Back to Archived Journals » Advances and Applications in Bioinformatics and Chemistry » Volume 17
Pyrazoline B-Paclitaxel or Doxorubicin Combination Drugs Show Synergistic Activity Against Cancer Cells: In silico Study
Authors Wiraswati HL ![]() , Bashari MH
, Bashari MH ![]() , Alfarafisa NM
, Alfarafisa NM ![]() , Ma'ruf IF, Sholikhah EN, Wahyuningsih TD, Satriyo PB, Mustofa M, Satria D, Damayanti E
, Ma'ruf IF, Sholikhah EN, Wahyuningsih TD, Satriyo PB, Mustofa M, Satria D, Damayanti E
Received 29 November 2023
Accepted for publication 10 February 2024
Published 27 February 2024 Volume 2024:17 Pages 33—46
DOI https://doi.org/10.2147/AABC.S452281
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Maria Miteva
Hesti Lina Wiraswati,1,2 Muhammad Hasan Bashari,1,2 Nayla Majeda Alfarafisa,1,2 Ilma Fauziah Ma’ruf,3 Eti Nurwening Sholikhah,4 Tutik Dwi Wahyuningsih,5 Pamungkas Bagus Satriyo,4 Mustofa Mustofa,4 Denny Satria,6 Ema Damayanti7
1Department of Biomedical Sciences, Faculty of Medicine, Universitas Padjadjaran, Bandung, Indonesia; 2Oncology and Stem Cells Working Group, Faculty of Medicine, Universitas Padjadjaran, Bandung, Indonesia; 3Research Center for Pharmaceutical Ingredients and Traditional Medicine, National Research and Innovation Agency, Bogor, Indonesia; 4Department of Pharmacology and Therapy, Faculty of Medicine Public Health and Nursing, Universitas Gadjah Mada, Yogyakarta, Indonesia; 5Department of Chemistry, Faculty of Mathematics and Natural Sciences, Universitas Gadjah Mada, Yogyakarta, Indonesia; 6Department of Pharmaceutical Biology, Faculty of Pharmacy, Universitas Sumatera Utara, Medan, Indonesia; 7Research Center for Food Technology and Processing, National Research and Innovation Agency, Gunungkidul, Indonesia
Correspondence: Hesti Lina Wiraswati, Department of Biomedical Science, Faculty of Medicine, Universitas Padjadjaran, Bandung, 40161, Indonesia, Tel +6285795183426, Fax +62222037823, Email [email protected]
Background: Multidrug resistance in various cancer types is a major obstacle in cancer treatment. The concept of a single drug molecular target often causes treatment failure due to the complexity of the cellular processes. Therefore, combination chemotherapy, in which two or more anticancer drugs are co-administered, can overcome this problem because it potentially have synergistic efficacy besides reducing resistance, and drug doses. Previously, we reported that pyrazoline B had promising anticancer activity in both in silico and in vitro studies. To increase the efficacy of this drug, co-administration with established anticancer drugs such as doxorubicin and paclitaxel is necessary.
Materials and Methods: In this study, we used an in silico approach to predict the synergistic effect of pyrazoline B with paclitaxel or doxorubicin using various computational frameworks and compared the results with those of an established study on the combination of doxorubicin-cyclophosphamide and paclitaxel-ascorbic acid.
Results and Discussion: Drug interaction analysis showed the combination was safe with no contraindications or side effects. Furthermore, molecular docking studies revealed that doxorubicin-pyrazoline B and doxorubicin-cyclophosphamide may synergistically inhibit cancer cell proliferation by inhibiting the binding of topoisomerase I to the DNA chain. Moreover, the combination of pyrazoline B-paclitaxel may has synergistic activity to cause apoptosis by inhibiting Bcl2 binding to the Bax fragment or inhibiting cell division by inhibiting α-β tubulin disintegration. Paclitaxel-ascorbic acid had a synergistic effect on the inhibition of α-β tubulin disintegration.
Conclusion: The results show that this combination is promising for further in vitro and in vivo studies.
Keywords: doxorubicin, in silico study, pyrazoline B, paclitaxel, synergistic effect
Introduction
Chemotherapy is the standard therapeutic option for cancer treatment. Drug resistance, toxicity, and tumor heterogeneity frequently restrict their efficacy. The emergence of multidrug-resistant cancer cells in response to various anticancer drug regimens, such as doxorubicin and paclitaxel, has been a severe problem for a long time.1,2 Combination chemotherapy, in which two or more medications were administered concurrently, is a promising strategy for addressing these problems because two single-target therapies may synergize via linked biological processes.3 Drug combination therapy could exhibit synergistic efficacy and reduce resistance.4,5 Combination therapy can also minimize harmful side effects by lowering the drug doses6,7 (Figure 1).
 |
Figure 1 Visualization of advantages of combination chemotherapy. |
Pyrazoline derivatives are heterocyclic compounds that have been reported to have promising pharmacological activities against cancer cells, such as cell division inhibition and apoptosis induction.8–12 N-Phenyl pyrazoline derivatives successfully synthesized in our laboratory showed anticancer activity against MCF-7 and T47D breast cancer cell lines.13–15 This compound also induces poor survival, reduces tumor sphere size, inhibits cell growth, and reduces CD133 levels in HeLa cervical cancer cell lines.14,16 We successfully showed a decrease in EGFR (Epidermal Growth Factor Receptor) levels in HeLa cells induced by an N-phenyl pyrazoline derivate.13,16,17 Therefore, the N-phenyl pyrazoline derivative is a promising anticancer drug and potential to be combined with other anticancer drugs to increase their effectiveness. We previously reported that a compound belonging of N-phenyl pyrazoline derivates, Pyrazoline B (methoxyphenyl)-1-phenyl-3-(thiophen-2-yl) −4,5-dihydro-1H-pyrazole), exerts promising anticancer activity based on in silico18 and in vitro19 studies. This compound may potential to be combined with established anticancer drugs such as doxorubicin or paclitaxel.
