")
Back to Journals » Journal of Inflammation Research » Volume 17
Pyroptosis-Related Gene Signature Predicts Prognosis and Response to Immunotherapy and Medication in Pediatric and Young Adult Osteosarcoma Patients
Received 17 September 2023
Accepted for publication 21 December 2023
Published 20 January 2024 Volume 2024:17 Pages 417—445
DOI https://doi.org/10.2147/JIR.S440425
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tara Strutt
Chaofan Guo,1,2,* Xin Yang,3,* Lijun Li1,2
1Department of Orthopedics, Shanxi Provincial People’s Hospital, Taiyuan, Shanxi Province, People’s Republic of China; 2Department of Spine Surgery, Fifth Hospital of Shanxi Medical University, Taiyuan, Shanxi Province, People’s Republic of China; 3Department of Neurosurgery, Chongqing Fourth People’s Hospital, Chongqing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Lijun Li, Department of Orthopedics, Shanxi Provincial People’s Hospital, Shuangtasi Street No. 29, Taiyuan, 030012, People’s Republic of China, Tel +86 0351-4960081, Email [email protected]
Purpose: Pyroptosis, a new form of inflammatory programmed cell death, has recently gained attention. However, the impact of the expression levels of pyroptosis-related genes (PRGs) on the overall survival (OS) of osteosarcoma patients remains unclear. This study aims to investigate the impact of the expression levels of PRGs on the OS of pediatric and young adult patients with osteosarcoma.
Patients and Methods: Transcriptome matrix datasets of normal muscle or skeletal tissues from the Genotype-Tissue Expression (GTEx) project and osteosarcoma specimen the National Cancer Institute’s (NCI) Therapeutically Applicable Research to Generate Effective Treatments (TARGET) database were used to identify pyroptosis-related genes (PRGs) associated with prognosis. The National Center for Biotechnology Information’s (NCBI) GSE21257 dataset was employed to validate the predictive value of the pyroptosis-related signature (PRS). Additionally, reverse transcription polymerase chain reaction (RT-qPCR) experiment was performed in normal and osteosarcoma cell lines.
Results: The study identified 18 differentially expressed PRGs (DEPRGs) between normal muscle or skeletal tissues and tumor samples. Multiple machine learning techniques were used to select PRGs, resulting in the identification of four hub PRGs. A PRS-score was calculated for each sample based on the expression of these four hub PRGs, and samples were categorized into low and high PRS-score level groups. It was confirmed that metastatic status and PRS-score level are independent prognostic predictors. A nomogram model for predicting OS of osteosarcoma patients was constructed. Single-cell RNA-sequencing data display the expression patterns of the hub PRGs. RT-qPCR data results were found to be consistent with the differential expression analysis performed on TARGET and GTEx samples.
Conclusion: The study developed a novel pyroptosis-related gene signature that can stratify pediatric and young adult osteosarcoma patients into different risk groups, thus predicting their response to immunotherapy and chemotherapy.
Keywords: TARGET, nomogram model, immune microenvironment, drug sensitivity
Introduction
Osteosarcoma, the most common primary malignant bone tumour among all ages, often occurs in children and adolescents. It accounts for around 5% of all childhood cancers.1 The most frequent primary sites of osteosarcoma are distal femur, proximal tibia, and proximal humerus.2 When the cancer is diagnosed, 15–20% of patients already have shown macroscopic evidence of metastases, most (85–90%) of which occur in their lungs.3,4 Before the 1970s, surgical resection is the primary treatment for osteosarcoma. Notably, the introduction of chemotherapy since the 1970s has tremendously improved long-term survival rates of patients with osteosarcoma, which have risen from less than 20% to 65–70%.3
Programmed cell death (PCD) is an essential part of human development and tissue homeostasis enabling the removal of unwanted cells.5 Pyroptosis, a newly-discovered type of PCD,6 is a highly inflammatory process in which cells lose their cytomembrane intactness.7 This form of cell death, characterized by swelling, bubble-like protrusions, and shedding of membrane vesicles, leads to the release of intracellular contents and entry of annexin V.5,8 Recent studies have revealed the critical role of pyroptosis in tumour biology, particularly in osteosarcoma, where it has been associated with patient survival and disease progression. Additionally, the role of pyroptosis in osteosarcoma development, although not fully understood, is becoming increasingly recognized, emphasizing the need for further research in this area.9,10 Pyroptosis is mediated by gasdermin (GSDM) proteins, triggering immune cell activation and infiltration.11–13 The expression of GSDMD, in particular, is essential for the anti-tumour function of CD8+ T cells,14 and GSDME may act as a tumor suppressor.15 Evidence shows that pyroptosis plays a pivotal role in the pathogenesis and progression of osteosarcoma, influencing responses to chemotherapy.16
Given these advancements in molecular biology and bioinformatics, the identification of diagnostic and prognostic genomic biomarkers, including pyroptosis-related genes (PRGs), offers new opportunities for research.17 The exact mechanism by which PRGs influence the development of osteosarcoma remains an area of active investigation. Accurate models predicting outcomes in children with osteosarcoma are still lacking. Considering the limitations of existing osteosarcoma therapies, novel targeted therapeutics, particularly those addressing pyroptosis pathways, should be explored to enhance clinical outcomes.
Aimed at developing a nomogram-based prognostic model utilising PRGs for pediatric and young adult patients with osteosarcoma, this study conducted a comprehensive analysis of PRG expression in pediatric and young adult patients with osteosarcoma in this population. The results indicate that the pyroptosis-related signature (PRS) can serve as an independent prognostic factor for these patients. Additionally, this study explored variations in the immune microenvironment, in silico sensitivities, and responses to immunotherapy and chemotherapy, offering potential insights for osteosarcoma treatment.
Materials and Methods
Dataset Source
The datasets for the study were obtained from various databases. Whole-transcriptome sequencing [total RNA sequencing (RNA-Seq)] data of pediatric and young adult patients with osteosarcoma combined with their clinical characteristics were downloaded from National Cancer Institute’s (NCI) Therapeutically Applicable Research to Generate Effective Treatments (TARGET) database (platform: Illumina HiSeq 2000) via the Genomic Data Commons (GDC) portal (https://portal.gdc.cancer.gov/). The TARGET set was used as a training set and the creation of the PRS and the predictive model was performed on it.
In this research, Genotype-Tissue Expression (GTEx) project was employed as a reference resource of gene expression levels from “normal”, non-diseased tissues18 in this research. The expression data of normal muscle and skeletal samples (n = 803) within the Developmental GTEx (dGTEx) collection of RNA-seq experiments (platform: Illumina HiSeq 2000) were obtained from the GTEx portal (https://gtexportal.org/home/).
In order to externally validate the prognostic accuracy of the PRS established in the TARGET cohort, an independent validation set was used. The dataset GSE21257 (platform: GPL10295, Illumina human-6 v2.0 expression beadchip) consisted of expression data and survival information of patients with osteosarcoma was retrieved from National Center for Biotechnology Information’s (NCBI) Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo). To gain further insights into the OS tumour microenvironment and cellular heterogeneity, GSE162454 dataset comprising single-cell RNA sequencing (scRNA-seq) data was also downloaded from the GEO database.
Study Population
Cases whose survival information was not clearly recorded or OS time was less than 30 days were excluded from the training cohort and validation set. The final study population included a total of 137 patients, with 84 patients in the training set (the TARGET cohort) and 53 patients in the validation set. In the analysis of scRNA-seq data, six primary tumor samples were obtained from six patients with osteosarcoma.
Cell Culture and RT-qPCR
The differential expression of hub PRGs was detected in two cancer cell lines compared to the normal cell line using the reverse transcription polymerase chain reaction (RT-qPCR) experiment. Human normal osteoblast cell line hFOB1.19 and osteosarcoma cell lines, 143B and U-2 OS were used for this study. hFOB 1.19 cells were obtained from Procell Life Science & Technology Co., Ltd. (Wuhan, China), 143B cells from iCell Bioscience Inc. (Shanghai, China), and U-2 OS from National Collection of Authenticated Cell Cultures (Shanghai, China). hFOB 1.19 cell line was grown in Dulbecco’s modified Eagle medium (DMEM) (Hyclone, Logan, USA) (1:2) supplemented with 10% fetal bovine serum (FBS) (Gibco, Brazil), 0.3mg/mL G418 disulfate solution, and 1% penicillin/streptomycin, 143b in DMEM (1:2–1:3) supplemented with FBS, U-2 OS cell line in Roswell Park Memorial Institute-1640 (RPMI-1640) (Hyclone, Logan, USA) (1:2–1:3) supplemented with FBS. The hFOB1.19 cell line is cultured in a humidified atmosphere consisting of 95% air and 5% CO2 at a temperature of 34 °C. The 143B and U-2 OS cells are cultured with the same humidity and CO2 concentration, but at a temperature of 37 °C.
Total RNA was isolated from individual cell lines with TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. The integrity of the RNA was assessed by agarose gel-electrophoresis. The expression levels of mRNA encoding hub genes were determined by RT-qCR with GAPDH transcripts as a reference. cDNA was synthesised from 1 μg of total RNA with PrimeScrip II Reverse Transcriptase (200 U/μL) (Takara, Japan) in accordance with the protocol recommended by the supplier. qPCR reactions of 10 μL of SYBR FAST qPCR Master Mix (Kapa Biosystems, Woburn, MA, USA), 1 μL of template, 8 μL of ddH2O (DNase/RNase-Free), and 0.5 μM of forward and 0.5 μM of reverse primers specific for GAPDH and for each hub genes encoding cDNA. After initial denaturation at 94 °C for 3 min, cDNA was amplified by 40 PCR-cycles (denaturation at 95 °C for 5s, primer pair-dependent annealing at 56 °C for 10s, and elongation at 72 °C for 25s) followed by final elongation at 65 °C for 5s. Each cell line was tested using three samples, and each sample was tested in triplicate for all experiments. The primer pairs are presented in Table 1.
 |
Table 1 Reverse Transcription Polymerase Chain Reaction (RT-qPCR) Primer Sequences |
Statistical Analysis
The Kaplan–Meier (K–M) plot of survival and the Cox hazard ratio (HR) regression model are widely used to identify factors associated with a time-to-event response variable. In this study, overall survival (OS) was used as the endpoint. HR values from K–M survival curves or Cox HR regression model were used to represent the relative difference between the exposed group and the reference group. A hazard ratio of 1 indicates a lack of association with risk, an HR value greater than 1 suggests an increased risk, and an HR value below 1 indicates a smaller risk. The K–M survival difference between subgroups was compared using Log rank test.
Statistical analyses were performed with R v. 4.2.2 (R Foundation for Statistical Computing, Vienna, Austria). All p-values are two-sided. 95% confidence interval (CI) of hazard ratio (HR) values, Harrell’s concordance index (C-index) values, area under the curve (AUC) values of the time-dependent receiver operating characteristic (ROC) curve and integrated Brier score (IBS) were adopted. A signature with an AUC value of ROC curve above 0.6 and a p-value from the survival analysis below 0.05 was considered predictive.
Preprocessing of Clinical Information, RNA-Seq Data, and scRNA-Seq Data
For the training set and validation cohort, the cutoff of age was 13 y/o, which was determined using the “surv_cutpoint” function from the “survminer” package.19 As osteosarcomas often occur in lower extremities,20 the tumour primary site variable was classified into two subgroups: lower extremities and other sites (other sites or unknown for the GEO dataset).
The duplicate ensembles/probes mapping to the same gene symbol were averaged by the “avereps” function from the “limma” package.21 The log2-transformed tables of (fragments per kilobase of transcript per million fragments mapped [FPKM] + 1) and (transcripts per kilobase million [TPM] + 1) for the TARGET samples, as well as the data of normalised probe intensity for the GEO cases, were each subjected to quantile normalization using the “normalizeBetweenArrays” function from the “limma” package.21 When the GTEx read count matrix and the TARGET table were merged, only the shared gene symbols in the two datasets were retained. In addition, the “edgeR” package was used to standardised read counts of GTEx cohort and TARGET set, filter out lowly expressed genes, and correct the batch effect.
The scRNA-seq data quality control was performed using the “Seurat” package22 and the batch effects between samples were corrected using the “harmony” package.23 Filtering of the expression matrices was also done to ensure high-quality scRNA-seq data. Genes that expressed in less than 10 cells and cells that individually had less than 200 detected genes were excluded. The raw read counts obtained from single-cell RNA sequencing (scRNA-seq) were first transformed into log-normalized values employing the “NormalizeData” function, followed by scaling using the “ScaleData” function. Both of these functions are integral components of the “Seurat” package.22 The “SCTransform” method from the “sctransform” package24 and Gamma-Poisson generalised linear model (GLM) regression from the “glmGamPoi” package25 were then used to further normalise the data.
