")
Back to Journals » OncoTargets and Therapy » Volume 11
RACK1 induces chemotherapy resistance in esophageal carcinoma by upregulating the PI3K/AKT pathway and Bcl-2 expression
Authors Liu B, Wang C, Chen P, Cheng B, Cheng Y
Received 28 September 2017
Accepted for publication 28 November 2017
Published 4 January 2018 Volume 2018:11 Pages 211—220
DOI https://doi.org/10.2147/OTT.S152818
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ingrid Espinoza
Bowen Liu,1 Cong Wang,1 Pengxiang Chen,1 Bo Cheng,2 Yufeng Cheng1
1Department of Radiation Oncology, Qilu Hospital of Shandong University, Jinan, Shandong, People’s Republic of China; 2Department of Radiation Oncology, Shandong Provincial Cancer Hospital, Jinan, Shandong, People’s Republic of China
Introduction: Accumulating evidence indicates that RACK1 is involved in the progression of tumors. We aimed to evaluate the function of RACK1 in esophageal squamous cell carcinoma (ESCC) and its role in the mechanism of chemotherapy resistance.
Materials and methods: Transfected ESCC cell lines with plasmids expressed shRACK1 or open reading frame (ORF) targeting RACK1 and established stable cell lines. We then examined the effects of RACK1 on cell proliferation and chemotherapy resistance in ESCC cell lines, and the expression of AKT, pAKT, ERK1/2, Bcl-2, and Bim was introduced to further detect the association between RACK1 and chemotherapy resistance.
Results: The proliferation ability of ESCC cells was improved in the overexpression RACK1 groups (P<0.001) and decreased in the transfected shRACK1 groups (P<0.001) compared with the control ones. Meanwhile, upregulation of RACK1 significantly suppressed cisplatin-induced apoptosis in Eca109 and EC9706 cells, while downregulation of RACK1 promoted the sensitivity compared to the control group (Eca109: P<0.001 for shRACK1, P<0.01 for shNC, and P<0.001 for overexpression group; EC9706: P<0.001 for shRACK1, P<0.001 for shNC, and P<0.05 for overexpression group). Furthermore, we found that RACK1 could activate the PI3K/AKT pathway and increase the expression level of Bcl-2 in ESCC, which leads to the enhancement of chemoresistance in ESCC.
Conclusion: RACK1 promotes proliferation and chemotherapy resistance in ESCC by activating the PI3K/AKT pathway and upregulating the Bcl-2 expression.
Keywords: RACK1, ESCC, chemotherapy resistance, PI3K/AKT pathway, Bcl-2
Introduction
Esophageal cancer is one of the most common gastrointestinal malignant tumors with dismal prognosis, even when diagnosed in early stages. In Asia, esophageal squamous cell carcinoma (ESCC) occupies for ~90% of esophageal carcinomas.1 Although surgical techniques and perioperative treatments have been advanced, the prognosis of ESCC still remains poor on account of the local recurrence and distant metastasis.2 Most patients with ESCC are diagnosed at advanced or late stages, and can only receive chemotherapy, radiotherapy, and adjuvant treatments. However, only responders benefit from chemotherapeutic drugs, while resisters remain a major concern in the clinical applications. Therefore, it is imperative to discover the key markers and elementary mechanism affecting the chemotherapy response.
In 1989, RACK1 was first cloned from a chicken genomic DNA clusters and human B-lymphoblastoid cell line cDNA library,3 which was sharing high homology with the beta subunit of G-proteins (Gβ). RACK1 is a member of the tryptophan-aspartate (WD40) repeat protein family, which adopts a highly conserved 7-bladed beta-propeller structure.4,5 RACK1 plays an important role in anchoring proteins at particular locations, shuttling proteins from one cellular compartment to another, participating in transcriptional and translational events, and modulating binding protein activity.6 It serves as a scaffold protein that is able to interact simultaneously with many protein kinases and membrane-bound receptors, and it also plays a crucial role in a large quantity of biological processes, including cell growth, development, adhesion, migration, differentiation, and immune response.7 Increased RACK1 expression has been observed in various kinds of carcinomas and regarded as an independent biomarker of breast cancer,8,9 non-small-cell lung cancer (NSCLC),10,11 hepatocellular carcinoma (HCC),12 oral squamous carcinoma,13 and so on. We have already confirmed that RACK1 predicted poor prognosis in ESCC with promoting tumor progression and Epithelial-Mesenchymal Transition (EMT).14 It is confirmed that RACK1 can regulate several apoptosis-related genes and drug-related genes and then lead to the occurrence of chemotherapy resistance.15,16 Furthermore, recent study showed that the expression level of RACK1 in tumor tissue was correlated with the chemoresistance of HCC.12 In the current study, we aimed to evaluate the role of RACK1 in proliferation and its involvement in the underlying mechanism of chemotherapy resistance in ESCC.
Materials and methods
Cell culture and transfection
The human esophageal carcinoma cell lines Eca109 and EC9706 were purchased commercially from American Type Culture Collection (ATCC) (Manassas, VA, USA). Eca109 and EC9706 cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium with 10% fetal bovine serum, 100 μg/mL streptomycin, and 100 U/mL penicillin in a 5% CO2 atmosphere at 37°C.