For cancer patients, selecting the proper medical combination can considerably reduce their pain and boost the efficiency of their treatment.5 However, personalized cancer therapies that use pharmacological combinations with a synergistic impact are appealing but extremely difficult to implement.20 Drug combinations used during clinical trials are costly, prone to errors, and may harm patients.21 Therefore, a computational framework simplifies the screening process before conducting in vitro, in vivo, or clinical trials. In this study, we aimed to estimate the synergistic anticancer effects of pyrazoline B with paclitaxel and doxorubicin using various web servers and software.
Materials and Methods
Drug–Drug Interaction Prediction (DDI)
Drug–drug interaction prediction was performed using the DDI-pred server (https://www.way2drug.com/ddi/). In this severity class, the adverse effects of the combined drug and overlapping P450 activities of each drug were estimated.22
Ligand-Protein Docking
Three-dimensional structures of the ligands were built using the MarvinSketch software, and the structures were minimized using a Dreiding force field. The ligands used in this study are pyrazoline B, cyclophosphamide, ascorbic acid, doxorubicin, and paclitaxel. Topoisomerase I (1A31) and topoisomerase II (6ZY5) were used as target enzymes of doxorubicin. α-β tubulin (6E7B) and Bcl2 (2XA) were used as enzyme targets of paclitaxel. The protein structures were downloaded from the Protein Data Bank (https://www.rcsb.org/).23 Ligand-protein docking was performed using UCSF Chimera24 and AutoDock Vina.25 A blinded docking approach was used in the present study. The topoisomerase I gridbox was set as follows: center_x = 20.00, center_y = 10.00, center_z = 20.00, and size_x = 70.00. size_y = 70.00, size_z = 70.00. The topoisomerase 2 grid box was set as follows: center_x = 165.00, center_y = 160.00, center_z = 130.00, size_x = 90.00, size_y = 100.00, and size_z = 100.00. The α-tubulin grid box was set as follows: center_x = 100.00, center_y = 32.00, center_z = 28.00, size_x = 60.00, size_y = 70.00, and size_z = 90.00. The grid box of Bcl2 was set as follows: center_x = 35.00, center_y = ‒15.00, center_z = ‒15.00, size_x = 35.00, size_y = 30.00, and size_z = 30.00. Protein and ligand visualizations were performed using the UCSF chimera24 and ligplot plus.26 The in silico synergistic effect of cyclophosphamide-doxorubicin27 on topoisomerase I and II and ascorbic acid-paclitaxel28 on α-β tubulin and Bcl2 was used as positive controls because the drug combination has been proven to have anticancer effects in vitro and in vivo.
Protein–Protein and Protein–DNA Docking
Macromolecular docking was performed to observe the binding interactions between the proteins using the HDOCK web server http://hdock.phys.hust.edu.cn/.29 Using this server, the binding affinities of Topoisomerase I and II without ligand, Topoisomerase I and II in complex with pyrazoline B, doxorubicin, pyrazoline B-doxorubicin, cyclophosphamide, and cyclophosphamide-doxorubicin with the DNA chain were measured. Binding affinity measurements were also performed for α-tubulin without the ligand, Bcl2 in complex with pyrazoline B, paclitaxel, pyrazoline B-paclitaxel cyclophosphamide, and cyclophosphamide-doxorubicin with β-tubulin. Binding interactions of Bcl2 without ligand, Bcl2 in complex with pyrazoline B, paclitaxel, pyrazoline B-paclitaxel cyclophosphamide, and cyclophosphamide-doxorubicin with the Bax fragment were also observed.
Results and Discussion
Drug–Drug Interaction Prediction
The Way2Drug web platform divides drug–drug interactions into five classes: class 1 (contraindicated), class 2 (provisionally contraindicated), class 3 (conditional), class 4 (minimal risk), and class 5 (no interaction).22 This server defines the probability in two terms: the probability of being active (Pa) and inactive (Pi). The probability is in the range of 0–1.30 The combination of doxorubicin and cyclophosphamide had a DDI severity class of class 4 (minimal risk), with a probability of 0.215. The combination of doxorubicin and pyrazoline B had a DDI severity class of class 4 (minimal risk), with a probability of 0.482. Tables 1 and 2 also show that this combination is considered safe because it has a high probability of being inactive, causing several adverse effects such as arrhythmia, bradycardia, hypertension, hypotension, qt_interval_prolongation, and tachycardia.
