice daily). The primary endpoint was change from baseline in morning pre-dose trough forced expiratory volume in 1 second (FEV1) on Day 8. Secondary endpoints included FEV1 area under the curve from 0 to 2 hours (AUC0–2) and peak change from baseline in FEV1 on Days 1 and 8 and forced vital capacity AUC0–2 on Day 8. Safety was also assessed. ClinicalTrials.gov Identifier: NCT03256552; http://www.ClinicalTrials.gov.
Results: Sixty-six patients were randomized and 62 were included in the modified intent-to-treat population (mean age 67.5 years). All three GP MDI doses significantly improved change from baseline in morning pre-dose trough FEV1 on Day 8 compared with placebo MDI (least squares mean differences 108–131 mL; all p<0.0001). Significant improvements in secondary efficacy endpoints were also observed for all three GP MDI doses compared with placebo MDI (all p<0.0001). Dose–response plateaued at GP MDI 14.4 µg. No significant safety findings were observed with any GP MDI dose or placebo MDI.
Conclusions: The results of this study suggest that GP MDI 14.4 µg (7.2 µg per inhalation) is the most appropriate dose for use in Phase III studies in Japanese patients with moderate-to-severe COPD.
Keywords: bronchodilator agents, dose–response relationship, forced expiratory volume, metered dose inhalers, COPD
Introduction
Globally, COPD is one of the leading causes of morbidity and mortality.1–5 Reports suggest that the prevalence of COPD in Japan is in the range of 7%–11%,6,7 with the economic burden in 2004 estimated to be an average annual total cost of ¥435,876 ($3,694 USD) per patient with moderate/severe COPD.8 Given the high burden of COPD in Japan, it is vital to continue to develop treatment options.
Bronchodilators, such as long-acting anti-muscarinic antagonists (LAMAs) and long-acting β2-agonists (LABAs), are the foundation of pharmacologic treatment for patients with COPD.4,9 When used in combination, LAMAs and LABAs improve the extent of bronchodilation compared with either monocomponent used alone, while also being well tolerated.10 In Japan, LAMA/LABA fixed-dose combinations approved for the maintenance treatment of adult patients with COPD are available as dry powder inhalers and a soft mist inhaler, but not in a pressurized metered dose inhaler (MDI). As a patient’s preference for inhaler device can impact on treatment adherence and effectiveness,11,12 having different devices available for administration of pharmacologic COPD therapies may be advantageous in order for patients to have a device that meets their individual requirements.
In the USA, GFF MDI (Bevespi Aerosphere™, AstraZeneca, Wilmington, DE, USA), a fixed-dose combination of the LAMA, glycopyrronium (GP; 14.4 μg, equivalent to glycopyrrolate 18 μg), and the LABA, formoterol fumarate dihydrate (FF; 10 μg, equivalent to formoterol fumarate 9.6 μg), formulated using innovative co-suspension delivery technology,13 is approved for twice-daily (BID) long-term maintenance treatment of airflow obstruction in patients with COPD.14 A series of Phase IIb studies in predominately Western patients with COPD determined that GP 14.4 μg was the most appropriate dose to combine with FF for the evaluation of GFF MDI in Phase III trials (NCT01350128, NCT01566773,15 NCT01349803, NCT01349816, NCT01587079,16 and NCT0108504517). However, no studies have yet explored the bronchodilator dose–response of GP MDI in Japanese patients with COPD. Here, we report the efficacy and safety data of three doses of GP MDI versus placebo MDI in Japanese patients with moderate-to-severe COPD.
Methods
Patient population
Key inclusion criteria
Male and female patients, 40–80 years of age with moderate-to-severe COPD, as defined by Japanese Respiratory Society (JRS) Guidelines,9 were enrolled. Patients were required to have a pre- and post-bronchodilator forced expiratory volume in 1 second (FEV1)/forced vital capacity (FVC) ratio of <70% and post-bronchodilator FEV1 ≥30% and <80% of predicted normal (calculated using JRS reference equations9) at screening, and a pre-bronchodilator FEV1/FVC ratio of <70% and pre-bronchodilator FEV1 <80% of predicted normal at baseline. Current or former smokers (smoking history ≥10 pack-years) were eligible for inclusion.