Plasmid with shRNA, nonsense shRNA for negative control (shNC), or open reading frame (ORF) targeting RACK1 gene was transfected into Eca109 and EC9706 cell lines with Lipofectamine® 2000 Reagent (Thermo Fisher Scientific, Waltham, MA, USA). Cells were transfected when the plating cells were 70%–90% confluent and harvested for subsequent assays within 48–72 h after transfection. Stably transfected cells were selected with 400 μg/mL of G418 antibiotic for 2 weeks and maintained in 300 μg/mL of G418 culture medium.
shRNA with pGPU6/GFP/Neo plasmid (shRACK1) was generated by GenePharma (Shanghai, China). The sequence of shRNA targeting RACK1 gene was as follows: sense 5′-CACCGCATGTATGCATGTGACTTATTTCAAGAGAATAA GTCACATGCATACATGCTTTTTG-3′; antisense 5′-GATCCAAAAAAGCATGT ATGCATGTGACTTATTCTCTTGAAATAAGTCACATGCATACTGC-3′. The sequence of shNC was as follows: sense 5′-CACCGTTCTCCGAACGTGTCACGTTTCAAGAGAACGTG ACACGTTCGGAGAATTTTTTG-3′; antisense 5′-GATCCAAAAAATTCTCCGA ACGTGTCACGTTCTCTTGAAACGTGACACGTTCGGAGAAC-3′.
The overexpressed RACK1 was transfected with pEZ-M13/CMV/Neo/c-flag plasmid, which was generated by GeneCopoeia (Montgomery, MD, USA). The ORF targeting the human RACK1 (GNB2L1) gene sequence was as follows: forward 5′-GCGGTAGGCGTGTACGGT-3′; reverse 5′-GTGGCACCTTCCA GGGTC-3′.
Colony formation assay
Cells were trypsinized into single-cell suspensions with the treatment of 0.25% trypsin. Approximately 500 cells per well were plated into six-well plates with the density of 250/mL cells. After incubating cells in a humidified environment at 37°C containing 5% CO2 for 14 days, the cells were fixed in anhydrous ethanol for 10 min and then stained with 0.1% crystal violet for half an hour. Colonies consisting of >50 cells were counted, and relative colony numbers were obtained. The ability of colony forming was evaluated by the ratio comparing the number of colonies formed in transfected cells with that in control group, multiplied by a hundred.
Chemotherapy resistance assay
To assess the response to chemotherapy drugs, Eca109 and EC9706 cell lines were cultured in 96-well plates with the density of 1×104 cells/well and, then, cells were treated with a series of different concentrations of cisplatin (DDP) or fluorouracil (5-FU) for 48 or 72 h. The CCK-8 assay was used to detect the cell viability. Approximately 1×105 cells per well were plated into six-well plates with the density of 5×104/mL cells for 24 h, and then, they were treated with cisplatin (1–320 μmol/L) or 5-FU (1–10 μmol/L). After 24, 48, and 72 h, cells were harvested for detecting the expression of mRNA and protein changes.
Cell proliferation and cytotoxicity assay: CCK-8 assay
Cell proliferation was assessed with CCK-8 (Cell Counting Kit-8) assay. In brief, cells transfected with the shRACK1 plasmid, control plasmid, and overexpressed plasmid were seeded into 96-well culture plates. The plates were preincubated for 24 h in a humidified incubator. Then, cells were treated with different concentrations of cisplatin or 5-FU for 48 or 72 h. Spectrometric absorbance at the 450 nm wavelength was measured on a microplate reader (BioRad Laboratories Inc., Hercules, CA, USA) after incubation with 10 μL CCK-8 reagents for 1 h. The cell viability was calculated with the quantitative value multiplied by 100%. The proliferation rate was recorded based on the optical density (OD) value.
RNA extraction, reverse transcription, and real-time PCR
Total RNA was extracted from Eca109 and EC9706 cell lines with the TRIzol reagent (Thermo Fisher Scientific) following the manufacturer’s protocol. The extractive RNA was dissolved with the RNase-free water, and equal amounts of RNA (1 μg) from each sample were used for cDNA synthesis. Then, real-time PCR was applied with a Real-Time PCR System (Bio-Rad) to detect the expression of RACK1 at the mRNA level. Expression data were normalized to the geometric mean of the housekeeping gene β-actin. Specific primers for β-actin were as follows: forward primer (F) 5′-GATCATTGCTCCTCCTGAGC-3′ and reverse primer (R) 5′-ACTCCTGCTTGCTGATCCAC-3′. Specific primers for RACK1 were as follows: F 5′-TTCTCCTCTGACAACCGGCA-3′ and R 5′-GCCATCCTTGCCTCCAGAA-3′. The efficiency of overexpression and knockdown was also evaluated. All amplifications were performed in three parallel samples.
Western blotting
Cells were lysed by cold RIPA buffer, lysates were centrifuged at 12,000× g for 10 min at 4°C, and protein concentrations were determined using the bicinchoninic acid (BCA) Protein Quantification Assay Kit. The protein extracts were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and electrotransferred to polyvinylidene fluoride (PVDF) membranes. After blocking in 5% defatted milk for 2 h at room temperature, proteins on the membranes were incubated with the indicated primary antibody overnight at 4°C, followed by incubation with horseradish peroxidase-conjugated immunoglobulin G (IgG) for 2 h. Detection was performed using the reagents with an enhanced chemiluminescence reaction kit (Thermo Fisher Scientific). The relative band intensity was determined by the ImageJ 1.47v software (NIH, Bethesda, MD, USA). The primary antibodies (Abs) used in the Western blots were as follows: protein kinase B (AKT) (1:2,000 dilution, No 2920; Cell Signaling Technology, Danvers, MA, USA); Phospho-AKT (Ser473) (1:1,000 dilution, No 12694; Cell Signaling Technology); Phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204) (1:2,000 dilution, No 4370; Cell Signaling Technology); RACK1 (1:5,000 dilution, No 610177; BD Biosciences, San Jose, CA, USA); Bcl-2 (1:1,000 dilution, No 15071; Cell Signaling Technology); Bim (1:500 dilution, ab32158; Abcam, Cambridge, UK); and actin (1:1,000 dilution, sc-58673; Santa Cruz Biotechnology Inc., Dallas, TX, USA). Secondary Abs used for immunodetection were as follows: horseradish peroxidase-conjugated goat antimouse IgG and goat antirabbit IgG (BD Biosciences).