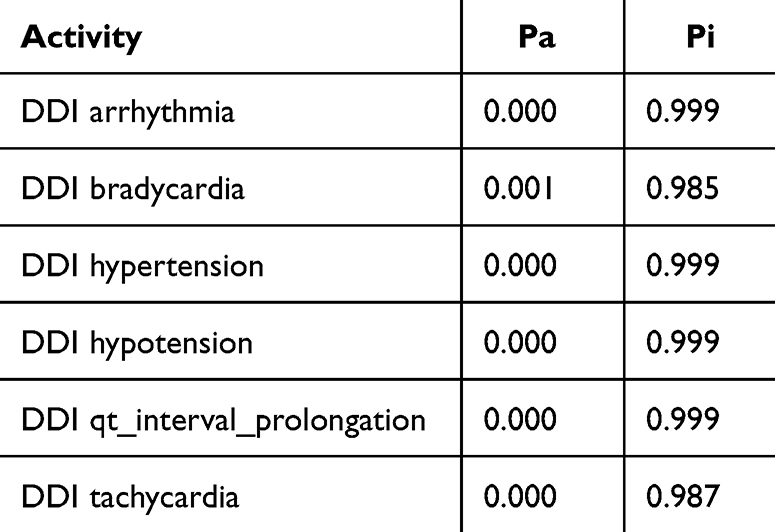 |
Table 1 Adverse Effect of Doxorubicin + Cyclophosphamide Combination. Probability Range: 0–1 |
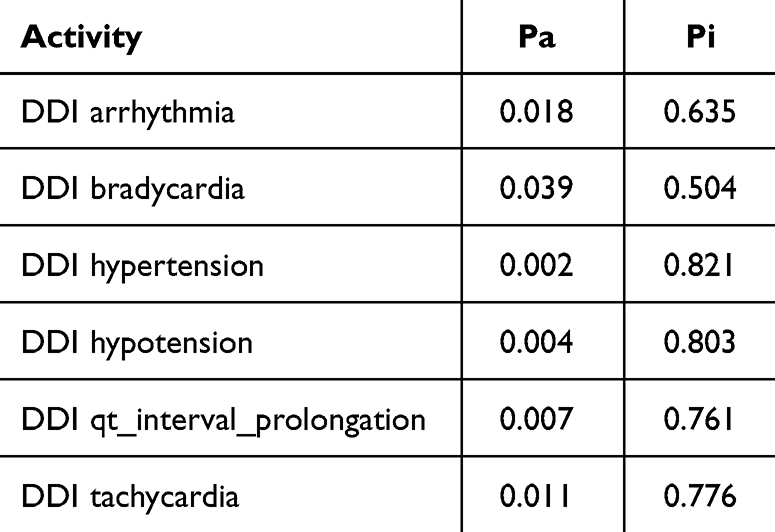 |
Table 2 Adverse Effect of Doxorubicin + Pyrazoline B Combination. Probability Range: 0–1 |
Cytochrome P450 (CYP) is a superfamily of heme-containing oxidizing enzymes involved in the metabolism of many drugs, xenobiotics, and endogenous substances. Most approved drugs are metabolized by five CYPs (1A2, 2C9, 2C19, 2D6, and 3A4). CYPs mainly mediate adverse drug–drug interactions, leading to major cause of premature drug development and medication removal from the market.31 Therefore, the overlap of CYPs activity in the drug combinations was also estimated. As shown in Table 3, the drug combination is safe because pyrazoline B is not considered a CYPs inducer, inhibitor, or substrate. Meanwhile, the combination of paclitaxel and ascorbic acid had a DDI severity class of class 4 (minimal risk) with a probability of ‒0.119. The combination of paclitaxel and pyrazoline B has a DDI severity class of class 2 (provisionally contraindicated) with a probability of 0.237, but this prediction is plausible because of the low probability. Moreover, this combination is also considered safe since it has high probability of being inactive, causing several adverse effects such as arrhythmia, bradycardia, hypertension, hypotension, qt_interval_prolongation, and tachycardia (Tables 4 and 5), and both paclitaxel and pyrazoline B are not considered as CYPs inducers/inhibitors/substrates (Table 6).
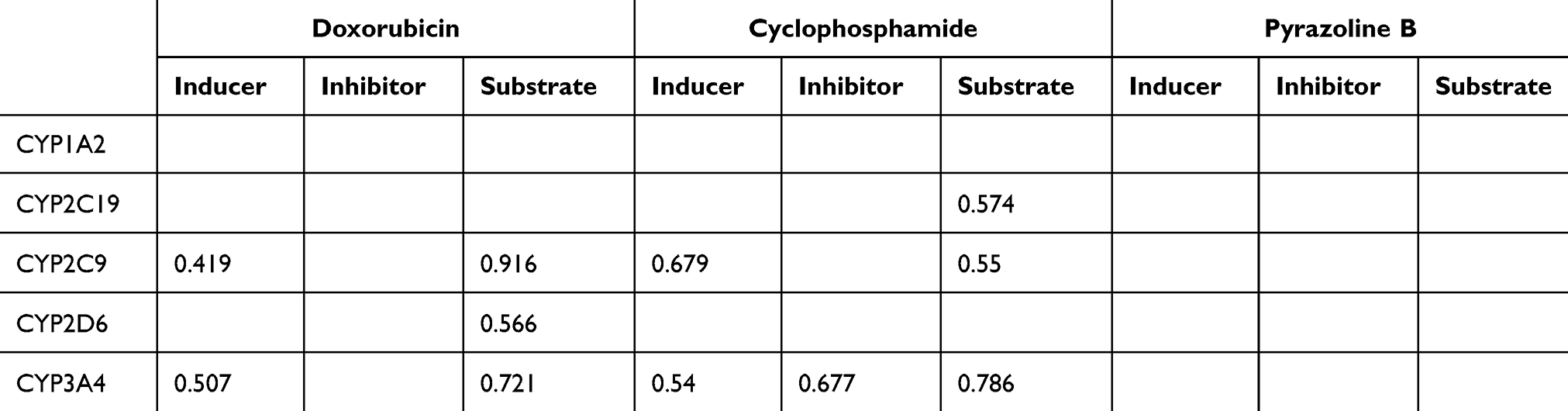 |
Table 3 Overlapping of P450 Activities of Doxorubicin, Cyclophosphamide, Pyrazoline B. Probability Range: 0–1 |
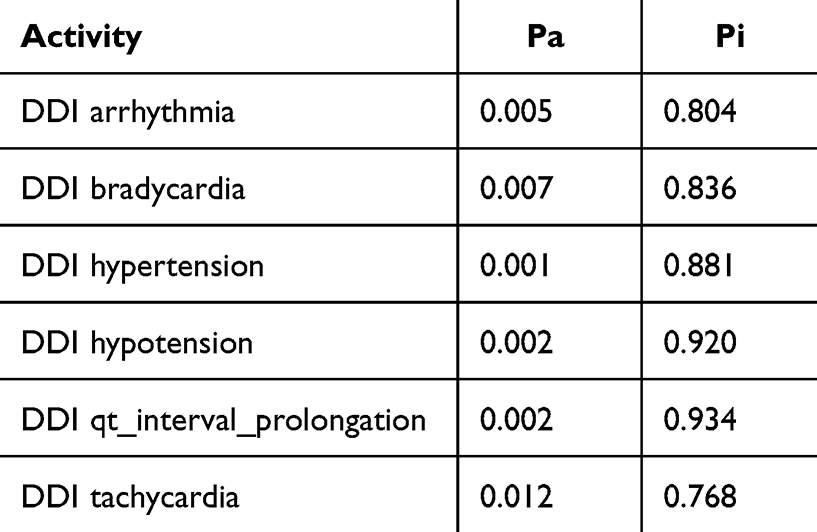 |
Table 4 Adverse Effect of Paclitaxel + Ascorbic Acid Combination. Probability Range: 0–1 |
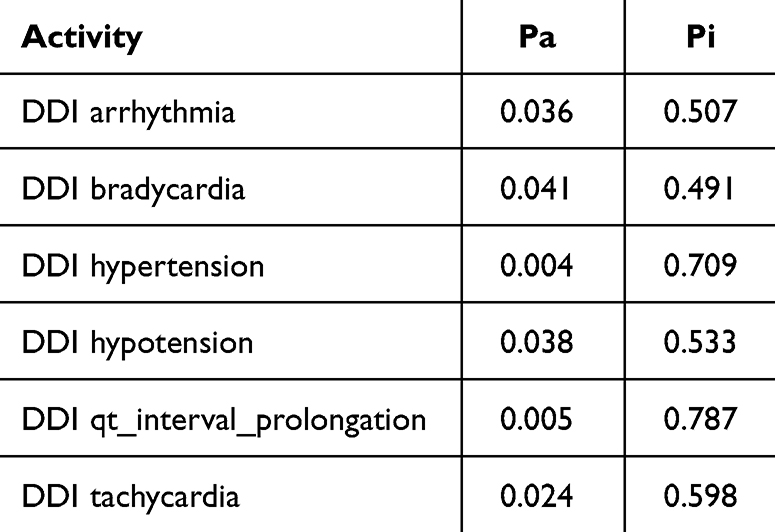 |