Key exclusion criteria
Patients were excluded if they had: poorly controlled COPD (acute worsening of COPD that required treatment with parenteral or oral corticosteroids or antibiotics) within 6 weeks prior to screening or during the screening period; hospitalization due to COPD within 3 months or lower respiratory tract infections that required antibiotics within 6 weeks, prior to, or during, the screening period; a change in smoking status (ie, start/stop smoking), or initiation of a smoking cessation program up to 6 weeks prior to, or throughout, the screening period; long-term oxygen therapy required for >12 hours/day; or a primary diagnosis of asthma. Patients with a history of asthma were eligible if COPD was their current primary diagnosis. Inhaler device training was conducted at screening, and as required at randomization and each visit, but patients who required the use of a spacer device with an MDI to compensate for poor hand-to-breath coordination were excluded from the study.
Patients taking prohibited medications (oral β2-agonists; LABAs, LAMAs, and corticosteroid/LABA combinations; cromoglycate or nedocromil inhalers; leukotriene antagonists; and phosphodiesterase [PDE] inhibitors and PDE-4 inhibitors) were switched to ipratropium bromide MDI (20 μg/inhalation) maintenance therapy during screening and washout periods. Ipratropium bromide was taken in accordance with the local package insert or as directed by the investigator; one or two inhalations four times per day could be reduced if symptoms were under control and stabilized, but at least one inhalation BID was to be administered. If they had been receiving an inhaled corticosteroid (ICS) as part of a fixed-dose combination, patients were switched to the corresponding ICS monotherapy plus ipratropium bromide, providing they had been maintained on a stable dose for ≥28 days. During the study, patients were permitted to use rescue salbutamol sulfate MDI (120 μg salbutamol sulfate corresponding to 100 μg salbutamol base/inhalation), as required.
Study design
PT001004 was a Phase IIb, multicenter, 7-day, randomized, double-blind, crossover study conducted at 20 study sites across Japan, from 28 January to 5 September 2015, which investigated the efficacy and safety of three doses of GP MDI (28.8, 14.4, and 7.2 μg; equivalent to 36, 18, and 9 μg glycopyrrolate, respectively) relative to a matching placebo MDI, all administered as two inhalations BID (Figure 118).
 | Figure 1 Study design. |
Endpoints and assessments
The primary efficacy endpoint was the change from baseline in morning pre-dose trough FEV1 on Day 8. Secondary efficacy endpoints included FEV1 area under the curve from 0 to 2 hours (AUC0–2) on Days 1 and 8, peak change from baseline in FEV1 on Days 1 and 8, and FVC AUC0–2 on Day 8. Other efficacy endpoints included change from baseline in average daily rescue medication use over the treatment period.
All pulmonary function tests were performed in accordance with American Thoracic Society criteria.19 All sites were provided with identical spirometry systems (KoKo Spirometer®; nSpire Health, Inc., Louisville, CO, USA) and all the study staff responsible for performing pulmonary function tests received standardized training. Spirometry assessments were performed 60 and 30 minutes pre-dose and 15, 30, 60, and 120 minutes post-dose on Days 1 and 8 of each treatment period. The average of the 60- and 30-minute pre-dose assessments was used to establish test-day baseline FEV1 and FVC. Patients recorded their total number of puffs of rescue medication use in a diary that they completed twice a day.
Safety assessments included electrocardiograms (ECGs), vital sign measurements, clinical laboratory tests, adverse events (AEs), and serious AEs. AEs of special interest, that is, those associated with inhalation from an MDI, were paradoxical bronchospasm and dry mouth. Vital signs were monitored and 12-lead ECGs were performed for up to 2 hours post-dose on Days 1 and 8.
Statistical analyses
The intent-to-treat (ITT) population (all patients who were randomized and received ≥1 dose of study treatment) was analyzed according to the treatment assigned through the randomization process. The safety population (also defined as all patients who were randomized and received ≥1 dose of study treatment) was analyzed by the treatment actually received. The modified ITT (mITT) population (primary population for efficacy analyses) was a subset of the ITT population who had ≥1 pulmonary function test assessment from ≥2 treatment periods and no major protocol deviations documented prior to unblinding.