Cell apoptosis: flow cytometry analysis
Esophageal carcinoma cells were harvested and washed by phosphate buffer saline (PBS) twice and then incubated with phycoerythrin (PE) and 7-amino-actinomycin D (7-AAD) staining buffer at 25°C for 15 min. The apoptotic percentage of samples was determined on a BD FACSCalibur (BD Biosciences), and data were analyzed by the FlowJo software (Tree Star Inc., Ashland, KY, USA).
Statistical analysis
The SPSS 19.0 software (IBM Corporation, Armonk, NY, USA) was used to perform statistical analyses. The data were expressed as the mean ± SD and compared using analysis of variance (ANOVA). Statistical comparisons were made using Student’s t-test and one-way ANOVA. P-values <0.05 were considered statistically significantly different. All the experimental data were repeated for at least three independent tests.
Results
Upregulation and downregulation of RACK1 mRNA and protein levels by transfection in ESCC cell lines
To investigate the importance of RACK1 in ESCC, we initially constructed two transfected cell lines, transfected Eca109 and EC9706 cells with a plasmid expressing shRACK1 or shNC or RACK1-overexpression, and established stable cell lines as described by G418 selection. Quantitative real-time PCR confirmed the mRNA levels of RACK1 in the overexpression, control, shNC, and shRACK1 ESCC cell lines. It was upregulated in overexpression Eca109 cells (6.76±1.46, P=0.0209) and EC9706 cells (7.76±0.31, P=0.0007) compared to control cells. In contrast with control cells, mRNA expression levels of RACK1 in the shRACK1 cells decreased to 0.44±0.06 (P=0.0037) and 0.33±0.07 (P=0.0041). Moreover, RACK1 protein expression levels in Eca109 and EC9706 cells were detected by Western blot. The RACK1 expression level increased in both clone E109-overexpression and E9706-overexpression groups compared to the control cells. Additionally, after transfection by shRACK1, RACK1 protein level reduced to 65 and 44% compared to the control group in both Eca109 and EC9706 cells (Figure 1A and B).
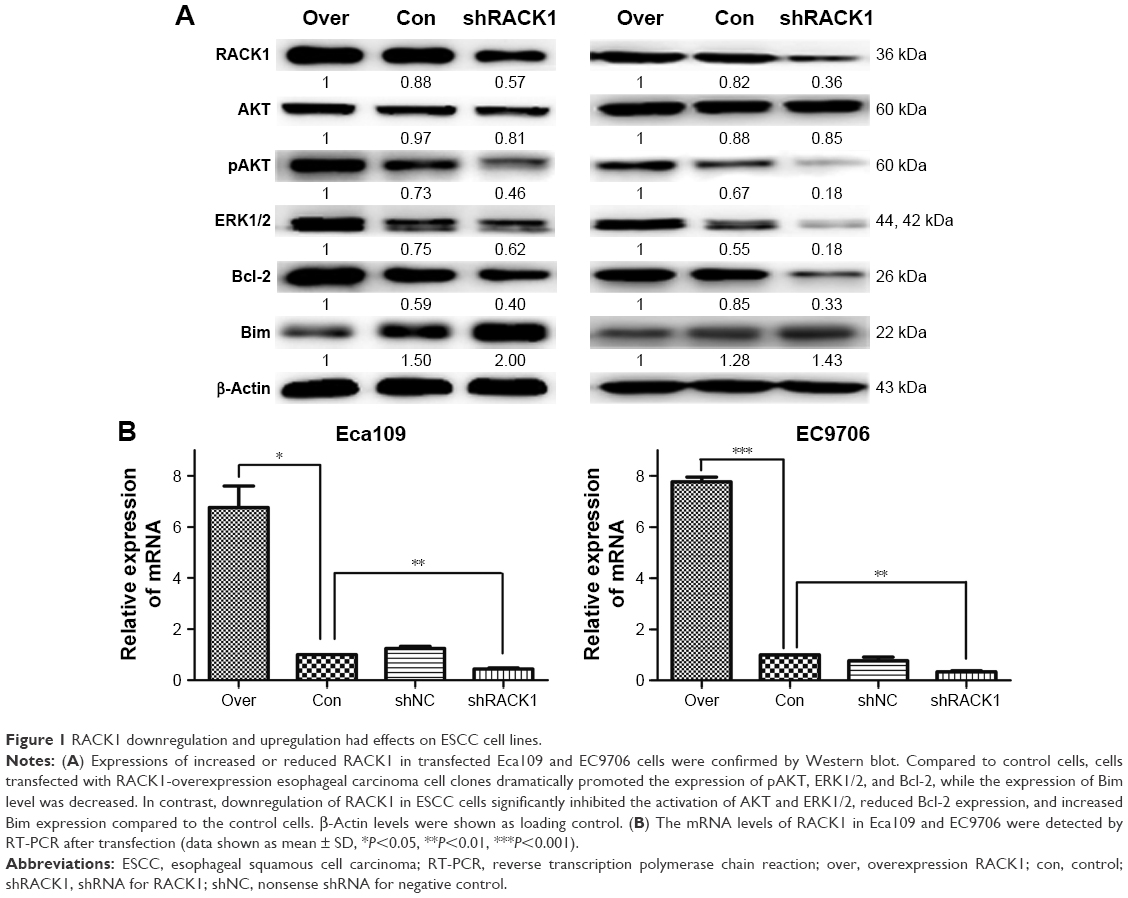 | Figure 1 RACK1 downregulation and upregulation had effects on ESCC cell lines. |
Downregulation of RACK1 inhibited cell colony formation ability of ESCC cells in vitro
To investigate the roles of RACK1 in the colony formation properties of esophageal carcinoma cells, we then conducted colony formation assay in vitro. The results showed that downregulation of RACK1 by shRACK1 transfection significantly suppressed cell proliferation in human esophageal carcinoma cells (Eca109: 254±11 vs 343±15, P=0.0013; EC9706: 187±23 vs 353±7, P=0.0003, Figure 2A and B). Moreover, the number of formation colony increased in clone E109-overexpression (393±14 vs 343±15, P=0.0148) and E9706-overexpression (369±12 vs 353±7, P=0.0280) groups compared with the control one (Figure 2A and B).
