Back to Journals » Infection and Drug Resistance » Volume 16
Quinolone Antibiotics: Resistance and Therapy
Received 16 December 2022
Accepted for publication 3 February 2023
Published 10 February 2023 Volume 2023:16 Pages 811—820
DOI https://doi.org/10.2147/IDR.S401663
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Héctor Mora-Montes
Kai Tang, Heng Zhao
Fujian Provincial Key Laboratory of Innate Immune Biology, Fujian Normal University, Fujian, People’s Republic of China
Correspondence: Heng Zhao, Fujian Provincial Key Laboratory of Innate Immune Biology, Fujian Normal University, Fujian, People’s Republic of China, Tel +86-17689970104, Email [email protected]
Abstract: The clinical application of quinolone antibiotics is particularly extensive. In addition to their high efficiency in infectious diseases, the treatment process brings multiple hidden dangers or side effects. In this regard, drug resistance becomes a major challenge and is almost unavoidable in the clinical application of quinolones. Both genetic and phenotypic variations contribute to bacterial survival resistance under antibiotic therapy. This review is focusing on the drug discovery history, compound structure, and bactericidal mechanism of quinolone antibiotics. Recent studies bring a more in-depth insight into the research progress of quinolone antibiotics in the causes of death, drug resistance formation, and closely related SOS response after disease treatment at this stage. Combined with the latest clinical studies, we summarize the clinical application of quinolone antibiotics and further lay a theoretical foundation for the mechanism study of resistant or sensitive bacteria in response to quinolone treatment.
Keywords: quinolone antibiotics, disease treatment, SOS response, lethality, bacteria
Introduction
The earliest quinolone antibiotics were discovered in the 1960s.1 Their basic structures consist of bicyclic skeletons, carboxylic acids, and keto groups at positions 3 and 4, respectively, which are necessary for their pharmacological activity2,3 (Figure 1). To obtain broad-spectrum antibiotics, the so-called fluoroquinolones (FQs) can be synthesized by introducing fluorine atoms into the chemical structure of quinolone. For instance, norfloxacin was discovered in 1979, followed by a series of fluorine-containing new quinolones that emerged as FQs. And FQs are one of the most widely prescribed broad-spectrum antibiotics and show sufficient antibacterial activity against both Gram-negative and Gram-positive bacteria.4
 |
Figure 1 Core structure of quinolone antibiotics. (A) Six important modification positions include R1, R5, R6, R7, R8, and X for improving the drug activity. X=C defines quinolones and X=N defines naphthenes. (B) Each modification position has different effects such as antibacterial, affecting pharmacokinetics, enhancing activity and enzymatic binding. |
Mechanism of Quinolone Action
The term quinolone is often used in a general sense to categorize a class of compounds including quinolones, nalididones, quinazolines, isothiazoquinolones, etc. Mechanistically, quinolones can target two basic bacterial enzymes, DNA helicase (topoisomerase II)5 and DNA topoisomerase IV,6 both of which contain four subunits (2 GyrA or ParC and 2 GyrB or ParE). The enzymes can transfer one region of double-stranded DNA to another, thereby forming the enzyme-quinolones-DNA complexes.7–11 Quinolones mainly target helicases and topoisomerases that are critical for regulating the superhelix of genomic DNA in the process of replication and transcription in bacterias.12,13 These complexes prevent the movement of replication forks and transcriptional complexes in vitro, thereby inhibiting bacterial growth.14–16 Quinolones capture DNA helicase and DNA topoisomerase IV on DNA in the form of complexes, therefore leading to DNA break. However, their effects can be limited by proteins such as drug-enzyme-DNA complexes that reversibly inhibit DNA replication or fragment bacterial chromosomes. In one pathway, chromosome fragmentation stimulates excessive accumulation of highly toxic reactive oxygen species (ROS)-related compounds that are the main causes of cell death.17 DNA double-strand break (DSB) is a potentially fatal DNA damage, while FQs contribute to DSB by binding to the active site of type II topoisomerase after DNA cleavage to form a quinolone-enzyme complex that disrupts the reconnection of lysed DNA.18
Quinolones are divided into first, second, third, and fourth generations according to their discovery history and their antibacterial properties. At present, third-generation antibiotics are most commonly used in clinical practice. The types, structural changes, and characteristics of each generation of antibiotics are shown in Figure 2. In general, the further optimization or refinement of the synthesis approaches of drugs can enhance their bactericidal effects, but drug resistance may also occur after the structural modifications, which should be further investigated in the future study.
 |
Figure 2 Classification and features of quinolone antibiotics. Quinolone antibiotics are developed from generations to generations, and a wider spectrum of activity is obtained by adding different substituents at different positions in the core structure. Substituent positions and groups are outlined in red. |
Recently, the discovery of various new antibiotics has been seriously hindered by the occurrence of drug-induced resistance. The challenge of drug resistance caused by quinolone antibiotics needs to be addressed urgently. This review focuses on the formation mechanism of quinolone antibiotic-resistant bacteria, summarizes the research progress in this field, and provides a more in-depth understanding of the research progress of quinolone antibiotics in infectious disease therapy.
