Back to Journals » Drug Design, Development and Therapy » Volume 15
Quaternary Lidocaine Derivatives: Past, Present, and Future
Authors Wang Q ![]() , Zhang Y
, Zhang Y ![]() , Liu J, Zhang W
, Liu J, Zhang W ![]()
Received 9 November 2020
Accepted for publication 25 December 2020
Published 14 January 2021 Volume 2021:15 Pages 195—207
DOI https://doi.org/10.2147/DDDT.S291229
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Georgios Panos
Qi Wang,1 Yujun Zhang,1 Jin Liu,1,2 Wensheng Zhang1,2
1Department of Anesthesiology, Laboratory of Anesthesia and Critical Care Medicine, Translational Neuroscience Centre, West China Hospital, Sichuan University, Chengdu, People’s Republic of China; 2National-Local Joint Engineering Research Center of Translational Medicine of Anesthesiology, West China Hospital, Sichuan University, Chengdu, People’s Republic of China
Correspondence: Wensheng Zhang
Laboratory of Anaesthesia & Critical Care Medicine, Translational Neuroscience Center, National-Local Joint Engineering Research Center of Translational Medicine of Anesthesiology, Sichuan University West China Hospital, Chengdu 610041, People’s Republic of China
Email [email protected]
Abstract: Local anesthetics have the advantage of complete analgesia with fewer side effects compared to systemic analgesics. However, their clinical use is limited due to their short duration of action. Thus, local anesthetics with fast onset, long duration of action, selective nociceptive block, and low local and systemic toxicity are highly desirable. In the past electrophysiological studies, quaternary lidocaine derivatives (QLDs) showed these characteristics. Here, we review electrophysiological properties of QLDs and their pharmacodynamic characteristics to shed light on potential problems.
Keywords: lidocaine, local anesthetics, derivatives, QX-314
Introduction
During the past century, local anesthesia has been commonly applied in clinical practice and lidocaine (LIDO) was the first amino-amide local anesthetic. Seventy years after its discovery, LIDO remains the most common local anesthetic worldwide. However, its short duration of action limits its application in many situations. In 1963, bupivacaine (BUP), a long-acting anesthetic (2–3-fold longer duration of action than LIDO), was first introduced for clinical application. However, in subsequent research and clinical reports, BUP was confirmed to be serious cardiac toxicity with a difficult resuscitation, which led to the development of levobupivacaine (LBUP) and ropivacaine (ROP). The duration of action of LBUP and ROP exceeds that of LIDO, but they do not still meet various clinical expectations.
Presently used local anesthetics block voltage-gated sodium channels in all motor and sensory fibers. A selective blockade of sensory, but not of other axons, is desired. Therefore, novel local anesthetics are expected to be long-lasting and to exert selective sensory blockade. In the 1970s, quaternary lidocaine derivatives (QLDs) first started to attract research attention. At present, the predominant QLDs include QX-222 (2-[(2,6-dimethylphenyl) amino]-N,N,N-trimethyl-2-oxoethaniminium chloride; Molecular weight: 256.77 g/mol), QX-572 (N,N-dimethyl-2-(phenylamino)-N-[2-(phenylamino)-2-oxoethyl)-2-oxoethanaminium chloride; Molecular weight: 347.84 g/mol), QX-314 (N-(2,6 dimethylphenylcarbamoylmethyl) triethylammonium bromide; Molecular weight: 343.30 g/mol), and QX-OH (2-(2,6-dimethylphenylamino)-N,N-diethyl-N-(2-hydroxyethyl)-2-oxoethanaminium bromide; Molecular weight: 359.30 g/mol) (Figure 1). In the past century, QLDs were frequently used in electrophysiological studies. In the early 21st century, research on QLDs focused on aspects such as long-lasting and selective effects, local and systemic toxicities, and on the mechanisms underlying local anesthesia. Several decades of research helped identify distinct advantages of QLDs compared with clinically used local anesthetics at present. For better understanding of QLDs, we summarize past research findings and the current state of research. More importantly, we revisit the shortcomings of QLDs and future challenges.
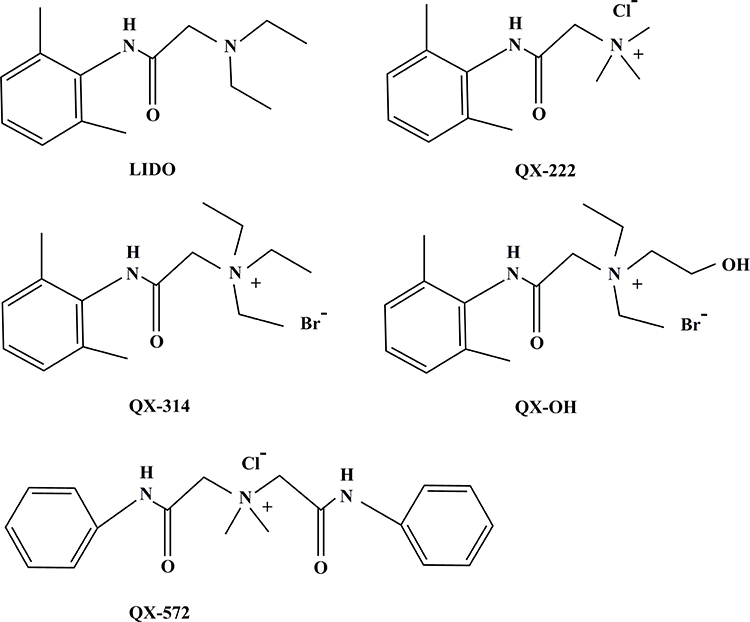 |
Figure 1 Molecular structure of LIDO, QX-222, QX-314, QX-OH, and QX-572. Abbreviation: LIDO, lidocaine. |
Past
LIDO has been applied as a local anesthetic and class I antiarrhythmic drug since it was developed. Similarly to LIDO, QLDs were used as antiarrhythmic drugs or a tool for research on electrophysiological effects in the initial stage. In the 1960s, QX-572 was reported to have the abilities to treat ventricular tachyarrhythmia with long-lasting effects.1–5 Regarding electrophysiology, various concentrations (12.5–50 mg/L) of QX-572 significantly increased the diastolic threshold and absolute refractory period, and it decreased conduction velocity and amplitude of contraction in isolated animal papillary muscles.2 By contrast, Ryden et al found that conduction of the atrioventricular node was shortened. The effective refractory period decreased in the atrium, atrioventricular, and ventricle conduction systems after administration of 8 mg/kg QX-572.6 The reason for this difference is multifaceted. Firstly, there are differences in many aspects of the experimental design, such as the subject and the QX-572 dosage. After slow injection with 8 mg/kg QX-572, the heart was exposed to a concentration of approximately 10 mg/L for at least 15 min.7 However, on isolated heart muscle preparations, no effect on normal electrical activity was observed at this concentration (10 mg/L). At concentrations exceeding 20 mg/L, QX-572 reduced the maximum rising velocity and decreased action potential amplitude and duration.8 Secondly, in addition to the direct effects of QX-527, cardiac electrophysiology is also affected by sympathetic and parasympathetic activity.
QX-572 was predominantly considered an antiarrhythmic drug for electrophysiological research on myocardial cells or on the heart. However, electrophysiological effects on nerve cells were the main focus of research for QX-222 and QX-314. In myelinated nerves, sodium currents were inhibited by QLDs with a manner dependent upon the voltage and frequency of depolarizing pulses.9 The frequency-dependent block and the voltage-dependent binding reaction between QX-314 and sodium channels were modulated by sodium inactivation mechanism.10 External QLDs hardly weakened membrane current. However, sodium currents were inhibited by 90% when QLDs were present in the axoplasm (<0.5 mM).9 Thus, the results gave us an explanation that QLDs enter opened sodium channels from the axoplasmic side and then block the channels.