Table 5 Adverse Effect of Paclitaxel + Pyrazoline B Combination. Probability Range: 0–1 |
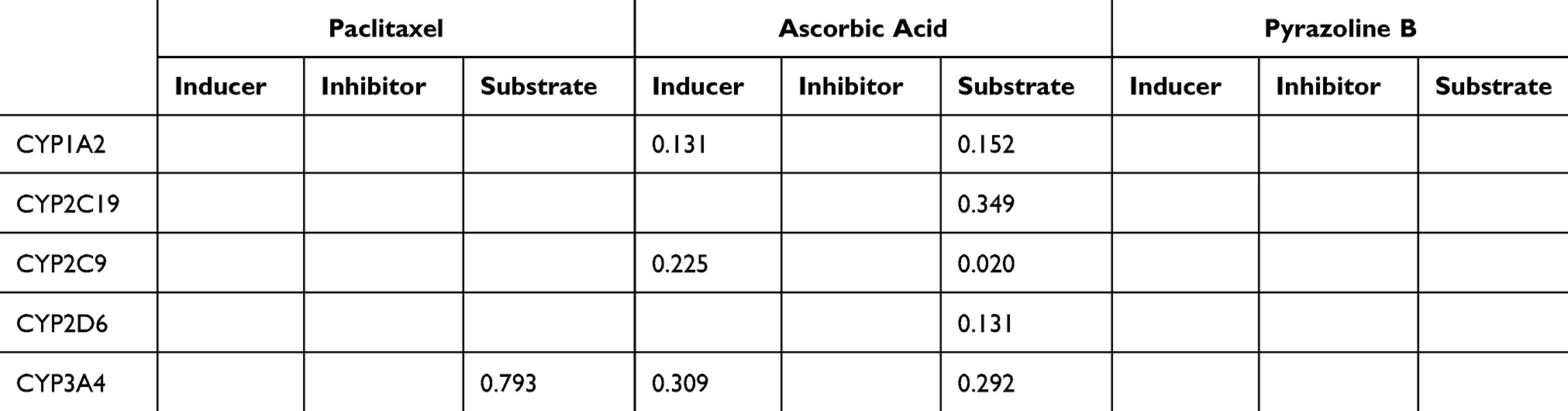 |
Table 6 Overlapping of P450 Activities of Paclitaxel, Ascorbic Acid, Pyrazoline B. Probability Range: 0–1 |
Molecular Docking Study
Molecular docking was performed against the native target of each drug to determine the synergistic effects of the drug combination. Doxorubicin exerts its molecular action by targeting DNA-associated enzymes, such as topoisomerase I and II.32 Doxorubicin nanoprodrug have been applied for treatment of various types of cancer, such as leukemia, lung, ovarian, breast etc.33 Therefore, in this study, molecular docking of doxorubicin and pyrazoline B was performed and compared to the synergistic effect of cyclophosphamide-doxorubicin, which has been proven to have anticancer effects and does not induce hepatotoxicity.27 Blind docking results in Figure 2A show that pyrazoline B binds to the DNA-binding site of topoisomerase I in a non-competitive manner with doxorubicin, similar to cyclophosphamide (Figure 2B). Doxorubicin, pyrazoline B, and cyclophosphamide have binding affinity of ‒8.4, ‒7.0, and ‒5.1 kcal/mol, respectively (Table 7). pyrazoline B forms one hydrogen bond with Thr501 (Figure 2C), Cyclophosphamides form four hydrogen bonds involving Asn491, Thr501, Gly503, and Ser506 (Figure 2D), whereas Doxorubicin forms seven hydrogen bonds with topoisomerase I involving Lys484, Leu485, Ala486, Arg590, and Val626 (Figure 2E). The van der Waals interaction of pyrazoline B appears to influence its affinity for the topoisomerase I receptor, where ten contact residues are involved (Figure 2C), compared to cyclophosphamide, which only interacts with seven contact residues (Figure 2D). This result indicated that pyrazoline B-doxorubicin and cyclophosphamide-doxorubicin might synergistically inhibit topoisomerase I.
