Back to Journals » Journal of Inflammation Research » Volume 19
R788 is Associated with Neuroprotective Effects in Experimental Ischemic Stroke Models via Modulation of the STAT1/Nrf2/GPX4 Axis
Authors Yang Y, Wang J, Zhou H, Jiang C, Zhu Z, Wang Y ![]()
Received 1 December 2025
Accepted for publication 19 May 2026
Published 25 June 2026 Volume 2026:19 580772
DOI https://doi.org/10.2147/JIR.S580772
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Dharmappa Krishnappa
Yong Yang,1,* Jing Wang,2,* Heng Zhou,3 Cailei Jiang,3 Zhansheng Zhu,1 Yamei Wang4
1Department of Neurosurgery, the First Affiliated Hospital of Yangtze University, the First People’s Hospital of Jingzhou, Jingzhou, Hubei, 434000, People’s Republic of China; 2Department of Health Management Centre, the First Affiliated Hospital of Yangtze University, the First People’s Hospital of Jingzhou, Jingzhou, Hubei, 434000, People’s Republic of China; 3Department of Stomatology and Regenerative Medicine, Renmin Hospital of Wuhan University, Wuhan, Hubei, 430000, People’s Republic of China; 4Department of Neurology, the First Affiliated Hospital of Yangtze University, the First People’s Hospital of Jingzhou, Jingzhou, Hubei, 434000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zhansheng Zhu; Yamei Wang, Email [email protected]; [email protected]
Background: Ischemic stroke, the most prevalent form of cerebrovascular disease, stands as a leading global cause of both disability and mortality. Oxidative stress damage is a critical pathological mechanism in stroke, and regulated cell death may be an effective therapeutic target for treating ischemic stroke. This work provides the first evidence that R788, a small-molecule inhibitor of spleen tyrosine kinase, exerts therapeutic efficacy in ischemic stroke, thereby unveiling a novel mechanistic basis and potential strategy for intervention.
Methods: The antioxidant defense and anti-apoptotic effects of R788 were assessed in a middle cerebral artery occlusion (MCAO) animal model and an H2O2-induced SH-SY5Y cell model of IS. A total of 24 male C57BL/6 mice were randomly divided into 4 groups (n=6). Then we measured the intracellular Fe2+ concentration and lipid peroxidation levels, and the expression of the STAT1/Nrf2/GPX4 pathway was evaluated in the MCAO animal model and the cell model.
Results: The results showed that both in the animal and cell models, the R788-treated group had an increased number of surviving SH-SY5Y cells compared to the control group. Additionally, the R788 group showed the inhibition of p-STAT1 and suppressed ferroptosis. We confirmed that R788 treatment alleviated oxidative stress damage by inhibiting ferroptosis-related pathways, thereby promoting cell survival. In summary, R788 demonstrated neuroprotective effects against ischemia-induced neural injury in both in vivo and in vitro models.
Conclusion: Our workss suggest that R788 alleviate localized ischemic injury and oxidative stress damage by regulating STAT1/Nrf2/GPX4 pathways. These findings indicate that R788 shows potential for the treatment of IS.
Keywords: R788, ischemic stroke, ferroptosis, STAT1
Introduction
Ischemic stroke is an acute cerebrovascular disease in clinical practice, seriously threatening human survival and health. It is primarily categorized into two subtypes: ischemic and hemorrhagic stroke.1,2 Current evidences indicate that ischemic stroke constitutes 85% of stroke cases.3 It is caused by the narrowing or occlusion of cerebral arteries, leading to insufficient blood supply to the brain. The main manifestations are cerebral blood circulation disorders, localized necrosis or softening of brain tissue, and neurological dysfunction resulting from ischemia and hypoxia. Rep reperfusion treatment with medication or surgery remains the most effective.4 However, the limited time window, coupled with low recanalization rates and numerous thrombolysis contraindications,5 restricts the clinical application of reperfusion therapy. In addition, the sudden increase in reactive oxygen species (ROS) and the accumulation of lipid peroxides upon reperfusion of ischemic tissue can lead to tissue damage and exacerbation of inflammatory responses.6,7 These functional and structural changes are called ischemia/reperfusion injury (IRI).8 However, due to the unclear molecular mechanisms of neuronal damage following ischemic stroke, a cure has not yet been achieved. Therefore, finding potential effective therapeutic targets and related mechanisms is crucial for reducing stroke-related damage.5
IRI in the brain mainly involves pathophysiological processes such as increased reactive oxygen species (ROS), intracellular calcium overload, and excessive activation of inflammatory responses.9–11 These physiological processes are often closely related to ferroptosis.12–14 Ferroptosis, a non-apoptotic form of programmed cell death, is triggered by an over-accumulation of lipid hydroperoxides.1,15 Ferroptosis is defined by its dependence on iron, depletion of glutathione (GSH), inactivation of glutathione peroxidase 4 (GPX4), and abnormalities in mitochondrial structure. The interplay between amino acid, lipid, and iron metabolism plays a pivotal role in initiating and executing this process. Ferroptosis frequently damages to the intracellular antioxidant system, leading to cellular dysfunction.16,17 In ischemic stroke, ferroptosis exhibit a reciprocal relationship, collectively amplifying the production of reactive oxygen species (ROS). Elevated ROS levels, in turn, perpetuate the cycle by further promoting ferroptosis.18,19 Traditional neuroprotective strategies have largely failed in clinical trials, likely due to their inability to address this multifaceted pathology. Targeting ferroptosis offers a unique advantage by simultaneously curbing ROS accumulation and mitigating ischemia-reperfusion–induced cellular damage, representing a promising avenue for more effective stroke therapy.
R788 (Fostamatinib) is an oral spleen tyrosine kinase (SYK) inhibitor metabolized to the active compound R406, which blocks SYK-mediated immune signaling.20 Originally approved for chronic immune thrombocytopenia (ITP), it also shows promise in autoimmune, inflammatory, and neoplastic diseases.21–23 Platelet-mediated SYK signaling has been implicated in I/R-induced distal lung injury, and R788 administration significantly attenuates platelet activation and pulmonary damage.24 Moreover, R788 reduces MUC1 expression in I/R-related lung injury models, suggesting benefits against ALI/ARDS.25 SYK also aggravates hepatic I/R injury by driving PKM2–STAT3 signaling, NET formation, and NLRP3 inflammasome activation, highlighting SYK inhibition as a potential therapeutic strategy.26 Despite progress in other areas, the therapeutic potential of R788 in neurological disorders remains largely unexplored. SYK is expressed in CNS cells—including microglia, astrocytes, and neurons—but its role in neuroinflammation, ischemic injury, and neurodegeneration is unclear. Few studies have addressed whether SYK inhibition modulates blood–brain barrier integrity, microglial activation, or neuronal survival. To fill this gap, we examined the neuroprotective effects and underlying mechanisms of R788 in IS.
In this study, the role of R788 on blocking to ferroptosis was firstly assessed in vivo and in vitro models. It was confirmed that R788 dose-dependently reduced neuronal loss in ischemic brain regions, shrank cerebral infarct volume, lowered neurological deficit scores, and alleviated oxidative stress damage. In vitro, with the H2O2-induced SH-SY5Y cell injury model, DHE/C11-BODIPY staining, and Fe2⁺ detection showed R788 scavenged intracellular ROS, inhibited lipid peroxidation and iron accumulation, while upregulating anti-apoptotic protein Bcl2, downregulating pro-apoptotic proteins Bax/C-caspase3, and improving cell viability. Further mechanistic studies revealed R788 inhibited STAT1 phosphorylation, relieved its inhibitory effect on Nrf2 transcription, upregulated GPX4, and ultimately blocked ferroptosis. This study not only confirms R788’s neuroprotective effect on ischemic stroke for the first time but also uncovers its new mechanism of intervening ferroptosis via the STAT1/Nrf2/GPX4 pathway, providing a new potential drug target and intervention strategy for ischemic stroke treatment. However, our findings are at the preclinical stage and will need to be validated in subsequent clinical studies.
Materials and Methods
Animals and Ethics Statement
This study strictly followed the ARRIVE 2.0 guidelines (Animal Research: Reporting of In Vivo Experiments) for experimental design and reporting. All animal procedures were approved by the Animal Ethics Committee of the First People Hospital of Jingzhou (approval number: LL2024122) and conducted under animal license SYXK2022-0061. A total of 24 male C57BL/6 mice (wild-type, 9weeks old, 22–26 g) were obtained from Bestcell Model Animal Center (Wuhan, China). Animals were housed under standard conditions(temperature 22± 3°C, humidity 60± 5%, 12-h light/dark cycle)with ad libitum access to food and water. All surgical procedures were performed under appropriate anesthesia with meticulous care to minimize suffering, following the 3R principles (Replacement, Reduction, Refinement).