LIDO and its quaternary derivatives have different effects and characteristics between sodium channel subtypes. Quaternary membrane-impermeant derivatives of LIDO are difficult to block skeletal muscle and nerve channels when applied externally,9,11 whereas QX-314 blocks cardiac channels when applied from either side of the membrane.12 Therefore, it is possible that QLDs have a faster time of onset in cardiomyocytes than in nerve and skeletal muscle cells. QX-572 works rapidly on isolated heart muscle preparations,8 which supported the above interpretation to some extent. However, the exact molecular mechanisms remain to be clarified. The sodium channel selectivity filter interacts with the hydrophilic part of the drugs, which plays an important role in extracellular access to and escape from their binding site.13,14 The sodium channel α-subunit consists of four homologous domains (I–IV), each containing six transmembrane segments (S1–S6).15 Some studies found that in skeletal muscle and brain channels, access and escape of QLDs was limited because of their aromatic residues on the IP-loop, and Cys and Val on IVS6. However, QLDs can access the internal binding site from the outside and escape from the cytoplasm to the outside in the heart channel.16 This isoform difference was explained by an isoform-specific residue (Cys in the heart, Phe in the brain and Tyr in the skeletal muscle) in the P-loop of domain I (IP-loop). Evidence suggests that a more hydrophilic residue promotes QLDs access. This was similar to the efficacy of the QLDs block of Thr in heart > Cys in skeletal muscle > Val in brain for the IVS6 isoform-specific residues.16 Apart from differences in sodium channels between cardiac and non-cardiac tissues, other pathways remain to be studied, such as the protein pathway, because charged molecules can permeate slowly through proteins. The way through which QLDs enter cells and bind targets is an important factor regarding differences in pharmacokinetics and pharmacodynamics.
In squid giant axons, two phases (a fast phase and a slow phase) occurred during recovery from the use-dependent block of sodium channels when LIDO, QX-222 and QX-314 were internally applied to perfused axons. The time constant of the fast phase was several milliseconds. However, the constants of the slow phase were different between drugs (for example, at −80 mV): LIDO, 270.0 ms; QX-222, 4.4 s; and QX-314, 17.0 s.17 In the fast phase, the recovery time constant was decreased when the membrane was hyperpolarized, whereas that for the slow phase was increased for QLDs or unchanged for LIDO. For slow recovery, the voltage dependence of the time constants is consistent with the m-gate trapping hypothesis. When the channel is opened, the cationic form of drug molecule escapes the channel through the hydrophilic pathway. However, LIDO may escape through the closed channel quickly via the hydrophobic pathway after losing its proton.17 Therefore, recovery from QX-314 and QX-222 is considerably slower than that from LIDO (approximately 10–40 fold). Recovery from the QX-314 block is approximately 4–5 fold slower than that from QX-222. This difference is attributable to many reasons, which are predominantly physical and chemical properties such as molecular weight as small molecule can escape the channel faster.18 In addition, the overall recovery rate is closely associated with the lifetime of the cationic molecule, which is determined by its deprotonation rate constant.
Therefore, research findings of QLDs before the 21st century can be summarized as follows: (1) QLDs have the property of being permanently positive charge; (2) the main target of QLDs is the sodium ion channel which is voltage- and frequency-dependent; (3) in nerve cells and skeletal muscle cells, it is difficult for QLDs to enter cells from the outside to block the sodium ion channel or to cross the membrane from the inside. For cardiomyocytes, QLDs can enter the internal binding site from the outside and escape from the cytoplasm; (4) compared with QX-572 and QX-222, QX-314 has a longer time of action regarding sodium channel blockade.
Present
After decades of research, the clinical value of QLDs has been recognized. The local anaesthesia effect of QLDs has become a research hotspot, especially QX-314 because of its advantages in duration. Therefore, in this context, QX-314 occupies the research hotspot among LIDO derivatives.
QLDs Produce Long-Lasting Local Anesthesia
Long-acting and selective sensory block by local anesthetics is urgently required. In 2007, an article published in the journal Nature started the research boom. In this article, the authors found that (1) QX-314 alone had no effect on sodium channels when applied to the extracellular region of sensory neurons, but in presence of the TRPV1 agonist, QX-314 can block sodium channels; (2) QX-314 and capsaicin produced long-lasting analgesic effects when co-injected into the sciatic nerve, and there was no motor or tactile disturbance.19 Therefore, subsequent studies mainly focused on TRPV1, long-acting and selective sensory block. In Table 1, we summarize pharmacodynamic data of QLDs used in the normal animal models over the past two decades.
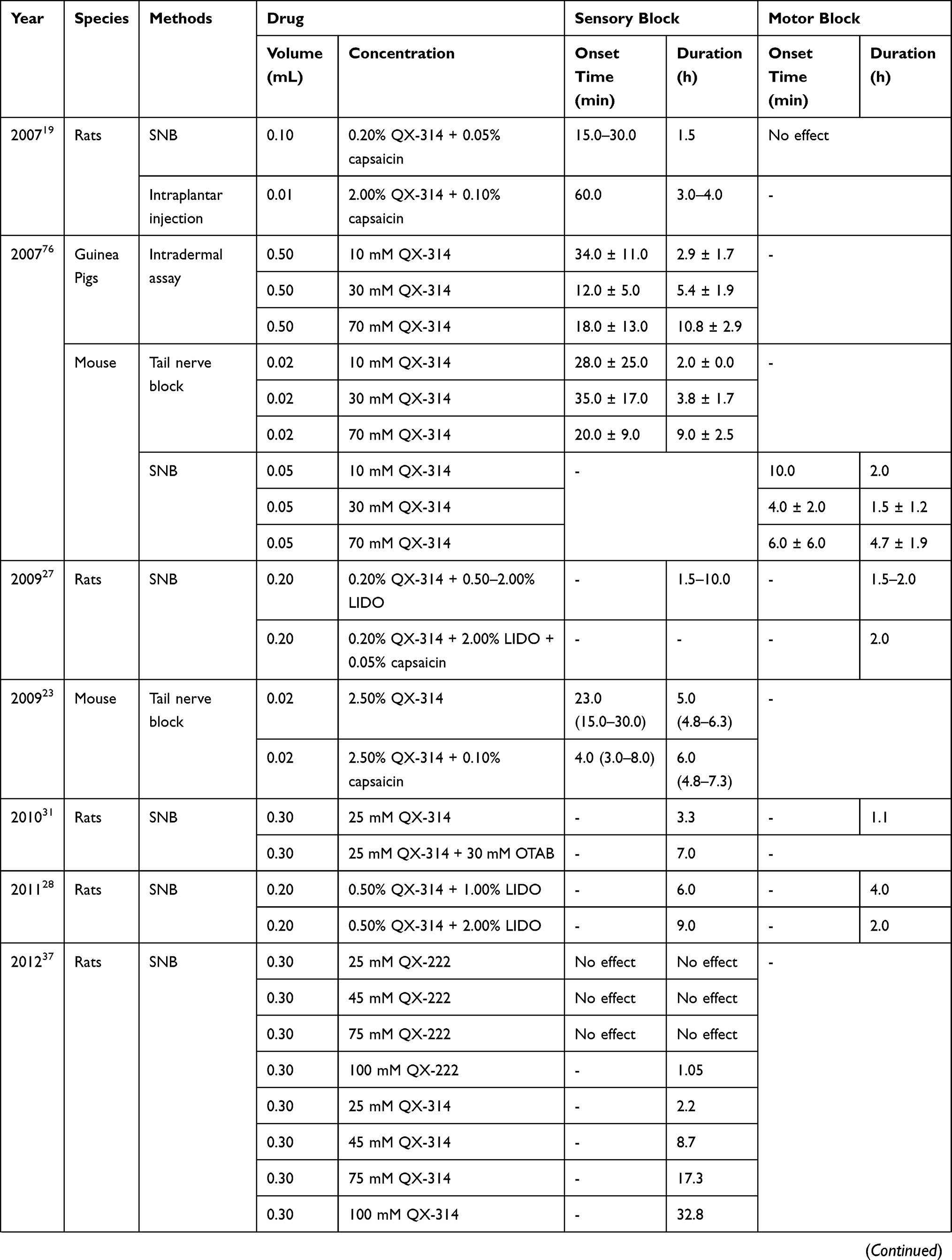 | 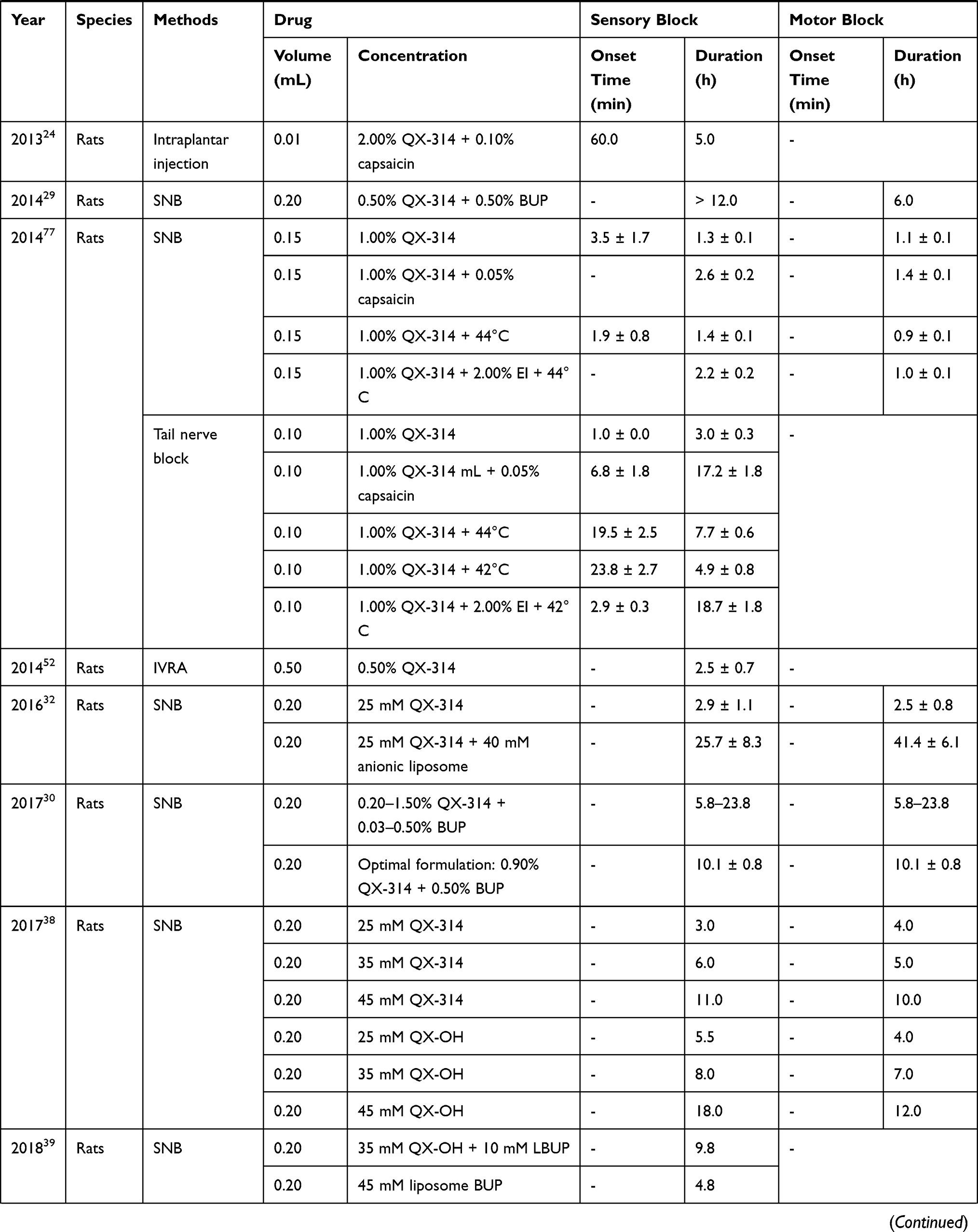 | 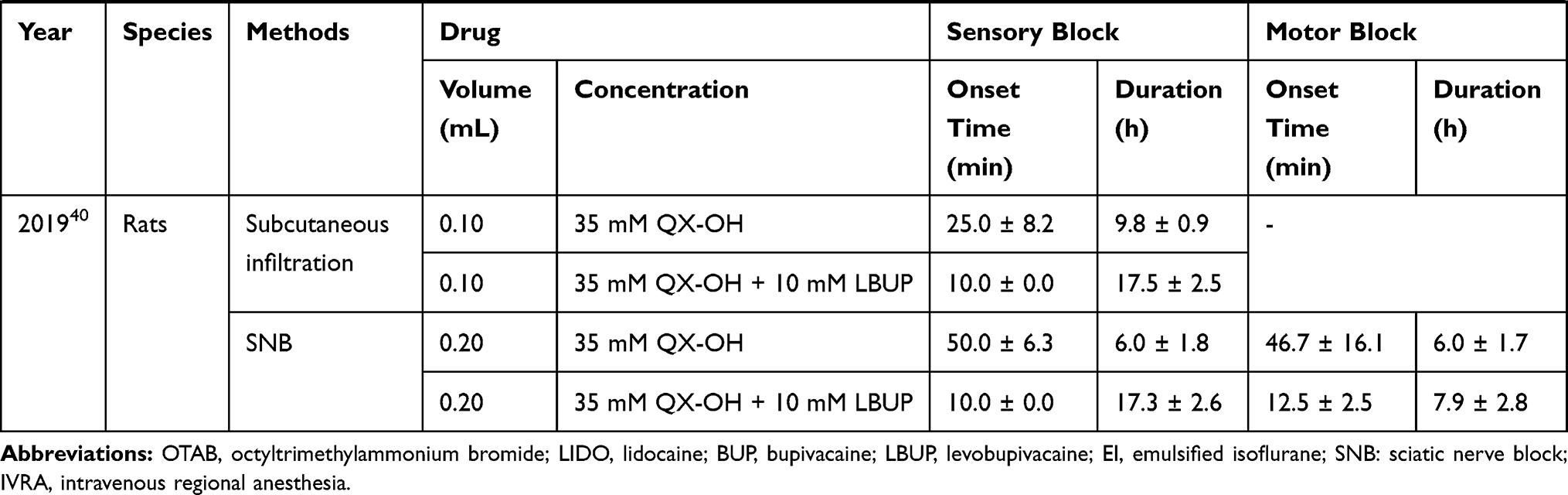 |