Comparison of Baseline Characteristics
The continuous non-normally distributed variable determined by the Shapiro–Wilk test is presented as the median [interquartile range (IQR)], and categorical variable as the number of cases (percentage). Bivariate associations between OS status and patients’ baseline characteristics (age, gender, tumour stage or grade, primary tumour site, metastatic status) in the TARGET cohort and GEO dataset were tested. The analysis of differences in categorical factors was conducted using Pearson’s chi-square test. However, if the expected count for any given cell was less than 5, Yates’ chi-square test was employed instead. A table of baseline characteristics was generated by “tableone” package.26
Identification, Biological Interaction Analysis, and Consensus Clustering of DEPRGs
A total of 79 PRGs, as detailed in Supplementary Table 1, were identified by summarizing previous studies27–33 and through the inclusion of a Gene Ontology (GO) term (GOBP_PYROPTOSIS) and a Reactome Pathway Database pathway (REACTOME_PYROPTOSIS) associated with pyroptosis. The gene lists of the GO terms and Reactome Pathways were downloaded from the MSigDB database (https://www.gsea-msigdb.org/gsea/msigdb). The differentially expressed PRGs (DEPRGs) between 803 normal muscle or skeletal samples in the GTEx cohort and 84 tumour tissues in the TARGET cohort were identified using a moderated independent sample t-test from the “limma” package.21 Genes with a false discovery rate (FDR) adjusted p-value < 0.05 and an absolute value of log2(fold-change) [log2(FC)] > 2 were considered significantly differentially expressed. The final DEPRG list is the intersection of the resultant DEG list and the PRG list. To visualise the results of differential expression analysis, heatmap and volcano plots of DEPRGs were generated by using the “ggplot2” package.34
Spearman rank correlation coefficients (r) calculated by the “Hmisc” package were used to quantify DEPRGs’ co-expression. p < 0.05 and |r| > 0.3 were used as cutoffs to indicate that two DEPRGs are co-expressed. All links between pairs of co-expressed DEPRGs were combined to construct a co-expression network. In addition to the co-expression network, the genes resulting from the differential expression analysis were also submitted to the Search Tool for the Retrieval of Interacting Genes database (STRINGdb, https://string-db.org/) version 12.0.35 This web tool includes protein–protein interactions (PPI) for the species Homo sapiens and was suitable for this study as the interactions were derived from systematic text-mining of a large amount of scientific literature. To fully investigate the PPI data, the exploration was configured to include all source parameters. PPIs with a confidence score of at least 0.400 were included in the network generation. The resulting PPI network was visualized using STRINGdb.36 Meanwhile, this repository was also used to perform a local network cluster (STRING) analysis. It is also noteworthy that transcription factors (TFs) are cis-acting “switches” that bind to TF binding sites (TFBSs) to modulate gene expression.37 The construction of a TF–mRNA regulatory network can help to clarify the complex regulatory relationships involved in various biological processes. Therefore, the ChIP-X Enrichment Analysis 3 database (https://maayanlab.cloud/chea3/) was utilised to conduct gene set enrichment analysis (GSEA) on DEPRGs against previously published ChIP-seq data (the literature ChIP-seq dataset) using Fisher’s exact test (FET).38 Among the set of TFs, cut-off FET p value was set to less than 0.05. The resultant TFs and the DEPRGs that are regulated by them were used to create a TF–DEPRG regulatory network. The DEPRG co-expression network and TF–DEPRG regulatory network were visualised with the assistance of the “ggraph” package.39
Then, the “ConsensusClusterPlus” package40 was utilised to generate consensus of samples according to the expression values of the DEPRGs and produce graphical plots indicating optimal number of groups (k). The K–M plot was visualised using the “survminer” package.19 The p value from the “survfit” object generated by the “survival” package41 was obtained using the “logrank_test” function from the “coin” package.42 A p-value in the survival analysis below 0.05 was considered significant.
Hub PRG Selection and Development of PRS–Score
Normalised log2(TPM+1) matrix was processed by the following machine learning (ML) methods to select the hub PRGs. Minimum redundancy maximum relevance (mRMR) method provided by the “mRMRe” package43 was applied to select 15 nonredundant genes into a subset. Uni- and multivariate Cox HR regression analyses and C-index value calculations were conducted with the “survival” package.41
Gene signatures with p < 0.05 and C-index > 0.6 at univariate analysis were considered as PRGs related to OS. These genes entered into a least absolute shrinkage and selection operator (LASSO)–penalised Cox HR regression (LASSO-Cox) model. At the same time, these signatures were also entered into a linear support vector machine (SVM) recursive feature elimination (SVM–RFE) regression model.44 In order to select the best hypermeter for the LASSO-Cox and linear SVM–RFE models, nested resampling cross-validation (CV) with a 5-fold inner loop for tuning hyperparameters was done; the outer loop also consisted of 5-folds with the held-out data used as a test set for unbiased performance estimation. In each inner loop for the LASSO-Cox model, an initial feature selection by survival C-index was also implemented to further reduce the feature space and tendency for overfitting. The optimal λ was determined using 5-fold CV. During each inner loop of the SVM–RFE model, a backward feature selection with 5-fold CV was executed under the guidance of the importance score ranking and the mean AUC value of subsets with the same number of variables. In addition, the significance of individual gene expression of PRGs related to OS toward the SVM classifier was determined by calculating the mean absolute Shapley additive explanation (SHAP) value of the feature across all samples (mean |ΔP|), where each SHAP value (ΔP) represents the contribution of a genetic variable toward the predicted probability of OS for an individual sample.45 SHAP values were calculated using 1000 Monte-Carlo sampling repetitions with the “fastshap” package46 and visualized as a bee swarm plot using the “shapviz” package.47 Finally, the LASSO-Cox and SVM–RFE models selected the most valuable features by applying optimal hyperparameters on the full TARGET dataset. Subsequently, the two sets were intersected to have a single subset of the genetic features. The two ML methods were done utilising the packages including “mlr3”,48 “mlr3proba”,49 “mlr3verse”,50 and “mlr3extralearners”.51
The multivariate Cox regression model relies on the assumption of proportional hazards (PH) across different variables.52 A multivariate Cox proportional hazards regression model was constructed based on the gene set selected by multiple ML methods. To test whether each feature in the model met the PH assumption, the “Cox’s.zph” function from the “survival” package41 was employed. Any gene that violated this assumption, as indicated by a p-value less than 0.05, was subsequently excluded from the model. A bi-directional stepwise procedure is a combination of forward selection and backward elimination.53 The screened variables from the last step were utilised to implement a multivariate Cox HR regression analysis with two-directional stepwise selection by the “StepReg” package.54 The stepwise approach started with the full model. Terms were automatically and sequentially removed and then reintroduced at each step of the algorithm, which was aimed at obtaining the minimum of corrected form of Akaike information criterion (AICc). Eventually, the genes in the resultant model were identified as hub PRGs.
The role of multiple hub PRGs in determining the risk of death in the TARGET cohort was modelled using multivariate Cox HR regression analysis. Each regression coefficient describes how a patient’s values of the corresponding predictor variable affects his/her death risk.55 To predict the risk in accordance with expression level [normalised log2(TPM+1)] of the hub PRGs, a continuous risk score, namely pyroptosis-related signature PRS-score, for each patient will be calculated as:

where PRS-scorei is the risk score for sample i, βij is the estimate (coefficient) computed from multivariate Cox HR regression models for sample i’s hub PRGj, and expij is the expression level of sample i’s hub PRG j. At the same time, in order to validate that the equation can accept FPKM RNA-seq, the PRS-score for each sample using normalised log2(FPKM+1) data as the hub PRGs’ expression level was also calculated.
The correlation coefficients between the expression levels [nomalised log2(TPM+1)] of hub PRGs and PRS-score in the TARGET cohort were calculated by the “Hsmic” package,56 and then was illustrated using the “circlize” package57 and “ComplexHeatmap” package.58
For the purpose of translating a continuous variable into a clinical decision, many medical researchers determined a cutoff point to stratify patients into two groups with each requiring a different kind of treatment.59 To divide the TARGET cohort into low and high PRS-score level groups, the “surv_cutpoint” function from the “survminer” package19 was utilised to determine the cutoff point for PRS-score variable. Samples with scores above the cutoff point were considered to have a high PRS-score level, while those below the cutoff point were considered to have a low PRS-score level. Independent t-test was also used to determine if there were differences in the expression level of hub genes between the high and low PRS-score level. Genes with an FDR adjusted p-value < 0.05 were identified as differentially expressed in the two groups. In the meanwhile, the PRS-score for each patient in GSE21257 cohort was calculated using the same coefficients and data of normalised probe intensity of hub PRGs. Since FPKM data and probe intensity totally differ from TPM data, the cutoff point to stratify the samples in the TARGET cohort based on the normalised log2(FPKM+1) table and the patients in GSE21257 cohort based on the normalised probe intensity matrix were recalculated using the same R function and arguments.
Evaluation of the Prognostic Accuracy of PRS–Score
The expression levels of hub PRGs between high and low PRS-score group of the TARGET cohort were compared using independent t-tests, and the p-values were FDR adjusted. Moreover, the prediction accuracy of the PRS-score was then internally and externally validated. Since the weighted expression levels of hub genes would continuously increase with the rank of patients’ risk, the performance of PRS-score in both the entire TARGET and GSE21257 cohorts was assessed and evaluated as the AUC values of 1-, 2-, 3-, 4-, and 5-year time-dependent ROC as implemented in the “timeROC” package.60 In order to compare the survival curves of the different PRS-score level groups in each cohort, K–M survival analysis was performed on the TARGET cohort and GEO. The overall survival of patients between the high- and low-risk level groups in each cohort was compared with the help of the “coin” package.42 A p-value of < 0.05 denoted a statistically significant result. Since the weighted expression levels of hub PRGs would continuously increase with the rank of patients’ risk, the performance of risk score in both the entire TARGET and GSE21257 cohorts was assessed and evaluated as the AUC values of 1-, 2-, 3-, 4-, and 5-year time-dependent ROC as implemented in the “timeROC” package.60 Moreover, to validate that the risk predictor can work well in predicting OS possibilities in subgroups stratified by demographic characteristics (ie, age and gender), 1-, 2-, 3-, 4-, and 5-year time-dependent ROC curves with their AUC values were also plotted.
The pyroptosis-related signature and other clinical feature(s) confirmed to be independent OS predictors were utilised to develop a nomogram-based prognostic model for predicting OS of pediatric and young adult osteosarcoma patients. To further investigate the association between PRGs and patients’ demographic or clinical information, differential analysis was performed on the expression level of each hub PRG and PRS-score against significant demographic or clinical factors in the multivariable Cox HR regression. Normally distributed data were compared using an independent t-test and non-normally distributed data were compared using a Wilcoxon rank test.
Additionally, nomogram-based predictive model performances were evaluated using the C-index for the model’s discriminatory power, IBS for mean squared prediction error, calibration curve for accuracy and calibration, and decision curve analysis (DCA) for utility. Similar to AUC of ROC, a C-index value of nomogram ranging from 0.5 to 1 assesses how well the model distinguishes between those with and without a dead outcome.61 The discrimination of the nomogram was evaluated on the training cohort and then validated on the validation set. IBS is an overall measure for the prediction of the model at all times.62 IBS of the nomogram model was computed using the “ipred” package63 with the parameter nbagg = 1 (a single survival tree). The upper and lower 95% CI were calculated by 1000-time bootstrap sampling using the “boot” package.64 In practice, a model with a C-index greater than 0.6 and an IBS below 0.25 was deemed useful. Meanwhile, calibration plots were generated based on 1000 bootstrap samples from both the TARGET and GSE21257 cohorts to compare the K–M method estimated and nomogram-predicted 1-, 2-, 3-, 4-, and 5-year OS rates.65 The calibration curve analyses in the training cohort and the validation set were carried out using the “rms” package.66 Time-dependent C-index values involving CV with 1000 bootstrap samples of each Cox regression model (Clinical model, PRS-score level model and nomogram model) were calculated and visualised utilising the “pec” package.67 Another novel discriminative performance measurement method, DCA, was simultaneously employed to internally validate the nomogram. Decision curves were constructed using the “stdca.R” script68 to evaluate the 1-, 2-, 3-, 4-, and 5-year performance of each model on the training set.
GO and KEGG Enrichment Analyses
Functional enrichment analyses were conducted with the objective of offering a functional interpretation for a group of genes sharing a common characteristic, such as differential expression.69 As such, significantly enriched GO terms and Kyoto Encyclopedia of Genes and Genomes (KEGG, https://www.kegg.jp/) pathways70 (FDR adjusted p-value < 0.05) were identified using hypergeometric distribution test [over-representation analysis (ORA)] and GSEA with the help of the “clusterProfiler” package.71 The GO database is categorised into three primary sections: cellular component (CC), molecular function (MF), and biological process (BP). The top 30 (if available) most significant terms in each GO category and pathways in the KEGG collection were ranked by risk ratio and visualised as lollipop charts using the “ggplot2” package.34 The KEGG pathway database comprises a set of pathway maps drawn by hand. For human species (Homo sapiens), these maps are divided into four distinct types: Environmental Information Processing, Cellular Processes, Organismal Systems, and Human Diseases. KEGG pathways that fall under the Human Diseases category were excluded from this analysis.
log2(FC) values between high and low PRS-score level groups were calculated from voom-transformed read count values with the “limma” package.21 After that, statistical significance of over-representation of the significantly down-regulated genes (log2(FC) < 0 and FDR adjusted p < 0.05) and up-regulated [log2(FC) > 0 and FDR adjusted p < 0.05] and between the two PRS-score level groups in GO terms and KEGG pathways was determined using ORA. From the top 30 significantly enriched KEGG pathways, those that involved the four hub genes or their gene family counterparts or those that were related to the skeleton were selected. Bayesian network diagrams were plotted using the “bngeneplot” function of the “CBNplot” package.72 The genes in these pathways serve as nodes, and expression levels serve as edges.