Lethality of Quinolone Antibiotics
The use of antimicrobial agents requires the rapid killing of pathogens, avoiding the induction of drug resistance, and improving the resolution of infectious diseases. Antimicrobial management also includes limiting the lethal effect, especially the protection of beneficial intestinal microbiota during therapy. Therefore, a better understanding of the process of cell death helps the manipulation of the bacterial flora.19 The earlier dogma that cell death was directly caused by specific types of stressors has been challenged, while a growing body of literature demonstrated that these lesions can stimulate the cellular accumulation of toxic substances, such as ROS under aerobic conditions and reactive nitrogen under anaerobic conditions. The first-generation compounds exerted a lethal effect via the accumulation of ROS. In the second-generation compounds, the effect of ROS is strongly dependent on the ROS concentration. For instance, the killing action of norfloxacin is related to the ROS at low/moderate concentrations but does not lead to cell death at high ROS concentrations. However, the effect of the third-generation compounds such as ciprofloxacin is not as lethal as that of previous generations, which may be due to the increased ability of chromosome fragments. The addition of Cmur8 methoxy in the drug structure can enhance the lethality of the fourth-generation compounds.20 The detection of various quinolone derivatives showed two killing modes including the accumulation of toxic ROS and the lethal DNA breaks. ROS could be easily detected and play a dominant role in the quinolone-mediated killing. In contrast, the quinolone-mediated DNA breakage may exert a significant effect due to deficiency in ROS detoxification or reducing ROS levels.21
DNA replication can be blocked within minutes after the addition of quinolones in bacterial cultures. The effect of cleaved complex correlates with the inhibition of DNA synthesis,22 while the concentration of quinolones that prevent DNA replication correlates with the bacteriostatic parameter MIC.23 DNA synthesis can be recovered after drug removal,24,25 and inhibition of DNA synthesis does not rapidly kill cells.25,26 Therefore, the cleaved complex is bacteriostatic rather than lethally damaging. Several species of bacteria are expressed two types of DNA topoisomerases including DNA helicases and DNA topoisomerase IV, while some bacteria such as Mycobacterium tuberculosis are only expressed helicases. These enzymes can change the DNA topology by introducing DSBs, traversing the other duplex (or another region of the same duplex), and then repairing the break. Quinolones can bind with helicase or topoisomerase IV and disrupt the DNA structure, allowing each 5ʹ end to be covalently bound to GyrA (helicase) or ParC (topoisomerase IV). When the cell metabolism is slowed during starvation or plateaus, compounds such as ciprofloxacin are lethal to large numbers of bacteria through inducing chromosome fragmentation.27 The addition of nutrients can stimulate the lethal activity of quinolones during the plateau phase.28 The presence of multiple lethal patterns suggests that drug concentration and latency are important factors and should be considered in data interpretation.29,30
Development of Resistance to Quinolone Antibiotics
Currently, the formation of bacterial resistance caused by the abuse of antibiotics leads to significant problems in public health. As a class of broad-spectrum antibiotics, quinolone antibiotics have a wide range of applications in the clinic due to their effective bactericidal functions, whereas their accompanying problems of drug resistance become a great challenge. According to the World Health Organization’s Global Antimicrobial Resistance and Use Surveillance System (GLASS) report, the main antibiotic-resistant bacteria in the world are Escherichia coli, Streptococcus pneumoniae, Klebsiella pneumoniae, Staphylococcus aureus, and Neisseria Gonorrhoeae (Figure 3).31 FQs have a very low minimum inhibitory concentration (MIC) for many bacterial strains. Therefore, most flora dies quickly due to the FQ treatment, whereas a small population of flora still survives (Figure 4). Before exposure to antibiotics, resting cells may randomly form during the culture process of bacteria.32–34 If helicases and topoisomerases are not active due to cell dormancy, persistent bacteria may also survive after FQ treatment.35 However, quinolone resistance also results from mutations that alter drug uptake, efflux, and structure.36–40 Recent studies suggest that the mechanisms underlying quinolone resistance are primarily mediated by chromosomal mutations and/or uptake of plasmid genes that alter topoisomerase targets, modify quinolones, and/or reduce uptake or increase efflux of accumulated drugs.3,41 Therefore, a single amino acid mutation in either of the two targeted enzymes of the quinolone (DNA helicase and topoisomerase IV) would impair their interactions with the quinolones, thereby reducing the response to the quinolone drug-like susceptibility. However, these mutations have been reported to be predominantly located in the amino-terminal regions of GyrA or ParC of these enzymes, and the most commonly mutant amino acids are serine residues and acidic residues (glutamic or aspartic acid).42 Ser83 and Asp87 mutations are the most common mutations associated with drug resistance of Escherichia coli gyrA, and there are similar mutations at the same positions in other species.43 This domain of these enzymes is responsible for anchoring the water-metal ion bridge, known as the quinolone resistance determining region (QRDR). Mutation of this QRDR disrupts the water-metal ion bridge, thereby reducing the drug affinity of the enzyme-DNA complex.44 Mutations of serines account for more than 90% of the mutation pool, followed by mutations of acidic residues.45 Single target mutations in DNA helicase and topoisomerase IV lead to increased extents of resistance.46