Table 1 Pharmacodynamic Data of QLDs in Normal Animal Models |
TRP channels belong to a class of protein channels widely distributed in the central and peripheral nervous systems. The TRP channel comprises six transmembrane proteins. The N- and C-terminus are located in the intracellular region, and the fifth and sixth transmembrane domains together form the channel pore region.20 These channels are regulated by temperature, pH, osmotic pressure, endogenous receptors, and exogenous receptors. At present, the TRP channel family can be divided into seven subfamilies, including TRPA, TRPC, TRPM, TRPML, TRPN, TRPP, and TRPV.20,21 Most TRP channels can mediate non-selective cationic inflow. Among TRP channels, the most important channel mediating the role of QX-314 is TRPV1, which is a non-selective channel for mono- and divalent cations. It is mainly expressed in primary sensory neurons, various brain nuclei, and in other non-neurological tissues.22 In afferent neurons, TRPV1 is distributed in the cell body, axon, and terminals. At the cellular level, TRPV1 is located on the plasma membrane, and its pores are large enough for QX-314 to enter the cell.19
Capsaicin (8-methyl-N-vanillyl-6-nonenamide), a secondary metabolite of chili peppers, can selectively activate TRPV1. Therefore, numerous studies began to assess and confirmed the local anesthetic effects of co-application of capsaicin and QX-314 in different animal models. The combination of QX-314 with capsaicin significantly shortened the onset time of QX-314.23 However, capsazepine, a transient receptor potential vanilloid receptor antagonist, decreases the efficacy of QX-314. Therefore, it was confirmed that capsaicin can accelerate the action of QX-314.23,24
Although it has been proven that capsaicin can promote the entry of QX-314 into sensory neurons, its application is limited due to its irritant effects and tissue toxicity.25 Thus, other agonists of TRPV1 channels as alternatives to capsaicin have been investigated. The current local anesthetic is the first choice, mainly because of the following points: (1) many existing local anesthetics can excite the TRPV1 channel;26 (2) the combination of local anesthetics and QX-314 can significantly shorten the onset time. When 0.2% QX-314 was injected with (0.5–2.0%) LIDO to the sciatic nerve in rats, the nociceptive blockade lasted for 1.5–10 hours, while the motor blockade continued for 1.5–2 hours.27 Therefore, co-application of LIDO and its quaternary derivative QX-314 produces a long-lasting, predominantly nociceptor-selective block, likely by facilitating QX-314 entry through TRPV1 channels. Considering the duration of action and the time of selective blockade, Roberson et al found that a combination of 2.0% LIDO and 0.5% QX-314 was optimal concentration ratio, which could produce 9 h sensory blockade with only 2h motion blockade.28 When 0.5% QX-314 was injected with 0.5% BUP in rat sciatic nerve injection model, the block duration persisted for longer than 12 h, whereas motor function deficits persisted for 6 h.29 Yin et al examined block time and local tissue toxicity and identified an optimal concentration ratio of 0.9% QX-314 plus 0.5% BUP.30
In addition to TRPV1 agonists, alternative methods of increasing the effect of QX-314 have been investigated. Chemical permeation enhancers can enhance transdermal penetration of drugs and increase drug flux across biological barrier. When 25 mM QX-314 was injected with octyltrimethylammonium bromide, a cationic surfactant, sensory blockade could reach 7 h, resulting in a marked predominance of sensory over motor blockade. QX-314 with anionic sodium octyl sulfate resulted in sensory block for approximately 15 h. Whereas co-injection of QX-314 and neutral polyoxyethylene (20) sorbitan monolaurate produced sciatic nerve block (SNB) only up to 5 h.31 Liposomes have been widely used for drug delivery with the aim of achieving slow release and prolonged effects, and their effectiveness and safety have been shown. When 25 mM QX-314 was injected with anionic liposomes, the duration of sensory and motor block was prolonged up to 25.7 ± 8.3 h and 41.4 ± 6.1 h, respectively, while cationic and neutral liposomes showed little effect.32 In conclusion, anionic liposomes and surfactants have a stronger effect of increasing QX-314. The reason may be that when coexisting with a negatively charged auxiliary agent, QX-314 can be easily attached through electrostatic interactions, thereby accelerating and increasing the compound’s transmission through cell membrane.
Recent studies further expanded the use of QX-314. In dry eye disease, TRPV1 played an important role in mediating enhanced nocifensive behavior.33 Pruritogen-mediated entry of QX-314 selectively inhibited conjunctival itching sensory fibers.34 When infected with methicillin-resistant Staphylococcus aureus (MRSA), three types of pore-forming toxins (hemolysin, phenol-soluble modulator and interleukin HlgAB) directly induced neuronal firing and produced spontaneous pain. QX-314 could block acute and chronic pain caused by MRSA infection through pores formed by neurons.35 Similar to LIDO, systemic application of QX-314 could also effectively relieve bone cancer pain through selective inhibition of TRPV1-expressing nerve fibers.36 Therefore, we believe that QX-314 will have a wide range of clinical applications.