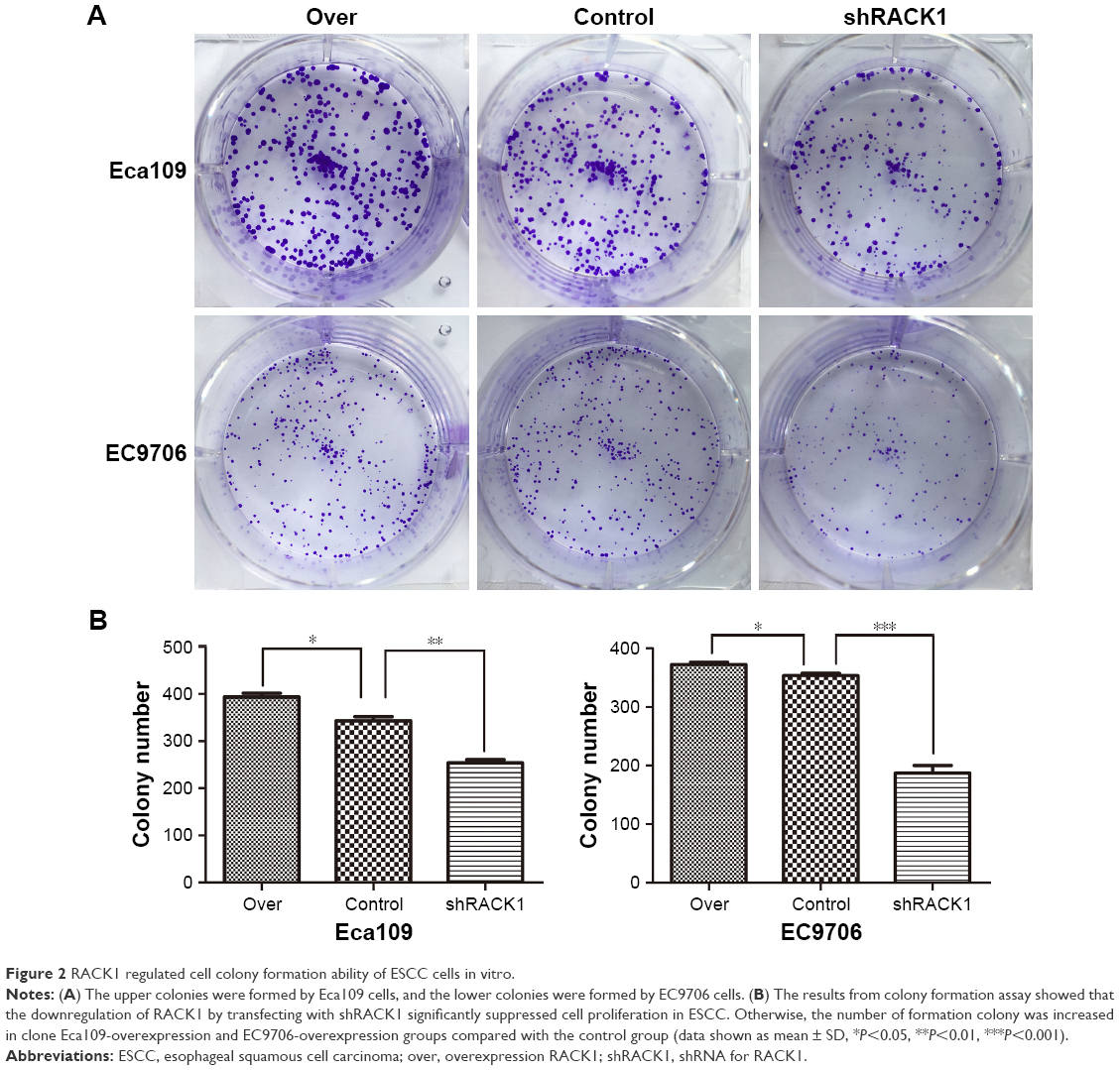 | Figure 2 RACK1 regulated cell colony formation ability of ESCC cells in vitro. |
RACK1 increased proliferation ability of esophageal carcinoma cells with the treatment of chemotherapy drugs
CCK-8 assay was conducted to validate the role of RACK1 on the proliferation ability of ESCC cells. Eca109 and EC9706 were exposed to different concentrations of cisplatin (0–320 μM) or 5-fluorouracil (Eca109, 0–10 μM; EC9706, 0–320 μM) for 48 and 72 h, respectively. The results showed that the proliferation ability of ESCC cells improved in the overexpression RACK1 groups (P<0.001) and decreased in the transfected shRACK1 groups (P<0.001) compared with the control group (Figure 3A and B).