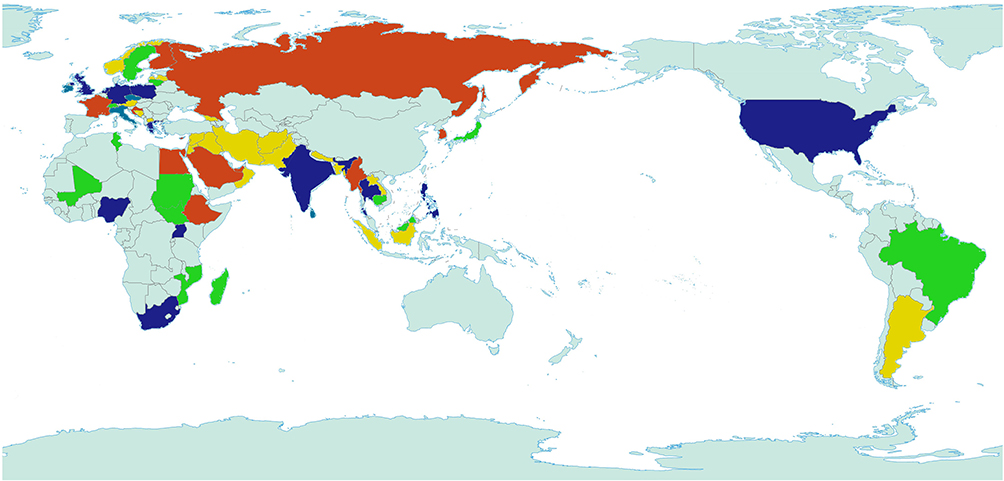 |
Figure 3 Global distribution of major quinolone resistant bacteria. Yellow represents Escherichia coli, green represents Klebsiella pneumoniae, red represents Staphylococcus aureus, blue represents Streptococcus pneumoniae and purple represents Neisseria Gonorrhoeae. Light green represents no data report. |
 |
Figure 4 MIC comparison of quinolones. The potency of each drug presented in the MIC90 (mg L−1) of different Gram-negative and Gram-positive strains. |
Since DNA helicases and topoisomerases are cytoplasmic enzymes, quinolones must pass through the bacterial envelope to exert killing effects. Therefore, the activity of quinolones is also influenced by their ability to penetrate cellular barriers and the effectiveness of efflux pumps to clear antibiotics from the cytoplasm. Currently, it is known that quinolones enter bacterial cells through porin and lipid pathways. Subsequently, drug resistance can be reduced not only by mutations of quinolone drug targets, but also by downregulation of porins, overexpression of efflux pumps, or changes in lipopolysaccharide (LPS) structure. Many quinolone-resistant bacterial strains have no mutation in the enzyme target QRDR47 and are less sensitive to independent compounds such as cyclohexane, salicylate, and tetracycline, indicating that drug resistance is related to the broad-spectrum efflux activity.48 It is known that the multiple antibiotic resistance (Mar) gene leads to resistance to many antibiotic compounds.49 The mutation of this gene leads to the overexpression of acrAB efflux pump and the decrease of OmpF (outer membrane protein F) porin expression. MarA is a positive regulator of acrAB transcription, which can be induced by MPPA gene mutation or exposure to salicylate and tetracycline. Therefore, salicylates and tetracyclines may stimulate resistance to quinolones. In addition, MarA can block the translation of OmpF gene and down-regulate OmpX expression due to porin expression, thus reducing the expression of many types of porins.50 Another gene, nfxB, can cause resistance to quinolones and other antimicrobials and change the functional expression of OmpF on the cell surface, thereby reducing the entry of quinolones.51 Quinolone resistance manners associated with efflux pumps include modification of the main promoter superfamily (MFS) or drug-resistant nodular division (RND) family of Gram-positive bacteria and multiple antibiotics and toxin excretion (Mate) and ATP binding cassette (ABC) of Gram-negative bacteria.50
In addition to the bacterial genomic mutations, plasmid-mediated mechanisms play an important role in the generation of quinolone resistance. Plasmids carrying quinolone resistance genes can cause serious clinical problems and reduce drug sensitivity by 10–250 times.52 The first plasmid family encodes Qnr, a pentapeptide repeat family protein. It is also reported that the qnr gene originates from the chromosomes of many aquatic bacteria and can compete with other qnr proteins from other sources, which can inhibit the entry of quinolones into the cleavage complex, reduce double-strand breaks on chromosomes, and reduce the chromosome toxicity of quinolones.53 More than 100 variants have been found in clinical isolates and can be divided into six subfamilies (qnrA, qnrB, qnrC, qnrD, qnrS, and qnrVC). These proteins are folded into a right quadrilateral β-helix and dimers to form a rod-like structure, whose size, shape, and electrostatic surface mimic the β-form of DNA.54 The second plasmid family coding the protein related to quinolone resistance is Aac (6’)-Ib-cr, which is a derivative of aminoglycoside acetyltransferase with Trp102Arg and Asp179Tyr mutations. The enzyme acetylates the unsubstituted nitrogen on the R7 piperazine ring, thereby inhibiting the activity of quinolones. Both mutations are necessary for this particular enzyme action. Trp102Arg mutations at the Asp179Tyr tyrosine aromatic ring result in its interaction with quinolones and fix it in this position.55,56 The third family associated with quinolone resistance is plasmid-mediated quinolone efflux pumps, including OqxAB and QepA. Their encoded genes do not directly lead to high levels of quinolone resistance but can promote topoisomerase mutations by the adaption of bacteria to low concentrations of quinolones.57,58
Relationship Between Quinolone Antibiotics and SOS Response