Apart from QX-314, there are relatively few reports on the local anesthesia effects of QX-222 and QX-572 as they have no obvious advantage over QX-314. When QX-222 at a concentration of 100 mM was used for SNB in rats, sensory blockade only lasted for 1.05 h, which was considerably shorter than the effect of the corresponding concentration of QX-314 (Table 1).37 In recent years, a novel type of QX-314 derivative has emerged, termed QX-OH, which was expected to be optimized in terms of toxicity and effectiveness. In a rat SNB model, different concentrations of QX-OH significantly prolonged predominant sensory block time compared to the corresponding concentration of QX-314 (Table 1), but they had the same onset time of approximately 30–60 min.38 For blocking motor functions, QX-OH also showed a longer duration than QX-314. A combination of 35 mM QX-OH and 10 mM LBUP significantly shortened the onset time and extended the sensory block time without prolonging motor block time compared with the application of 35 mM QX-OH alone (Table 1).39,40 Therefore, QX-OH resembles QX-314 regarding slow onset but long-lasting nerve block effects. QX-OH, as an analogue of QX-314, may have a similar mechanism and characteristics with QX-314. According to the studies cited above, QX-OH is slightly more effective than QX-314, which may be due to changes in other physical and chemical properties as the novel compound also remains permanent positive charge. Additional hydroxyl group makes QX-OH less lipophilic than QX-314.38 Reduced lipid solubility makes it more difficult for the drug to escape after it enters the cell, thereby achieving a longer block time. However, the onset time will not improve through this modification but may even be decelerated.
Local Toxicity Induced by QLDs
In 2010, Schwarz et al reported severe consequences of lumbar intrathecal QX-314 in mice. Two microliters 5 mM and 10 mM QX-314 caused marked irritation in six (100%) and five (83%) of six mice, respectively. Death occurred in one mice at 5 mM (17%) and in two at 10 mM (33%).41 QX-314 is a derivative of LIDO, and theoretically its neurotoxicity should be similar to that of LIDO. Severe neurotoxicity induced by LIDO mainly manifests as irreversible blockade and does not manifest as acute irritable or nocifensive behavior. Therefore, the mechanism by which QX-314 cause acute irritation cannot be explained by the corresponding effects of LIDO neurotoxicity. Further studies found that intrathecal injection of 0.2% QX-314 alone or concurrent injection with capsaicin caused abnormal behaviors in rats, including agitation, vigorous vocalization, running, circling, rolling, and jumping.42 However, no overt lesions or signs of increased neuroinflammation were detected in spinal cord sections, indicating that behavioral changes were not caused by overt lesions of the spinal cord. Thus, the specific reasons for these effects remain unclear. But it tells us that QX-314 may not be suitable for intrathecal injection.
With respect to local tissue toxicity, QLDs show concentration-dependent nerve and muscle toxicity. The damage to muscle cells appeared to include nuclear internalization, degeneration, regeneration, and inflammatory responses.37 Tissue damage and inflammation caused by QX-314 are more severe than those caused by QX-222 at the same concentrations. Studies showed that the tissue response caused by 25 mM QX-314 was similar to that caused by 0.5% BUP and was clinically acceptable.32 When QX-314 was used in combination with BUP and the concentration of QX-314 was lower than 1.2%, the degree of local tissue reaction was similar to that of 0.5% BUP.30 Regarding QX-OH at 25 mM, the tissue damage and inflammation scores were not significantly different from those caused by QX-314 at the same concentration.38 However, at concentrations of 35 or 45 mM, local toxicity of QX-OH was more slighter than that of QX-314. Local toxicity caused by 45 mM QX-OH was more severe than that of 23 mM BUP. Therefore, we conclude that local tissue toxicity of QLDs is concentration-dependent, and local toxicity of QX-314 is more severe than that of QX-222 and QX-OH.38,43 According to current research, QX-314 and QX-OH are relatively safe at concentrations of 35 mM and below. In terms of the mechanisms underlying local toxicity, QLDs differ from LIDO. QX-314 causes local tissue toxicity, mainly through activation of TRPV1 to mediate calcium influx.44,45 LIDO, like other local anesthetics, exerts local toxicity mainly through the destruction of outer cell membranes, activation of caspases, and destruction of mitochondrial membranes.46,47 Although LIDO can activate TRPV1, its cytotoxicity is not correlated with intracellular calcium levels.48 Therefore, the strong cytotoxicity of LIDO may not be mediated by TRP channels. In addition, unlike LIDO, which is easily cleared from the cell by diffusion, QLDs remain in the cell for longer periods of time due to their non-permeability to the membrane, thus enhancing the time of cytotoxic effects. Thus, mechanistic studies also require pharmacokinetic support at the global and cellular levels.