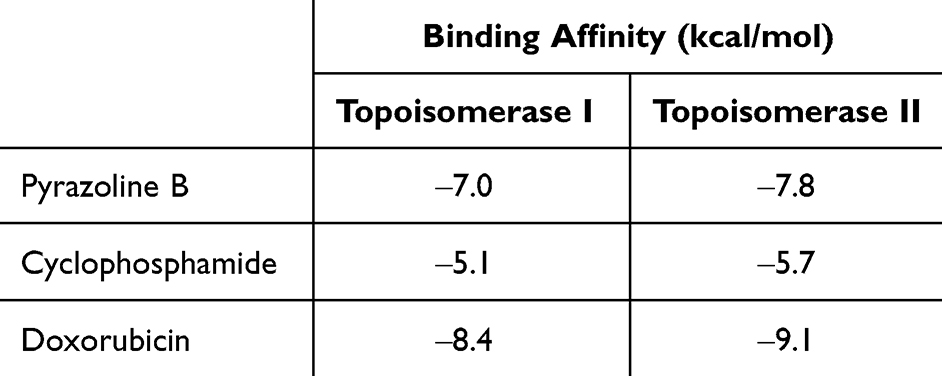 |
Table 7 Binding Affinity of Pyrazoline B and Doxorubicin Against Target of Doxorubicin (Topoisomerase I and II) |
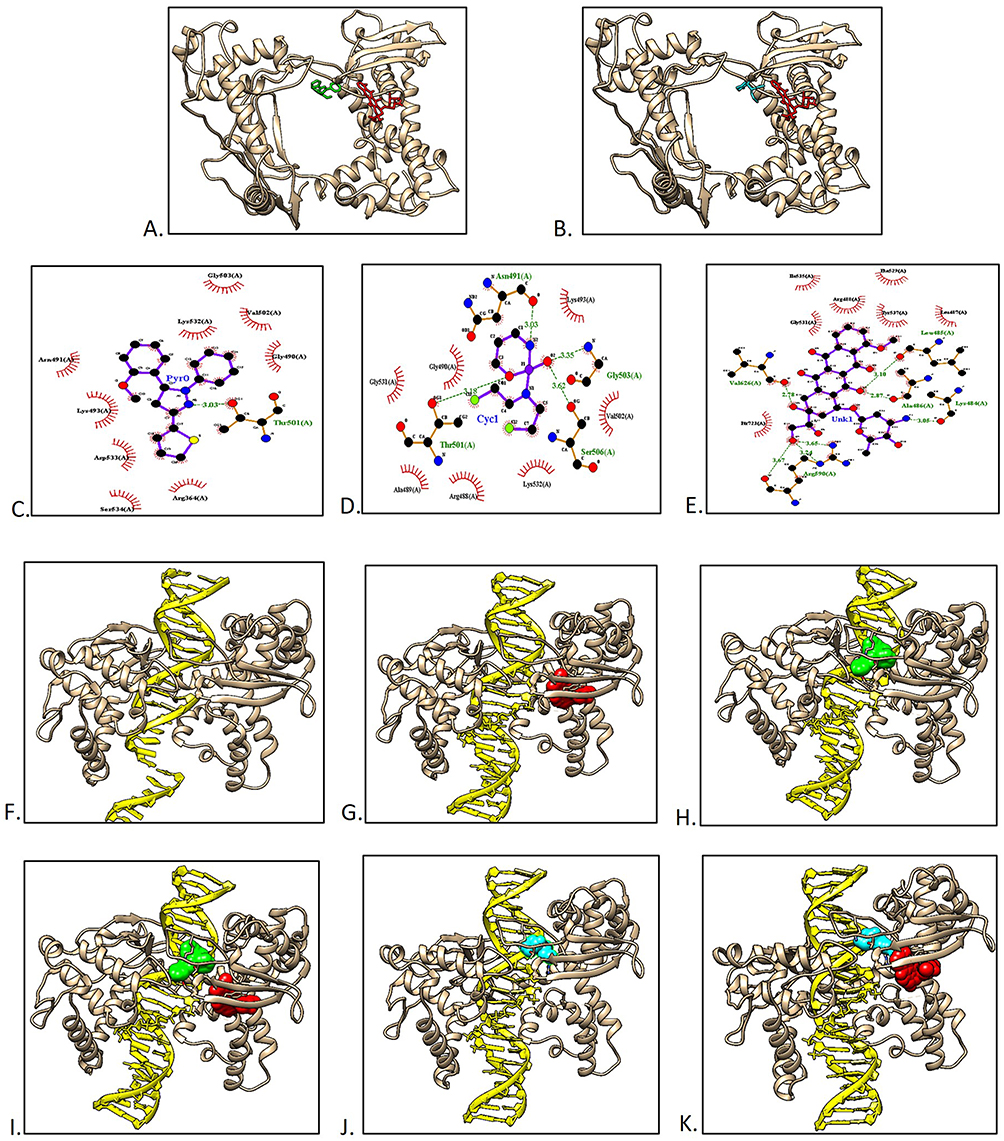 |
Figure 2 Protein-ligand docking and macromolecular docking of topoisomerase I. (A) The binding of topoisomerase I and pyrazoline B (green) or doxorubicin (red). (B) The binding of topoisomerase I and cyclophosphamide (cyan) or doxorubicin (red). (C) Pyrazoline B-topoisomerase I interaction. (D) Cyclophosphamide-topoisomerase I interaction. (E) Doxorubicin-topoisomerase I interaction. (F) Topoisomerase I without ligand. (G) Topoisomerase I-doxorubicin. (H) Topoisomerase I-pyrazoline B. (I) Topoisomerase I-Doxorubicin-pyrazoline B. (J) Topoisomerase I cyclophosphamide. (K) Topoisomerase I-doxorubicin-cyclophosphamide. |
To prove this hypothesis further, a docking simulation between topoisomerase I and the DNA chain was performed in the absence and presence of ligand(s) (Table 8 and Figure 1F–K). In the absence of the ligand, topoisomerase I had a docking score of ‒509.60 with a DNA chain and Figure 2F show the native binding mode between topoisomerase I and DNA chain. In the presence of doxorubicin, the binding affinity of topoisomerase I to the DNA chain was reduced, with a docking score of ‒499.54 (Figure 2G). Interestingly, although pyrazoline B had a lower affinity than doxorubicin for topoisomerase I, this compound exhibited more significant binding inhibition, with a docking score of ‒299.96 (Figure 2H). This analysis showed a synergistic effect of the pyrazoline B-doxorubicin combination, as this combination showed higher inhibition than single docking with doxorubicin, with a docking score of ‒301.81 (Figure 2I). This result showed that the drug combination