Quinolones bind to the cleavage-junction active site of the DNA-cleavage complex. This binding produces cleavage complexes at a stable concentration and disrupts the replication process, leading to the stable cleavage complex colliding with the DNA replication system (replication fork, transcriptional complex, and targeting system). Therefore, the binding of quinolones results in chromosome breakage. In response to the effect of quinolones on DNA damage, SOS response and other DNA repair pathways are activated in bacteria. The downstream actions of SOS response include lengthening cell filaments by expressing LexA inhibitors and activating toxin-antitoxin modules to induce programmed cell death. E. coli efficiently repair DSBs through a series of enzymatic reactions related to homologous recombination and replication.59 Treatment of double-strand breaks induces the SOS response, which is a complicated network of more than 40 genes.60,61 Most of these genes are essential for the efficient repair of various DNA lesions, including DSBs.62,63 It is known that SOS regulation is primarily controlled by the LexA and recA gene products in E. coli. Single-stranded DNA (ssDNA) regions are formed when DNA nucleotides are destroyed by genotoxins such as ultraviolet light and mitomycin. RecA wraps ssDNA and then triggers the SOS response. Additionally, the RecA/ssDNA interaction stimulates the degradation of LexA (an inhibitor of RecA during normal repair). Subsequently, the inactivation of LexA affects other genes (such as dinI) directly involved in the SOS response and downstream genes (such as rpoS) related to DNA replication, cell division, and mutation.64 RecA and RecBC are essential for double-stranded repair in Escherichia coli. Based on the literature, Yamanaka et al summarized the gene network diagram of SOS response65 (Figure 5), suggesting that the SOS response is intricate. Under the treatment of FQ antibiotics, the bacteria that can survive without developing genetic resistance are often referred to as “persistent bacteria”, and their survival is dependent upon the SOS gene network. Studies have verified mutations in all known SOS-regulated genes. To determine the functions necessary for tolerance in E. coli, studies found that cell survival was decreased after the loss of the dinG and UvrD helicases and the Holliday ligation processing enzymes (including ruvA and ruvB). And analysis of the corresponding mutants showed that bacterial tolerance can be induced not only by repairing double-strand breaks but also by the repair of collapsed replication forks and stalled transcriptional complexes. And mutations in the RecF gene lead to increased survival, suggesting that RecAF recombination is a poisoning mechanism that is not previously associated with FQ lethality. DinG plays a critical role in the upstream of SOS system to promote its induction, while RuvAB only participates in repair. UvrD directly promotes all repair processes initiated by FQ-induced damage and prevents RecF-dependent error repair, making it one of the key SOS functions required for tolerance.64,66,67
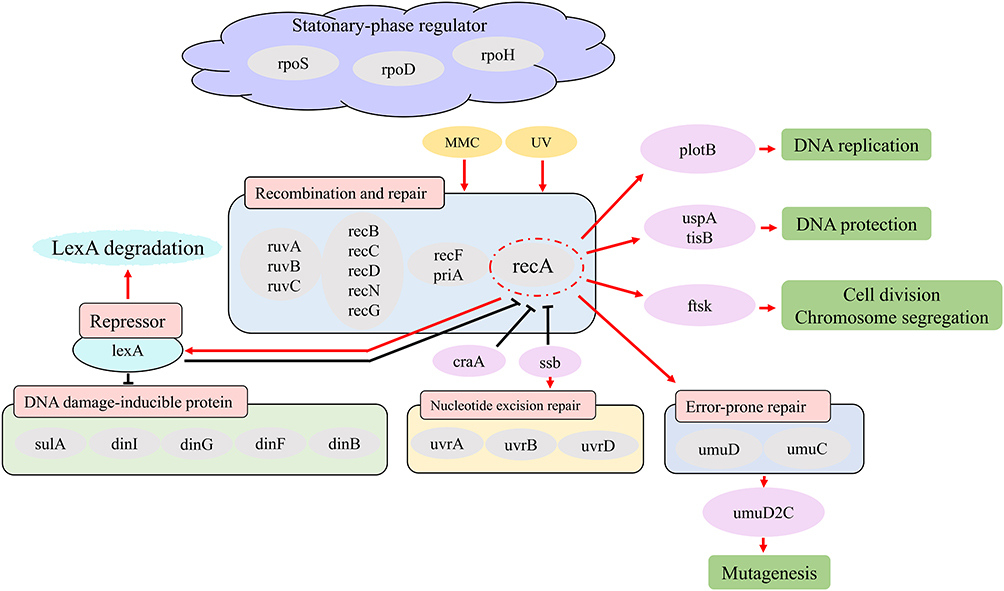 |
Figure 5 Schematic of a gene interaction network related to the SOS response in E. coli based on literature. The figure represents the inducible genes/proteins in the SOS response for repairing DNA damage. Black lines indicate pathways during normal repair and red lines indicate arrows activated/induced due to exposure to damaging substances. Recombination and repair, DNA damage-inducible protein, nucleotide excision repair, error-prone repair, and stationary-phase regulator have family molecules in each box. |
FQ ciprofloxacin causes phase transition of respiration and membrane potential of Escherichia coli, which is significantly related to its concentration. In the first stage, cells maintain their metabolic activity and membrane potential, accelerate K+ absorption, and produce low levels of superoxide and H2O2. However, the second stage is negatively correlated with the concentration of ciprofloxacin and depended on the SOS response. The second stage is also characterized by respiratory inhibition, membrane depolarization, leakage of K+ and glutathione, and cessation of glucose consumption, which can be considered cell death. Knockout of atpA, gshA, or kefBkefC gene disrupts the flux of proton and K +, thereby changing the degree and duration of respiratory inhibition and potassium retention. The deletion of K+ outflow channels, KefB and KefC, increases the sensitivity of Escherichia coli to ciprofloxacin.68
Clinical Application of Quinolone Antibiotics
Currently, quinolone is mainly used in the treatment of infectious diseases such as respiratory tract infection, gastrointestinal infection, gynecological infection, sexually transmitted diseases, urinary tract infection and prostatitis, bone and joint infection, skin and soft tissue infection, and other diseases. Oral or intravenous quinolones can successfully be used to treat a variety of infectious diseases. The clinical efficacy of quinolones has been verified in respiratory tract infections including acute bacterial exacerbation of chronic bronchitis, community-acquired pneumonia, hospital-acquired pneumonia, and bacterial sinusitis. Some quinolones are also useful in the treatment of immunocompromised patients with febrile neutropenia.69–71 It should be noted that not all FQs are approved for the treatment of all these infections, but the exchange and use of all FQs are discouraged in the clinic.