Systemic Toxicity Induced by QLDs
Systemic toxicity of local anesthetics mainly occurs in the central nervous system and cardiovascular system. With respect to the effects of QX-314 on respiratory rhythm generation in brainstem-spinal cord preparations, the extracellular application of QX-314 (200 μM) decreased the C4 burst rate, amplitude and slope during the initial rising phase, and the effects slowly developed with a half-decay time of approximately 20 min. However, the combination of capsaicin (10 or 100 μM) and QX-314 (100 μM) showed no additional effect.49 The reason may be that capsaicin exerts a biphasic response to respiratory rhythm (a transient decrease followed by an increase in the C4 rate).50 In addition, QX-314 also exerts biphasic effects on the TRPV1 channels, inhibiting the capsaicin-evoked TRPV1 currents at lower (μM) concentrations and activating TRPV1 channels at higher (mM) concentrations.51 Therefore, the micromolar concentration of QX-314 may antagonize the effects of capsaicin to open TRPV1 channels.
As noted above, when QLDs are applied alone, they may not easily pass through the peripheral nerve cell membrane, but they can enter and exit myocardial cells, which may explain cardiotoxic effects of QX-314. Intravenous injection with 5 or 10 mg/kg QX-314 caused no significant central nervous system symptoms and changes in heart rate in rats. However, at a dosage of 20 mg/kg, four out of ten rats developed tonic-clonic seizures and exhibited a significant decrease in heart rate, and six out of ten rats died.52 After intravenous injection of mice with QX-314, the ED50 for central nervous system and cardiac toxicity were 10.7 mg/kg and 10.6 mg/kg, respectively, which were significantly lower than the respective ED50 of LIDO (19.5 mg/kg and 21.2 mg/kg, respectively).53 In other words, systemic toxicity of QX-314 was significantly higher than that of LIDO. However, the underlying mechanisms remain unclear. We suspect that pharmacokinetic differences between QX-314 and LIDO may in part explain these effects, such as the inherent property of QX-314 to diffuse more slowly owing to its lipophobic nature. Secondly, cardiotoxicity of local anesthetics is associated with increased membrane fluidity.54 This effect can be seen in differences between enantiomers of local anesthetics.55 Therefore, further research is needed to clarify whether variations in the molecular structure of LIDO have caused QX-314 to have a stereospecificity in pharmacology with LIDO.56 Although QX-OH is slightly more hydrophilic than QX-314, the ED50 of QX-OH for cardiac toxicity is not significantly increased compared to that of QX-314 (10.4 vs 10.0 mg/kg). However, both ED50 were significantly higher than the ED50 of BUP (2.97 mg/kg).57 Therefore, we believe that potential systemic toxicity will not impede future clinical applications of QLDs.
Future
In summary, QLDs, and particularly QX-314 series compounds, exert considerable effects regarding the blockade of peripheral nerves. Despite decades of development, further researches are still required for the mechanism of action, selective pain block and optimization of local anesthesia effect.
Mechanism of Action of QLDs
Research on the mechanism of QLDs is important to better understand their characteristics. Local anesthetics frequently enter cells in the form of bases and subsequently block sodium channels in the form of cations inside the cell. Therefore, it is difficult for QLDs to penetrate through the nerve cell membrane because of their permanent positive charge, which has been confirmed in previous electrophysiological studies. In this context, the importance of the TRPV1 channel in the role of local anesthesia in QLDs was discovered. QX-314 directly pass through the “standard” pores formed by the TRPV1 channel without requiring expansion or activation of additional downstream channels.58 Surprisingly, QX-314 can exert biphasic effects on TRPV1 channels, which activate TRPV1 channels at higher (mM) concentrations and inhibit capsaicin-induced TRPV1 currents at lower (μM) concentrations.51,59 However, nerve block is always established at a higher (mM) concentration, at which TRPV1 is also activated. TRPV1 is mainly expressed in unmyelinated afferent nerve fibers (small-diameter C-fiber), which is also the main target for the action of QLDs. However, what about myelinated A fibers? Some studies showed that extracellular application of QX-314 and capsaicin only reduces the action potential of C fibers and the sodium ion current of C fiber neurons, but does not affect A fibers.60–62 Other studies showed that TRPV1 is still expressed on Aδ-fiber neurons.63,64 Aδ-fiber (but not Aα, β-fiber) components of compound action potential have been shown to be significantly reduced by capsaicin.65,66 TRPV1-positive A-fibers also play an important role in the induction of ectopic long-term pain enhancement.67 Therefore, QLDs can still inhibit the transmission of nociception to thermal stimulation by blocking the TRPV1 channel on the Aδ-fiber.
TRPV1 is the main channel for QLDs to exert their effects. In contrast to studies on TRPV1, fewer studies focused on the relationship between TRPA1 and QLDs. Initial studies reported that in rat models, TRPA1 and TRPM8 did not play a role for the transport of QX-314.24 However, it was reported in later studies that in human embryonic kidney 293 (HEK-293) cells, QX-314 can activate and penetrate the TRPA1 channel, thereby blocking sodium channels.29,44 The reason for the different results is likely that some functional and pharmacological properties of TRPA1 and TRPV1 have species-specific differences between human and rodent subtypes.68–70 Therefore, whether TRPA1 participates in the role of QLDs requires further in vivo and in vitro studies. With respect to TRPM8, it has been confirmed that activated TRPM8 can mediate cellular penetration of QX-314, thereby blocking the voltage-gated sodium channel.71 In in vivo studies, QX-314 exerted selective cold pain suppression through TRPM8.72 Taken together, the relationship between other TRP channels and QLDs has rarely been investigated; thus, further research on this aspect is needed.
In recent years, some studies have been conducted on the independence of QX-314 on TRP channels, but few studies have been published so far. At TRPV1-negative primary afferents, QX-314 also increased the excitatory postsynaptic current latency.73 Similarly, in TRPA1- or TRPV1/TRPA1-double-knockout models, the compound action potential of the mice sciatic nerve was also extended by QX-314, indicating a TRP-channel independent entry pathway.29 Activation of TLR5 also leads to QX-314 entering neurons, thereby selectively inhibiting Aβ-fiber conduction in naive and chemotherapy-treated mice without affecting conduction in Aα- and Aδ-fibers.61,74 In addition, the P2X receptor is concerned because it can penetrate ions of larger molecular mass. QX-314 can inhibit ATP-induced currents in P2X receptors in a concentration-dependent manner.75 The mechanism of action of QX-314 may be associated with P2X receptors; thus, further research is needed for confirmation. It is crucial to study the channels and mechanisms of QLDs during local anesthesia, because these channels may become targets for improving the pharmacological characteristics of QLDs.
Selective Nociceptive Blockade by QLDs
Initial research showed that QX-314 produced a complete selective nociceptive block.19 However, many subsequent studies confirmed that QLDs can also produce motor block with or without capsaicin, but the duration of motor block was significantly shorter than that of sensory block.32,38,76–79 Thus, QLDs are not complete sensory selective block, but partial sensory selective block. This block characteristic is similar to the sensorimotor separation produced by ROP. Therefore, the drug concentration plays a decisive role. According to Table 1, when the concentration of QX-314 exceeds 1.0%, selective sensory block is not obvious. When the concentration is reduced to 0.5% or 0.2%, it can produce a more obvious predominantly sensory-selective block. This concentration-dependency of QLDs again suggests that their function may not entirely depend on the TRP channels as they are primarily expressed in sensory neurons.22,80,81 Studies have found that application of QX-314 alone at high concentrations can increase its ability to enter the cell through the cell membrane.58 Therefore, in this case, it not only acts on sensory neurons through TRP channels or non-TRP channels, but also through certain unknown channels to block motor neurons.