KEGG pathway GSEA was also performed using the differential expression ranking metric method.73 Then, the “aPEAR” package74 was used to leverage similarities between the significantly enriched KEGG pathways, and the “ggraph” package39 was utilised to represent the result as a network of interconnected clusters.
Analysis of Tumour Immune Microenvironment and Prediction of Response to ICB Therapy
In order to investigate immune contexture in 84 osteosarcoma samples from the TARGET programme, cell-type identification by estimating relative subsets of RNA transcripts (CIBERSORT) algorithm75 was used to process the gene expression matrix built from TPM data using the CIBERSORT R script v. 1.04 (University of Stanford, Stanford, CA, USA) with 1000 permutations. The output result of the CIBERSORT analysis describes the expression fingerprints of 22 immune cell phenotypes, including different cell types and functional states. The analytical tool estimates the cell fractions using ν-support vector regression (ν-SVR).76 For each type of immune cell, Wilcoxon rank sum test provided by the “rstatix” package77 was used to calculate the statistical significance of the difference between the two groups. Moreover, association strengths between the expression levels of hub genes presented in TPM and the fraction of 22 immune cells that showed significant difference between two PRS-score level groups were explored utilising Spearman rank correlation analysis. p-values below 0.05 were defined as statistically significant.
Immune Cell Abundance Identifier (ImmuCellAI) is a tool that can predict the response to immune checkpoint blockade (ICB) therapy.78 The gene expression data of the TARGET cohort were analysed by the ImmuCellAI tool using the ImmuCellAI web application at http://bioinfo.life.hust.edu.cn/ImmuCellAI, following the developer’s instructions. Immunotherapy responses outcome (0 = negative and 1 = positive) and scores of TARGET patients were retrieved from the output table. The association between PRS-score and ICB therapy score was assessed using the Spearman rank correlation coefficient. A p-value below 0.05 was considered indicating a significant correlation between the two variables. Apart from that, a logistic regression analysis was conducted to investigate the relationship between PRS-score and the ICB therapy outcome. An ROC curve was generated from this model, and the AUC was calculated. An AUC greater than 0.6 indicated that PRS-score had good discriminatory power in predicting the ICB response outcome.
Analysis of scRNA-Seq Data
The “Seurat” package22 was used to perform Uniform Manifold Approximation and Projection (UMAP) analysis on cell-topic distributions to further reduce the dataset to two dimensions and then apply a density-based clustering method proposed by Macosko, et al79 to identify potential clusters. The clustering results from the scRNA-seq dataset are visualised using the “ggplot2” package.34 Subsequently, cell types were annotated based on the accurate clustering of the cells using the LM22 gene signature file for CIBERSORT immune cell types75 in the “MAESTRO” package.80 Additionally, markers from the research conducted by Zhou et al81 were also used. The minimum number of overlapped gene signature was set to 1, and if the score of all input signatures was less than 1, the cluster will be annotated as “Others”. Finally, the expression level of each hub PRG in main cell clusters was visualised by the “ggplot” function from the “ggplot2” package34 and “Dotplot” function provided by the “Seurat” package.22
In-Silico Drug Sensitivity Prediction
The “pRRophetic” package82 was used to fit models for baseline gene expression [log2(TPM+1)] and then yield in vivo drug sensitivity [also known as the half-maximal inhibitory concentration (IC50)] predictions.
Afterwards, differences in IC50 value of 251 drugs from the Cancer Genome Project (CGP) 2016 database83 between the high and low PRS-score level groups were statistically analysed with the help of Wilcoxon rank-sum test. A lower IC50 value [-log10(IC50)] means that the drug could work at a lower concentration. The co-relationship between predicted and measured drug sensitivity values was measured by Spearman correlation test. Two of the above-mentioned tests were performed using the “stats” package,84 and the drugs whose p-values from the two tests were both less than 0.001 could be candidates for the treatment of either low or high PRS-score level group.
RT-qPCR Data Analysis
RT-qPCR data were calculated by the comparative cycle threshold (Ct) (ΔΔCt) method, using GAPDH as the internal control. The relative expression level of each hub gene was shown as a 2–ΔΔCt value and was normalised to the control cell value, which was set to be 1. To analyze the differential expression of the hub gene in cancer and normal cell lines, pairwise independent t-tests were performed and p values were adjusted for multiple testing using FDR method by the “rstatix” package77 and visualised as a bee swarm plot using the “ggplot2” package34 and “ggbeeswarm” package.85
Results
Patient Demographics
Of the 84 pediatric and young adult patients diagnosed with osteogenic sarcoma in the TARGET cohort, the median age was 14 [IQR: 12–17] y/o. Fifty-five were alive with disease (65.48%), and 29 (34.52%) were died. The distribution of gender was almost equally distributed with 37 participants (44.05%) being female and 47 participants (55.95%) being male. Complete demographic and pathological information of the TARGET cohort (training set) and GSE21257 set (validation set) is displayed in Table 2.
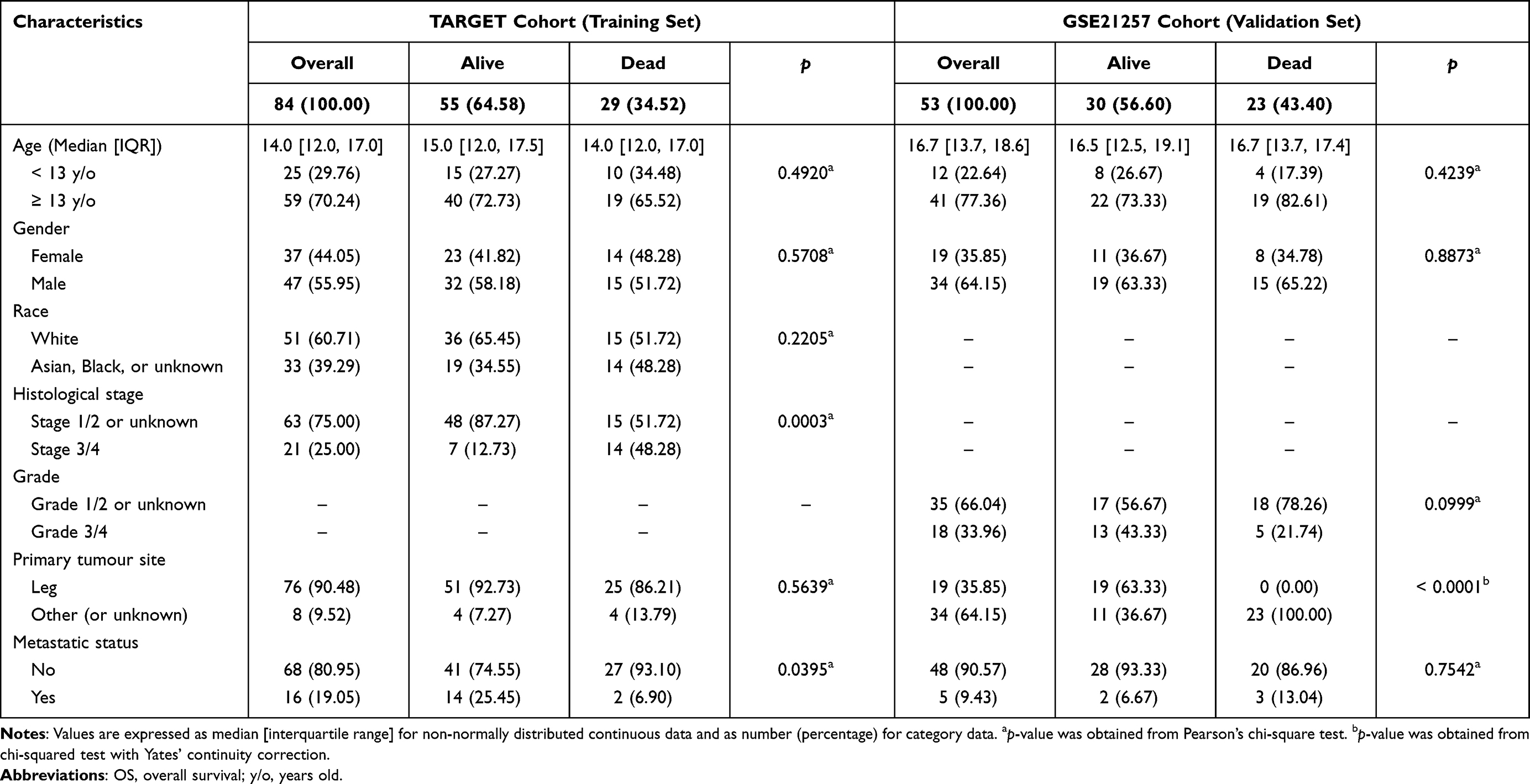 |
Table 2 Demographic and Clinical Information of the TARGET Cohort (Training Set) and GSE21257 Cohort (Validation Set) |
Identification, Construction of Biological Network, and Consensus Clustering of DEPRGs
Initially, to explore PRG dysregulation occurring in pediatric and young adult patients with osteosarcoma, a comparison of gene expression profiles between normal tissues from the GTEx (n = 803) cohort and malignant samples from the TARGET dataset (n = 84) was conducted. The results are presented in Supplementary Table 2 and Figures 1A–C. As illustrated in the Venn plot (Figure 1A) and the volcano plot (Figure 1B), the analysis of differential expression has revealed that 18 out of the 79 PRGs present in the combined RNA-seq dataset are associated with cancer, comprising 7 down-regulated PRGs [log2(FC) < −2, FDR adjusted p < 0.05, sky blue scatters in Figure 1B] and 11 up-regulated PRGs [log2(FC) > 2, FDR adjusted p < 0.05, tomato red scatters in Figure 1B]. The cluster heatmap (Figure 1C) based on the expression of DEPRGs also displays similar information on the whole. Steel blue colour means down-regulation and red colour signifies up-regulation. The rows of RNA-seq heat map represent PRGs, and the columns represent samples. Each cell is colorised based on the level of expression of that gene in that sample. Gene symbols (steel blue: down-regulated PRGs, red: up-regulated PRGs) with corresponding statistical significance symbols are shown on the right part of the illustration.
 |
Figure 1 Identification, construction of biological network, and consensus clustering of differentially expressed pyroptosis-related genes (PRGs) (DEPRGs). (A) Venn diagrams showing polygenic overlap between down-regulated PRGs (sky blue), up-regulated PRGs (tomato red), and PRGs (gold). DEPRGs (down-regulated and up-regulated genes) were determined in the TARGET osteosarcoma specimen (n = 84) vs GTEx normal biopsies (n = 803) according to |log2(fold change[FC]) and -log10(false discovery rate [FDR] adjusted p-value) with a |log2(FC)| threshold of 2 and an FDR adjusted p-value threshold of 0.05. (B) Volcano plot showing that 18 PRGs are significantly different between the tumour specimen and normal biopsies (dark blue dot: down-regulated PRG, red dot: up-regulated PRG, grey dot: non-significant PRG). Each DEPRG is labelled with its symbol. (C) Cluster heatmap visualising the 18 DEPRGs identified in TARGET (n = 84) tissues vs GTEx samples (n = 803). (D) Network visualisation of co-expression of DEPRGs (blue circle: down-regulated PRG, red circle: up-regulated PRG, circle size: degree, sky blue line: negative correlation, tomato red line: positive correlation). (E) Protein–protein interaction (PPI) network of 18 DEPRGs from the STRING database. The circle nodes in the diagram represent DEPRGs (node colour: InterPro term that the gene are involved; sky blue halo: down-regulated expression, tomato red halo: up-regulated expression), and the edges represent node interactions (confidence cutoff = 0.400, interaction network p < 0.001). (F) Heatmap plot of enriched local network clusters (STRING) (FDR adjusted p-value < 0.05) and involved proteins from the STRING database. (G) DEPRG–transcription factor (TF) (mRNA–TF) regulatory network. 12 interacting TFs of DEPRGs were identified by the ChEA3 tool [blue circle: down-regulated PRG, red circle: up-regulated PRG, green circle: transcriptional activators (OR > 1), circle size: degree, sky blue line: negative correlation, pink line: upregulated correlation]. (H) Consensus matrix (CM) plot for k = 2 (k = 2–6). CM plots can be used to find the “cleanest” cluster partition where items nearly always either cluster together giving a high consensus (dark blue colour) or do not cluster together giving a low consensus (white colour). (I) Cumulative distribution function (CDF) plot for each k (k = 2–6). CDF plot is used to determine the value of k at which the distribution approaches its approximate maximum. (J) Kaplan–Meier (K–M) plot displaying a significant difference in OS probability between the two clusters. *p < 0.05, ***p < 0.001. Abbreviations: PUB, peptide:N-glycanase; UBA, UBX-containing proteins; DAPIN, domain in apoptosis and inflammation; TRAIL, tumour necrosis factor-related apoptosis-inducing ligand signaling. |
The co-expression network of DEPRGs was constructed using the outcome of Spearman correlation test in accordance with the selection criteria (p < 0.05 and |r| > 0.3). As shown in Figure 1D and Supplementary Table 3, the resulting network contains 13 DEPRGs (3 down-regulated PRGs represented by blue circles and 10 up-regulated PRGs represented by red circles) connected by 26 expression interactions. Two of the total connections are negative, indicated by sky blue lines, while 24 are positive, represented by orange lines. In the DEPRG co-expression network, the genes with the most co-expression partners are the GSDMA, IL1B, and TREM2, each of which has six positively correlated pairs.