Middle Cerebral Artery Occlusion (MCAO) and Drug Treatments
A total of 24 male C57BL/6 mice were randomly assigned to four experimental groups (n=6 per group) using a computer-generated randomization sequence to minimize selection bias. All experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) and performed in accordance with the ARRIVE guidelines and AVMA Guidelines for the Euthanasia of Animals. Mice were anesthetized via intraperitoneal administration of sodium pentobarbital (30 mg/kg, i.p). with physiological parameters strictly monitored. Body temperature was continuously maintained at 36.5–37.5°C using a homeothermic heating pad (Harvard Apparatus, Model 50–7221). Through a midline cervical incision, the right common carotid artery (CCA), external carotid artery (ECA), and internal carotid artery (ICA) were surgically exposed and meticulously isolated. Hemostasis was achieved through bipolar electrocoagulation of the superior thyroid and occipital arteries.
Focal cerebral ischemia was established using a modified Longa method of transient middle cerebral artery occlusion (MCAO). Briefly, transient occlusion was induced by advancing a 0.24-mm-diameter monofilament nylon suture from the ICA bifurcation to obstruct the origin of the middle cerebral artery (MCA). After a 60-minute occlusion period, the nylon suture was carefully removed to facilitate blood reperfusion. Mice in the control group underwent the same surgical procedure but without the insertion of the nylon suture. Throughout the entire surgical process, the body temperature of the mice was strictly maintained at 37.0±0.5°C to mimic physiological conditions.
The experimental design comprised four cohorts (n=6 per group): 1. Sham operation (The surgical area was exposed and sutured) + PBS (100 μL, i.p).; 2. MCAO model + PBS (100 μL, i.p).; 3. MCAO + R788 (7.5 mg/kg, i.p).; 4. MCAO + R788 (15 mg/kg, i.p). R788 (MCE, HY-13038A, purity >98%, USA) was administered immediately post-reperfusion via intraperitoneal injection. All surgical interventions were conducted under aseptic conditions with postoperative analgesia provided.
All sacrifices were performed 5 days after surgery. After the mice were anesthetized, they were perfused with 10 mL of PBS followed by 10 mL of 4% paraformaldehyde (PFA, Sigma-Aldrich, St. Louis, MO, USA). Subsequently, the tissues were harvested and fixed for subsequent experiments.
Euthanasia at the experimental endpoint: Mice were induced to a deep anesthesia level with 3–5% isoflurane (Baxter), followed by administration of an overdose of pentobarbital sodium (150 mg/kg, intraperitoneally; Dorminal®) for euthanasia. All anesthesia and euthanasia procedures were performed in accordance with the ARRIVE guidelines and the AVMA Guidelines for the Euthanasia of Animals.
Hematoxylin Eosin (H&E) Staining and Nissl Staining
After deparaffinizing, the sections were stained using a H&E staining kit, observed under a light microscope, and the resulting images were captured for further analysis.
For Nissl staining, the sections were immersed in Nissl staining solution and incubated at 37°C for 3 minutes. Subsequently, they were rinsed with distilled water and then treated with 95% ethanol to remove excess stain. After dehydration and clearing processes, the slides were mounted for observation. The stained sections obtained were examined and photographed using a light microscope to acquire visual data. ImageJ 1.52a counts the number of Nis-positive neurons per unit area. Actual field of view diameter (mm) = Eyepiece field (FN, mm)/Objective magnification
Neurological Scoring
Neurological deficit scores were evaluated by two independent, trained investigators who were blinded to group allocation at 24 hours after reperfusion, according to a modified 5‑point scale, as previously described.25 The scoring system was as follows:
0 points: Normal neurological function;
1 point: Mild neurological dysfunction (left front leg flexion when tail is raised);
2 points: Moderate neurological dysfunction (turning to the left when walking);
3 points: Moderate neurological dysfunction (leaning to the left);
4 points: No spontaneous walking, decreased consciousness;
5 points: Death related to ischemia.
Infarct Volume Measurement
After anesthetizing the mice, their brains were promptly extracted and sectioned into 2-mm slices. These fresh slices were stained with 2% 2,3,5-triphenyltetrazolium chloride (TTC; Sigma-Aldrich, St. Louis, MO, USA) at 37°C for 15 minutes, followed by fixation in 4% formaldehyde at 4°C overnight. Post-TTC staining, red regions represented non-infarcted tissue, while white regions indicated infarcted areas. Red areas indicated viable tissue, and white areas represented infarcted regions.Blinded investigators manually outlined infarct areas, and quantitative analysis was performed using ImageJ 1.52a. Infarct volume percentage was calculated as:Cerebral infarct volume (%) = (Infarct volume / Contralateral hemisphere volume) × 100%.
Immunohistochemical Staining
We fixed mouse brain tissue samples with formaldehyde fixative, sliced the paraffin-embedded tissue into 4–6μm thin sections, and sequentially immersed the slides on the glass in xylene and a series of decreasing concentration alcohol solutions to remove paraffin and rehydrate the samples. The slides were incubated at 4°C overnight with the primary antibody solution and washed with PBS. The slides were incubated with the secondary antibody labeled with HRP for 2 hours, and the substrate solution was added. All IHC image acquisition and quantitative analyses (positive cell density and integrated optical density) were performed by blinded investigators using ImageJ 1.52a to avoid observer bias.
Measurement of MDA, LDH, and GSH Activities
After transcardial perfusion with ice-cold saline (4 °C), brains were carefully excised, and the tissues were homogenized in phosphate-buffered saline (PBS). The enzymatic activities of LDH, GSH, and MDA were subsequently quantified using commercially available assay kits (Nanjing Jiancheng Corp., China).
SH-SY5Y Cell Culture and Drug Treatment
SH-SY5Y cells were obtained from Wuhan Procell Biotechnology Co., Ltd. (Wuhan, China) and cultured in DMEM/F12 medium (Thermo Fisher Scientific, USA) supplemented with 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA, USA) under standard conditions (37°C, 5% CO2). Cells were seeded into 24-well plates at a density of 1×105 cells/well. For pretreatment, cells were exposed to 100μM hydrogen peroxide (H2O2) for 12 hours, while control cells received an equivalent volume of physiological saline. Following pretreatment, the culture medium of the experimental groups was replaced with phosphate-buffered saline (PBS) containing R788 at concentrations of 0.25μM, 0.5μM, or 1μM for 24 hours. Afterward, we cultured cells for 12h and then collected them for analysis. To ensure the accuracy of the results, the in vitro experiment was repeated three times.
Measurement of Intracellular Fe2⁺ Levels
Intracellular Fe2⁺ was detected using FerroOrange (1µM, Dojindo, Japan). Cells were incubated with the respective probes at 37 °C for 30 min, washed with PBS, and fluorescence intensity was measured using a microplate reader.
Oxidative Stress and Lipid Peroxidation Assays
Intracellular ROS production was assessed using dihydroethidium (DHE, Sigma-Aldrich, USA). Cells were incubated with 5µM DHE in serum-free medium at 37 °C for 30 min in the dark, washed three times with PBS, and fluorescence signals were detected using a fluorescence microscope (Ex 518 nm/Em 605 nm). Mean fluorescence intensity (MFI) was quantified with ImageJ software.
Lipid peroxidation was evaluated using C11-BODIPY 581/591 (Thermo Fisher Scientific, USA). Cells were loaded with 2µM probe in serum-free medium at 37 °C for 30 min in the dark and washed twice with PBS. Images were acquired with a confocal microscope, and the fluorescence intensity was calculated using ImageJ. All assays were performed under light-protected conditions.
Real Time qPCR
Total RNA was extracted using TRIzol kit (15596–018, Beijing Solarbio Technology Co., Ltd., Beijing, China) according to the instructions, and then RNA concentration was determined by Nanodrop. First-strand complementary DNA (cDNA) was synthesized from total RNA using the SuperScript™ III First-Strand Synthesis System for RT-PCR (Thermo Fisher Scientific, USA). Relative gene expression levels were determined using the ΔCt method, with GAPDH employed as the internal reference gene. The detailed primers were presented in Table S1 Each experiment was repeated three times. The above steps are also applicable to animal experiments.