At present, the most commonly used FQs in the United States are ciprofloxacin, levofloxacin, gatifloxacin, and moxifloxacin.72 Ciprofloxacin has been approved for simple and complex urinary tract infections (including cystitis, pyelonephritis and chronic bacterial prostatitis, genitourinary and rectal gonorrhea without complications), skin and other soft tissue infections, bone and joint infections, infectious diarrhea and typhoid, intra-abdominal infections (when used with metronidazole), sinusitis and hospital-acquired pneumonia, etc. Ciprofloxacin is also approved for empirical treatment in patients with febrile neutropenia, prevention and treatment of anthrax, and lower respiratory tract infections (including acute bacterial deterioration of chronic bronchitis, pneumonia (except pneumococcal pneumonia), hospital-acquired pneumonia, etc). Levofloxacin is approved for the treatment of simple and complex urinary tract infections (including pyelonephritis and chronic bacterial prostatitis), skin and skin structure infections, acute maxillary sinusitis, acute bacterial exacerbation of chronic bronchitis, community-acquired pneumonia (including pneumonia caused by penicillin) drug-resistant Streptococcus pneumoniae and multidrug-resistant Streptococcus pneumoniae and hospital-acquired pneumonia.73 Gatifloxacin is approved for the treatment of simple and complex urinary tract infections (including pyelonephritis), simple genitourinary gonorrhea, simple skin and skin structure infections, acute sinusitis, chronic bronchitis, and acute bacterial exacerbation of community-acquired pneumonia.74 Moxifloxacin is approved for the treatment of acute bacterial sinusitis, simple skin and skin structure infections, chronic bronchitis, and community-acquired pneumonia.75 Gemifloxacin is approved for the treatment of acute bacterial exacerbation of chronic bronchitis and mild to moderate-severe community-acquired pneumonia.
The possible danger caused by the use of quinolones is mainly associated with unreasonable drug use. In view of the existing problems, the specific measures are as follows: 1) Strictly follow the doctor’s instructions, formulate medication rules based on the pharmacological characteristics of quinolones and the actual application in the hospital, and strengthen the guidance on the scope of application, dosage, course of treatment, adverse reaction treatment, and combination medication of quinolones; 2) Provide health education for clinical medical staff, publicize the pharmacological characteristics, usage and matters needing attention of quinolones through lectures, guide rational drug use, and open a consultation hotline in the pharmacy department, and promptly answer the questions of patients, their families and clinical medical staff in the process of disease treatment; 3) Dynamically understand the medication situation of clinicians, especially the unreasonable situation in the selection process of quinolones, and judge whether the condition analysis in the prescription meets the applicable standards of quinolones. At the same time, the problems (whether the dosage is appropriate, the frequency is reasonable, or the combination drug is correct) should be addressed.
Conclusions and Future Perspectives
By reviewing the association of quinolones with drug resistance and SOS response, we found that the current research on quinolones mainly focuses on two aspects including direct targeting action leading to lesions and indirect accumulation of poisons and cell death. Regarding the formation of drug resistance, the methods mainly focus on three aspects including mutations targeting enzyme site, antibiotic efflux, or the carrying plasmid. Studies have shown that quinolone antibiotics are closely related to the SOS response, and multiple genes are involved in the regulation of this process. Although our understanding of quinolone antibiotics is gradually becoming clearer, some issues are still existed and deserved our consideration. For instance, the binding of quinolone antibiotics to the target is a reversible reaction, so does it necessarily mean that its sterilization depends primarily on the accumulation of toxins after treatment, and there is a possibility of other sites of action. On the other hand, the ROS generation contributes to the quinolone-related toxin. However, the unanswered questions remain whether other important toxins are involved, how the repair-related protein recF regulates and kills bacteria through the cytotoxic mechanism, and how the toxin expression is balanced in the body. Furthermore, quinolones have a significant bactericidal effect and are widely used in infectious disease treatment. The precautions for their use are needed to reduce the risk of drug abuse.