To further estimate the connections of typical DEPRGs, protein–protein association data of 18 DEPRGS were extracted from STRING v. 12 (Figure 1E and Supplementary Table 4). The mined PPI result consisted of a total of 44 PPIs as shown in Figure 1E (p < 0.001). The colored halos around the protein bubbles are displayed in a color gradient from sky blue to tomato red according to the log2(FC) from the differential expression analysis. Sky blue represents a down-regulated expression, and tomato red signifies an up-regulated expression. Manual curation of the PPI data revealed that two out of 18 proteins individually had >6 interacting partners in the network. AIM2 has nine partners, and CASP5 has eight. Additionally, two members of the chromatin modifying protein (CHMP) family, namely CHMP2A and CHMP4C, were found to interact with each other, and they together form a separate interaction pair. Additionally, the TP63 gene did not interact with any other genes. To further elucidate the functional implications of these DEPRGs, protein enrichment analysis was simultaneously performed. As shown in Figure 1F, eight DEPRGs are involved in four local network clusters (STRING). The first three clusters are closely related to the inflammasome complex, indicating a strong association of these genes with inflammatory response and pyroptosis. The presence of GSDM and domain in apoptosis and inflammation (DAPIN) domain proteins in the first two clusters suggests a role in pore formation and cell death execution. The fourth cluster is associated with tumour necrosis factor (TNF)-related apoptosis-inducing ligand signaling (TRAIL), which is known to be involved in apoptosis, and possibly indicates a cross-talk between apoptosis and pyroptosis pathways.
Analyzing the biological circuitry of TF–DEPRG (TF–mRNA) interactions can uncover the molecular mechanisms driving osteosarcoma progression at the molecular level. GSEA was performed using the ChEA3 tool. Twelve TFs (JUN, RELA, STAT4, ETV2, STAT3, VDR, GBX2, MNX1, KLF5, TCF21, BACH1, and SALL4) that are believed to regulate the DEPRG expression were identified (Figure 1G and Supplementary Table 5). Notably, all of these TFs are transcriptional activators (OR > 1). The TF–mRNA regulatory network is shown in Figure 1G.
After the establishment of biological networks, the consensus clustering (CC) method was used to process the expression matrix of DEPRGs. The consensus matrix (CM) plots (Figure 1H) and consensus cumulative distribution function (CDF) plot (Figure 1I) thereby generated identified the optimal cluster number was two. Consequently, the samples were divided into two different clustering subtypes. The K–M plot (Figure 1J) indicates that there is a significant difference in OS probability between the two clusters (Cluster 2 vs Cluster 1 HR = 2.562 [95% CI: 1.215–5.403], p = 0.0118).
Establishment of Pyroptosis-Related Signature
A series of ML techniques were employed to select the PRGs of interest. Initially, mRMR criterion based on mutual information was used to maximize the relevancy of a subset of 15 PRGs (NLRP2, IL6, CASP8, GSDMC, CHMP4A, TREM2, CHMP4C, ELANE, GZMA, TP63, PYDC1, GBP5, CASP5, GSDMA, and TNFRSF21) while minimizing the redundancy among them.
Afterwards, univariate Cox HR analysis indicated that five genes (TNFRSF21, GSDMA, CHMP4C, TREM2, and GZMA) individually have a possible effect on the patient’s OS (each p < 0.05 and C-index > 0.6). The results are shown in Supplementary Table 6.
Both ML methods including an internal 5-fold CV step were adopted to further screen genetic variables. To avoid over-fitting of the final multivariate model, LASSO-Cox regression was performed. Five genes (GSDMA, TNFRSF21, GZMA, CHMP4C, and TREM2) with non-zero coefficients at the LASSO penalization level (λ = 0.042) that minimises partial likelihood deviance (PLD) were selected as the effective sets. Figure 2A shows the PLD from the LASSO-Cox fit, and Figure 2B indicates the estimated coefficients. In a LASSO-Cox regression analysis, the sign of the coefficient for each variable reflects whether it is a risk factor or a protective factor for survival outcomes. In this analysis, the coefficient for CHMP4C was positive, indicating that its presence may decrease the likelihood of OS. On the other hand, the coefficients for GSDMA, TNFRSF21, GZMA, and TREM2 were negative, indicating that their presence may increase the likelihood of OS.
 |
Figure 2 Hub PRG selection and development of pyroptosis-related signature (PRS)–score. (A) Plot indicating the optimal λ selection by 5 cross-validated (CV) partial likelihood deviance (PLD) of the least absolute shrinkage and selection operator (LASSO)–Cox regression. 5-fold CV PLD was plotted against ln(λ). (B) Plot of the estimated coefficients from the LASSO–Cox regression against ln(λ). Finally, 5 genetic features being non-zero were selected at optimal λ = 0.042. (C) Plot of the support vector machine (SVM) recursive feature elimination (SVM–RFE) regression model information criteria as a function of the subsets generated by RFE algorithm. 5-fold CV mean AUC was plotted against number of selected variables. The red dot represents that the optimal number of selected variables is five. (D) Lollipop plot showing the normalised importance value of each of the five selected features in the linear–SVM model. (E) Shapley additive explanations (SHAP) values using 1000 Monte-Carlo sampling repetitions for the linear–SVM model. The y-axis signifies genes and the x-axis represents the corresponding SHAP values for each data instance. (F) Forest plot showing multivariable Cox regression coefficient of each hub PRG. (G) Chord diagram presenting the pairwise correlation between expression level of hub PRGs and PRS-score. The connecting chords are displayed in a colour gradient from light green to bluish green in accordance with the Spearman correlation coefficients ranging from −1 to 1, and their widths are also proportional to the |r|. |
Also, a feature subset selection comprising five genes (CHMP4C, GSDMA, TNFRSF21, GZMA, and TREM2) with the maximum mean AUC was obtained from the SVM–RFE algorithm (Figures 2C and D). Notably, features with larger absolute SHAP values are more important for prediction about their positive or negative effects on the endpoint depending on its sign.86 From Figure 2E, it can be inferred that TREM2, CHMP4C, GASM, and GZMA were mostly overexpressed in osteosarcoma patients when predicting death outcomes. By contrast, TNFRSF21 showed a reverse pattern, with mostly under-expression when important for the model.
Then, the PRGs in the intersection subset of LASSO-COX selected features and SVM–RFE selected markers were entered into bi-directional multivariate Cox HR analysis to establish the polygenetic predictor for patients’ OS. Eventually, TNFRSF21, GSDMA, GZMA, and CHMP4C were selected to be the hub PRGs. The results are presented in Supplementary Table 7.
The multivariate Cox HR model established in the TARGET cohort was used to estimate the coefficients associated with each patient’s death risk. A PRS-score was assigned to each patient in the TARGET cohort using a linear weighted combination of the expression level of the hub PRGs:

As indicated in the equation, TNFRSF21, GSDMA, and GZMA showed a favourable effect on prognosis, while CHMP4C indicated an adverse association. The coefficients were displayed using a forest plot (Figure 2F). Subsequently, a chord diagram (Figure 2G) was used to present pairwise inter-relationship between expression levels of hub PRGs and PRS-score. Each variable is represented by a scaled arc on the outer part of the circular layout. The connecting chords are displayed in a colour gradient from cyan to red in accordance with the Spearman correlation coefficients ranging from −1 to 1, and their widths are also proportional to the |r|. As expected, each of the hub PRGS was strongly connected with PRS-score. The following steps will assess and evaluate whether the four-PRG-based signatures can enhance the prediction performance when combined with clinical prognostic factors such as metastatic status. The forest plot in Figure 2F shows the coefficients of the multivariate Cox HR regression for the four hub PRGs.
Confirmation of the Predictive Value of PRS
The PRS-score of each sample in the training set was calculated using the coefficients in the equitation and normalised log2(TPM+1) data. The coefficient and formula remained unchanged when evaluating the cases in GSE21257 dataset. To classify samples into specific PRS-score level groups, thresholds of PRS-score in the training cohort (cutoff = −2.32431) and the validation set (cutoff = −7.28264) were, respectively, determined using the “survminer” package. For each dataset, the samples with PRS-score less than the threshold were divided into lower PRS-score level group, and the others high PRS-score level group. The distribution of risk scores ordered from low to high against the rank of patients in the TARGET set and GSE21257 cohort and the corresponding cutoffs are shown in Figure 3A and F.
 |
Figure 3 Association between the pyroptosis-related signature (PRS) and overall survival (OS) of the TARGET and GSE21257 patients. For the TARGET cohort, the PRS was constructed using the normalized value of log2(TPM+1); for GSE21257 dataset, the PRS was constructed using the normalized value of probe intensity. The distribution of each patient’s PRS-score ordered from low to high in (A) the TARGET cohort and (F) GSE21257 dataset. Patients in each dataset were individually divided into two PRS-score level groups based on the cutoffs calculated by the “survminer” package. Scatter diagram of the OS time against the patients’ rank of PRS-score in (B) the TARGET cohort and (G) GSE21257 dataset. Boxplot depicting the expression level of each hub gene was significantly different between the PRS-score groups in (C) the TARGET cohort and (H) GSE21257 dataset. Low PRS-score tissues marked in green and high PRS-score samples marked in Orange. The independent sample t-test was applied for comparing the differences, and the p-values were FDR adjusted. Kaplan–Meier (K–M) plot demonstrating elevation in OS probability in the TARGET patients with low PRS-score in (D) the TARGET cohort and (I) GSE21257 dataset. p-value was from a Log rank test. Plots of the time-dependent receiver operating characteristic (ROC) curves for the PRS-score prognostic model for 1-, 2-, 3-, 4-, and 5-year OS and the corresponding AUC values in (E) the TARGET cohort and (J) GSE21257 dataset. Each AUC value is represented in the legend as the estimated value [95% CI]. ROC curves demonstrating elevation in OS probability in each gender subgroups of the TARGET cohort: (K) Female and (L) Male, with low PRS-score. Plots of the time-dependent ROC curves for the PRS-score prognostic model in each age subgroups of the TARGET cohort: (M) < 13 and (N) ≥ 13, for 1-, 2-, 3-, 4-, and 5-year OS and the corresponding AUC values. The AUC value is represented in the legend as the estimated value [95% confidence interval (CI)]. *p < 0.05, **p < 0.01, ***p < 0.001. |
The simplest plots indicating the discrimination of a model are probably a scatter diagram of the OS time against the patients’ rank of PRS-score. Figure 3B indicates that the discrimination of the pyroptosis-related predictive model in the TARGET cohort is strong, and Figure 3G confirms that the model has a good ability to discriminate between high- and low-risk patients in the validation cohort. Moreover, all the four hub genes were significantly differentially expressed between high and low PRS-score level group osteosarcoma samples. As shown in the boxplot of hub PRG expression levels [normalised log2(TPM+1)] (Figure 3C), CHMP4C (FDR-adjusted p-value < 0.001) exhibited significantly higher expression in the high PRS-score group (n = 31) compared to the low-risk group (n = 53), whereas TNFRSF21, GSDMA, and GZMA (all FDR-adjusted p-value < 0.001) showed lower expression in the high-risk group. The results of differential gene expression analysis of the four hub genes in the validation set (Figures 3H) were completely consistent with their differential trends in the training set (the low-risk group: n = 16, the high PRS-score group: n = 37).
Also, the performance of the PRS-score was evaluated by K–M analysis. As shown in Figure 3D and I, the risk predictor works well in predicting OS possibilities in both TARGET (High vs Low HR = 3.870 [95% CI: 1.745–8.583], p < 0.001) and GSE21257 cohorts (High vs Low HR = 2.694 [1.151–6.306], p = 0.0463), as OS was significantly shorter for the patients in the high PRS-score level group compared with samples in the low PRS-score level group. Furthermore, the predictive ability is assessed with the AUC of time-dependent ROC curves at 1-, 2-, 3-, 4-, and 5-year. Figures 3E and J indicate that the AUC values for the PRS-score prognostic model for 1-, 2-, 3-, 4-, and 5-year OS in the two datasets were greater than 0.6. The accuracy of the pyroptosis-related signature was also validated on the FPKM data, and the results are shown in Supplementary Figure 1. Moreover, the results of time-dependent ROC analyses in various clinical subgroups (Figures 3K–N) indicated that the PRG–based prognostic signature has a good discriminatory capacity in different demographic phenotypes of osteosarcoma patients.