Transfection of siRNA and Plasmid
Cells were seeded into six-well plates and cultured to 50–60% confluency prior to transfection. Transfection procedures were performed using Lipofectamine™ 2000 transfection reagent (Invitrogen, USA) according to the manufacturer’s specifications, with either small interfering RNA (siRNA) or plasmid constructs. The full-length STAT1 coding sequences were subcloned into pcDNA3.1 expression vectors (constructed by Qinke Bioteck, China). For siRNA transfection experiments, a final concentration of 100 nM was maintained throughout the procedure. Following a 6-hour transfection period, the transfection medium was replaced with fresh complete growth medium. R788 (R788) treatment was then initiated and maintained for specified durations as outlined in experimental designs. Cellular samples from both treatment and control groups were collected for subsequent analytical processing. The si-RNA sequences were given (Table S2).
Dual Luciferase Reporter Gene Assay
To assess the transcriptional activity of STAT1, a luciferase reporter assay was conducted using SH-SY5Y cells. Cells were seeded in 24-well plates at a density of 1×105 cells per well and cultured overnight under standard conditions (37 °C, 5% CO2). The following day, cells were co-transfected with a firefly luciferase reporter plasmid pGL3-NRF2 (Table S3), and a Renilla luciferase plasmid (pRL-TK) as an internal control to normalize for transfection efficiency. For NRF2 biding sites detection, the promoter sequences were shown in Table S4. Transfection was performed using Lipofectamine™ 3000 in accordance with the manufacturer’s instructions. After 24–48 h post-transfection, cells were harvested and lysed using Passive Lysis Buffer (Promega). Luciferase activity was measured using the Dual-Luciferase® Reporter Assay System (Promega) on a microplate luminometer (eg., GloMax® Discover, Promega). The firefly luciferase activity was normalized to the corresponding Renilla luciferase activity to account for variation in transfection efficiency and cell viability.
All experiments were performed in triplicate and repeated independently at least three times. Data were presented as mean ± standard deviation (SD) of relative luciferase units (RLU). Statistical significance was determined as described in the Data Analysis section.
Chromatin Immunoprecipitation (CHIP) Assay
A total of 107 SH-SY5Y cells, either with or without STAT1 overexpression, were initially crosslinked with 1% formaldehyde for 10 minutes to preserve protein-DNA interactions. The crosslinking reaction was subsequently quenched by incubation with 0.2 g glycine for 5 minutes. Cells are lysed, and nuclei are isolated to extract chromatin. The DNA-protein complexes are fragmented into smaller (100 to 300 bp) pieces, typically by sonication (230 W, 2 × 5-second pulses over 15 minutes). The resulting supernatant was reserved for pre-clearing.
Lysates were incubated overnight at 4 °C with either anti-STAT1 or anti-FLAG antibodies, with normal IgG serving as a negative control. On the following day, Protein A/G magnetic beads were added to capture the immune complexes, followed by a 30-minute incubation at room temperature. The complexes were subsequently washed with standard wash buffer and TE buffer to remove nonspecific bindings. After elution and reversal of crosslinking, DNA was precipitated and purified for downstream analysis. Quantitative PCR (qPCR) was performed to assess the enrichment of target genomic regions (Table S5).
Western Blot Analysis
The injury site tissues and cultured cells (1×107 /group) were lysed, and each group were quantified using the BCA assay (Beyotime, China) to ensure equal protein loading. then separated of the protein samples using 6%–12% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis. These samples were then transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA, USA). Next, the membranes were incubated overnight at 4°C with the corresponding primary antibodies. After a 1-hour blocking step, the membranes were incubated with secondary antibodies. The immunoreactive bands were detected using enhanced chemiluminescence (ECL) reagents. The primary antibodies utilized are detailed in Table S6. Western blot band quantification and densitometric analysis were performed by independent investigators blinded to group allocation using ImageJ 1.52a software. Target protein expression was normalized to β‑actin or GAPDH.
Statistical Analysis
All statistical analyses were conducted using SPSS 22.0 (IBM Corp., Armonk, NY, USA). Quantitative data were expressed as mean ± standard deviation (SD). For comparisons among multiple groups, one-way analysis of variance (ANOVA) was employed, followed by Bonferroni post hoc tests for pairwise multiple comparisons to control for the family-wise error rate and mitigate the risk of Type I error. Statistical significance was defined as a two-tailed P value < 0.05.
Results
R788 Alleviates Oxidative Stress Damage and Apoptosis in vitro
As shown in Figure 1A, the chemical structural formula of R788 was presented. First, we determined the optimal concentration of R788 in the SH-SY5Y cell model and found that cell viability was highest at concentrations ranging from 0.25 to 1μM. Therefore, the gradient of 0, 0.25, 0.5, and 1 μM was selected for subsequent experiments (Figure 1B). The IC50 was accessed as 5.37 μM. DHE and C11-BODIPY staining revealed that hydrogen peroxide treatment induced a significant increase in intracellular ROS and lipid peroxidation, whereas R788 treatment reduced the fluorescence intensity of both probes, indicating effective clearance of harmful ROS and lipid peroxides (Figure 1C–E). Upon exposure to 100 μM H2O2, SH-SY5Y cells exhibited extensive cell death, whereas treatment with R788 (0.25, 0.5, 1μM) markedly attenuated cell death, with the most pronounced protective effect observed at 1μM (Figure 1F). These findings suggest that R788 enhances intracellular antioxidant activity, alleviates oxidative stress, and reduces H2O2–induced oxidative damage.
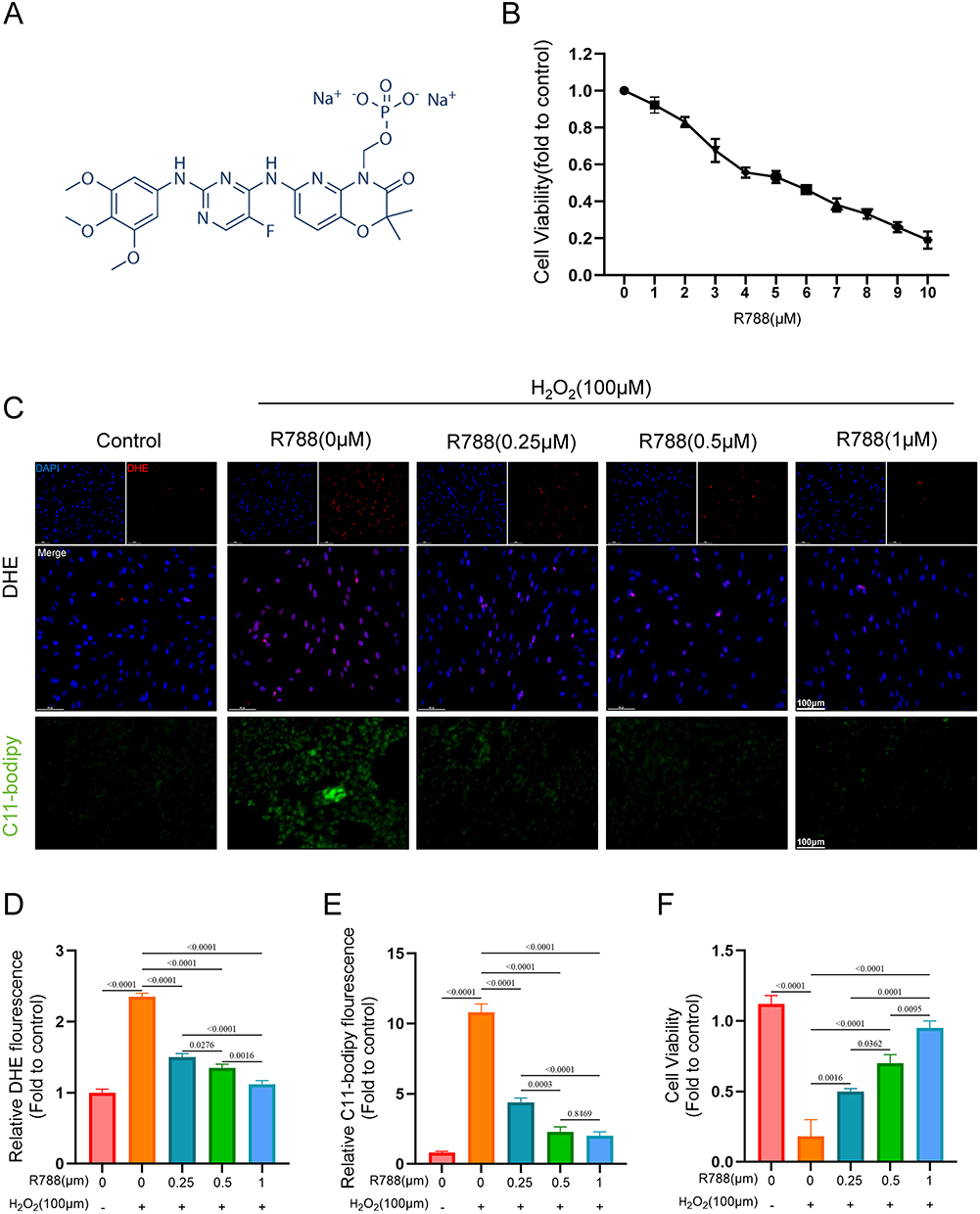 |
Figure 1 Biological performance testing of R788. (A) Source and chemical structure of R788. (B) Screening of the optimal concentration of R788 for SH-SY5Y cells. (C–E) Detection of DHE and C11-bodipy, fluorescence microscope images and fluorescence intensity per unit area analysis. The data are presented as the mean ± SD (n = 6). Scale bar:50μm. (F) Cell viability test of SH-SY5Y cells (Exposure time: 60min, 37°C, 5% CO2). The data are presented as the mean ± SD (n = 6). |
R788 Ameliorated Iron Accumulation and Ferroptosis in SH-SY5Y Cells
To evaluate whether R788 could alleviate the ferroptosis on, the SH-SY5Y cells were co-treated with RSL3 (a ferroptosis activator) and R788. As shown in Figure 2A, RSL3 treatment significantly increased the expression of nuclear factor kappa B (NF-κB), while suppressing the expressions of glutathione peroxidase 4 (GPX4) and nuclear factor erythroid 2-related factor 2 (Nrf2). With the increase in R788 concentration, this trend of protein expression was reversed. Furthermore, intracellular Fe2⁺, GSH, LDH, and MDA levels were measured, demonstrating that R788 treatment decreased Fe2⁺ (Figure 2B), LDH (Figure 2C), and MDA (Figure 2E) concentrations while increasing GSH levels (Figure 2D). Calcein-AM staining showed a marked loss of mitochondrial fluorescence under stress conditions, indicating MPTP opening, whereas R788 treatment preserved calcein signals, suggesting inhibition of MPTP opening (Figure 2F and G). Taken together, these data suggest that R788 may protect neurons from RSL3-induced injury by inhibiting ferroptosis.