Abbreviations
FQ, fluoroquinolones; ROS, reactive oxygen species; DSB, double-strand break; MIC, minimum inhibitory concentration; QRDR, quinolone resistance determining region.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Emmerson AM, Jones AM. The quinolones: decades of development and use. J Antimicrob Chemother. 2003;51(Suppl 1):13–20.
2. Bush NG, Diez-Santos I, Abbott LR, Maxwell A. Quinolones: mechanism, lethality and their contributions to antibiotic resistance. Molecules. 2020;25(23):E5662.
3. Pham TDM, Ziora ZM, Blaskovich MAT. Quinolone antibiotics. MedChemComm. 2019;10(10):1719–1739.
4. Hooper DC. Mechanisms of action of antimicrobials: focus on fluoroquinolones. Clin Infect Dis. 2001;32(Suppl 1):S9–S15.
5. Gellert M, Mizuuchi K, O’Dea MH, Itoh T, Tomizawa JI. Nalidixic acid resistance: a second genetic character involved in DNA gyrase activity. Proc Natl Acad Sci U S A. 1977;74(11):4772–4776.
6. Kato J, Nishimura Y, Imamura R, Niki H, Hiraga S, Suzuki H. New topoisomerase essential for chromosome segregation in E. coli. Cell. 1990;63(2):393–404.
7. Sugino A, Peebles CL, Kreuzer KN, Cozzarelli NR. Mechanism of action of nalidixic acid: purification of Escherichia coli nalA gene product and its relationship to DNA gyrase and a novel nicking-closing enzyme. Proc Natl Acad Sci U S A. 1977;74(11):4767–4771.
8. Peng H, Marians KJ. Decatenation activity of topoisomerase IV during oriC and pBR322 DNA replication in vitro. Proc Natl Acad Sci U S A. 1993;90(18):8571–8575.
9. Mizuuchi K, Fisher LM, O’Dea MH, Gellert M. DNA gyrase action involves the introduction of transient double-strand breaks into DNA. Proc Natl Acad Sci U S A. 1980;77(4):1847–1851.
10. Kreuzer KN, Cozzarelli NR. Formation and resolution of DNA catenanes by DNA gyrase. Cell. 1980;20(1):245–254.
11. Hiasa H, DiGate RJ, Marians KJ. Decatenating activity of Escherichia coli DNA gyrase and topoisomerases I and III during oriC and pBR322 DNA replication in vitro. J Biol Chem. 1994;269(3):2093–2099.
12. Wang JC. Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol. 2002;3(6):430–440.
13. Drlica K, Zhao X. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol Mol Biol Rev. 1997;61(3):377–392.
14. Willmott CJ, Critchlow SE, Eperon IC, Maxwell A. The complex of DNA gyrase and quinolone drugs with DNA forms a barrier to transcription by RNA polymerase. J Mol Biol. 1994;242(4):351–363.
15. Wentzell LM, Maxwell A. The complex of DNA gyrase and quinolone drugs on DNA forms a barrier to the T7 DNA polymerase replication complex. J Mol Biol. 2000;304(5):779–791.
16. Hiasa H, Yousef DO, Marians KJ. DNA strand cleavage is required for replication fork arrest by a frozen topoisomerase-quinolone-DNA ternary complex. J Biol Chem. 1996;271(42):26424–26429.
17. Drlica K, Hiasa H, Kerns R, Malik M, Mustaev A, Zhao X. Quinolones: action and resistance updated. Curr Top Med Chem. 2009;9(11):981–998.
18. Wohlkonig A, Chan PF, Fosberry AP, et al. Structural basis of quinolone inhibition of type IIA topoisomerases and target-mediated resistance. Nat Struct Mol Biol. 2010;17(9):1152–1153.
19. Drlica K, Zhao X. Bacterial death from treatment with fluoroquinolones and other lethal stressors. Expert Rev Anti Infect Ther. 2021;19(5):601–618.
20. Zhao X, Xu C, Domagala J, Drlica K. DNA topoisomerase targets of the fluoroquinolones: a strategy for avoiding bacterial resistance. Proc Natl Acad Sci. 1997;94(25):13991–13996.
21. Hong Y, Li Q, Gao Q, et al. Reactive oxygen species play a dominant role in all pathways of rapid quinolone-mediated killing. J Antimicrob Chemother. 2020;75(3):576–585.
22. Snyder M, Drlica K. DNA gyrase on the bacterial chromosome: DNA cleavage induced by oxolinic acid. J Mol Biol. 1979;131(2):287–302.
23. Chow RT, Dougherty TJ, Fraimow HS, Bellin EY, Miller MH. Association between early inhibition of DNA synthesis and the MICs and MBCs of carboxyquinolone antimicrobial agents for wild-type and mutant [gyrA nfxB(ompF) acrA] Escherichia coli K-12. Antimicrob Agents Chemother. 1988;32(8):1113–1118.
24. Mustaev A, Malik M, Zhao X, et al. Fluoroquinolone-Gyrase-DNA Complexes. J Biol Chem. 2014;289(18):12300–12312.
25. Goss WA, Deitz WH, Cook TM. Mechanism of action of nalidixic acid on Escherichia coli.Ii. inhibition of deoxyribonucleic acid synthesis. J Bacteriol. 1965;89:1068–1074.