Univariable and multivariable Cox HR regression was performed to assess for variables significantly associated with OS, and a summary of the results is displayed in the forest plot (Figure 4A). Because patients’ gender and age were not found to be statistically significant in univariate analyses, these variables did not enter the final models. In the multivariable analyses, metastasis occurring (HR = 4.020 [95% CI: 1.911–8.459], p < 0.001) and high PRS-score level (HR = 3.371 [95% CI: 1.565–7.259], p < 0.001) were significantly associated with worse OS in the TARGET dataset.
 |
Figure 4 Development, and internal and external validation of pyroptosis-related gene (PRG)-based prognosis model. (A) Forest plot of uni- and multivariable Cox hazard ratio (HR) model for OS in the TARGET cohort. The diagram reports the HR value and the corresponding 95% CI for each covariate (blue circle and strap: HR and 95% CI band from univariable Cox HR model, red square and strap: HR and 95% CI band from multivariable Cox HR regression). (B) A nomogram for predicting the 1-, 2-, 3-, 4-, and 5-year OS possibilities in the pediatric and young adult osteosarcoma patients relying on the TARGET population. To use the nomogram, the value for each predictor (metastatic status and PRS-score level) is determined by drawing a line upward to the point reference line, the points are summed, and a line is drawn downward from the total points line to find the predicted 1-, 2-, 3-, 4-, and 5-y OS possibilities. The diagram also prints how many linear predictor units there are per point and the number of points per unit change in the linear predictor (lp). Rain cloud plots demonstrating significant differences in (C) TNFRSF21 expression, (D) GZMA expression, and (E) PRS-score between different metastatic status. (F) Time-dependent c-index curves for PRS-score level, clinical characteristic, and nomogram models. Calibration curves of K–M vs nomogram predicted (G) 1-, (H) 2-, (I) 3-, (J) 4-, and (K) 5-year OS for the original patient (TARGET) cohort and GSE21257 dataset. DCA for the Clinical, PRS-score level, and Nomogram models built to predict the (L) 1-, (M) 2-, (N) 3-, (O) 4-, and (P) 5-year OS probability based on records of patients in the training cohort (Orange bar: range where the Nomogram model outperformed than others, deep blue bar: range not relevant to the Nomogram model). ** p < 0.01; *** p < 0.001. |
The nomogram to estimate 1-, 2-, 3-, 4-, and 5-year OS probabilities was built using metastatic status and PRS-score level variables of the TARGET dataset. The corresponding point scales are shown in Figure 4B. The steps for using the nomogram are to (1) determine the patient’s value for each predictor, (2) draw a straight line upwards from each predictive value to the top point reference line, (3) sum the points from each predictive variable, (4) locate the sum on the total points reference line, and (5) draw a straight line from total points line down to the bottom probability lines to obtain the patient’s likelihood of 1-, 2-, 3-, 4-, and 5-year OS. Figures 4C–E indicated significant differences in TNFRSF21 expression (p < 0.001), GZMA expression (p = 0.0282), and PRS-score (p = 0.0019) between different metastatic states.
The discrimination of the PRS-score as a continuous variable was compared with another genetic risk predictor proposed by other scholars. The coefficients and gene symbols were obtained from the article of Cao et al,87 and then the risk score was calculated by the sum of these genes’ expression level [normalised log2(TPM+1)] weighted by the coefficients. In this research, the PRS-score prognostic model showed better predictive performance, as its C-index (0.728 [95% CI: 0.686–0.771]) was higher compared to that of predictors of Cao et al (0.714 [95% CI: 0.666–0.762]).
The nomogram-based prognostic model incorporated the two identified independent predictive factors demonstrated excellent discrimination and calibration. Based on the training cohort, the C-index was 0.776 [95% CI: 0.737–0.816] (IBS: 0.138 [95% CI: 0.106–0.170]) for the model; with regard to the test dataset, the result remained consistent, with a c-index of 0.757 [95% CI: 0.707–0.806] (IBS: 0.073 [95% CI: 0.054–0.092]). Slope of calibration curve evaluating the agreement between observed and predicted values with values closer to 1 demonstrates an ideal performance.61
In order to internally validate that the nomogram model has better predictive accuracy than any other Cox HR regression models established using a single predictor (metastatic status or PRS-score level group), calibration plots, discrimination curves, and DCA curves for the three models were plotted. Also, medical research articles typically evaluate the C-index at the maximum follow-up time in the cohort study and by ignoring unusable pairs. In fact, it would be preferable to truncate the concordance index at each earlier time point than the maximum follow-up time.88 Figure 4F shows the estimated time-independent C-index of the three models over time using the 1000-repetition bootstrap CV procedure. The estimated C-index of Nomogram model (represented by green line) was greater than other models (clinical model represented by blue line and PRS-score level model represented by blue line) at any prediction horizon, indicating that it has the best discrimination. The 1000-resampling calibration plots demonstrate good performance of the predictive model for the training cohort and the validation set (Figures 4G–K) at 1-, 2-, 3-, 4-, and 5-year, as there are only small deviances from the 45° reference line. In a DCA plot, clinical usefulness is displayed where a model has greater net benefit than other types of intervention.89 Decision curves in Figures 4L–P show that the nomogram model established on the TARGET cohort (represented by red line) were greater than the any other options [“All (intervention for all patients)” represented by black line, “None (intervention for none)” represented by black line, “PRS-score level” represented by cyan line, or “Clinical” represented by green line], since it had the highest net benefit across that range to predict the 1-, 2-, 3-, 4-, and 5-year OS probability. The orange bar in each DCA plot represents a threshold probability range where the nomogram model outperforms than other models. The value of the Nomogram model was sometimes lower than that of PRS-score level or clinical model, which was due to random noise.89
Enriched GO Terms and KEGG Pathways
To investigate biologic features shared by the two PRS-score level groups, over-representation of significantly 3575 down-regulated [log2(FC) < 0 and FDR adjusted p < 0.05] genes and 3453 up-regulated genes [log2(FC) > 0 and FDR adjusted p < 0.05] in GO terms and KEGG pathways was tested. These enriched terms and pathways may play a key role in tumour cell pyroptosis.
In the gene functional enrichment ORA analysis, DEGs between low and high risk level groups from the training set (the TARGET cohort) were employed to determine gene ontologies and significant pathways. Figures 5A–C display the top 30 most enriched terms in the three GO categories. These DEGs genes were enriched in several pathways, predominantly related to immune response regulation. Some of the key BP terms (Figure 5A) include the production of molecular mediators of immune response, regulation of lymphocyte activation, immune response-regulating signaling pathway, immunoglobulin production, and activation of immune response. In terms of CC ontology (Figure 5B), these genes were found to be associated with the external side of the plasma membrane, immunoglobulin complex, receptor complex, T cell receptor complex, and plasma membrane signaling receptor complex, among others. With regard to MF ontology (Figure 5C), these DEGs were involved in antigen binding, immune receptor activity, MHC protein complex binding, peptide antigen binding, and cytokine receptor binding. These findings suggest that DEGs are intricately linked to the orchestration of the immune response in the context of osteosarcoma, potentially influencing patient prognosis. Research has shown that approximately 15% to 20% of all cancer cases are preceded by infection, chronic inflammation, or autoimmunity in the same tissue or organ site.90
 |
Figure 5 Diagrams of functional enrichment analysis outcomes of 7028 differentially expressed genes (DEGs) in TARGET patients with low vs high pyroptosis-related signature (PRS)–score level. The size of dot in each plot represents the number of DEGs which are involved in the corresponding Gene Ontology (GO) term or Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway. GO lollipop plots showing fold enrichment of the top 20 (if available) mostly significantly overrepresented gene terms with a false discovery rate (FDR) p-value threshold of < 0.05 in various ontologies: (A) biological processes (BP), (B) cellular components (CC), and (C) molecular functions (MF). (D) KEGG lollipop plots showing fold enrichment of the top 20 (if available) mostly significantly overrepresented pathways with an FDR p-value threshold of < 0.05. The Bayesian network plots depicted the linkages of genes and three representative enriched KEGG pathways: (E) cytokine-cytokine receptor interaction, (F) tumour necrosis factor (TNF) signaling pathway, and (G) osteoclast differentiation. (H) Clustering network of significantly enriched KEGG pathways in the GSEA analysis. The nodes represent the significant KEGG pathways and the edges represent similarity between them and are coloured by normalised enrichment score (NES). The lines connected similar pathways are coloured by similarity. |
Figures 5D display the enriched terms in the three KEGG categories. The ORA–KEGG enrichment analysis points to a multifaceted role of immune signaling pathways and cellular processes in the disease state, with a strong emphasis on the interaction between the tumor and the immune system. In addition, the “Osteoclast differentiation” pathway, directly tying into the skeletal system and suggesting a link between the immune response and bone remodeling or degradation, which is a critical aspect of osteosarcoma pathology.
The Bayesian network plots (Figures 5E–G) depicted the linkages of genes and three representative enriched KEGG pathways as networks. The genes that belong to these pathways are clearly present in the diagram. As indicated by Figures 5E and F, TNFRSF21 and its gene family counterparts were implicated in the regulation of certain pathways among the most significantly enriched in the KEGG analysis. Specifically, TNFRSF21 and numerous members of the TNFRSF family are involved in the cytokine–cytokine receptor interaction pathway (Figure 5E). In addition, several TNFRSF family members have been identified as participants in the TNF signaling pathway (Figure 5F). Moreover, genes differentially expressed between groups with high and low PRS-score level may also play roles in the regulation of the osteoclast differentiation pathway (Figure 5G).
In addition, the significant KEGG pathways in GSEA analysis were subjected to clustering analysis to group the pathways with similar enrichment patterns as shown in Figure 5H. Clusters identified from the GSEA results included “Antigen processing and presentation”, “Leukocyte transendothelial migration”, and “Phospholipase D signaling pathway”. The network graph highlights several pathways with relevance to immune function and skeletal biology. For instance, pathways like “Cell adhesion molecules” and “Hematopoietic cell lineage” were connected, suggesting a coordinated regulation in the context of bone health and immune response. The network graph represents KEGG pathway interactions, emphasizing the interconnected nature of immune response and its relevance to skeletal biology in the context of osteosarcoma.
Immune Cell Landscape and ICB Therapy Response Prediction
Multiple enrichment analyses indicated that the differentially expressed genes (DEGs) between the low and high PRS-score level group were enriched in terms of pathways associated with inflammation and immune response. In addition, the importance of a range of PRGs in anti-tumour immune response has been confirmed.13,91 The involvement of various immune functions in pyroptosis events in pediatric and young adult patients with osteosarcoma should be further investigated. As such, a comparative tumour microenvironment analysis was performed with the aim of exploring differences in immune cell landscape between low and high PRS-score tumours.
CIBERSORT was applied to the gene expression matrix in order to infer the relative abundance of 22 tumour-infiltrating immune cells for each sample in the TARGET cohort (Figure 6A). The results from the comparison of high (n = 31) vs low PRS-score (n = 53) for all 22 immune cells are shown in Figure 6B. The Wilcoxon rank-sum test confirmed that a decrease in abundance of plasma cells (p = 0.0494), T cells CD8 (p = 0.0064), and T cells CD4 memory activated (p = 0.0020) was found in high PRS-score tumours. This comparison also showed that high PRS-score samples had a higher fraction of macrophages M0 (p = 0.0123) and mast cells (MCs) activated (p = 0.0139).
 |
Figure 6 Immune cell composition in osteosarcoma tumour samples of the TARGET patients. (A) Barplot showing composition of 22 infiltrating immune cells in low (n = 53) and high pyroptosis-related signature (PRS)–score samples (n = 31). Fraction values of CIBERSORT immune cells were determined for each patient; each bar represents one patient. (B) Vioplot illustrating CIBERSORT‐derived relative abundance of 22 distinct immune cells based on PRS-score level. Median values and IQR for each cell subset were calculated for each patient group and compared the two groups using the Wilcoxon rank sum test. (C) Correlation heat map of expression level of hub genes and fraction of immune cells that differs in two PRS-score level group. (D) Scatter plot of the PRS-score against immune checkpoint blockade (ICB) response score calculated by the ImmuCellAI tool. The plot illustrates a positive correlation between PRS-score and ICB response score, indicating a potential relationship between genetic risk and ICB response. (E) Logistic curve with jitters predicting ICB response status as a function of PRS-score. (F) The logistic receiver operating characteristic (ROC) curve plot showing the performance of PRS-score in classifying positive and negative ICB response outcomes. The AUC value is represented in the legend as the estimated value [95% confidence interval (CI)]. ns not significance, * p < 0.05, ** p < 0.01, *** p < 0.001. Abbreviations: NK, nature killer; OR, odds ratio. |
The association between expression levels of hub PRGs and the fraction of immune cell types that differs between two PRG level groups in the TARGET cohort was also explored (Figure 6C). Increased TNFRSF21 expression was correlated with higher MCs activated signature (r = −0.306, p = 0.0047). High GSDMA expression was associated with increased T cells CD8 signature (r = 0.258, p = 0.0180) signature. GZMA expression showed negative association with Macrophages M0 (r = −0.466, p < 0.001) and MCs activated (r = −0.251, p = 0.0212), and positive relation with T cells CD4 memory activated (r = 0.239, p = 0.0288). There is no significant correlation between CHMP4C expression and the abundance of the five immune cell types.
The ImmuCellAI tool was used to predict the response to ICB immunotherapy in 84 patients from the TARGET cohort (Supplementary Table 8). The Spearman correlation analysis results showed a significant positive correlation between the ICB immunotherapy score and the PRS-score (r = 0.240, p = 0.0276) (Figure 6D). Furthermore, the logistic regression model (OR = 9.346 [95% CI: 2.099–81.642], p = 0.0118) (Figure 6E) for the effect of risk score on immune therapy response status had an ROC AUC greater than 0.6, indicating that the risk score has a good discriminatory ability for ICB immune response (Figure 6F).