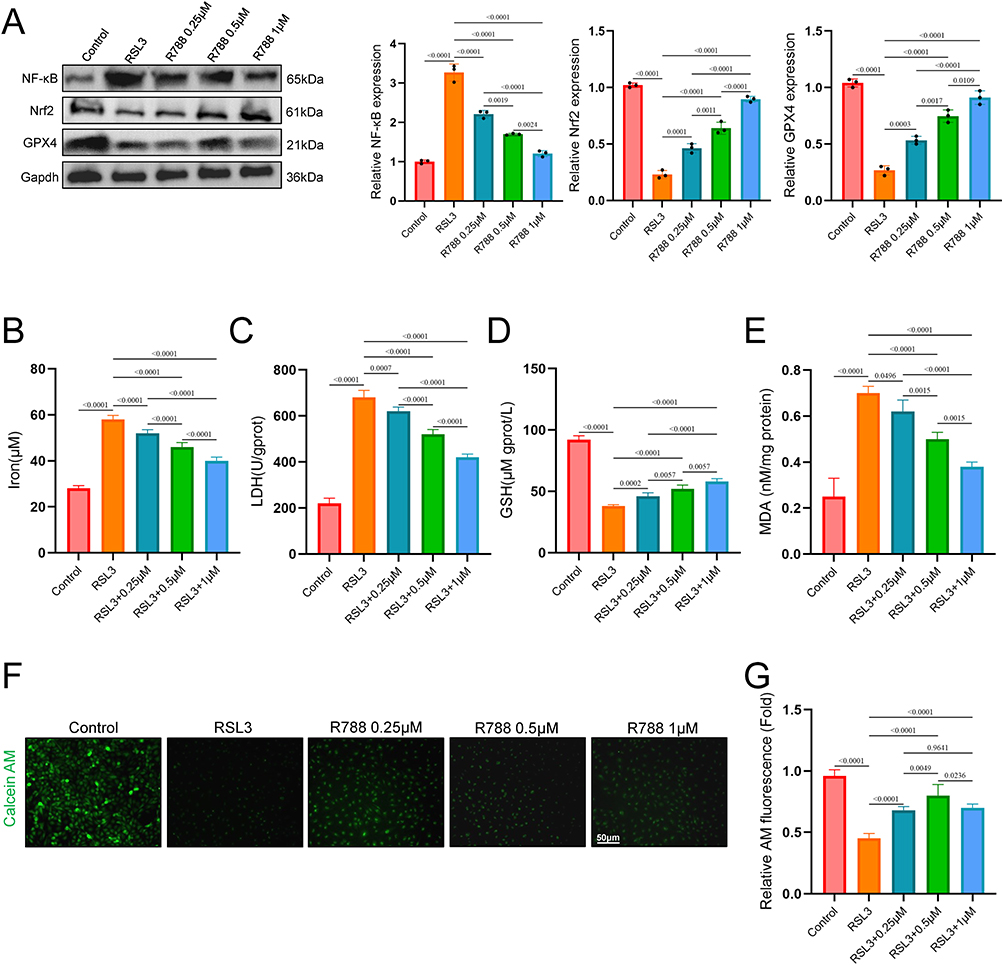 |
Figure 2 The effect of R788 on oxidative stress in vitro. (A) Western blot of Nrf2, GPX4, and NF-kB. The data are presented as the mean ± SD (n = 6). (B–E) Analysis of intracellular iron, LDH, GSH and MDA concentrations. Data are presented as the mean ± SD (n = 6). (F and G) Assessment of mitochondrial permeability transition pore (MPTP) opening by Calcein-AM staining. Data are presented as the mean ± SD (n = 6). Scale bar: 50μm. |
R788 Reduced Ischemic Infarction and Improved Neurological Functional Outcome
To evaluate whether R788 provided neuroprotective effect from IS, the mice were undergone with MCAO and given with different treatment strategies. The infarct area of the mice using TTC staining. As shown in Figure 3A, compared with Sham group, the infarct volume of MCAO was significantly increased. In mice treated with R788 (7.5mg/kg, 15mg/kg) was significantly reduced after MCAO, compared to the MCAO group (Figure 3B). The ND scores of MCAO mice was significantly increased, while was reduced with R788 (7.5mg/kg, 15mg/kg) treatment (Figure 3C). These data strongly indicate that R788 can protect the brain injury from IRI and enhance neurological function.