Analyses of the primary efficacy endpoint and average daily rescue medication use were conducted using a mixed model with baseline covariate (FEV1 or rescue medication use), period, and treatment as fixed effects. Secondary efficacy endpoints were analyzed using a mixed model with repeated measures, with baseline value (FEV1 or FVC), treatment, day (Day 1 or Day 8), period, and treatment-by-day interaction as fixed effects. Both primary and secondary analyses included patient as a random effect to model correlation within a patient across the entire study. The models did not include treatment sequence as it was determined to be nonsignificant in the primary model. For the secondary efficacy endpoints, the covariance structure for patient within period was unstructured. For the primary endpoint, the family-wise type I error was not controlled for multiplicity. All doses of GP MDI were tested relative to placebo MDI.
Assuming that 60 patients would be randomized, a drop-out rate of 15%, and a within-subject SD of 130 mL, the study was ~80% powered to demonstrate a difference between any two treatments of 75 mL for the primary endpoint, using a 2-sided alpha level of 0.05.
Ethics approval and informed consent
This study was conducted in accordance with Good Clinical Practice guidelines, including the International Conference on Harmonisation, the Japan Ministerial Ordinance on Standards for the Implementation of Clinical Studies on Pharmaceutical Product, and the Declaration of Helsinki. Institutional review boards (Box S1) approved the protocol and informed consent form, and the investigator obtained written informed consent from patients prior to screening (ClinicalTrials.gov Identifier: NCT03256552; http://www.ClinicalTrials.gov).
Results
Study population
A total of 66 patients were included in the ITT/safety population, 61 (92.4%) of whom completed treatment in all four treatment periods (Figure 2). The mean age of the mITT population (n=62) was 67.5 years; 44 (71.0%) were ≥65 years of age, and 59 (95.2%) were male (Table 1). The mean number of pack-years smoked was 51.3, and 37 (59.7%) were current smokers. Most of the mITT population (n=51 [82.3%]) had moderate COPD and the mean duration of COPD was 4.8 years. The majority of patients (n=53 [85.5%]) did not use an ICS at baseline.
 | Figure 2 Patient disposition. |
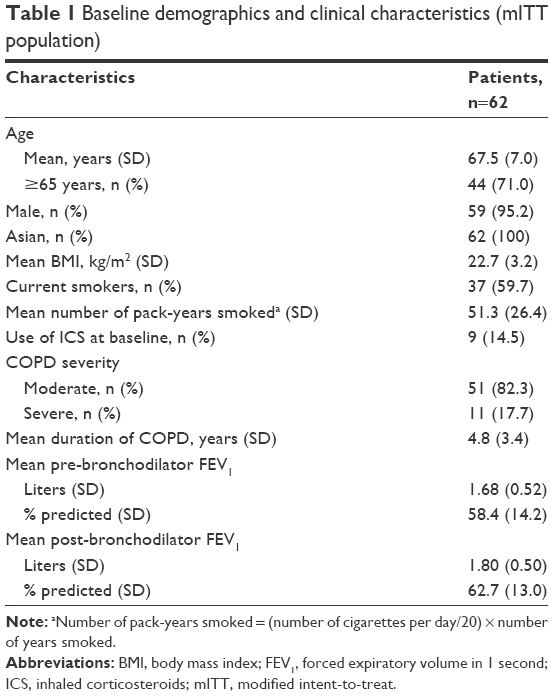 | Table 1 Baseline demographics and clinical characteristics (mITT population) |
Lung function
For the primary endpoint of change from baseline in morning pre-dose trough FEV1 on Day 8, all three doses of GP MDI significantly increased lung function compared with placebo MDI (least squares mean [LSM] differences 131, 129, and 108 mL for GP MDI 28.8, 14.4, and 7.2 μg, respectively; all p<0.0001; Table 2). GP MDI 28.8 and 14.4 μg demonstrated numerically greater improvements compared with GP MDI 7.2 μg.