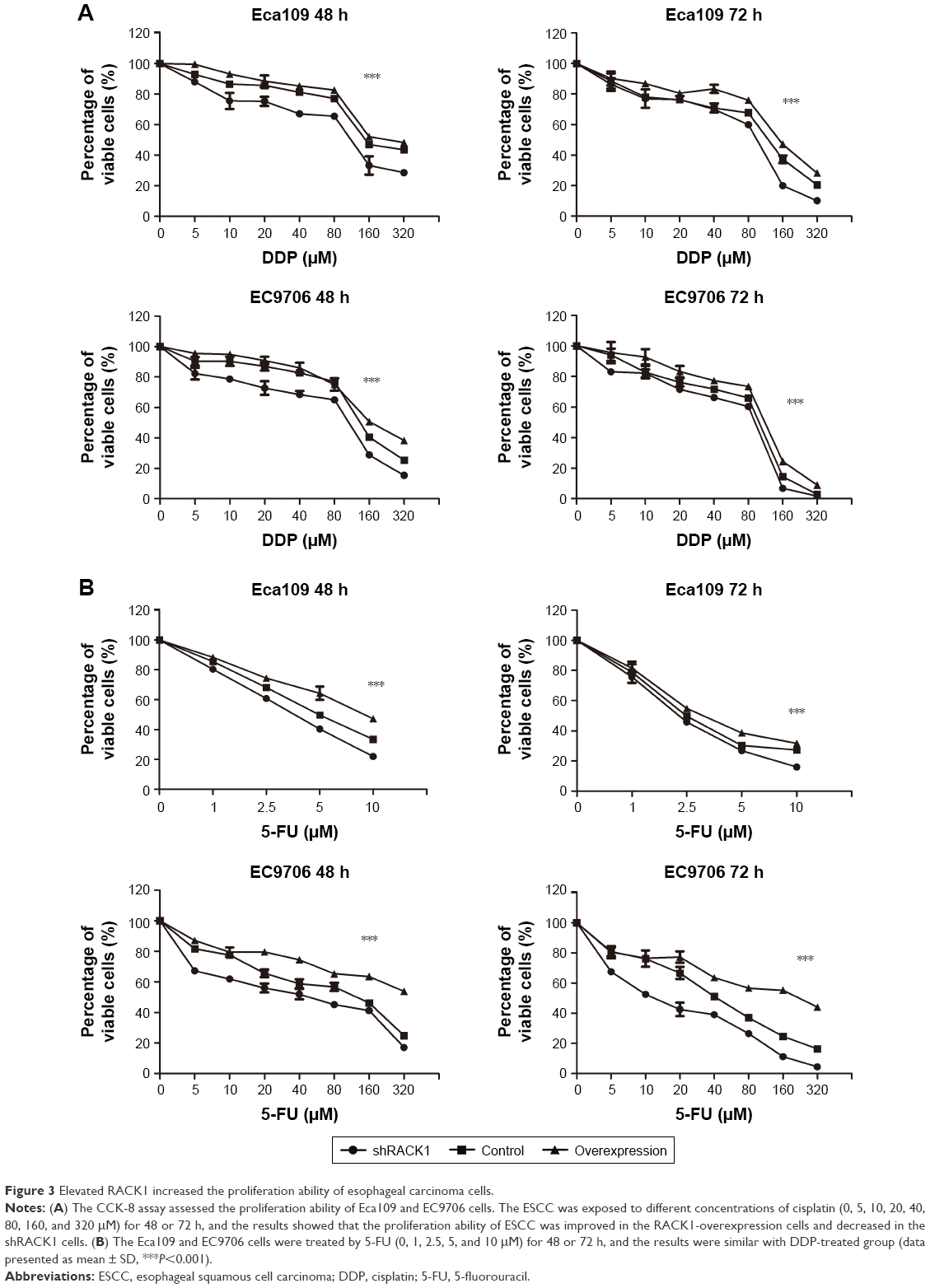 | Figure 3 Elevated RACK1 increased the proliferation ability of esophageal carcinoma cells. |
RACK1 regulated the chemosensitivity of ESCC in vitro
Since RACK1 was found to be capable of regulating the apoptotic response to chemotherapy in breast cancer and HCC, we then detected the impacts of RACK1 in the chemosensitivity of ESCC in vitro. As shown in Figure 4, upregulation of RACK1 significantly suppressed cisplatin-induced apoptosis in Eca109 and EC9706 cells, while downregulation of RACK1 promoted the sensitivity compared to the control group (Eca109: P<0.001 for shRACK1, P<0.01 for shNC, and P<0.001 for overexpression group; EC9706: P<0.001 for shRACK1, P<0.001 for shNC, and P<0.05 for over, respectively, Figure 4A). We observed the similar results in 5-fluorouracil-treated Eca109 and EC9706 cells (Eca109: P<0.001 for shRACK1, P<0.01 for shNC, and P<0.05 for over; EC9706: P<0.001 for shRACK1, P<0.001 for shNC, and P<0.05 for over, Figure 4B and C). These data indicate that RACK1 plays an important regulatory role in the chemosensitivity of ESCC in vitro.