produced better topoisomerase I binding inhibition to the DNA chain. Topoisomerase I inhibitors have molecular action in inhibiting cancer cell proliferation by inhibiting DNA replication, inducing DNA damage, and stimulating cell-cycle arrest.34 Comparison with cyclophosphamide also showed that the binding of cyclophosphamide or cyclophosphamide-doxorubicin reduced the binding affinity of topoisomerase I to the DNA chain (docking scores ‒382.41 and ‒385.16, respectively) (Figure 2J and K).
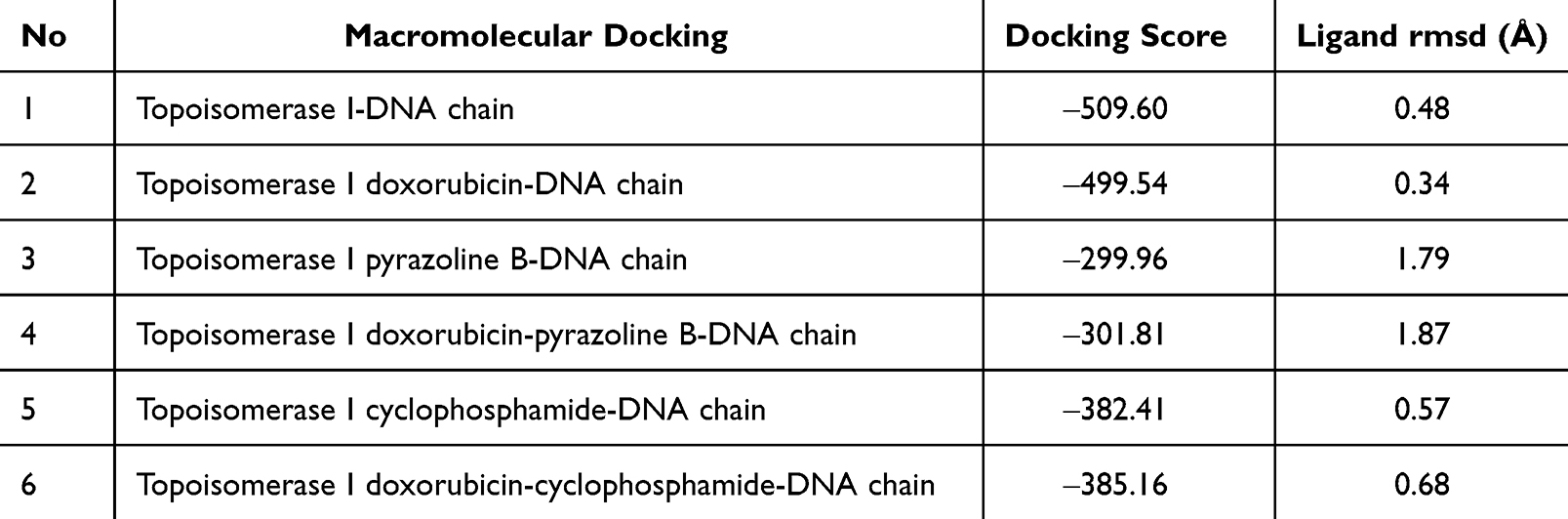 |
Table 8 Results of Topoisomerase I-DNA Chain Docking |
Figure 3A shows that pyrazoline B binds to topoisomerase II noncompetitively with doxorubicin similar with cyclophosphamide (Figure 3B). However, these two compounds did not bind to the topoisomerase II DNA-binding site. Doxorubicin, Cyclophosphamide and pyrazoline B have binding affinity of ‒9.1, −5.7 and ‒7.8 kcal/mol, respectively (Table 7). The binding of pyrazoline B involved only van der Waals interactions (Figure 3C). Cyclophosphamide only formed van der Waals interactions (Figure 3D). Meanwhile, Doxorubicin formed 12 hydrogen bonds involving Thr620, Ser621, Lys622, Asn774, Asn782, Gly788, Gln789, and Asn894 (Figure 3E). The macromolecular docking between topoisomerase II and the DNA chain is in line with previous results showing that Topoisomerase II-doxorubicin, Topoisomerase II-pyrazoline B, Topoisomerase II-doxorubicin-pyrazoline B, Topoisomerase II-cyclophosphamide, and Topoisomerase II-doxorubicin-cyclophosphamide did not reduce the binding affinity toward the DNA chain, indicating that pyrazoline B, doxorubicin, and cyclophosphamide do not bind to the DNA binding site of topoisomerase II (Table 9). These results align with another report that higher doses of doxorubicin-cyclophosphamide benefit patients with HER-2-amplified tumors, and do not correlate with topoisomerase II.35