 | Table 2 Primary and secondary endpoints: LSM treatment difference versus placebo MDI (mITT population) |
Treatment with each of the three GP MDI doses significantly increased the LSM FEV1 AUC0–2 on Days 1 and 8 relative to placebo MDI (all p<0.0001; Table 2). The results for the peak change from baseline in FEV1 were consistent with the FEV1 AUC0–2 data, with significant improvements versus placebo MDI observed for all three doses of GP MDI on both Days 1 and 8 (all p<0.0001; Table 2). All three doses of GP MDI also significantly increased the LSM FVC AUC0–2 on Day 8 versus placebo MDI (all p<0.0001; Table 2). For all the secondary lung function endpoints on Day 8, the improvements in lung function with GP MDI 28.8 and 14.4 μg were similar and both were numerically greater than GP MDI 7.2 μg (Table 2).
Rescue medication
The baseline rescue medication use was low in this study population (mean 0.2 puffs/day for each treatment). GP MDI 28.8 and 7.2 μg significantly reduced the change from baseline in average daily rescue medication use by more than placebo MDI (−0.22 and −0.16 puffs/day, respectively; both p<0.05). GP MDI 14.4 μg reduced average daily rescue medication use numerically compared with placebo MDI (−0.14 puffs/day; p=0.0570).
Safety
The majority of treatment-emergent AEs (TEAEs) were considered mild or moderate, with only one patient who experienced a severe TEAE of worsening COPD following treatment with placebo MDI. Only two TEAEs (nasopharyngitis and rash) occurred in more than one patient with any particular treatment (Table 3). One patient experienced serious TEAEs of pneumonia and worsening of COPD following treatment with placebo MDI, which led to study discontinuation (Table 3). There were no deaths reported in this study (Table 3). No paradoxical bronchospasms were reported in this study and only one patient (GP MDI 28.8 μg) reported dry mouth. Clinical laboratory data demonstrated no substantial changes from baseline with GP MDI or placebo MDI. ECG and vital signs assessments did not reveal any clinically relevant treatment-related effects.
 | Table 3 Summary of treatment-emergent adverse events (safety population) |
Discussion
This study examined the efficacy and safety of a dose range of GP MDI 28.8, 14.4, and 7.2 μg relative to placebo MDI in Japanese patients with moderate-to-severe COPD. For the primary endpoint of change from baseline in morning pre-dose trough FEV1 on Day 8, the improvements with all doses of GP MDI versus placebo MDI exceeded 100 mL, which suggests that they were clinically meaningful differences.20 GP MDI 28.8 and 7.2 μg decreased rescue medication use compared with placebo MDI. However, as the use of rescue medication at baseline was low (0.2 puffs/day), the treatment effects observed were small.
In a Phase IIb study in Western patients with COPD, Fabbri et al reported that GP MDI 14.4 and 7.2 μg showed superiority to placebo MDI with regards to change from baseline in morning pre-dose trough FEV1 (both p<0.001) and peak change from baseline in FEV1 (both p<0.0001) after 14 days of chronic dosing.15 In another Western Phase IIb study, Reisner et al demonstrated that GP MDI 28.8 μg showed superiority to placebo MDI with regards to change from baseline in morning pre-dose trough FEV1 and peak change from baseline in FEV1 after 7 days of chronic dosing (all p<0.01).17 Furthermore, a pooled analysis of Western Phase IIb studies showed that improvements in lung function with GP MDI 28.8 and 14.4 μg doses were similar.21 Therefore, across the dose range examined, findings were comparable in Japanese and Western patients with COPD.
Overall, the results of six randomized Phase IIb studies in Western patients with COPD established GP MDI 14.4 μg as the optimum dose to take forward into the Phase III trials that confirmed the efficacy and safety of the GP 14.4 μg dose, both as a monotherapy and in combination with FF 10 μg, compared with placebo MDI (PINNACLE-1 [NCT01854645] and PINNACLE-2 [NCT01854658]22). The results of this present dose-ranging study demonstrate that the 14.4 μg dose of GP MDI is also appropriate for further clinical development for Japanese patients with COPD.