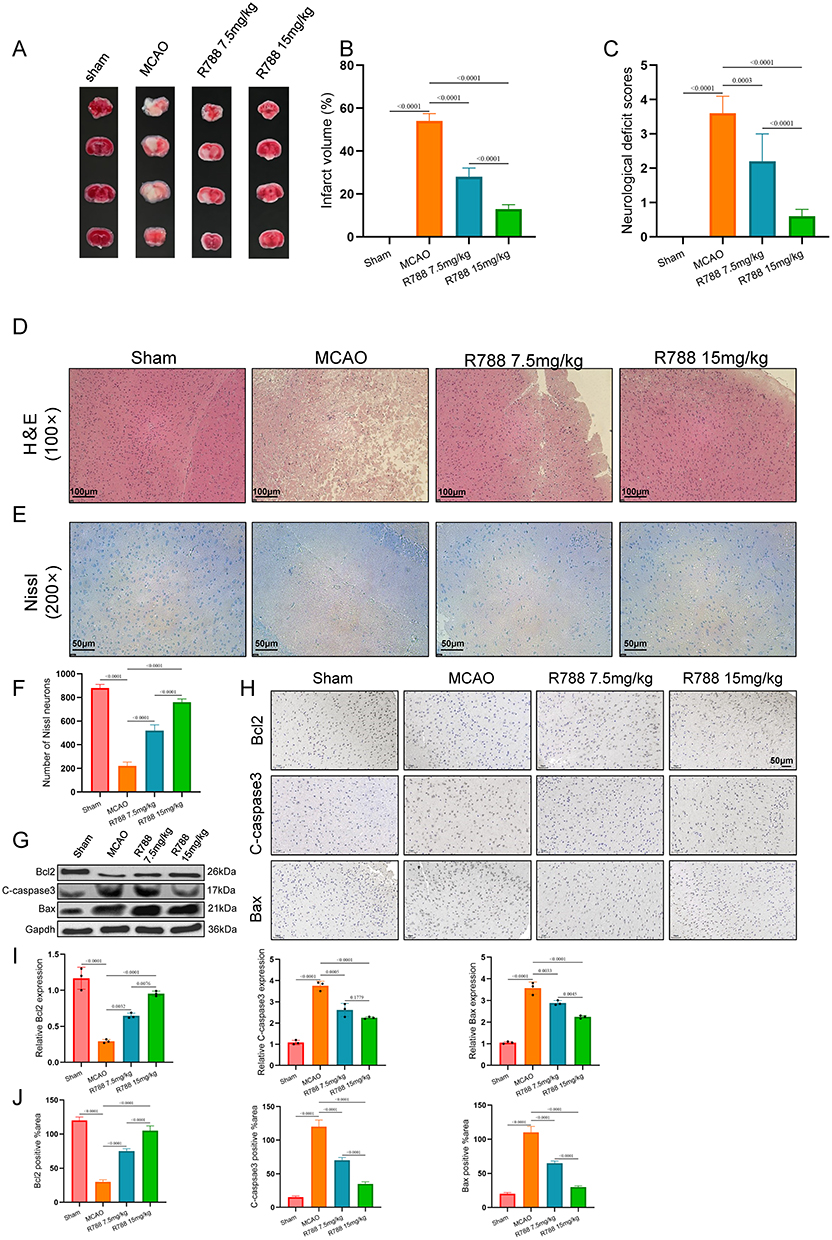 |
Figure 3 R788 significantly reduced cerebral infarction area in middle cerebral artery occlusion (MCAO) mice through apoptotic inhibition. (A) Representative images of brain tissue from MCAO mice showing ischemic lesions and neuronal injury compared with sham controls. (B and C) Assessment of Infarct volume (%) and Neurological deficit score of the mice. All data shown are the means ± standard error of the mean (n = 6). (D–F) H&E staining and Nissl staining, and the number of Nissl neurons. The data are presented as the mean ± SD (n = 6), *P<0.05; **P<0.001. (D) Scale bar: 100μm. (E) Scale bar: 50μm. (G and I) Western blot detection of Bcl2, C-caspsae3, and Bax. The data are presented as the mean ± SD (n = 6). (H and J) The expression of Bcl2, C-caspsae3, and Bax was detected by immunohistochemical staining. The data are presented as the mean ± SD (n = 6) Scale bar: 50μm. |
Further, H&E and Nissl staining showed that the number of Nissl neurons in the R788 treated group was more than that in the MCAO group (Figure 3D–F). Western blot analysis of apoptosis-related proteins showed that R788 treatment upregulated Bcl‑2 expression and downregulated Bax and cleaved caspase‑3 levels, with the most pronounced effects observed at 15 mg/kg (Figure 3G and I). Immunohistochemical staining at the lesion site confirmed similar changes: higher Bcl‑2 and lower Bax and caspase‑3 levels in R788-treated groups, especially at 15 mg/kg (Figure 3H and J). Collectively, these data indicate that R788 exerts significant neuroprotection against ischemic stroke by inhibiting neuronal apoptosis.
R788 Inhibits Ferroptosis in vivo
Hence R788 showed ferroptosis inhibition against RSL3 in vitro, the inhibitory effect of R788 in vivo was also detected confirmed. Previous studies have found that ferroptosis is closely related to ischemia-reperfusion injury (IRI), and the Nrf2/GPX4 pathway plays an essential anti-inflammatory role.27 Results of Western Blot (Figure 4A) and immunohistochemical staining (Figure 4B) showed that, the Nrf2/GPX4 pathway was significantly inhibited in the MCAO group compared with the control group. In contrast, the expression of the inflammation-related pathway NF-κB was upregulated. However, after R788 treatment, the expression of Nrf2/GPX4 increased, indicating that cellular ferroptosis was inhibited, and the expression of NF-κB decreased, indicating that the inflammatory response was suppressed (Figure 4C–H).
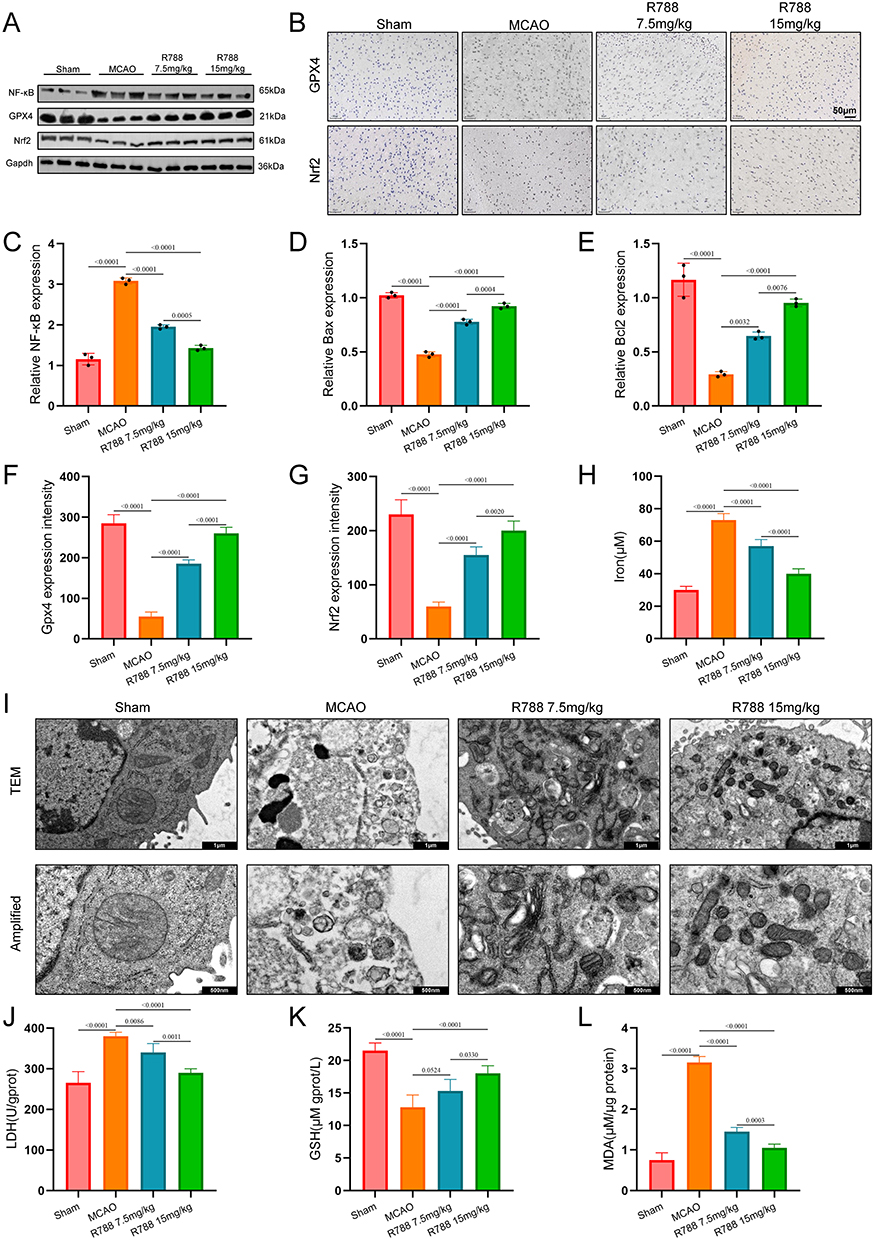 |
Figure 4 R788 contributes the protection of neuron by inhibition of ferroptosis. (A, C–E) Western blot detection of Nrf2, GPX4, and NF-kB in cerebral of mice. The data are presented as the mean± SD (n = 6). (B, F–H) The expression of Nrf2, GPX4, and NF-kB was detected by immunohistochemical staining. The data are presented as the mean ± SD (n = 6). Scale bar: 50μm. (I) Representative transmission electron microscopy images of mouse neural tissue showing neuronal and mitochondrial ultrastructure in sham and MCAO groups. Scale Bar: 1μm, 500nm. (J, K and L) Detection of LDH, GSH and MDA in the brain tissue of MCAO mice. The data are presented as the mean ± SD (n = 6). |
Electron microscopy revealed swollen mitochondria, disrupted cristae, and lipid peroxide accumulation in MCAO brain tissue, whereas R788 treatment alleviated tissue damage and preserved mitochondrial integrity (Figure 4I). Consistently, LDH, GSH, and MDA measurements showed results similar to those observed in vitro, further confirming the antioxidative effect of R788 against oxidative stress injury (Figure 4J–L). Together, the above results indicate that R788 inhibits ferroptosis by Nrf2/GPX4 Pathway Activations
R788 Upregulates Nrf2/GPX4 Pathway via Suppressing the Transcriptional Activity of STAT1
Previous study has indicated the association between STAT1 activation and ferroptosis. Hence, we further detect the effect of R788 on STAT1. As shown in Figure 5A and B, in both in vitro and in vivo models, we observed an increase in STAT1 phosphorylation under MCAO or oxidative stress conditions, whereas this effect was reversed by R788 treatment (Figure 5C and D). Then, the phosphorylation level of STAT1 was detected by immunohistochemical staining. Similar with the results of Westerm blot, R788 significantly inhibited the phosphorylation of STAT1 in MCAO mice (Figure 5E and F). These findings suggest that STAT1 may be involved in the regulation of ferroptosis by R788.