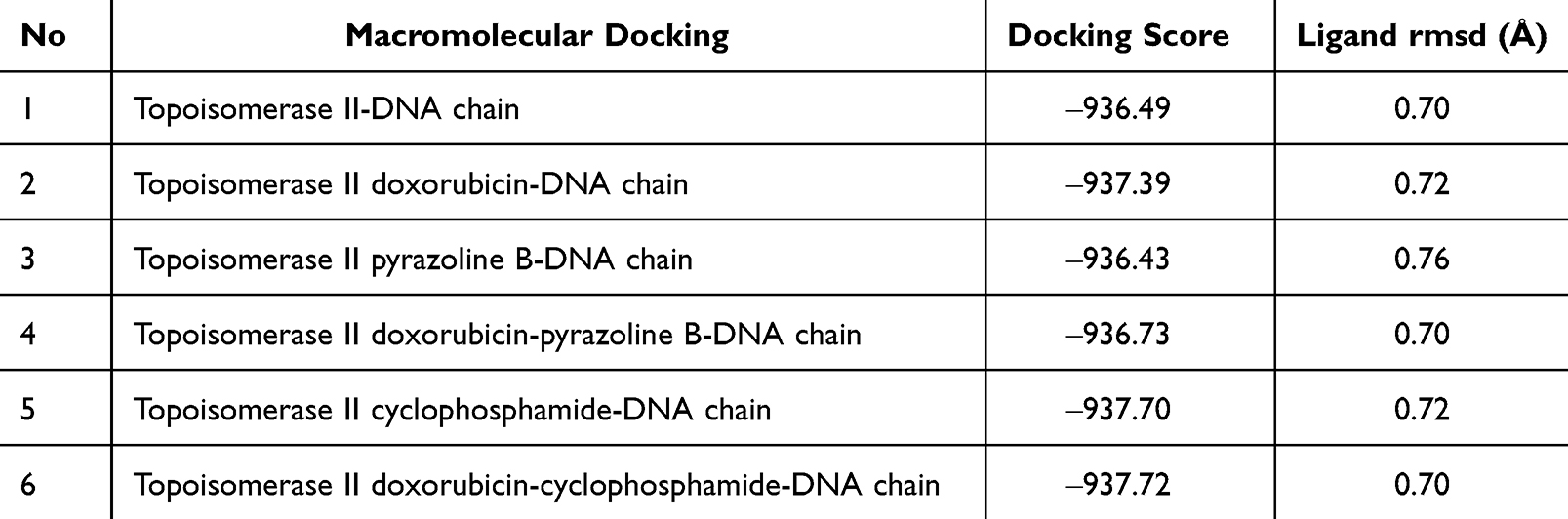 |
Table 9 Results of Topoisomerase II-DNA Chain Docking |
 |
Figure 3 Docking of topoisomerase II and ligands. (A) The binding of topoisomerase II and pyrazoline B (green) or doxorubicin (red). (B) The binding of topoisomerase II and cyclophosphamide (cyan) or doxorubicin (red). (C) Pyrazoline-topoisomerase II interaction. (D) Cyclophosphamide-topoisomerase II interaction. (E) Doxorubicin-topoisomerase II interaction. |
We investigated the binding of pyrazoline B-paclitaxel, to α-tubulin. Paclitaxel exerts its molecular action by stabilizing bundles and preventing microtubule disintegration during cell division. As a result, cell growth is reduced by stopping the cell cycle at the metaphase/anaphase border and forming an incomplete metaphase plate of chromosomes, which is triggered by microtubule dynamics stabilization.36 Therefore, in this study, molecular docking of pyrazoline B and paclitaxel was performed and compared to the synergistic effect of ascorbic acid-paclitaxel, which has been proven to alleviate the anticancer effect of paclitaxel.28 Binding affinity of paclitaxel, pyrazoline B and ascorbic acid were −8.1, −6.9 and −6.7 kcal/mol, respectively (Table 10). Figure 4A reveals that pyrazoline B and Paclitaxel competitively bind to the binding site between α-tubulin and β-tubulin similar with ascorbic acid (Figure 4B). pyrazoline B possesses only van der Waals interaction (Figure 4C). Ascorbic acid forms six hydrogen bonds involving Glu71, Ala99, Ala100, Thr145, Gly146, and Lys252 (Figure 4D). Paclitaxel forms three hydrogen bonds with α-β tubulin, involving Thr225 and Arg320 (Figure 4E).