SC Transcriptional Profiling in Osteosarcoma
Aimed to characterize cellular heterogeneity in resected tumour biospecimens from patients with osteosarcoma, sc-RNA seq data analysis was performed. Twenty-five main cell clusters were presented in the UMAP plot (Figure 7A). The clusters were then annotated using markers for characterising cellular heterogeneity in resected tumour biospecimens from patients with osteosarcoma. The two-dimensional UMAP representation of all sequenced cells (Figure 7B) reveals 17 main cell clusters: osteoblastic cell, osteoclasts, chondroblastic cells, fibroblasts, myofibroblasts, myeloid cells, endothelial cells, resting dendritic cells (DCs), activated plasmacytoid DCs (pDCs), activated mast cells (MCs), naive B cells, experienced T cells (CD8 Tex), CD8 T cells, T cells expressing the marker of ki67 (TMKI67), plasma cells, monocytes, and others. Osteoblastic cell (18%) was found to dominate the tumour immune microenvironment (Figure 7C). Further analysis of gene expression in different cell types revealed interesting findings. As shown in Figures 7D–H, TNFRSF21 was found to be highly expressed in activated MCs, activated pDCs, and resting DCs, GZMA was highly expressed in CD8 Tex cells and CD8 T cells, CHMP4C was highly expressed in chondroblastic cells and other cells, while GSDMA was generally distributed among all tumour microenvironment types.
 |
Figure 7 Single-cell (SC) transcriptional profiling in osteosarcoma of hub pyroptosis-related genes (PRGs). (A) Two-dimensional uniform manifold approximation and projection (UMAP) representation of all sequenced cells reveals 25 main cell clusters. Colors represent the cell clusters. (B) Two-dimensional UMAP representation of all sequenced cells reveals 17 main CIBERSORT cell clusters. Colors represent the cell types. (C) The immune cell composition in osteosarcoma samples illustrated as a ring chart. Violin plots showing hub (D) TNFRSF21, (E) GSDMA, (F) GZMA, and (G) CHMP4C in the nine main cell clusters on the single-cell level. (H) Dot plot depicting the expression levels of hub genes in each cell type. The color of each dot represents the hub PRG’s expression level across the cell type (average expression), while the size of each dot corresponds to the percentage of cells in the cluster expressing the gene (percent expressed). Abbreviations: MCs, mast cells; dendritic cells; pDCs, plasmacytoidDCs; CD8 Tex, experienced T cells; TMKI67, T cells expressing the marker of ki67. |
These findings provide important insights into the cellular heterogeneity of osteosarcoma tumours and identify the dominant cell types in the tumour immune microenvironment. Furthermore, the results also have implications for the development of new therapeutic strategies targeting specific cell types in the tumour microenvironment. However, further studies are needed to validate these findings and determine their clinical significance.
Candidate Drugs for Low and High PRS-Score Level Patients
The possibility that each group might have specific drug sensitivity to 251 drugs from the CGP 2016 was investigated. The identification of candidate drugs applied a phenotype prediction ML tool to matching cell line chemotherapeutic response to baseline tumour gene expression.92
It is imperative to continue the search for effective drugs for osteosarcomas. As illustrated in Figure 8, low PRS-score group shows greater sensitivity to AZD7762, BEZ235, bortezomib, CGP-60474, JNK Inhibitor VIII, lapatinib, MG-132, TGX221, trametinib, and Z-LLNle-CHO, compared to the high PRS-score group, and those patients could benefit from the treatment. The analysis also found MP470 and SB52334 could be potentially more effective for high PRS-score group.
 |
Figure 8 Prediction results of drug sensitivity tests in different PRS–score level subtypes from the TARGET cohort. Drug sensitivity was predicted for each case in the TARGET dataset. Ten therapeutic agents were found to show significantly greater sensitivity [lower log10(IC50)] in low PRS-score subpopulation: (A) AZD7762, (B) BEZ235, (C) bortezomib, (D) CGP-60474, (E) JNK inhibitor VIII, (F) lapatinib, (G) MG-132, (H) TGX221, (I) trametinib, (J) Z-LLNle-CHO. Two therapeutic agents were detected to indicate significantly greater sensitivity in high PRS-score subgroup: (K) MP470, (L) SB52334. ***p < 0.001. |
Difference in Relative Expression Level of Hub PRGs in Cell Lines
As shown in Figure 9, the relative expression level of all hub PRGs in each of the two cancer cell lines (143B and U-2 OS) was significantly higher than that in the normal cell line (hFOB1.19). This is consistent with the results of differential expression analysis performed on TARGET and GTEx samples. The aforementioned differences arise from biological experiments suggesting the importance of understanding hub PRG expression patterns.
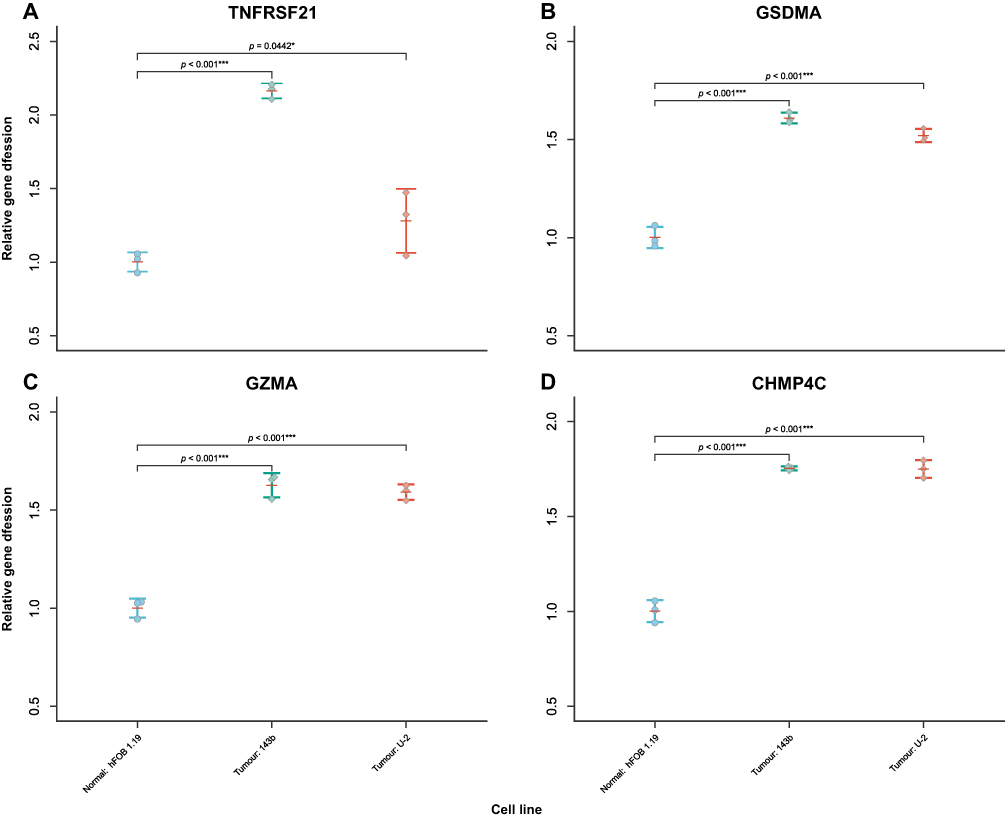 |
Figure 9 Beeswarm plot of relative hub gene expression in the normal cell line (hFOB1.19) and osteosarcoma cell lines (143B and U-2 OS). The RNA transcription levels of hub genes were evaluated by using the ΔΔCt method. (A) TNFRSF21, (B) GSDMA, (C) GZMA, and (D) CHMP4C were upregulated in two cancer cell lines compared to the normal cell line. GAPDH was used as internal control. Error bars indicate SD. *p < 0.05, ***p < 0.001. |
Discussion
Pyroptosis represents a type of cell death that ends with the loss of plasma membrane intactness and is induced by activation of so-called inflammasome sensors.93 Pyroptosis is an important event in cancer formation, and relevant therapeutic approaches have been developed in the hope of positively influencing outcomes among cancer patients.94 However, presently, there still remains very little knowledge about how PRGs affect young and OS of patients with osteosarcoma.
In this study, we developed a PRG–based signature using the entire TARGET dataset. The genetic predictive signature consisting of four hub PRGs, among which are TNFRSF21, GSDMA, GZMA, and CHMP4C. Pyroptosis is a type of necrotic and inflammatory PCD induced by inflammatory caspases.95 TNFRSF21, also known as DR6, is a member of the TNF receptor superfamily (TNFRSF) and has been implicated in various immune responses. Besides, overexpression of TNFRSF21 in neurons can lead to caspase activation and cell death.96 GZMA is one among five different types of granzymes, namely the serine proteases used by cytotoxic T lymphocytes (CTL) and NK cells to kill their target cells via caspase-independent apoptosis.97 GSDMA, also referred to as “GSDM1”, is a member of gasdermin family, and it is considered a possible tumour suppressor.98 In this study, GSDMA expression was found to be independently associated with good prognosis in osteosarcoma, which is in line with prior studies. CHMP4C belongs to the chromatin-modifying protein (CHMP) family. The transcription of CHMP4C s modulated by the P53 protein, thus promoting exosome production.99 These transcriptional signals are involved in cell interaction and immune activation.100
The samples in the training cohort and the validation set were, respectively, divided into two PRS-score levels. The predictive accuracy of the genetic predictor is internally and externally validated. These results demonstrate that the models are capable of accurately predicting 1-, 2-, 3-, 4-, and 5-year OS in osteosarcoma patients based on the clinical variables involved and the PRS-score level. Prognostic accuracy analysis for the demographic subgroups in the training cohort was also conducted. The results indicated that PRS-score is also applicable to different gender and age populations. PRS-based signature was confirmed to be acceptable to TPM, FPKM, and microarray data, and it performs well in predicting the prognosis of osteosarcoma patients.
Subsequently, PRS-score level variable was employed to establish a nomogram predicting OS in pediatric and young adult patients with osteosarcoma. The employment of validated nomogram has become more popular in clinical practice due to the model’s precision and usability.101 In the TARGET cohort study of osteosarcoma patients, two significant independent risk factors, namely metastasis status and PRS-score level, were identified and were used to construct validated nomograms for predicting 1-, 2-, 3-, 4-, and 5-year OS rates. Previous studies3,4 implied that 15–20% of osteosarcoma patients have detectable metastasis at diagnosis, 85–90% of which occurs in the lungs. Furthermore, lung metastasis from osteosarcomas is a dominant cause of death.102 Surveillance, Epidemiology, and End Results (SEER) Program population-based research also implies the risk of death is higher for those patients whose tumours have extended to distant sites.101
Enriched functional gene sets of DEGs between high PRS-score level and low PRS-score level groups from the training cohort (the TARGET cohort) were then investigated. A noteworthy facet of the results lies in the significant enrichment of terms or pathways related to immune response regulation. The results of GO–ORA and KEGG–ORA suggested that the immune system’s ability of pediatric and young adult osteosarcoma patients to detect and respond to tumour cells could be a pivotal factor in osteosarcoma pathophysiology and could have prognostic implications. Indeed, the correlation between cancer and preceding inflammatory conditions, as indicated by prior research, may reflect the importance of immune surveillance mechanisms in the disease context.
Further emphasizing the role of the immune system, the involvement of TNFRSF21 and related family genes in cytokine interactions and TNF signaling pathways aligns with the known importance of these pathways in immune modulation and inflammation. Given the complex network of cytokines involved in immune cell communication, these pathways could be influential in shaping the tumor microenvironment, which in turn could affect tumor growth and patient survival.
Bayesian network plots illustrate the interconnectivity of genes and pathways, shedding light on the potential molecular interactions that might govern disease processes. The osteoclast differentiation pathway’s connection to these gene networks underscores the dual nature of osteosarcoma as both a malignancy of bone and an immune-influenced disease. Osteoclast differentiation is crucial for bone remodeling, and its dysregulation can lead to bone destruction—a hallmark of osteosarcoma. Thus, the regulation of this pathway by genes associated with PRS-score could provide insights into the mechanisms of bone degradation in osteosarcoma.
The clustering of significant KEGG pathways from the GSEA analysis further refines our understanding of the disease. The identification of clusters related to antigen processing, leukocyte migration, and phospholipase D signaling implies a coordinated interaction of these processes in osteosarcoma. The network graph’s portrayal of interconnected pathways, particularly those related to cell adhesion and hematopoietic lineage, underscores the interplay between bone integrity and immune function, a relationship that is particularly relevant in bone cancers such as osteosarcoma.
The findings of gene enrichment analysis osteosarcoma’s aggressive behaviour and the subsequent prognosis of patients could be significantly influenced by immune-related gene expressions and their involvement in specific signaling pathways. The interrelation of immune response, cytokine signaling, and bone remodeling pathways offers important implications in the context of osteosarcoma’s complexity. Further investigations should be performed to validate these observations and unravel the potential mechanisms involved.
Based on the enlightenment provided by the previous functional enrichment analysis, the difference in the fraction of immune cell types in two distinct PRG level groups was examined. High PRS-score tumours were found to be associated with a decrease in the abundance of certain immune cells, such as plasma cells, T cells CD8, and T cells CD4 memory activated. In contrast, high PRS-score samples had a higher fraction of macrophages M0 and mast cells activated. The observed associations between the PRS-score level and tumour-infiltrating immune cells may have important implications for cancer diagnosis, prognosis, and treatment. For example, a decrease in the abundance of T cells CD8 and CD4 memory activated is known to be associated with poorer clinical outcomes in various types of cancer, while an increase in the fraction of macrophages M0 and MCs activated has been reported to promote tumour growth and metastasis. These results may be used to identify with osteosarcoma patients who are likely to benefit from specific immunotherapeutic interventions, such as ICBs or adoptive T cell therapy.