26. Chen CR, Malik M, Snyder M, Drlica K. DNA gyrase and topoisomerase IV on the bacterial chromosome: quinolone-induced DNA cleavage. J Mol Biol. 1996;258(4):627–637.
27. Malik M, Hussain S, Drlica K. Effect of anaerobic growth on quinolone lethality with Escherichia coli. Antimicrob Agents Chemother. 2007;51(1):28–34.
28. Gutierrez A, Jain S, Bhargava P, Hamblin M, Lobritz MA, Collins JJ. Understanding and sensitizing density-dependent persistence to quinolone antibiotics. Mol Cell. 2017;68(6):1147–1154.e1143.
29. Wu X, Wang X, Drlica K, Zhao X. A toxin-antitoxin module in Bacillus subtilis can both mitigate and amplify effects of lethal stress. PLoS One. 2011;6(8):e23909.
30. Liu Y, Liu X, Qu Y, Wang X, Li L, Zhao X. Inhibitors of reactive oxygen species accumulation delay and/or reduce the lethality of several antistaphylococcal agents. Antimicrob Agents Chemother. 2012;56(11):6048–6050.
31. Organization WH. Global antimicrobial resistance surveillance system (GLASS) report: early implementation 2020. 2020.
32. Keren I, Kaldalu N, Spoering A, Wang Y, Lewis K. Persister cells and tolerance to antimicrobials. FEMS Microbiol Lett. 2004;230(1):13–18.
33. Keren I, Shah D, Spoering A, Kaldalu N, Lewis K. Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli. J Bacteriol. 2004;186(24):8172–8180.
34. Shah D, Zhang Z, Khodursky A, Kaldalu N, Kurg K, Lewis K. Persisters: a distinct physiological state of E. coli. BMC Microbiol. 2006;6:53.
35. Lewis K. Persister cells, dormancy and infectious disease. Nat Rev Microbiol. 2007;5(1):48–56.
36. Jacoby GA. Mechanisms of resistance to quinolones. Clin Infect Dis. 2005;41(Suppl 2):S120–126.
37. Park CH, Robicsek A, Jacoby GA, Sahm D, Hooper DC. Prevalence in the United States of aac(6’)-Ib-cr encoding a ciprofloxacin-modifying enzyme. Antimicrob Agents Chemother. 2006;50(11):3953–3955.
38. Poole K. Efflux-mediated resistance to fluoroquinolones in gram-positive bacteria and the mycobacteria. Antimicrob Agents Chemother. 2000;44(10):2595–2599.
39. Poole K. Efflux-mediated resistance to fluoroquinolones in gram-negative bacteria. Antimicrob Agents Chemother. 2000;44(9):2233–2241.
40. Morgan-Linnell SK, Becnel Boyd L, Steffen D, Zechiedrich L. Mechanisms accounting for fluoroquinolone resistance in Escherichia coli clinical isolates. Antimicrob Agents Chemother. 2009;53(1):235–241.
41. Vázquez X, Fernández J, Hernáez S, Rodicio R, Rodicio MR. Plasmid-Mediated Quinolone Resistance (PMQR) in two clinical strains of Salmonella enterica serovar corvallis. Microorganisms. 2022;10(3):579.
42. Hopkins KL, Davies RH, Threlfall EJ. Mechanisms of quinolone resistance in Escherichia coli and Salmonella: recent developments. Int J Antimicrob Agents. 2005;25(5):358–373.
43. Price LB, Vogler A, Pearson T, Busch JD, Schupp JM, Keim P. In vitro selection and characterization of Bacillus anthracis mutants with high-level resistance to ciprofloxacin. Antimicrob Agents Chemother. 2003;47(7):2362–2365.
44. Redgrave LS, Sutton SB, Webber MA, Piddock LJV. Fluoroquinolone resistance: mechanisms, impact on bacteria, and role in evolutionary success. Trends Microbiol. 2014;22(8):438–445.
45. Hiramatsu K, Igarashi M, Morimoto Y, Baba T, Umekita M, Akamatsu Y. Curing bacteria of antibiotic resistance: reverse antibiotics, a novel class of antibiotics in nature. Int J Antimicrob Agents. 2012;39(6):478–485.
46. Hooper DC, Jacoby GA. Topoisomerase inhibitors: fluoroquinolone mechanisms of action and resistance. Cold Spring Harb Perspect Med. 2016;6(9):a025320.
47. Lee S, Hinz A, Bauerle E, et al. Targeting a bacterial stress response to enhance antibiotic action. Proc Natl Acad Sci. 2009;106(34):14570–14575.
48. McMurry LM, Oethinger M, Levy SB. Overexpression of marA, soxS, or acrAB produces resistance to triclosan in laboratory and clinical strains of Escherichia coli. FEMS Microbiol Lett. 1998;166(2):305–309.
49. Goldman JD, White DG, Levy SB. Multiple antibiotic resistance (mar) locus protects Escherichia coli from rapid cell killing by fluoroquinolones. Antimicrob Agents Chemother. 1996;40(5):1266–1269.
50. Correia S, Poeta P, Hébraud M, Capelo JL, Igrejas G. Mechanisms of quinolone action and resistance: where do we stand? J Med Microbiol. 2017;66(5):551–559.