In this Japanese study and the Western GP MDI dose-ranging study,15 the overall incidences of TEAEs were similar following administration of active treatments or placebo MDI, with no dose-related effects of GP MDI on TEAEs observed. The most frequent TEAE in this study, nasopharyngitis (3.2% of patients receiving GP MDI 14.4 μg), was reported by less than 2% of patients receiving GP MDI 14.4 or 28.8 μg in the Western Phase IIb studies.15–17 Dry mouth was the most commonly reported TEAE in the studies in Western patients,15–17 while this TEAE was reported by only one Japanese patient following treatment with GP MDI 28.8 μg. In both Japanese and Western patients with COPD, no clinically important changes in clinical laboratory results, vital signs, or ECGs were observed.15–17 To the best of our knowledge, there are no studies that report differences in efficacy or safety outcomes in Japanese or Western (Caucasian) patients with COPD following administration of a LAMA or LABA.
Limitations that apply to the interpretation of the results from this study include the small number of patients with COPD involved and the short (7 days) treatment duration. However, the systemic exposure of GP reaches a steady state within 2–3 days of repeated dosing and, therefore, will have reached its maximum level before the Day 8 assessments in this study.14
Conclusions
All three doses of GP MDI were associated with statistically significant improvements in lung function for both the primary and secondary endpoints, relative to placebo MDI. The improvements in the primary endpoint exceeded the minimal clinically important difference of 100 mL for all three doses of GP MDI versus placebo MDI. Dose-dependent improvements in lung function plateaued at the GP MDI 14.4 μg dose. Overall, no significant safety findings were observed with GP MDI treatment at any dose level relative to placebo MDI. The results of this study support the use of GP MDI 14.4 μg BID in Phase III trials in Japanese patients with COPD.
Data sharing statement
All relevant data analyzed during this study are included in this published article.
Acknowledgments
This study was supported by Pearl – a member of the AstraZeneca Group. The authors thank all the patients and their families and the team of investigators, research nurses, and operations staff involved in these studies. The authors would also like to thank Chad Orevillo (former employee of Pearl) for his valuable contribution to this work and Everest Clinical Research, who performed the statistical analyses for this study. Medical writing support, under the direction of the authors, was provided by Thomas Owens, PhD, of CMC CONNECT, a division of Complete Medical Communications Ltd, Manchester, UK, funded by AstraZeneca, Cambridge, UK, in accordance with Good Publication Practice (GPP3) guidelines.23 The sponsor did not place any restriction on authors about the statements made in the final article.
The research described in this manuscript was presented as a poster at the Japanese Respiratory Society 2017 Congress in Tokyo, Japan. Study results are also posted on http://www.ClinicalTrials.gov (ClinicalTrials.gov Identifier: NCT03256552).
Author contributions
All authors contributed toward conception and design, data acquisition, or data analysis and interpretation, critically revising and providing final approval of the manuscript, and agree to be accountable for all aspects of the work.
Disclosure
ESR and AM are employees of Pearl – a member of the AstraZeneca Group. SS is an employee of AstraZeneca. CR is an employee of Pearl – a member of the AstraZeneca Group and an employee of AstraZeneca. The other authors report no conflicts of interest in this work.