This study also focused on the intricate interplay between the expression levels of hub PRGs and the distribution of various immune cell types in the TARGET cohort. The negative association between TNFRSF21 expression and activated MCs suggests a potential role for this receptor in modulating MC activation and function. In contrast, GSDMA expression exhibited a positive association with CD8 T cell signature, highlighting a possible link between pyroptosis and the recruitment or activation of cytotoxic T cells in the tumour microenvironment. In addition, GZMA expression showed a complex pattern of associations, being negatively correlated with both Macrophages M0 and MCs activated. Conversely, the positive relation with CD4 memory activated T cells pointed towards a supportive role in adaptive immune memory. Interestingly, CHMP4C expression did not show a significant correlation with any of the five immune cell types, which varied between two risk score level groups.
Pyroptosis can affect not only the tumour microenvironment but also the immune response.103 As such, this study also predicted the response results of ICB treatment in the TARGET cohort. Generally, osteosarcoma is considered to be a “cold” tumor, which may hardly benefit from the treatment of immune checkpoint inhibitors. Osteosarcoma is generally classified as a “cold” tumour,104 which may be hardly responsive to ICBs.105 Nevertheless, this study yielded inspiring outcomes concerning the response to ICB treatment. The correlation analysis between the PRS-score and immune response score and the ROC AUC of the logistic regression model indicated, the PRS-score can be considered as a useful tool for identifying patients who are more likely to respond to ICB immunotherapy, and for tailoring treatment strategies accordingly. This information may be particularly valuable in the context of precision medicine, where treatment decisions are increasingly guided by biomarkers that predict treatment response.
In addition to predicting the response to immunotherapy, this study also analysed the difference in drug sensitivity between the high and low PRS-score patients. The drug sensitivity test showed that the PRS-score be a useful tool for identifying patients with osteosarcoma who are more likely to respond to specific drugs, and for tailoring treatment strategies accordingly. The identification of effective drugs for osteosarcomas is of utmost importance, as this type of cancer is associated with poor outcomes and limited treatment options. These findings will be conducive in improving patients’ overall treatment outcomes.
Pyroptosis has been confirmed to be a double-edged sword in cancer progression. Nonetheless, development of anti-tumour formulars utilising pyroptosis mechanisms and relevant clinical trials are currently underway. In silico study provides important insights into the potential use of chemistry therapies for osteosarcoma patients based on their genetic profiles. Further studies are needed to validate our findings and to identify drugs that may be effective for high PRS-score level patient population.
scRNA seq is a revolutionary method used in cancer research to identify tumor cell composition and analyze differences in gene expression at single-cell resolution. A detailed characterization of the immune cell composition in osteosarcoma samples is likely to help clinicians enrich their knowledge of prognostic biomarkers. This study found that the dominant cell type in the tumor immune microenvironment was osteoblastic cells, accounting for 18% of the total cell population. This finding suggests that osteoblastic cells are closely associated with osteosarcoma and highlights the importance of these cell types in the tumor microenvironment. The analysis of single-cell RNA sequencing data has revealed several interesting findings regarding the expression of specific genes in different cell types within the tumor microenvironment. The expression of TNFRSF21 was found to be highly expressed in activated MCs, activated pDCs, and resting DCs, suggesting that it plays a role in regulating these cell types in the tumor microenvironment. GZMA was highly expressed in CD8 Tex cells and CD8 T cells, indicating that this gene may be involved in cytotoxic T-cell-mediated killing of cancer cells. CHMP4C was highly expressed in chondroblastic cells and other cells, while GSDMA was generally distributed among all tumor microenvironment types. Overall, these findings provide valuable insights into the cellular and molecular heterogeneities of osteosarcoma tumors and could have important implications for the development of new therapeutic strategies targeting specific cell types and gene expression patterns in the tumor microenvironment. However, further studies are needed to validate the findings of this study and determine their clinical significance.
Most importantly, the consistency between in vitro cell biology experiments and in silico analysis of gene expression indicates that the risk prognosis model constructed for this bioinformatics study of PRG is based on genes that are relevant to cancer development and exhibit distinct expression patterns in cancer cells. This further supports the generalizability and reliability of the model, as it is constructed based on known genes related to cancer and their expression patterns in cancer cells. It also suggests that the model can serve as an effective prognostic tool for assessing the risk of patients with osteosarcoma and predicting the likelihood of disease progression. Furthermore, this study provides enlightenment for further research on changes in hub gene expression patterns and the mechanisms of cancer occurrence.
The study still has some deficiencies. The primary limitation was the small number of cases in the training cohort. In addition, the exact mechanism of the hub PRGs in osteosarcoma and the patients’ sensitivity to the candidate drugs need to be further investigated in vivo and in vitro. Moreover, future research should perform an independent and external validation of the PRS-score on a larger cohort of patients compared with GSE21257 dataset. Nonetheless, this work still highlights the importance of TARGET genomic resource that would expand clinicians’ understanding of this lethal disease.
Conclusion
Presently, there are only a few studies that focused on role of PRGs in osteosarcoma. In this research, four hub PRGs, including TNFRSF21, GSDMA, GZMA, and CHMP4C, were identified, and these genes may work as key executors of pyroptosis in TARGET pediatric and young adult osteosarcoma patients. A novel PRG-related gene signature was subsequently developed using the sum of weighted expression of hub PRGs. Additionally, the predictive efficacy of the gene signature was externally validated in population belonging to GSE21257. Despite the study’s limitation of sample size, the nomogram-based OS prediction tool established with the pyroptosis-related signature and clinical feature in the training set demonstrate good accuracy in internal validation. The nomogram can serve as a counselling and clinical decision aid to patients for clinicians. Functional enrichment analyses and tumour microenvironment analyses established a theoretical framework for future research on the connection between immunity and PRGs in osteosarcoma. Moreover, the potential therapeutic implications of candidate drugs and ICB therapy are also provided. Overall, this study provides enlightenment for the treatment of cancer in the context of precision medicine.
Data Sharing Statement
The datasets supporting the conclusions of this article are available in the following database: GDC portal (https://portal.gdc.cancer.gov/); the GTEx portal (https://gtexportal.org/home/); and the GEO database (https://www.ncbi.nlm.nih.gov/geo). R code scripts for the downstream analysis of the datasets: GTEx, TARGET-Osteosarcoma, GSE21257, and GSE162454, and the data generated during this study are available from the corresponding author on reasonable request.
Ethics Approval
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Ethics Committee of Shanxi Provincial People’s Hospital [Protocol Code (2023) Sheng Yike Lun Shen Zi No. 457 of 28 January 2023].
Acknowledgments
We would like to thank the TARGET program, the GTEx project, and the GEO database for their free use.
Funding
This work is supported by Science and Technology Innovation Talent Team Project of Shanxi Province (Grant No. 202204051002035). The funders had no role in study design, data collection or analysis, preparation of the manuscript, or the decision to publish.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Hameed M, Mandelker D. Tumor syndromes predisposing to osteosarcoma. Adv Anat Pathol. 2018;25(4):217–222. doi:10.1097/PAP.0000000000000190
2. Ritter J, Bielack SS. Osteosarcoma. Ann Oncol. 2010;21:vii320–vii325. doi:10.1093/annonc/mdq276
3. Isakoff MS, Bielack SS, Meltzer P, et al. Osteosarcoma: current treatment and a collaborative pathway to success. J Clin Oncol. 2015;33(27):3029–3035. doi:10.1200/JCO.2014.59.4895
4. Meazza C, Scanagatta P. Metastatic osteosarcoma: a challenging multidisciplinary treatment. Expert Rev Anticancer Ther. 2016;16(5):543–556. doi:10.1586/14737140.2016.1168697
5. Miao EA, Rajan JV, Aderem A. Caspase-1-induced pyroptotic cell death. Immunol Rev. 2011;243(1):206–214. doi:10.1111/j.1600-065X.2011.01044.x
6. Robinson N, Ganesan R, Hegedűs C, et al. Programmed necrotic cell death of macrophages: focus on pyroptosis, necroptosis, and parthanatos. Redox Biol. 2019;26:101239. doi:10.1016/j.redox.2019.101239
7. Bortoluci KR, Medzhitov R. Control of infection by pyroptosis and autophagy: role of TLR and NLR. Cell Mol Life Sci. 2010;67(10):1643–1651. doi:10.1007/s00018-010-0335-5
8. Li J, Anraku Y, Kataoka K. Self‐boosting catalytic nanoreactors integrated with triggerable crosslinking membrane networks for initiation of immunogenic cell death by pyroptosis. Angew Chem. 2020;132(32):13628–13632. doi:10.1002/ange.202004180
9. Yang M, Zheng H, Su Y, et al. Novel pyroptosis-related lncRNAs and ceRNAs predict osteosarcoma prognosis and indicate immune microenvironment signatures. Heliyon. 2023;9(11):e21503. doi:10.1016/j.heliyon.2023.e21503
10. Zhang J, Deng J, Ding R, et al. Identification of pyroptosis-related genes and long non-coding RNAs signatures in osteosarcoma. Cancer Cell Int. 2022;22(1):322. doi:10.1186/s12935-022-02729-1
11. Kovacs SB, Miao EA. Gasdermins: effectors of pyroptosis. Trends Cell Biol. 2017;27(9):673–684. doi:10.1016/j.tcb.2017.05.005
12. Kolb R, Liu GH, Janowski AM, et al. Inflammasomes in cancer: a double-edged sword. Protein Cell. 2014;5(1):12–20. doi:10.1007/s13238-013-0001-4
13. Zhang Z, Zhang Y, Xia S, et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature. 2020;579(7799):415–420. doi:10.1038/s41586-020-2071-9
14. Xi G, Gao J, Wan B, et al. GSDMD is required for effector CD8+ T cell responses to lung cancer cells. Int Immunopharmacol. 2019;74:105713. doi:10.1016/j.intimp.2019.105713
15. De Schutter E, Croes L, Ibrahim J, et al. GSDME and its role in cancer: from behind the scenes to the front of the stage. Int J Cancer. 2021;148(12):2872–2883. doi:10.1002/ijc.33390
16. Han J, Hu Y, Ding S, et al. The analysis of the pyroptosis-related genes and hub gene TP63 ceRNA axis in osteosarcoma. Front Immunol. 2022;13:974916. doi:10.3389/fimmu.2022.974916
17. Ternès N, Rotolo F, Michiels S. Empirical extensions of the LASSO penalty to reduce the false discovery rate in high-dimensional Cox regression models. Stat Med. 2016;35(15):2561–2573. doi:10.1002/sim.6927
18. GTEx Consortium. Genetic effects on gene expression across human tissues. Nature. 2017;550(7675):204–213. doi:10.1038/nature24277
19. Kassambara A, Kosinski M, Biecek P, et al. Package ‘survminer’. Boston, MA: CRAN; 2021. Available from: https://cran.r-project.org/web/packages/survminer/survminer.pdf.
20. Campanacci M, Bertoni F, Bacchini P. Bone and Soft Tissue Tumors. New York, NY: Springer-Verlag; 1990.
21. Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi:10.1093/nar/gkv007
22. Hao Y, Hao S, Andersen-Nissen E, et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184(13):3573–3587.e29. doi:10.1016/j.cell.2021.04.048
23. Korsunsky I, Millard N, Fan J, et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods. 2019;16(12):1289–1296. doi:10.1038/s41592-019-0619-0
24. Choudhary S, Satija R. Comparison and evaluation of statistical error models for scRNA-seq. Genome Biol. 2022;23(1):27. doi:10.1186/s13059-021-02584-9
25. Ahlmann-Eltze C, Huber W. glmGamPoi: fitting Gamma-Poisson generalized linear models on single cell count data. Bioinformatics. 2021;36(24):5701–5702. doi:10.1093/bioinformatics/btaa1009
26. Yoshida K, Bartel A. tableone: create”Table 1” to describe baseline characteristics with or without propensity score weights; 2022. Available from https://github.com/kaz-yos/tableone.
27. Wang Q, Liu Q, Qi S, et al. Comprehensive pan-cancer analyses of pyroptosis-related genes to predict survival and immunotherapeutic outcome. Cancers. 2022;14(1):237. doi:10.3390/cancers14010237
28. Karki R, Kanneganti T-D. Diverging inflammasome signals in tumorigenesis and potential targeting. Nat Rev Cancer. 2019;19(4):197–214. doi:10.1038/s41568-019-0123-y
29. Xia X, Wang X, Cheng Z, et al. The role of pyroptosis in cancer: pro-cancer or pro-“host”? Cell Death Dis. 2019;10(9):650. doi:10.1038/s41419-019-1883-8
30. Wang B, Yin Q. AIM2 inflammasome activation and regulation: a structural perspective. J Struct Biol. 2017;200(3):279–282. doi:10.1016/j.jsb.2017.08.001
31. Man SM, Kanneganti T-D. Regulation of inflammasome activation. Immunol Rev. 2015;265(1):6–21. doi:10.1111/imr.12296
32. Dong Z, Bian L, Wang M, et al. Identification of a pyroptosis-related gene signature for prediction of overall survival in lung adenocarcinoma. J Oncol. 2021;2021:6365459. doi:10.1155/2021/6365459
33. Ye Y, Dai Q, Qi H. A novel defined pyroptosis-related gene signature for predicting the prognosis of ovarian cancer. Cell Death Discov. 2021;7(1):71. doi:10.1038/s41420-021-00451-x
34. Gómez-Rubio V. ggplot2-elegant graphics for data analysis. J Stat Softw. 2017;77:1–3.
35. Szklarczyk D, Kirsch R, Koutrouli M, et al. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023;51(D1):D638–D646. doi:10.1093/nar/gkac1000
36. Szklarczyk D, Gable AL, Nastou KC, et al. The STRING database in 2021: customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021;49(D1):D605–D612. doi:10.1093/nar/gkaa1074
37. Charles S, Natarajan J. Integrated regulatory network based on lncRNA-miRNA-mRNA-TF reveals key genes and sub-networks associated with dilated cardiomyopathy. Comput Biol Chem. 2021;92:107500. doi:10.1016/j.compbiolchem.2021.107500
38. Keenan AB, Torre D, Lachmann A, et al. ChEA3: transcription factor enrichment analysis by orthogonal omics integration. Nucleic Acids Res. 2019;47(W1):W212–W224. doi:10.1093/nar/gkz446
39. Pedersen TL. A grammar of graphics for relational data; 2023. Available from https://ggraph.data-imaginist.com/.
40. Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010;26(12):1572–1573. doi:10.1093/bioinformatics/btq170
41. Therneau TM, Grambsch PM. The Cox model. In: Modeling Survival Data: Extending the Cox Model. New York: Springer New York; 2000:39–77.