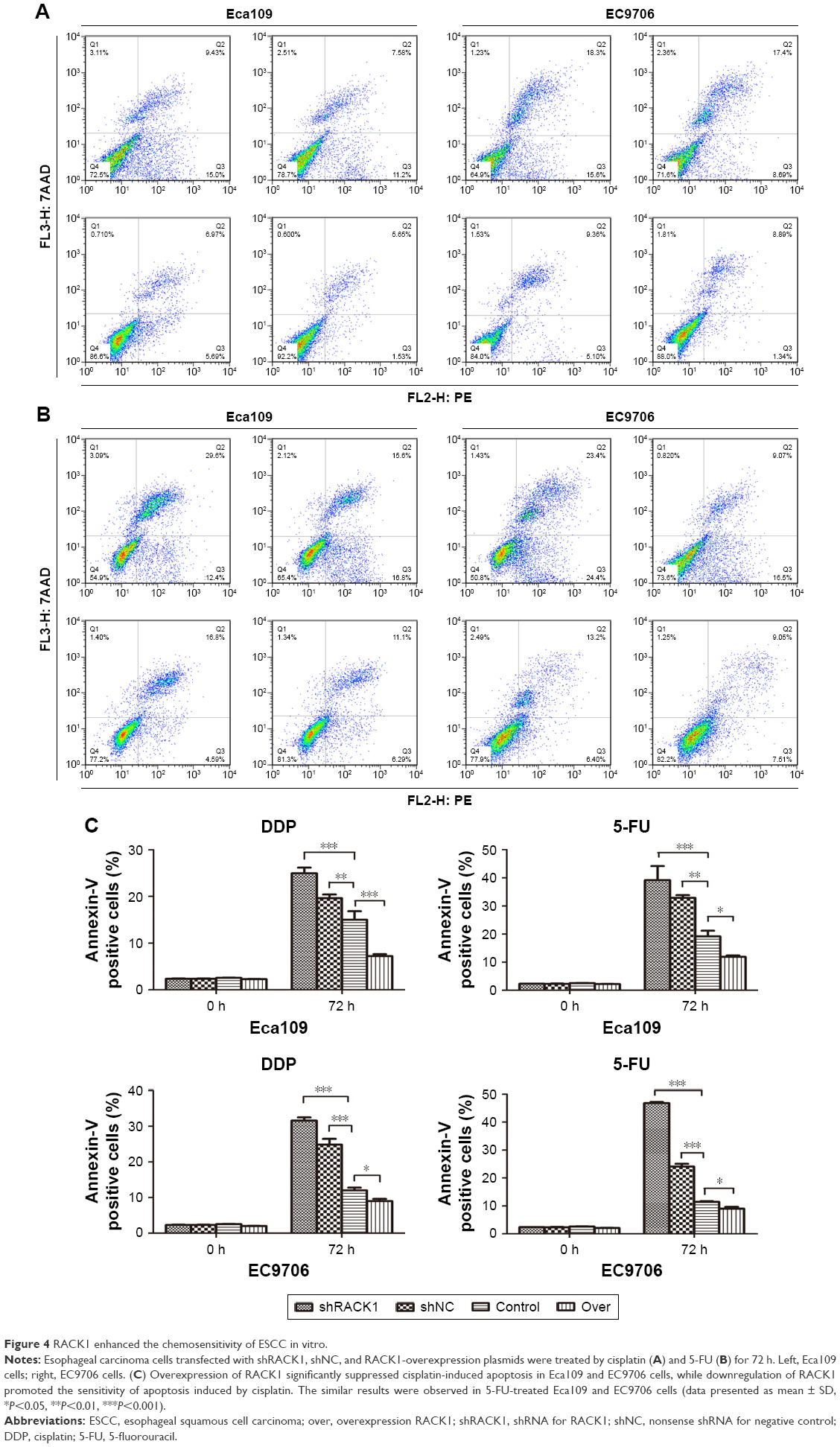 | Figure 4 RACK1 enhanced the chemosensitivity of ESCC in vitro. |
Elementary mechanism involved in the regulation of cell chemosensitivity by RACK1 in vitro
To explore the preliminary mechanism of RACK1 regulation on cell chemosensitivity in ESCC cells, in the present study, we focused on the AKT, ERK1/2, Bcl-2, and Bim molecules by Western blot (Figures 1A and 5). Our data found notable changes in the Phospho-AKT levels and expression levels of cell antiapoptotic protein Bcl-2 and proapoptotic protein Bim. Afterward, we examined the effects of cisplatin on the expression of AKT, pAKT, ERK1/2, Bcl-2, and Bim. As shown in Figure 1, downregulation of RACK1 in ESCC cells significantly inhibited the activation of AKT and ERK1/2, reduced Bcl-2 expression, and increased Bim expression compared to the control groups. In contrast, upregulation of RACK1 in Eca109 and EC9706 cells promoted the activation of AKT and ERK1/2, facilitated Bcl-2 expression, and suppressed Bim expression. After 24, 48, and 72 h treatment with cisplatin, proteins were extracted and analyzed by Western blot, respectively. Compared to control cells, we observed that esophageal carcinoma cells transfected by shRACK1 led to a significant suppression of expression levels of pAKT and Bcl-2, while the expression of Bim was observably increased. In addition, the expression level of Bim became increasingly higher with the extension of effect time. These results provided the clues of RACK1 targeting Bcl-2 and Bim to promote chemoresistance of the esophageal carcinoma cell through phosphoinositide 3-kinase (PI3K) pathway activation.