Optimizing Local Anesthesia Effects of QLDs
At present, there are mainly the following ways to enhance the efficacy of local anesthetics: (1) combined application of different local anesthetics; (2) the application of auxiliary drugs, such as epinephrine, dexmedetomidine, hormones and so on; (3) controlled release technology, such as using liposomes and nanomaterials loaded with local anesthetics. Except for capsaicin, there are few reports on the use of other adjuvants in combination with QLDs. The clinical dosage of dexmedetomidine can enhance SNB by QX-314.82 Sustained-release technology has significant effects on the prolongation of local anesthesia from QLDs, which has great prospects in the future.83 With respect to combination of QLDs and other local anesthetics, the theoretical basis for the combination of commonly used local anesthetics and QLDs is that local anesthetics can activate the TRP channel, thereby shortening the onset time and prolonging the effect. However, 3 mM BUP and 10 mM LIDO caused QX-314 to accumulate in cells that did not express TRPV1 or TRPA1 channels, including non-neuronal cells.29 So far, the specific intake mechanism is unclear, and there is a lack of pharmacokinetic data on QX-314, which would be helpful for explaining some of its characteristics.
QX-OH is a derivative of QX-314 and should thus have pharmacokinetic properties similar to those of QX-314. Zhang et al investigated metabolic kinetic parameters of drugs in local tissues and plasma when 0.2 mL 35 mM QX-OH alone or combined with 10 mM LBUP were used for rat SNB. When QX-OH was applied alone, QX-OH absorption was considerably fast. After administration, the concentration of QX-OH peaked at 0.7 h in plasma, at 0.5 h in muscle tissue, and at 0.7 h in sciatic nerve. However, only 0.46–0.54% of QX-OH would be absorbed into the blood circulation. The elimination rate of QX-OH from the sciatic nerve was slower than elimination from muscle tissue and plasma. The half-lives in plasma, muscle tissue and in sciatic nerve were 2.75 h, 3.52 h, and 4.28 h, respectively.84 Similar to QX-OH was administered alone, the half-lives of QX-OH when combined with LBUP in plasma, muscle tissue, and sciatic nerve were 2.64 h, 3.20 h, and 3.79 h, respectively.85 Thus, the long half-life in nerve tissue and cationic properties of QX-OH can provide theoretical support for its slow-acting and long-lasting pharmacodynamic characteristics. When QX-OH was combined with LBUP, the rate of QX-OH absorption into the blood was still as fast as when used alone. The peak concentration of QX-OH in blood plasma occurred significantly sooner than that of BUP (0.71 ± 0.06 h vs 4.11 ± 0.39 h). In other words, LBUP does not accelerate or decelerate the absorption of QX-OH into the blood. Interestingly, when combined with QX-OH, the peak time of LBUP in plasma increased by 4-fold compared with LBUP alone (4.11 ± 0.39 h vs 1.07 ± 0.16 h), which indicated that QX-OH delayed absorption of LBUP from the injection site into the circulation.85 This may be a potential mechanism extending the blocking time. Numerous factors can affect the rate of drug absorption into the blood, such as physical and chemical properties of the drug, local blood flow, and drug interactions. In the case of certain drugs, interactions between local blood flow and drugs can be the main influencing factor. Theoretically, LBUP tends to exert relaxing effects on peripheral blood vessels at concentrations commonly used under clinical settings, and it therefore increases local tissue blood flow.86 However, LBUP does not accelerate QX-OH absorption into the blood. The specific reason for this is unclear. In contrast, QX-OH significantly reduced the rate of absorption of LBUP into the blood. At present, no studies are available on the effect of QLDs on peripheral blood vessels, however, which should be investigated. Moreover, QLDs differ from traditional local anesthetics because of the positive charge they carry, and whether they interact with the charge of LBUP remains to be investigated. In the cellular drug uptake assay, LBUP can also promote the entry of QX-OH into cells, increasing the intake of intracellular drugs, whereas the intake of LBUP is not affected.
In general, the extended action time of combination of QLDs and LBUP can be explained by the positive charge characteristics of QLDs, longer elimination half-life of QLDs in nervous tissue, delayed absorption of BUP by QLDs, and increased cellular intake of QLDs induced by LBUP.
Conclusions
Initial electrophysiological studies showed that QX-314 has more characteristics as a long-acting local anesthetic, and subsequent studies confirmed this insight. However, some shortcomings prevent this compound from being an ideal long-acting local anesthetic, such as slow onset, incomplete sensory selective block, and intraspinal toxicity. QX-OH has not sufficiently improved these deficiencies. Therefore, on the basis of QLDs, we still need to find targets to further improve its action characteristics from drug structure, drug development, and mechanism of action.
Funding
This work was supported by the National Science and Technology Major Project, Ministry of Science and Technology of the People’s Republic of China (No.2014ZX09101001-003), Beijing, China.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Katz RL. Antiarrhythmic action of N, N-Bis (Phenylcarbamoylmethyl) dimethyl ammonium chloride (Qx-572) in cat and dog. Anesthesiology. 1964;25(3):291–296. doi:10.1097/00000542-196405000-00005
2. Covino BG, Rachwall P. Comparative cardiac effects of quinidine and N,N-Bis (Phenylcarbamoylmethyl)-dimethylammonium chloride (Qx-572), a new antiarrhythmic agent. J New Drugs. 1964;4(1):30–37.
3. Katz RL. Antiarrhythmic and neuromuscular effects Of Qx-572 in man. Acta Anaesthesiol Scand. 1965;9:73–81.
4. Ryden L, Hjalmarson A, Wasir H, Werko L. Effects of a long-acting antiarrhythmic agent–QX-572–on therapy resistant ventricular tachyarrhythmias. Br Heart J. 1974;36(8):811–821.
5. Ryden L, Hjalmarson A, Waldenstrom A. Effects of the quaternary ammonium compound OX-572 on ventricular tachyarrhythmias complicating acute myocardial infarction. Br Heart J. 1975;37(4):426–437.
6. Ryden L, Olsson B, Kvasnicka J. Electrophysiological effects of the antiarrhythmic agent QX-572 in the human heart with special reference to rate-induced changes in effective refractory periods. Cardiovasc Res. 1975;9(1):81–94.
7. Ryden L, Berlin A, Treiber L. Plasma concentrations and urinary excretion of the antiarrhythmic quaternary ammonium compound QX-572 in man. Eur J Clin Pharmacol. 1975;8(3–4):277–282.
8. Schipperheijn JJ, de Klerk G, Olsson B. Electrophysiological effect of QX 572, a lidocaine derivative, on isolated heart muscle preparations. Acta Pharmacol Toxicol. 1975;37(2):97–105.
9. Strichartz GR. The inhibition of sodium currents in myelinated nerve by quaternary derivatives of lidocaine. J Gen Physiol. 1973;62(1):37–57.
10. Yeh JZ. Sodium inactivation mechanism modulates QX-314 block of sodium channels in squid axons. Biophys J. 1978;24(2):569–574.
11. Frazier DT, Narahashi T, Yamada M. The site of action and active form of local anesthetics. II. Experiments with quaternary compounds. J Pharmacol Exp Ther. 1970;171(1):45–51.
12. Alpert L, Fozzard H, Hanck D, Makielski J. Is there a second external lidocaine binding site on mammalian cardiac cells? Am J Physiol. 1989;257(1):H79–H84.
13. Sunami A, Dudley SC, Fozzard HA. Sodium channel selectivity filter regulates antiarrhythmic drug binding. Proc Nat Acad Sci. 1997;94(25):14126–14131.