References
Burney PG, Patel J, Newson R, Minelli C, Naghavi M. Global and regional trends in COPD mortality, 1990–2010. Eur Respir J. 2015;45(5):1239–1247. | ||
Lozano R, Naghavi M, Foreman K, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380(9859):2095–2128. | ||
GBD 2015 Chronic Respiratory Disease Collaborators. Global, regional, and national deaths, prevalence, disability-adjusted life years, and years lived with disability for chronic obstructive pulmonary disease and asthma, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Respir Med. 2017;5(9):691–706. | ||
Global Initiative for Chronic Obstructive Lung Disease. Global Strategy for the Diagnosis, Management and Prevention of COPD [updated 2018]. Available from: http://www.goldcopd.org. Accessed February 22, 2018. | ||
GBD 2016 Causes of Death Collaborators. Global, regional, and national age-sex specific mortality for 264 causes of death, 1980–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet. 2017;390(10100):1151–1210. | ||
Landis SH, Muellerova H, Mannino DM, et al. Continuing to confront COPD International Patient Survey: methods, COPD prevalence, and disease burden in 2012–2013. Int J Chron Obstruct Pulmon Dis. 2014;9:597–611. | ||
Fukuchi Y, Nishimura M, Ichinose M, et al. COPD in Japan: the Nippon COPD Epidemiology study. Respirology. 2004;9(4):458–465. | ||
Nishimura S, Zaher C. Cost impact of COPD in Japan: opportunities and challenges? Respirology. 2004;9(4):466–473. | ||
Japanese Respiratory Society. Japanese Respiratory Society Guidelines for the Diagnosis and Treatment of COPD [updated 2009]. Available from: http://www.jrs.or.jp/. Accessed February 22, 2018. | ||
Cazzola M, Page CP, Calzetta L, Matera MG. Pharmacology and therapeutics of bronchodilators. Pharmacol Rev. 2012;64(3):450–504. | ||
Bonini M, Usmani OS. The importance of inhaler devices in the treatment of COPD. COPD Res Pract. 2015;1:9. | ||
Braido F, Chrystyn H, Baiardini I, et al; Respiratory Effectiveness Group. “Trying, but failing” – The role of inhaler technique and mode of delivery in respiratory medication adherence. J Allergy Clin Immunol Pract. 2016;4(5):823–832. | ||
Vehring R, Lechuga-Ballesteros D, Joshi V, Noga B, Dwivedi SK. Cosuspensions of microcrystals and engineered microparticles for uniform and efficient delivery of respiratory therapeutics from pressurized metered dose inhalers. Langmuir. 2012;28(42):15015–15023. | ||
AstraZeneca Pharmaceuticals LP. Bevespi Aerosphere™ Prescribing Information [updated 2017]. Available from: http://www.azpicentral.com/bevespi/bevespi_pi.pdf. Accessed February 5, 2018. | ||
Fabbri LM, Kerwin EM, Spangenthal S, et al. Dose-response to inhaled glycopyrrolate delivered with a novel Co-Suspension™ Delivery Technology metered dose inhaler (MDI) in patients with moderate-to-severe COPD. Respir Res. 2016;17(1):109. | ||
Tashkin DP, Martinez FJ, Rodriguez-Roisin R, et al. A multicenter, randomized, double-blind dose-ranging study of glycopyrrolate/formoterol fumarate fixed-dose combination metered dose inhaler compared to the monocomponents and open-label tiotropium dry powder inhaler in patients with moderate-to-severe COPD. Respir Med. 2016;120:16–24. | ||
Reisner C, Fabbri LM, Kerwin EM, et al. A randomized, seven-day study to assess the efficacy and safety of a glycopyrrolate/formoterol fumarate fixed-dose combination metered dose inhaler using novel Co-Suspension™ Delivery Technology in patients with moderate-to-very severe chronic obstructive pulmonary disease. Respir Res. 2017;18(1):8. | ||
Williams EJ. Experimental designs balanced for the estimation of residual effects of treatments. Aust J Sci Res B. 1949;2(2):149–168. | ||
Miller MR, Hankinson J, Brusasco V, et al; ATS/ERS Task Force. Standardisation of spirometry. Eur Respir J. 2005;26(2):319–338. | ||
Jones PW, Beeh KM, Chapman KR, Decramer M, Mahler DA, Wedzicha JA. Minimal clinically important differences in pharmacological trials. Am J Respir Crit Care Med. 2014;189(3):250–255. | ||
Reisner C, Orevillo C, Fernandez C, et al. Pooled analyses of five phase 2b studies support dose selection of glycopyrrolate-formoterol fumarate (GFF) MDI (PT003) 18/9.6 μg for phase III development. Eur Respir J. 2013;42(Suppl 57):P4153. | ||
Martinez FJ, Rabe KF, Ferguson GT, et al. Efficacy and safety of glycopyrrolate/formoterol metered dose inhaler formulated using co-suspension delivery technology in patients with COPD. Chest. 2017;151(2):340–357. | ||
Battisti WP, Wager E, Baltzer L, et al; International Society for Medical Publication Professionals. Good publication practice for communicating company-sponsored medical research: GPP3. Ann Intern Med. 2015;163(6):461–464. |
Supplementary material
 | Box S1 Institutional review boards (IRBs) |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.