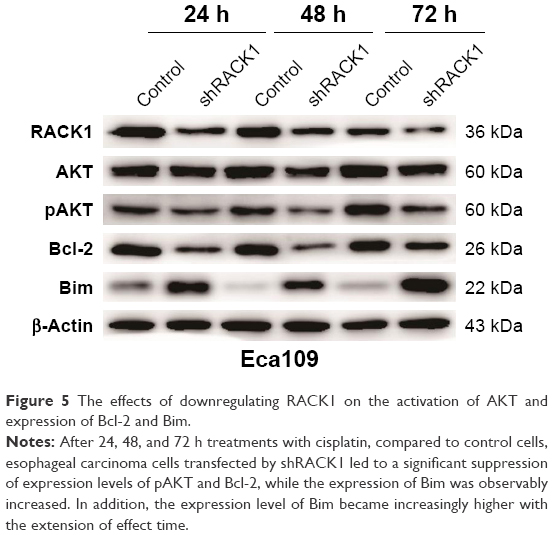 | Figure 5 The effects of downregulating RACK1 on the activation of AKT and expression of Bcl-2 and Bim. |
Discussion
RACK1 was first identified as a crucial anchoring protein for protein kinase C (PKC);17 it plays a pivotal role in numerous biological processes including cell growth, development, adhesion, migration, and tumor progression via interaction with different partners.18,19 Chemotherapy is a critical treatment strategy for esophageal carcinoma.20 We have already confirmed that RACK1 predicted poor prognosis in ESCC with promoting tumor progression and participating in tumor EMT.14 However, the role of RACK1 in the chemoresistance of ESCC cells and the underlying mechanism have not been uncovered. Recently, Ruan et al12 showed that aberrant expression of RACK1 involved in chemoresistance in vitro as well as tumor growth of HCC in vivo. Our research proved to be consistent with these previous results.
In the current study, we found that RACK1 promoted cell proliferation ability as an oncogenic gene in ESCC. Besides, overexpression of RACK1 resulted in the enhancement of the resistance to apoptosis induced by cisplatin or 5-FU, while downregulation of RACK1 enhanced the sensitivity to cisplatin or 5-FU-induced apoptosis. Our findings also showed that RACK1 could activate PI3K/AKT pathway and upregulate the expression of Bcl-2, which subsequently led to chemotherapy resistance. Inhibition of RACK1 largely abolished the chemoresistance. In brief, our findings show that RACK1 may be available as a marker of the chemotherapy resistance and poor survival in ESCC ultimately. Therefore, the treatment of RACK1 provides ways to improve the efficacy of chemotherapy in ESCC.
Analyzing the sophisticated molecular pathways regulating chemotherapy sensitivity and the correlation mechanism of chemoresistance is essential to discover new targeted agents, conquer chemoresistance, and improve outcome of chemotherapy. There is accumulating evidence showing that aberrant activation of the protein kinase B (AKT) pathway plays a pivotal role in tumor development, progression, and chemoresistance.21–23 RACK1 has been reported to be involved in the regulation of the activity of AKT.24–26 We detected the activation of critical members in the PI3K/AKT pathway and several cell apoptosis-related proteins through Western blot to investigate the underlying mechanism. In the current study, we found that overexpression of RACK1 significantly promoted the phosphorylation of AKT and, as well, it enhanced the phosphorylation of ERK. Moreover, we demonstrated that knockdown of RACK1 inhibited the phosphorylation of AKT and ERK in the Eca109 and EC9706 cells to a certain extent. In addition, apoptosis is a significant mechanism in chemotherapy-induced cell death. However, failure to initiate the apoptotic process represents a key mechanism of chemoresistance in tumor cells. RACK1 could exert antiapoptotic functions effectively via interaction with different partners.27,28 The expression of Bcl-2 could be upregulated by phosphorylating AKT. The balance among proteins of the Bcl-2 family plays an important role in the apoptotic pathways caused by several extraneous stimuli. Bcl-2 is an antiapoptotic member of the Bcl-2 family, while Bim is a proapoptotic protein belonging to Bcl-2 family.29,30 Furthermore, we found that the overexpression of RACK1 could facilitate the expression of Bcl-2, restrain the expression of Bim, and lead to the resistance to chemotherapy in ESCC. As mentioned earlier, our study is the first to uncover an oncogenic role of RACK1 and to illuminate the underlying mechanism of chemoresistance in ESCC. Thus, PI3K/AKT and Bcl-2 may be effective anticancer therapeutic targets in RACK1 overexpression-induced chemoresistance ESCC patients.
To our knowledge, this is the first comprehensive report concerning the role of RACK1 in chemotherapy resistance in ESCC. Although we have a more profound understanding about the impact of RACK1 in ESCC, it is possible that RACK1 may lead to the chemotherapy resistance through other mechanisms except for the PI3K/AKT and Bcl-2 pathways; further investigations are certainly necessary to understand the complete mechanism.
Conclusion
This present study not only provides new insights into the relationship between RACK1 and chemoresistance in ESCC but also demonstrates the key mechanism of the RACK1-induced chemoresistance. This research provides a promising chemoresistance therapeutic target for ESCC.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (81572958) and the Independent Innovation Project of Universities in Jinan City (201401252).