14. Qu Y, Rogers J, Tanada T, Scheuer T, Catterall WA. Molecular determinants of drug access to the receptor site for antiarrhythmic drugs in the cardiac Na+ channel. Proc Nat Acad Sci. 1995;92(25):11839–11843.
15. Catterall WA. Cellular and molecular biology of voltage-gated sodium channels. Physiol Rev. 1992;72(suppl_4):S15–S48.
16. Sunami A, Glaaser IW, Fozzard HA. A critical residue for isoform difference in tetrodotoxin affinity is a molecular determinant of the external access path for local anesthetics in the cardiac sodium channel. Proc Nat Acad Sci. 2000;97(5):2326–2331.
17. Yeh JZ, Tanguy J. Na channel activation gate modulates slow recovery from use-dependent block by local anesthetics in squid giant axons. Biophys J. 1985;47(5):685–694.
18. Courtney K. Structure-activity relations for frequency-dependent sodium channel block in nerve by local anesthetics. J Pharmacol Experimental Therapeutics. 1980;213(1):114–119.
19. Binshtok AM, Bean BP, Woolf CJ. Inhibition of nociceptors by TRPV1-mediated entry of impermeant sodium channel blockers. Nature. 2007;449(7162):607–610.
20. Ferreira G, Raddatz N, Lorenzo Y, González C, Latorre R. Biophysical and molecular features of thermosensitive TRP channels involved in sensory transduction. TRP Channels Sensory Transduction. 2015;1–39.
21. Li H. TRP channel classification. Adv Exp Med Biol. 2017;976:1–8.
22. Szallasi A, Cortright DN, Blum CA, Eid SR. The vanilloid receptor TRPV1: 10 years from channel cloning to antagonist proof-of-concept. Nat Rev Drug Discov. 2007;6(5):357–372.
23. Ries CR, Pillai R, Chung CC, Wang JT, MacLeod BA, Schwarz SK. QX-314 produces long-lasting local anesthesia modulated by transient receptor potential vanilloid receptors in mice. Anesthesiology. 2009;111(1):122–126.
24. Nakagawa H, Hiura A. Comparison of the transport of QX-314 through TRPA1, TRPM8, and TRPV1 channels. J Pain Res. 2013;6:223–230.
25. Chard P, Bleakman D, Savidge J, Miller R. Capsaicin-induced neurotoxicity in cultured dorsal root ganglion neurons: involvement of calcium-activated proteases. Neuroscience. 1995;65(4):1099–1108.
26. Leffler A, Fischer MJ, Rehner D, et al. The vanilloid receptor TRPV1 is activated and sensitized by local anesthetics in rodent sensory neurons. J Clin Invest. 2008;118(2):JCI32751.
27. Binshtok AM, Gerner P, Oh SB, et al. Coapplication of lidocaine and the permanently charged sodium channel blocker QX-314 produces a long-lasting nociceptive blockade in rodents. Anesthesiology. 2009;111(1):127–137.
28. Roberson DP, Binshtok AM, Blasl F, Bean BP, Woolf CJ. Targeting of sodium channel blockers into nociceptors to produce long‐duration analgesia: a systematic study and review. Br J Pharmacol. 2011;164.
29. Brenneis C, Kistner K, Puopolo M, et al. Bupivacaine‐induced cellular entry of QX‐314 and its contribution to differential nerve block. Br J Pharmacol. 2014;171(2):438–451.
30. Yin Q, Li J, Zheng Q, et al. The quaternary lidocaine derivative QX-314 in combination with bupivacaine for long-lasting nerve block: efficacy, toxicity, and the optimal formulation in rats. PLoS One. 2017;12:3.
31. Sagie I, Kohane DS. Prolonged sensory-selective nerve blockade. Proc Nat Acad Sci. 2010;107(8):3740–3745.
32. Yin Q, Ke B, Chen X, et al. Effects of liposomes charge on extending sciatic nerve blockade of N-ethyl bromide of lidocaine in rats. Sci Rep. 2016;6:38582.
33. Bereiter DA, Rahman M, Thompson R, Stephenson P. H S. TRPV1 and TRPM8 channels and nocifensive behavior in a rat model for dry eye. Invest Ophthalmol Vis Sci. 2018;59(8):3739–3746.
34. Huang CC, Yang W, Guo C, et al. Anatomical and functional dichotomy of ocular itch and pain. Nat Med. 2018.
35. Blake KJ, Baral P, Voisin T, et al. Staphylococcus aureus produces pain through pore-forming toxins and neuronal TRPV1 that is silenced by QX-314. Nat Commun. 2018;9(1):37.
36. Fuseya S, Yamamoto K, Minemura H, Yamaori S, Kawamata T. Systemic QX-314 reduces bone cancer pain through selective inhibition of transient receptor potential vanilloid subfamily 1–expressing primary afferents in mice anesthesiology ASA publications. Anesthesiology. 2016;125(1):204.
37. Shankarappa SA, Sagie I, Tsui JH, et al. Duration and local toxicity of sciatic nerve blockade with coinjected site 1 sodium-channel blockers and quaternary lidocaine derivatives. Reg Anesth Pain Med. 2012;37(5):483–489.
38. Zhang Y, Yang J, Yin Q, Yang L, Liu J, Zhang W. QX-OH, a QX-314 derivative agent, produces long-acting local anesthesia in rats. Eur J Pharm Sci. 2017;105:212–218.
39. Zhao W, Yang J, Zhang Y, Liu J, Zhang W. QX-OH/Levobupivacaine: fixed-dose combination to provide a long-acting postoperative pain of knee surgery in rodents. Eur J Pharm Sci. 2018;111:418–424.
40. Yin Q, Zhang Y, Lv R, et al. A fixed-dose combination, QXOH/ Levobupivacaine, produces long-acting local anesthesia in rats without additional toxicity. Front Pharmacol. 2019;10:243.
41. Schwarz SKW, Cheung MC, Ries CR, Sang ML, Macleod BA. Lumbar intrathecal administration of the quaternary lidocaine derivative, QX-314, produces irritation and death in mice. Anesthesiology. 2010;113(2):438–444.
42. Shen J, Fox LE, Cheng J. Differential effects of peripheral versus central coadministration of QX-314 and capsaicin on neuropathic pain in rats. Anesthesiology. 2012;117(2):365.
43. Werdehausen R, Fazeli S, Braun S, et al. Apoptosis induction by different local anaesthetics in a neuroblastoma cell line. Br J Anaesth. 2009;(5):5.
44. Stueber T, Eberhardt MJ, Hadamitzky C, Jangra A, Leffler A. Quaternary lidocaine derivative QX-314 activates and permeates human TRPV1 and TRPA1 to produce inhibition of sodium channels and cytotoxicity. Anesthesiology. 2016;124(5):1.
45. Stock K, Kumar J, Synowitz M, et al. Neural precursor cells induce cell death of high-grade astrocytomas through stimulation of TRPV1. Nat Med. 2012;18(8):1232.
46. Werdehausen R, Braun S, Essmann F, et al. Lidocaine induces apoptosis via the mitochondrial pathway independently of death receptor signaling. Anesthesiology. 2007;107(1):136–143.
47. Onizuka S, Tamura R, Yonaha T, et al. Clinical dose of lidocaine destroys the cell membrane and induces both necrosis and apoptosis in an identified Lymnaea neuron. J Anesth. 2012;26(1):54–61.
48. Doan LV, Eydlin O, Piskoun B, et al. Despite differences in cytosolic calcium regulation, lidocaine toxicity is similar in adult and neonatal rat dorsal root ganglia in vitro. Anesthesiology. 2014;120(1):50–61.
49. Takahashi K, Hayakawa C, Onimaru H. Effects of a quaternary lidocaine derivative, QX-314, on the respiratory activity in brainstem-spinal cord preparation from newborn rats. Neurosci Lett. 2016;619:121–125.
50. Tani M, Kotani S, Hayakawa C, et al. Effects of a TRPV1 agonist capsaicin on respiratory rhythm generation in brainstem-spinal cord preparation from newborn rats. Pflugers Archiv. 2017;469(2):327–338.