51. Hooper DC, Wolfson JS, Souza KS, Tung C, McHugh GL, Swartz MN. Genetic and biochemical characterization of norfloxacin resistance in Escherichia coli. Antimicrob Agents Chemother. 1986;29(4):639–644.
52. Strahilevitz J, Jacoby GA, Hooper DC, Robicsek A. Plasmid-mediated quinolone resistance: a multifaceted threat. Clin Microbiol Rev. 2009;22(4):664–689.
53. Tran JH, Jacoby GA. Mechanism of plasmid-mediated quinolone resistance. Proc Natl Acad Sci U S A. 2002;99(8):5638–5642.
54. Vetting MW, Hegde SS, Wang M, Jacoby GA, Hooper DC, Blanchard JS. Structure of QnrB1, a plasmid-mediated fluoroquinolone resistance factor. J Biol Chem. 2011;286(28):25265–25273.
55. Sánchez MB, Hernández A, Rodríguez-Martínez JM, Martínez-Martínez L, Martínez JL. Predictive analysis of transmissible quinolone resistance indicates Stenotrophomonas maltophilia as a potential source of a novel family of Qnr determinants. BMC Microbiol. 2008;8(1):148.
56. Vetting MW, Park CH, Hegde SS, Jacoby GA, Hooper DC, Blanchard JS. Mechanistic and structural analysis of aminoglycoside N -acetyltransferase AAC(6′)-Ib and its bifunctional, fluoroquinolone-active AAC(6′)-Ib-cr variant. Biochemistry. 2008;47(37):9825–9835.
57. Aldred KJ, McPherson SA, Turnbough CL, Kerns RJ, Osheroff N. Topoisomerase IV-quinolone interactions are mediated through a water-metal ion bridge: mechanistic basis of quinolone resistance. Nucleic Acids Res. 2013;41(8):4628–4639.
58. Li J, Zhang H, Ning J, et al. The nature and epidemiology of OqxAB, a multidrug efflux pump. Antimicrob Resist Infect Control. 2019;8:44.
59. Kowalczykowski SC, Dixon DA, Eggleston AK, Lauder SD, Rehrauer WM. Biochemistry of homologous recombination in Escherichia coli. Microbiol Rev. 1994;58(3):401–465.
60. Courcelle J, Khodursky A, Peter B, Brown PO, Hanawalt PC. Comparative gene expression profiles following UV exposure in wild-type and SOS-deficient Escherichia coli. Genetics. 2001;158(1):41–64.
61. Fernández De Henestrosa AR, Ogi T, Aoyagi S, et al. Identification of additional genes belonging to the LexA regulon in Escherichia coli. Mol Microbiol. 2000;35(6):1560–1572.
62. Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. Washington, DC, USA: ASM Press; 2005.
63. Radman M. SOS repair hypothesis: phenomenology of an inducible DNA repair which is accompanied by mutagenesis. Basic Life Sci. 1975;5A:355–367.
64. Theodore A, Lewis K, Vulic M. Tolerance of Escherichia coli to fluoroquinolone antibiotics depends on specific components of the SOS response pathway. Genetics. 2013;195(4):1265–1276.
65. Yamanaka T, Toyoshiba H, Sone H, Parham FM, Portier CJ. The TAO-Gen algorithm for identifying gene interaction networks with application to SOS repair in E. coli. Environ Health Perspect. 2004;112(16):1614–1621.
66. Feliciello I, Zahradka D, Zahradka K, Ivanković S, Puc N, Đermić D. RecF, UvrD, RecX and RecN proteins suppress DNA degradation at DNA double-strand breaks in Escherichia coli. Biochimie. 2018;148:116–126.
67. Górecka KM, Krepl M, Szlachcic A, Poznański J, Šponer J, Nowotny M. RuvC uses dynamic probing of the Holliday junction to achieve sequence specificity and efficient resolution. Nat Commun. 2019;10(1):4102.
68. Smirnova GV, Tyulenev AV, Muzyka NG, Peters MA, Oktyabrsky ON. Ciprofloxacin provokes SOS-dependent changes in respiration and membrane potential and causes alterations in the redox status of Escherichia coli. Res Microbiol. 2017;168(1):64–73.
69. Andriole CL, Andriole VT. Are all quinolones created equal. Mediguide Infect Dis. 2002;21:1–5.
70. Iannini PB, Niederman MS, Andriole VT. Treatment of respiratory infections with quinolones. In: The Quinolones. Elsevier; 2000:255–284.
71. Weigelt J, Brasel K, Faro S. Use of quinolones in surgery and obstetrics and gynecology. In: The quinolones. Elsevier; 2000:285–301.
72. DiCarlo RP, Martin DH. Use of the quinolones in sexually transmitted diseases. In: The quinolones. Elsevier; 2000:227–254.
73. Nicolle LE. Use of quinolones in urinary tract infection and prostatitis. In: The Quinolones. Elsevier; 2000:203–225.
74. Hamer DH, Gorbach SL. Use of the quinolones for treatment and prophylaxis of bacterial gastrointestinal infections. In: The quinolones. Elsevier; 2000:303–323.
75. Karchmer AW. Use of the quinolones in skin and skin structure (Osteomyelitis) and other infections. In: The Quinolones. Elsevier; 2000:371–395.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.