 |
Figure 5 R788 inhibits phosphorylated level of STAT1. (A and C) Western blot detection of STAT1 and p-STAT1 in SH-SY5Y cells. The data are presented as the mean± SD (n = 6). (B and D) Western blot detection of STAT1 and p-STAT1 in MCAO mice. The data are presented as the mean± SD (n = 6). (E and F) The expression of STAT1 and p-STAT1 was detected by immunohistochemical staining. The data are presented as the mean ± SD (n = 6). Scale bar: 50μm. |
R788 May Binds with STAT1 to Inhibit the Transcriptional Activity
To further elucidate the molecular mechanism by which R788 modulates ferroptosis through the Nrf2/GPX4 pathway, the interction between R788 and STAT1 were then subjected to molecular docking with with AutoDockto establish distinct binding patterns and energies (Figure 6A). Then we performed siRNA-mediated knockdown and plasmid-based overexpression of STAT1 in SH-SY5Y cells (Figure 6B). The Nrf2/GPX4 expression was significantly inhibited with the overexpression of STAT (Figure 6C), the si-STAT1 exerted a reversed effect (Figure 6D). In addition, the cell viability of SH-SY5Y cells was determined (Figure 6E). Hence, STAT1 was account for the transcriptional level of Nrf2 and GPX4. The mRNA expression of Nrf2/GPX4 was detected and the expression trends of similar with Western blot (Figure 6F and G).
 |
Figure 6 The inhibition effect of R788 on activity of STAT1. (A) Autodocking of STAT1 and R788 by using CB-dock2 (https://cadd.labshare.cn/cb-dock2/php/index.php). (B–G) After transfecting SH-SY5Y cells with either STAT1 overexpression plasmids or siRNA, the expression levels of Nrf2 and GPX4 were quantitatively analyzed by qPCR and Western blot. The data are presented as the mean ± SD (n = 6). (H–K) Western blot analysis was performed to detect the expression of Nrf2 and GPX4 in SH-SY5Y cells with STAT1 overexpression or knockdown following R788 treatment. The data are presented as the mean± SD (n = 6). |
Furthermore, we transfected with pcDNA3.1-STAT1/si-STAT1 with the treatment of R788 and RSL3 in SH-SY5Y cells. The Western blot analyses revealed that the overexpression of STAT1 significantly reversed the R788 on the expression of Nrf2 and GPX4 (Figure 6H and J). And si-STAT1 significantly contribute to the increased expression of Nrf2 and GPX4 by R788 (Figure 6I and K). Above all, these results indicated that R788 may interacts with STAT1 and unfreezes the transcriptional inhibition of Nrf2/GPX4 pathway.
The Bioactive Effect of R788 Relies on STAT1
We used the JASPAR bioinformatics database (http://www.jaspar.generate.net) to predict transcription factor binding sites. Analysis revealed potential STAT1-binding motifs within the Nrf2 promoter region (Figure 7A). To validate transcriptional regulation, dual-luciferase reporter assays were performed in SH‑SY5Y cells. Luciferase activity was significantly lower in the pc‑STAT1+Nrf2 group than in the pcDNA3.1+Nrf2 group, confirming that STAT1 negatively regulates Nrf2 transcription. R788 further reduced luciferase activity in pcDNA3.1+Nrf2 cells and strengthened STAT1‑mediated suppression of Nrf2 (Figure 7B). As expected, the transfection of si-STAT1+Nrf2 inhibited promoter activity of Nrf2, and R788 treatment contribute to the effect (Figure 7C). Due to the presence of multiple predicted binding sites, some of which are in close proximity, a total of 5 pairs of ChIP primers were designed (Table S6). ChIP-qPCR verified that these 5 sites in the Nrf2 promoter were significantly enriched, while R788 treatment could weaken their interaction (Figure 7D). In summary, these comprehensive results suggest that Nrf2 is a direct transcriptional target inhibited by Stat1, and this effect is further enhanced upon exposure to R788.
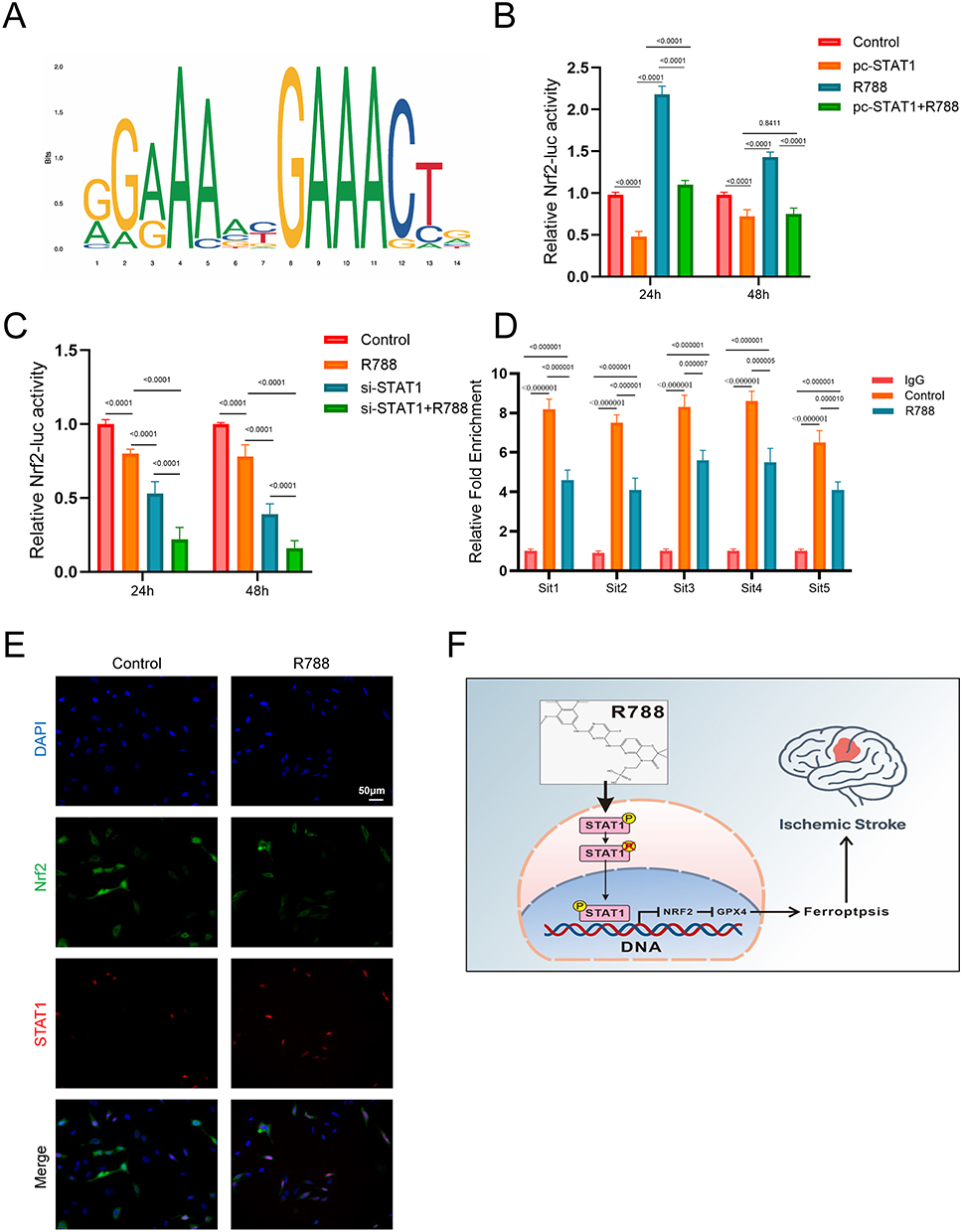 |
Figure 7 The bioactive effect of R788 relies on STAT1. (A) Stat1 binding motif provided by the JASPAR database. (B and C) Dual-luciferase reporter assay of STAT1 with or without R788 treatment. The data are presented as the mean ± SD (n = 6). (D) STAT1 binds to its binding sites in the Nrf2 promoter region as shown by the CHIP assay with or without the transfection of STAT1 overexpression plasmid. The data are presented as the mean ± SD (n = 6). (E) Representative images of immunofluorescence for p-Stat1 staining and Nrf2 staining in SH-SY5Y cells. (F) The mechanism diagram of R788 in treating stroke by regulating ferroptosis. |
Immunofluorescence results showed that Nrf2 was physiologically localized in the cytoplasm, with minimal presence in the nucleus. R788 significantly decreased the level of Nrf2, especially in the nucleus, indicating the inhibition of its nuclear translocation. Meanwhile, the level of pStat1 in the control group was low, but after treatment with R788, the level of pStat1 (Ser727) in the nucleus increased (Figure 7E). In summary, these results demonstrate that R788 induces ferroptosis by promoting the phosphorylation and nuclear translocation of Stat1, thereby inhibiting the expression and nuclear localization of Nrf2 (Figure 7F).