Disclosure
The authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Koshy M, Esiashvilli N, Landry JC, Thomas CR Jr, Matthews RH. Multiple management modalities in esophageal cancer: combined modality management approaches. Oncologist. 2004;9(2):147–159. | ||
Guillemot F, Billault A, Auffray C. Physical linkage of a guanine nucleotide-binding protein-related gene to the chicken major histocompatibility complex. Proc Natl Acad Sci U S A. 1989;86(12):4594–4598. | ||
Chang BY, Conroy KB, Machleder EM, Cartwright CA. RACK1, a receptor for activated C kinase and a homolog of the beta subunit of G proteins, inhibits activity of src tyrosine kinases and growth of NIH 3T3 cells. Mol Cell Biol. 1998;18(6):3245–3256. | ||
Ron D, Jiang Z, Yao L, Vagts A, Diamond I, Gordon A. Coordinated movement of RACK1 with activated betaIIPKC. J Biol Chem. 1999;274(38):27039–27046. | ||
Li JJ, Xie D. RACK1, a versatile hub in cancer. Oncogene. 2015;34(15):1890–1898. | ||
McCahill A, Warwicker J, Bolger GB, Houslay MD, Yarwood SJ. The RACK1 scaffold protein: a dynamic cog in cell response mechanisms. Mol Pharmacol. 2002;62(6):1261–1273. | ||
Cao XX, Xu JD, Liu XL, et al. RACK1: a superior independent predictor for poor clinical outcome in breast cancer. Int J Cancer. 2010;127(5):1172–1179. | ||
Cao XX, Xu JD, Xu JW, et al. RACK1 promotes breast carcinoma proliferation and invasion/metastasis in vitro and in vivo. Breast Cancer Res Treat. 2010;123(2):375–386. | ||
Shi S, Deng YZ, Zhao JS, et al. RACK1 promotes non-small-cell lung cancer tumorigenicity through activating sonic hedgehog signaling pathway. J Biol Chem. 2012;287(11):7845–7858. | ||
Clark AS, West KA, Blumberg PM, Dennis PA. Altered protein kinase C (PKC) isoforms in non-small cell lung cancer cells: PKCdelta promotes cellular survival and chemotherapeutic resistance. Cancer Res. 2003;63(4):780–786. | ||
Ruan Y, Sun L, Hao Y, et al. Ribosomal RACK1 promotes chemoresistance and growth in human hepatocellular carcinoma. J Clin Invest. 2012;122(7):2554–2566. | ||
Wang Z, Zhang B, Jiang L, et al. RACK1, an excellent predictor for poor clinical outcome in oral squamous carcinoma, similar to Ki67. Eur J Cancer. 2009;45(3):490–496. | ||
Wang N, Liu F, Cao F, et al. RACK1 predicts poor prognosis and regulates progression of esophageal squamous cell carcinoma through its epithelial-mesenchymal transition. Cancer Biol Ther. 2015;16(4):528–540. | ||
Ikebuchi Y, Ito K, Takada T, Anzai N, Kanai Y, Suzuki H. Receptor for activated C-kinase 1 regulates the cell surface expression and function of ATP binding cassette G2. Drug Metab Dispos. 2010;38(12):2320–2328. | ||
Ikebuchi Y, Takada T, Ito K, et al. Receptor for activated C-kinase 1 regulates the cellular localization and function of ABCB4. Hepatol Res. 2009;39(11):1091–1107. | ||
Ron D, Chen CH, Caldwell J, Jamieson L, Orr E, Mochly-Rosen D. Cloning of an intracellular receptor for protein kinase C: a homolog of the beta subunit of G proteins. Proc Natl Acad Sci U S A. 1994;91(3):839–843. | ||
Adams DR, Ron D, Kiely PA. RACK1, a multifaceted scaffolding protein: structure and function. Cell Commun Signal. 2011;9:22. | ||
Volta V, Beugnet A, Gallo S, et al. RACK1 depletion in a mouse model causes lethality, pigmentation deficits and reduction in protein synthesis efficiency. Cell Mol Life Sci. 2013;70(8):1439–1450. | ||
Nakajima M, Kato H. Treatment options for esophageal squamous cell carcinoma. Expert Opin Pharmacother. 2013;14(10):1345–1354. | ||
Li B, Li J, Xu WW, et al. Suppression of esophageal tumor growth and chemoresistance by directly targeting the PI3K/AKT pathway. Oncotarget. 2014;5(22):11576–11587. | ||
Falasca M. PI3K/Akt signalling pathway specific inhibitors: a novel strategy to sensitize cancer cells to anti-cancer drugs. Curr Pharm Des. 2010;16(12):1410–1416. | ||
LoPiccolo J, Blumenthal GM, Bernstein WB, Dennis PA. Targeting the PI3K/Akt/mTOR pathway: effective combinations and clinical considerations. Drug Resist Updat. 2008;11(1–2):32–50. | ||
Zhang X, Liu N, Ma D, et al. Receptor for activated C kinase 1 (RACK1) promotes the progression of OSCC via the AKT/mTOR pathway. Int J Oncol. 2016;49(2):539–548. | ||
Peng R, Jiang B, Ma J, et al. Forced downregulation of RACK1 inhibits glioma development by suppressing Src/Akt signaling activity. Oncol Rep. 2013;30(5):2195–2202. | ||
Wang F, Yamauchi M, Muramatsu M, Osawa T, Tsuchida R, Shibuya M. RACK1 regulates VEGF/Flt1-mediated cell migration via activation of a PI3K/Akt pathway. J Biol Chem. 2011;286(11):9097–9106. | ||
Subauste MC, Ventura-Holman T, Du L, et al. RACK1 downregulates levels of the pro-apoptotic protein Fem1b in apoptosis-resistant colon cancer cells. Cancer Biol Ther. 2009;8(23):2297–2305. | ||
Sang N, Severino A, Russo P, et al. RACK1 interacts with E1A and rescues E1A-induced yeast growth inhibition and mammalian cell apoptosis. J Biol Chem. 2001;276(29):27026–27033. | ||
Gomez-Bougie P, Bataille R, Amiot M. Endogenous association of Bim BH3-only protein with Mcl-1, Bcl-xL and Bcl-2 on mitochondria in human B cells. Eur J Immunol. 2005;35(3):971–976. | ||
Zhang W, Cheng GZ, Gong J, et al. RACK1 and CIS mediate the degradation of BimEL in cancer cells. J Biol Chem. 2008;283(24):16416–16426. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.