42. Hothorn T, Hornik K, van de Wiel MA, et al. A lego system for conditional inference. Am Stat. 2006;60(3):257–263. doi:10.1198/000313006X118430
43. De Jay N, Papillon-Cavanagh S, Olsen C, et al. mRMRe: an R package for parallelized mRMR ensemble feature selection. Bioinformatics. 2013;29(18):2365–2368. doi:10.1093/bioinformatics/btt383
44. Kuhn M. Package ‘caret’. Boston, MA: CRAN; 2020. Available from: https://cran.r-project.org/web/packages/caret/caret.pdf.
45. Lewis JE, Kemp ML. Integration of machine learning and genome-scale metabolic modeling identifies multi-omics biomarkers for radiation resistance. Nat Commun. 2021;12(1):2700. doi:10.1038/s41467-021-22989-1
46. Greenwell B. Package ‘fastshap’. Boston, MA: CRAN;2023. Available from: https://cran.r-project.org/web/packages/fastshap/fastshap.pdf.
47. Mayer M, Stando A. Package ‘shapviz’. Boston, MA: CRAN;2023. Available from: https://cran.r-project.org/web/packages/shapviz/shapviz.pdf.
48. Lang M, Binder M, Richter J, et al. mlr3: a modern object-oriented machine learning framework in R. J Open Source Softw. 2019;4(44):1903. doi:10.21105/joss.01903
49. Sonabend R, Király FJ, Bender A, et al. mlr3proba: an R package for machine learning in survival analysis. Bioinformatics. 2021;37(17):2789–2791.
50. Lang M, Schratz P. mlr3verse: easily install and load the ‘mlr3’ package family. 2023. Available from https://github.com/mlr-org/mlr3verse.
51. Sonabend R, Schratz P, Fischer S. mlr3extralearners; 2023. Available from https://github.com/mlr-org/mlr3extralearners.
52. Kuitunen I, Ponkilainen VT, Uimonen MM, et al. Testing the proportional hazards assumption in cox regression and dealing with possible non-proportionality in total joint arthroplasty research: methodological perspectives and review. BMC Musculoskelet Disord. 2021;22(1):489. doi:10.1186/s12891-021-04379-2
53. Smith G. Step away from stepwise. J Big Data. 2018;5(1):32. doi:10.1186/s40537-018-0143-6
54. Li J, Lu X, Cheng K, et al. Package ‘StepReg’. Boston, MA: CRAN; 2022. Available from: https://cran.r-project.org/web/packages/StepReg/StepReg.pdf.
55. Pavlou M, Ambler G, Seaman SR, et al. How to develop a more accurate risk prediction model when there are few events. BMJ. 2015;351:h3868. doi:10.1136/bmj.h3868
56. Frank E, Harrell J. Package ‘Hmisc’; 2023. Available from https://cran.r-project.org/web/packages/Hmisc/Hmisc.pdf.
57. Gu Z, Gu L, Eils R, et al. circlize implements and enhances circular visualization in R. Bioinformatics. 2014;30(19):2811–2812. doi:10.1093/bioinformatics/btu393
58. Gu Z. Complex heatmap visualization. iMeta. 2022;1(3):e43.
59. Budczies J, Klauschen F, Sinn BV, et al. Cutoff finder: a comprehensive and straightforward web application enabling rapid biomarker cutoff optimization. PLoS One. 2012;7(12):e51862. doi:10.1371/journal.pone.0051862
60. Blanche P, Dartigues J-F, Jacqmin-Gadda H. Estimating and comparing time-dependent areas under receiver operating characteristic curves for censored event times with competing risks. Stat Med. 2013;32(30):5381–5397. doi:10.1002/sim.5958
61. Semenkovich TR, Yan Y, Subramanian M, et al. A clinical nomogram for predicting node-positive disease in esophageal cancer. Ann Surg. 2021;273(6):e214–221. doi:10.1097/sla.0000000000003450
62. Kronek L-P, Reddy A. Logical analysis of survival data: prognostic survival models by detecting high-degree interactions in right-censored data. Bioinformatics. 2008;24(16):i248–i253. doi:10.1093/bioinformatics/btn265
63. Peters A, Hothorn T, Lausen B. ipred: improved predictors. R News. 2002;2(2):33–36.
64. Canty AJ. Resampling methods in R: the boot package. Newsl R Proj. 2002;2(3):2–7.
65. Lazic SE. Medical risk prediction models: with ties to machine learning. J R Stat Soc Ser a Stat Soc. 2022;185(1):425. doi:10.1111/rssa.12756
66. Frank E, Harrell J. rms: regression modeling strategies; 2023. Available from https://CRAN.R-project.org/package=rms.
67. Mogensen UB, Ishwaran H, Gerds TA. Evaluating random forests for survival analysis using prediction error curves. J Stat Softw. 2012;50(11):1–23. doi:10.18637/jss.v050.i11
68. Vickers AJ, Cronin AM, Elkin EB, et al. Extensions to decision curve analysis, a novel method for evaluating diagnostic tests, prediction models and molecular markers. BMC Medical Informatics and Decision Making. 2008;8(1):53. doi:10.1186/1472-6947-8-53
69. Cao J, Zhang S. A Bayesian extension of the hypergeometric test for functional enrichment analysis. Biometrics. 2014;70(1):84–94. doi:10.1111/biom.12122
70. Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28(1):27–30. doi:10.1093/nar/28.1.27
71. Wu T, Hu E, Xu S, et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation. 2021;2(3):100141. doi:10.1016/j.xinn.2021.100141
72. Sato N, Tamada Y, Yu G, et al. CBNplot: bayesian network plots for enrichment analysis. Bioinformatics. 2022;38(10):2959–2960. doi:10.1093/bioinformatics/btac175
73. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. doi:10.1073/pnas.0506580102
74. Ieva K, Juozas G. aPEAR: an R package for autonomous visualisation of pathway enrichment networks. bioRxiv. 2023. doi:10.1101/2023.03.28.534514
75. Newman AM, Liu CL, Green MR, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12(5):453–457. doi:10.1038/nmeth.3337
76. Finotello F, Trajanoski Z. Quantifying tumor-infiltrating immune cells from transcriptomics data. Cancer Immunol Immunother. 2018;67(7):1031–1040. doi:10.1007/s00262-018-2150-z
77. Kassambara A. Package ‘rstatix’. Boston, MA: CRAN;2020. Available from: https://cran.r-project.org/web/packages/rstatix/rstatix.pdf.
78. Miao YR, Zhang Q, Lei Q, et al. ImmuCellAI: a unique method for comprehensive T-cell subsets abundance prediction and its application in cancer immunotherapy. Adv Sci. 2020;7(7):1902880. doi:10.1002/advs.201902880
79. Macosko EZ, Basu A, Satija R, et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161(5):1202–1214. doi:10.1016/j.cell.2015.05.002
80. Wang C, Sun D, Huang X, et al. Integrative analyses of single-cell transcriptome and regulome using MAESTRO. Genome Biol. 2020;21(1):198. doi:10.1186/s13059-020-02116-x
81. Zhou Y, Yang D, Yang Q, et al. Single-cell RNA landscape of intratumoral heterogeneity and immunosuppressive microenvironment in advanced osteosarcoma. Nat Commun. 2020;11(1):6322. doi:10.1038/s41467-020-20059-6
82. Geeleher P, Cox N, Huang RS. pRRophetic: an r package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS One. 2014;9(9):e107468. doi:10.1371/journal.pone.0107468
83. Garnett MJ, Edelman EJ, Heidorn SJ, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483(7391):570–575. doi:10.1038/nature11005
84. R Core Team. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing;2022. Available from: https://www.R-project.org/.
85. Clarke E, Sherrill-Mix S, Dawson C. Package ‘ggbeeswarm’. Boston, MA: CRAN;2023. Available from: https://cran.r-project.org/web/packages/ggbeeswarm/ggbeeswarm.pdf.
86. Ramos B, Pereira T, Moranguinho J, et al. An interpretable approach for lung cancer prediction and subtype classification using gene expression. Annu Int Conf IEEE Eng Med Biol Soc. 2021;2021:1707–1710. doi:10.1109/embc46164.2021.9630775
87. Cao M, Zhang J, Xu H, et al. Identification and development of a novel 4-gene immune-related signature to predict osteosarcoma prognosis. Front Mol Biosci. 2020;7:608368. doi:10.3389/fmolb.2020.608368
88. Gerds TA, Kattan MW, Schumacher M, et al. Estimating a time-dependent concordance index for survival prediction models with covariate dependent censoring. Stat Med. 2013;32(13):2173–2184. doi:10.1002/sim.5681
89. Vickers AJ, Elkin EB. Decision curve analysis: a novel method for evaluating prediction models. Med Decis Making. 2006;26(6):565–574.
90. Stiller CA, Trama A, Serraino D, et al. Descriptive epidemiology of sarcomas in Europe: report from the RARECARE project. European Journal of Cancer. 2013;49(3):684–695. doi:10.1016/j.ejca.2012.09.011
91. Frank D, Vince JE. Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell Death Differ. 2019;26(1):99–114. doi:10.1038/s41418-018-0212-6
92. Geeleher P, Cox NJ, Huang RS. Clinical drug response can be predicted using baseline gene expression levels and in vitro drug sensitivity in cell lines. Genome Biol. 2014;15(3):R47. doi:10.1186/gb-2014-15-3-r47
93. Bertheloot D, Latz E, Franklin BS. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol Immunol. 2021;18(5):1106–1121. doi:10.1038/s41423-020-00630-3
94. Bittner ZA, Schrader M, George SE, et al. Pyroptosis and its role in SARS-CoV-2 infection. Cells. 2022;11(10):1717. doi:10.3390/cells11101717
95. Vande Walle L, Lamkanfi M. Pyroptosis. Curr Biol. 2016;26(13):R568–R572. doi:10.1016/j.cub.2016.02.019
96. Mi S, Lee X, Hu Y, et al. Death receptor 6 negatively regulates oligodendrocyte survival, maturation and myelination. Nat Med. 2011;17(7):816–821. doi:10.1038/nm.2373
97. Roufas C, Georgakopoulos-Soares I, Zaravinos A. Distinct genomic features across cytolytic subgroups in skin melanoma. Cancer Immunol Immunother. 2021;70(11):3137–3154. doi:10.1007/s00262-021-02918-3
98. Saeki N, Usui T, Aoyagi K, et al. Distinctive expression and function of four GSDM family genes (GSDMA-D) in normal and malignant upper gastrointestinal epithelium. Genes Chromosomes Cancer. 2009;48(3):261–271. doi:10.1002/gcc.20636
99. Yu X, Riley T, Levine AJ. The regulation of the endosomal compartment by p53 the tumor suppressor gene. FEBS J. 2009;276(8):2201–2212. doi:10.1111/j.1742-4658.2009.06949.x
100. Lespagnol A, Duflaut D, Beekman C, et al. Exosome secretion, including the DNA damage-induced p53-dependent secretory pathway, is severely compromised in TSAP6/Steap3-null mice. Cell Death Differ. 2008;15(11):1723–1733. doi:10.1038/cdd.2008.104
101. Huang Y, Wang C, Tang D, et al. Development and validation of nomogram-based prognosis tools for patients with extremity osteosarcoma: a SEER population study. J Oncol. 2022;2022:9053663. doi:10.1155/2022/9053663
102. Munajat I, Zulmi W, Norazman MZ, et al. Tumour volume and lung metastasis in patients with osteosarcoma. J Orthop Surg Res. 2008;16(2):182–185. doi:10.1177/230949900801600211
103. Lou X, Li K, Qian B, et al. Pyroptosis correlates with tumor immunity and prognosis. Commun Biol. 2022;5(1):917. doi:10.1038/s42003-022-03806-x
104. Wu CC, Beird HC, Andrew Livingston J, et al. Immuno-genomic landscape of osteosarcoma. Nat Commun. 2020;11(1):1008. doi:10.1038/s41467-020-14646-w
105. Li X, Luo L, Jiang M, et al. Cocktail strategy for ‘cold’ tumors therapy via active recruitment of CD8+ T cells and enhancing their function. J Control Release. 2021;334:413–426. doi:10.1016/j.jconrel.2021.05.002
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.