51. Rivera-Acevedo RE, Pless SA, Ahern CA, Schwarz SKW. The quaternary lidocaine derivative, QX-314, exerts biphasic effects on transient receptor potential vanilloid subtype 1 channels in vitro. Anesthesiology. 2011;114(6):1425–1434.
52. Zhao Y, Zhou C, Liu J, et al. The quaternary lidocaine derivative QX-314 produces long-lasting intravenous regional anesthesia in rats. PLoS One. 2014;9:6.
53. Cheung HM, Lee SM, MacLeod BA, Ries CR, Schwarz SK. A comparison of the systemic toxicity of lidocaine versus its quaternary derivative QX-314 in mice. Canadian J Anesthesia. 2011;58(5):443–450.
54. Tsuchiya H, Ueno T, Mizogami M, Takakura K. Local anesthetics structure-dependently interact with anionic phospholipid membranes to modify the fluidity. Chem Biol Interact. 2010;183(1):19–24.
55. Tsuchiya H, Mizogami M. R(+)-, Rac-, and S(-)-bupivacaine stereostructure-specifically interact with membrane lipids at cardiotoxically relevant concentrations. Anesth Analg. 2012;114(2):310–312.
56. Tsuchiya H. Membrane interactivity of charged local anesthetic derivative and stereoselectivity in membrane interaction of local anesthetic enantiomers. Local Reg Anesth. 2008;1(1):1–9.
57. Qi W, Qinqin Y, Jun Y, et al. Evaluation of the cardiotoxicity and resuscitation of rats of a newly developed mixture of a QX-314 analog and levobupivacaine. J Pain Res. 2017;10:737–746.
58. Puopolo M, Binshtok AM, Yao GL, Oh SB, Woolf CJ, Bean BP. Permeation and block of TRPV1 channels by the cationic lidocaine derivative QX-314. J Neurophysiol. 2013;109(7):1704–1712.
59. Rivera-Acevedo RE, Pless SA, Schwarz SKW, Ahern CA. Expression-dependent pharmacology of transient receptor potential vanilloid subtype 1 channels in Xenopus laevis oocytes. Channels. 2013;7(1):47–50.
60. Brenneis C, Kistner K, Puopolo M, et al. Phenotyping the function of TRPV1-expressing sensory neurons by targeted axonal silencing. J Neurosci. 2013;33(1):315–326.
61. Xu ZZ, Kim YH, Bang S, et al. Inhibition of mechanical allodynia in neuropathic pain by TLR5-mediated A-fiber blockade. Nat Med. 2015;21(11):1326–1331.
62. Youtian H, Xiaoyun Y, Shaoyong. QX-314 inhibits acid-induced activation of esophageal nociceptive C fiber neurons. Neurogastroenterol Motility. 2019.
63. Kissin I. Vanilloid-induced conduction analgesia: selective, dose-dependent, long-lasting, with a low level of potential neurotoxicity. Anesth Analg. 2008;107(1):271.
64. Ma QP. Expression of capsaicin receptor (VR1) by myelinated primary afferent neurons in rats. Neurosci Lett. 2002;319(2):0–90.
65. Petsche U, Fleischer E, Lembeck F, Handwerker HO. The effect of capsaicin application to a peripheral nerve on impulse conduction in functionally identified afferent nerve fibres. Brain Res. 1983;265(2):233–240.
66. Baranowski R, Lynn B, Pini A. The effects of locally applied capsaicin on conduction in cutaneous nerves in four mammalian species. Br J Pharmacol. 1986;89:2.
67. Henrich F, Magerl W, Klein T, Greffrath W, Treede RD, Capsaicin-sensitive C. A-fibre nociceptors control long-term potentiation-like pain amplification in humans. Brain. 2015;138(Pt 9):2505–2520.
68. Bianchi BR, Zhang X-F, Reilly RM, Kym PR, Yao BB, Chen J. Species comparison and pharmacological characterization of human, monkey, rat, and mouse TRPA1 channels. J Pharmacol Exp Ther. 2012;341(2):360–368.
69. Chen J, Kang D, Xu J, et al. Species differences and molecular determinant of TRPA1 cold sensitivity. Nat Commun. 2013;4.
70. De lR J, Eberhardt MJ, Klinger AB, et al. The molecular basis for species-specific activation of human TRPA1 protein by protons involves poorly conserved residues within transmembrane domains 5 and 6. J Biol Chem. 2013;288(28):20280–20292.
71. Mccoy DD, Palkar R, Yang Y, Ongun S, Mckemy DD. Cellular permeation of large molecules mediated by TRPM8 channels. Neurosci Lett. 2017;639:59–67.
72. Ongun S, Sarkisian A, Mckemy DD. Selective cold pain inhibition by targeted block of TRPM8-expressing neurons with quaternary lidocaine derivative QX-314. Communications Biol. 2018;1:1.
73. Hofmann ME, Largent-Milnes TM, Fawley JA, Andresen MC. External QX-314 inhibits evoked cranial primary afferent synaptic transmission independent of TRPV1. J Neurophysiol. 2014;112(11):2697–2706.
74. Alibrandi E. Neuronal circuitry for pain processing in the dorsal horn. Nat Rev Neurosci. 2010;11(12):823.
75. Okura D, Horishita T, Ueno S, et al. Lidocaine preferentially inhibits the function of purinergic p2x7 receptors expressed in xenopus oocytes. Anesth Analg. 2015;120(3):597–605.
76. Lim TK, MacLeod BA, Ries CR, Schwarz SK. The quaternary lidocaine derivative, QX-314, produces long-lasting local anesthesia in animal models in vivo. Anesthesiology. 2007;107(2):305–311.
77. Zhou C, Liang P, Liu J, et al. Emulsified isoflurane enhances thermal transient receptor potential vanilloid-1 channel activation–mediated sensory/nociceptive blockade by QX-314. Anesthesiology. 2014;121(2):280–289.
78. Hu S-P, Zhao -J-J, Wang W-X, et al. Coapplication of lidocaine and membrane-impermeable lidocaine derivative QX-222 produces divergent effects on evoked and spontaneous nociceptive behaviors in mice. Biomed Res Int. 2014;2014.
79. Shao CJ, Gao Y, Zhao L, Jin D, Wang D, Wang DQ. Co‐application of lidocaine and QX‐572 induces divergent pain behaviours in mice. J Pharmacy Pharmacol. 2015;67(9):1272–1278.
80. Sanchez J, Krause J, Cortright D. The distribution and regulation of vanilloid receptor VR1 and VR1 5ʹ splice variant RNA expression in rat. Neuroscience. 2001;107(3):373–381.
81. Kim H, Kim K, Li H, et al. Selectively targeting pain in the trigeminal system. Pain. 2010;150(1):29–40.
82. Zhao W, Yin, et al. Addition of dexmedetomidine to QX-314 enhances the onset and duration of sciatic nerve block in rats. Can J Physiol Pharmacol. 2018.
83. Kopach O, Zheng K, Dong L, et al. Nano-engineered microcapsules boost the treatment of persistent pain. Drug Deliv. 2018;25(1):435–447.
84. Zhang Y, Gong D, Zheng Q, Liu J, Zhang W. LC-MS/MS method for preclinical pharmacokinetic study of QX-OH, a novel long-acting local anesthetic, in sciatic nerve blockade in rats. J Pharm Biomed Anal. 2017;146:161–167.
85. Zhang Y, Yin Q, Gong D, Kang Y, Zhang W. The preclinical pharmacological study of a novel long-acting local anesthetic, a fixed-dose combination of QX-OH/Levobupivacaine, in rats. Front Pharmacol. 2019;10.
86. Newton DJ, Khan F, Belch JJF. Vasoactive characteristics of bupivacaine and levobupivacaine with and without adjuvant epinephrine in peripheral human skin. Br J Anaesth. 2005;5:5.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.