Discussion
Stroke is considered a leading cause of death and disability worldwide. It causes 5 million deaths and another 5 million permanent disabilities each year, posing a significant challenge to global public health.28 Stroke is a neurological disorder caused by insufficient cerebral circulation due to cerebral vascular infarction or hemorrhage.3,28 Depending on the pathological changes in cerebral blood vessels, stroke can be either ischemic stroke or hemorrhagic stroke, with ischemic stroke (IS) accounting for 85% of cases.3 Oxidative stress and inflammatory damage are recognized as critical contributors to the pathogenesis of IS. Due to its potent antioxidant and anti-inflammatory properties, R788 has been evaluated for its potential therapeutic effects in many diseases. However, the application of R788 in neurological disorders remains largely unexplored. Therefore, we investigated its therapeutic potential in IS.29
Our investigation into the therapeutic effects of R788 on IS, through both in vitro and in vivo models, has yielded promising results. R788 treatment has shown a remarkable ability to reduce neuronal apoptosis and decrease oxidative stress damage in the injured areas of MCAO mice. Furthermore, in the H2O2-induced SHSY5Y cell damage model, R788 has demonstrated its ability to inhibit H2O2-induced oxidative stress damage. These significant neuroprotective effects of R788, observed in both in vivo and in vitro experiments, instill confidence in its potential as a stroke treatment.
To further investigate the underlying mechanisms of R788 in the treatment of IS. We performed a series of assays focusing on ferroptosis, a critical pathological mechanism in IS. Reactive oxygen species (ROS) are by-products of cellular metabolism. Under normal physiological conditions, ROS maintain homeostasis and participate in cell signal transduction. However, under pathological conditions such as radiation exposure, chemical drug effects, and excessive transition metal ions, excessive ROS accumulation within cells leads to cell death. Notably, recent studies have revealed that the pathological effects of ROS are closely associated with their induction of lipid peroxidation, a process critically involved in inflammatory cascades during stroke and related diseases.30 The aberrant accumulation of lipid peroxides exacerbates ischemic injury by triggering ferroptosis. Characterized as an iron-dependent form of programmed necrosis, ferroptosis features lipid peroxidation-driven plasma membrane disintegration, distinguishing it mechanistically from classical apoptosis.31 Regarding its regulatory mechanisms, the Nrf2/GPX4 signaling axis has been identified as a central regulatory pathway: On one hand, the transcription factor Nrf2 activates downstream antioxidant genes in response to oxidative stress, thereby maintaining cellular redox homeostasis; on the other hand, its target molecule glutathione peroxidase 4 (GPX4) directly participates in scavenging lipid peroxides — suppression of GPX4 expression abolishes cellular repair capacity against lipid peroxidation damage, ultimately inducing ferroptosis.32,33 The discovery of this regulatory network not only elucidates the core molecular mechanism underlying ferroptosis but also identifies potential therapeutic targets for ischemic diseases.
Our results suggest a potential link between R788 and this mechanism. Compared with sham mice, MCAO mice showed downregulation of Nrf2/GPX4 expression, indicating ubiquitin-mediated degradation of Nrf2 and activation of ferroptosis. In the R788-treated group, Nrf2/GPX4 expression was upregulated compared to the MCAO group, suggesting that R788 may inhibits ferroptosis by upregulating the Nrf2/GPX4 pathway. Additionally, the inflammatory pathway NF-κB was downregulated, indicating that R788 may promote cell survival by inhibiting ferroptosis. STAT1 (Signal Transducer and Activator of Transcription 1) is an important transcription factor and a member of the STAT protein family. It primarily mediates the activation of interferon signaling pathways, particularly those involving IFN-α, IFN-β, and IFN-γ. STAT1 serves as a key regulatory factor in various biological processes, including antiviral responses, immune modulation, and apoptosis.34,35 Recent studies suggest that inhibition of STAT1 expression can suppress neuronal apoptosis, significantly reduce inflammatory responses at sites of spinal cord injury, and alleviate neurological dysfunction.36,37 Research by Lin et al demonstrated the presence of five overlapping binding sites between STAT1 and Nrf2. Phosphorylation of STAT1 at Ser727 activates it, allowing it to bind to the promoter region of Nrf2, thereby suppressing Nrf2 transcription and promoting ferroptosis. Based on this, we hypothesize that R788 may promote Nrf2 expression by inhibiting STAT1.38 Our results show that overexpression of STAT1 suppresses the expression of Nrf2 and GPX4, whereas STAT1 inhibition has the opposite effect. Moreover, treatment with R788 may alleviates STAT1-induced ferroptosis, suggesting a potential mechanism underlying its neuroprotective effect.
The application of R788 in stroke treatment is an exciting research area. However, many unresolved issues remain. First, our study demonstrates an association between R788, STAT1, and ferroptosis but lacks direct binding evidence. R788 may selectively modulate STAT1 phosphorylation or exert off-target effects, potentially affecting outcomes. What’s more, using SH-SY5Y cells instead of primary neurons limits generalizability, and the short observation period in animal experiments precludes evaluation of long-term functional recovery. To translate this therapeutic approach from the laboratory to clinical practice, further exploration of its therapeutic targets and mechanisms is necessary. Additionally, extensive preclinical studies are still required.
Conclusion
In summary, our experiments suggested the therapeutic effects of R788 on IS. In MCAO mice, R788 treatment reduced oxidative stress and apoptotic cells in the damaged area and upregulated Nrf2/GPX4 pathway. In the cell model, R788 downregulated STAT1 and activated Nrf2/GPX4 pathway, thereby inhibited RSL3-mediated ferroptosis in SH-SY5Y cells, alleviated oxidative stress damage, and promoted cell survival. Our research is the first to suggest that R788 may alleviate localized ischemic injury and oxidative stress damage caused by stroke by regulating ferroptosis, highlighting its promising application in the treatment of IS. However, our cellular and animal studies still have certain methodological limitations. The long-term efficacy of R788 remains uncertain, and its therapeutic potential for IS requires further validation through additional preclinical studies.
Data Sharing Statement
The datasets for this study are available on reasonable request from the corresponding authors.
Author Contributions
Yong Yang: Writing – original draft, Investigation. Jing Wang: Writing – original draft, Formal analysis. Heng Zhou: Writing – review & editing, Software. Cailei Jiang: Writing – review & editing, Software. Zhansheng Zhu: Writing – review & editing, Project administration, Funding acquisition. Yamei Wang: Writing – review & editing, Validation, Conceptualization. All authors gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research was supported by Jingzhou Scientific Research Fund Project under Grant No. 2024LHY21. Grant from the Doctoral Research Initiation Fund Project of the First People’s Hospital of Jingzhou, Grant No. 2026DIF03.
Disclosure
The authors declare that they have no conflicts of interest in this paper.
References
1. Stockwell BR, Angeli JP, Bayir H. et al. Ferroptosis: a Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell. 2017;171(2):273–17.
2. Washington HH, Glaser KR, Ifejika NL. CE: acute Ischemic Stroke. Am J Nurs. 2021;121(9):26–33. doi:10.1097/01.NAJ.0000790184.66496.1d
3. Benjamin EJ, Virani SS, Callaway CW, et al. Heart Disease and Stroke Statistics-2018 Update: a Report From the American Heart Association. Circulation. 2018;137(12):e67–e492. doi:10.1161/CIR.0000000000000558
4. Mu Q, Xue Y, Lu Y, et al. Advances in the therapy of cerebral ischemia-reperfusion injury with natural product-based nanoparticles. Nano TransMed. 2022;1(2–4):e9130009. doi:10.26599/NTM.2022.9130009
5. Tuo QZ, Zhang ST, Lei P. Mechanisms of neuronal cell death in ischemic stroke and their therapeutic implications. Med Res Rev. 2022;42(1):259–305. doi:10.1002/med.21817
6. Eltzschig HK, Eckle T. Ischemia and reperfusion--from mechanism to translation. Nat Med. 2011;17(11):1391–1401. doi:10.1038/nm.2507
7. Yan HF, Tuo Q-Z, Yin Q-Z, et al. The pathological role of ferroptosis in ischemia/reperfusion-related injury. Zool Res. 2020;41(3):220–230. doi:10.24272/j.issn.2095-8137.2020.042
8. Soares ROS, Losada DM, Jordani MC, et al. Ischemia/Reperfusion Injury Revisited: an Overview of the Latest Pharmacological Strategies. Int J Mol Sci. 2019;20(20):5034.
9. Farmer EE, Mueller MJ. ROS-mediated lipid peroxidation and RES-activated signaling. Annu Rev Plant Biol. 2013;64(1):429–450. doi:10.1146/annurev-arplant-050312-120132
10. Sekerdag E, Solaroglu I, Gursoy-Ozdemir Y. Cell Death Mechanisms in Stroke and Novel Molecular and Cellular Treatment Options. Curr Neuropharmacol. 2018;16(9):1396–1415. doi:10.2174/1570159X16666180302115544
11. Chen QS, Shen A, Dai J-W, et al. IL37 overexpression inhibits autophagy and apoptosis induced by hepatic ischemia reperfusion injury via modulating AMPK/mTOR/ULLK1 signalling pathways. Life Sci. 2021;276:119424. doi:10.1016/j.lfs.2021.119424
12. Sun MS, Jin H, Sun X, et al. Free Radical Damage in Ischemia-Reperfusion Injury: an Obstacle in Acute Ischemic Stroke after Revascularization Therapy. Oxid Med Cell Longev. 2018;2018(1):3804979. doi:10.1155/2018/3804979
13. Mao R, Zong N, Hu Y, et al. Neuronal Death Mechanisms and Therapeutic Strategy in Ischemic Stroke. Neurosci Bull. 2022;38(10):1229–1247.
14. Dugbartey GJ. Cellular and molecular mechanisms of cell damage and cell death in ischemia-reperfusion injury in organ transplantation. Mol Biol Rep. 2024;51(1):473.
15. Zhou Y, Liao J, Mei Z, et al. Insight into Crosstalk between Ferroptosis and Necroptosis: novel Therapeutics in Ischemic Stroke. Oxid Med Cell Longev. 2021;2021:9991001.
16. Cui Y, Zhang Y, Zhao X, et al. ACSL4 exacerbates ischemic stroke by promoting ferroptosis-induced brain injury and neuroinflammation. Brain Behav Immun. 2021;93:312–321. doi:10.1016/j.bbi.2021.01.003
17. Tuo QZ, Lei P, Jackman KA, et al. Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol Psychiatry. 2017;22(11):1520–1530. doi:10.1038/mp.2017.171
18. Orellana-Urzúa S, Rojas I, Líbano L, et al. Pathophysiology of Ischemic Stroke: role of Oxidative Stress. Curr Pharm Des. 2020;26(34):4246–4260. doi:10.2174/1381612826666200708133912
19. Tian J, Guo S, Chen H, et al. Combination of Emricasan with Ponatinib Synergistically Reduces Ischemia/Reperfusion Injury in Rat Brain Through Simultaneous Prevention of Apoptosis and Necroptosis. Transl Stroke Res. 2018;9(4):382–392. doi:10.1007/s12975-017-0581-z
20. Sae-Khow K, Charoensappakit A, Udompornpitak K, et al. Syk inhibitor attenuates lupus in FcγRIIb(-)(/-) mice through the Inhibition of DNA extracellular traps from macrophages and neutrophils via p38MAPK-dependent pathway. Cell Death Discov. 2025;11(1):63. doi:10.1038/s41420-025-02342-x
21. Paik J. Fostamatinib: a Review in Chronic Immune Thrombocytopenia. Drugs. 2021;81(8):935–943. doi:10.1007/s40265-021-01524-y
22. Morales-Torres J. The status of fostamatinib in the treatment of rheumatoid arthritis. Expert Rev Clin Immunol. 2012;8(7):609–615. doi:10.1586/eci.12.63
23. Duan QQ, Wang H, Su W-M, et al. TBK1, a prioritized drug repurposing target for amyotrophic lateral sclerosis: evidence from druggable genome Mendelian randomization and pharmacological verification in vitro. BMC Med. 2024;22(1):96. doi:10.1186/s12916-024-03314-1
24. Lapchak PH, Kannan L, Rani P, et al. Inhibition of Syk activity by R788 in platelets prevents remote lung tissue damage after mesenteric ischemia-reperfusion injury. Am J Physiol Gastrointest Liver Physiol. 2012;302(12):G1416–22. doi:10.1152/ajpgi.00026.2012
25. Alimova M, Sidhom E-H, Satyam A, et al. A High Content Screen for Mucin-1-Reducing Compounds Identifies Fostamatinib as a Candidate for Rapid Repurposing for Acute Lung Injury during the COVID-19 pandemic. bioRxiv. 2020;2020:1. doi:10.1101/2020.06.30.180380
26. Chen X, Jiang C, Chen M, et al. SYK promotes the formation of neutrophil extracellular traps by inducing PKM2 nuclear translocation and promoting STAT3 phosphorylation to exacerbate hepatic ischemia-reperfusion injury and tumor recurrence. Mol Med. 2024;30(1):146. doi:10.1186/s10020-024-00907-7
27. Hu Q, Zhang T, Yi L, et al. Dihydromyricetin inhibits NLRP3 inflammasome-dependent pyroptosis by activating the Nrf2 signaling pathway in vascular endothelial cells. Biofactors. 2018;44(2):123–136. doi:10.1002/biof.1395
28. Hankey GJ. Stroke. Lancet. 2017;389(10069):641–654. doi:10.1016/S0140-6736(16)30962-X
29. Patel M. Targeting Oxidative Stress in Central Nervous System Disorders. Trends Pharmacol Sci. 2016;37(9):768–778. doi:10.1016/j.tips.2016.06.007
30. Li L, Tan J, Miao Y, et al. ROS and Autophagy: interactions and Molecular Regulatory Mechanisms. Cell Mol Neurobiol. 2015;35(5):615–621. doi:10.1007/s10571-015-0166-x
31. Dixon SJ, Lemberg K, Lamprecht M, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi:10.1016/j.cell.2012.03.042
32. Deng X, Chu W, Zhang H, et al. Nrf2 and Ferroptosis: a New Research Direction for Ischemic Stroke. Cell Mol Neurobiol. 2023;43(8):3885–3896. doi:10.1007/s10571-023-01411-y
33. Wang Y, Wu S, Li Q, et al. Pharmacological Inhibition of Ferroptosis as a Therapeutic Target for Neurodegenerative Diseases and Strokes. Adv Sci. 2023;10(24):e2300325. doi:10.1002/advs.202300325
34. Clark DN, Begg LR, Filiano AJ. Unique aspects of IFN-γ/STAT1 signaling in neurons. Immunol Rev. 2022;311(1):187–204. doi:10.1111/imr.13092
35. Zuo Y, Feng Q, Jin L, et al. Regulation of the linear ubiquitination of STAT1 controls antiviral interferon signaling. Nat Commun. 2020;11(1):1146. doi:10.1038/s41467-020-14948-z
36. Wu Y, Yang L, Mei X, et al. Selective inhibition of STAT1 reduces spinal cord injury in mice. Neurosci Lett. 2014;580:7–11. doi:10.1016/j.neulet.2013.11.055
37. Wu YX, Gao CZ, Fan KL, et al. STAT1 inhibitor alleviates spinal cord injury by decreasing apoptosis. Genet Mol Res. 2016;15(1):7271. doi:10.4238/gmr.15017271
38. Guo L, Ma J, Xiao M, et al. The involvement of the Stat1/Nrf2 pathway in exacerbating Crizotinib-induced liver injury: implications for ferroptosis. Cell Death Dis. 2024;15(8):600. doi:10.1038/s41419-024-06993-z
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.