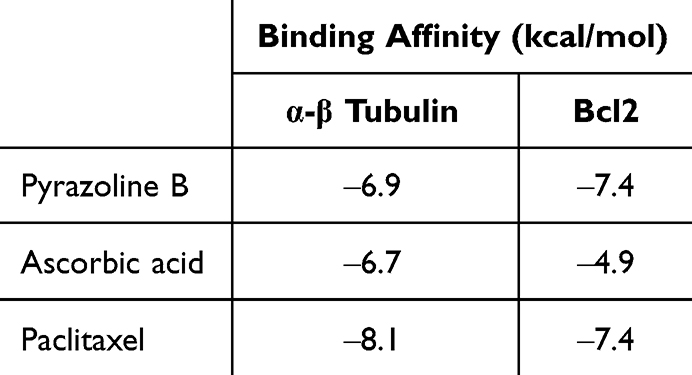 |
Table 10 Binding Affinity of Pyrazoline B and Paclitaxel Against Target of Paclitaxel (α-β Tubulin and Bcl2) |
 |
Figure 4 Protein-ligand docking and macromolecular docking of α tubulin. (A) Pyrazoline B (green) and Paclitaxel (Orange) binding to α-β tubulin. (B) Ascorbic acid (blue) and Paclitaxel (Orange) binding to α-β tubulin. (C) α-β tubulin-pyrazoline B. (D) α-β tubulin-ascorbic acid. (E) α- β tubulin-paclitaxel. (F) α-tubulin without ligand. (G) α tubulin-Paclitaxel. (H) α tubulin-pyrazoline B. (I) α tubulin-Paclitaxel-pyrazoline B. (J) α tubulin-ascorbic acid. (K) α tubulin-Paclitaxel-ascorbic acid. |
To determine whether the compound would increase or decrease the binding between α-tubulin and β-tubulin, macromolecular docking was performed. The results showed that the binding of paclitaxel, pyrazoline B, paclitaxel-pyrazoline B, ascorbic acid, and paclitaxel-ascorbic acid to α tubulin increased the binding affinity between α and β tubulin compared to α-β-tubulin without ligand (Table 11). The binding of α-βtubulin in the absence of ligand were 503.57 (Figure 4F). The binding of α-βtubulin in the presence of paclitaxel or pyrazoline B alone (docking score ‒600.39 and ‒572.05, respectively) (Figure 4G and H) was lower than in the presence of paclitaxel-pyrazoline B (docking score ‒619.53) (Figure 4I). The binding of α-β-tubulin in the presence in the presence of ascorbic acid only (docking score, ‒564.25) (Figure 4J) was lower than that of paclitaxel-ascorbic acid (docking score, ‒612.83) (Figure 4K). This phenomenon supports the hypothesis that paclitaxel, pyrazoline B, and ascorbic acid may have molecular mechanisms that increase the binding of α and β tubulin in cancer cells. Therefore, prevention of cell division occurs through the prevention of α-β-tubulin disintegration.
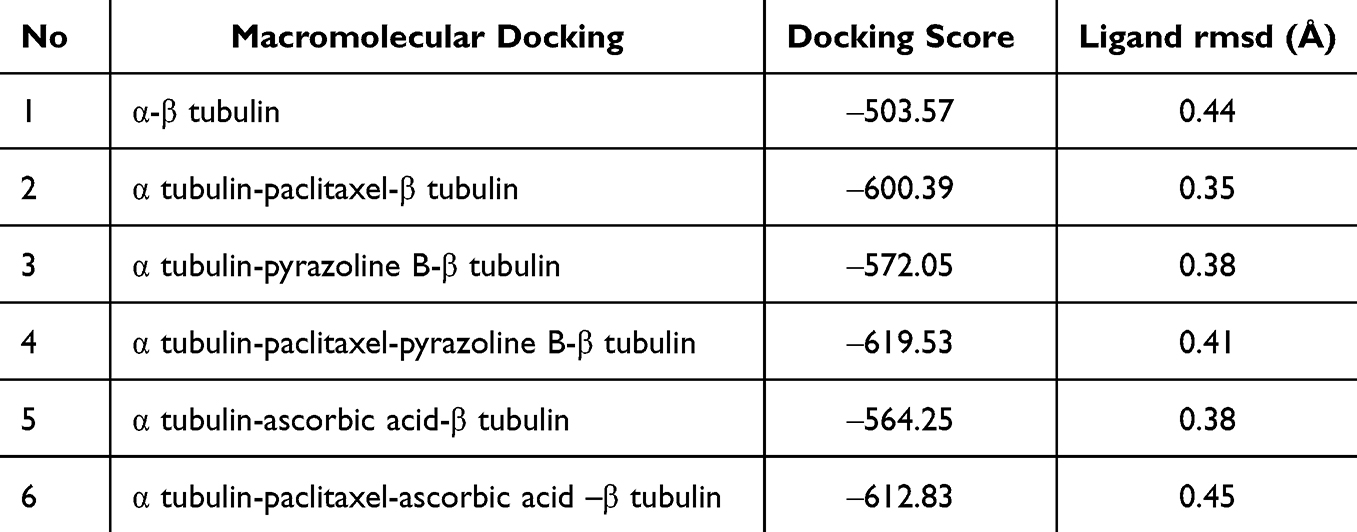 |
Table 11 Macromolecules Docking of α and β Tubulin |
Tubulin poisoning action of paclitaxel also killed cancer cells expressing Bcl2 at high concentrations; the drug inhibited the anti-apoptotic activity of Bcl2.37 PTX targets the loop domain of Bcl2, leading to apoptosis initiation. Molecular modeling showed a similarity between β-tubulin and Bcl2 binding sites.38,39 Molecular docking analysis revealed that the affinity energies of paclitaxel and pyrazoline B for Bcl2 were similar (‒7.4 kcal/mol) (Table 11), with close binding sites (Figure 5A), whereas binding site of ascorbic acid was far from paclitaxel (Figure 5B) with affinity −4.9 kcal/mol. Pyrazoline B only possesses van der Waals interactions (Figure 5C). Ascorbic acid formed four hydrogen bonds involving Arg127, Glu179, Arg183, and His184 (Figure 5D). Paclitaxel forms three hydrogen bonds with Bcl2, involving Tyr108 and Gln118 (Figure 5E). The van der Waals interactions of pyrazoline B appear to have a greater influence on its affinity for the Bcl2 receptor, where ten contact residues are involved (Figure 5C), compared to ascorbic acid, which interacts with only five contact residues (Figure 5D). In addition, the binding site of ascorbic acid is far from that of paclitaxel on Bcl2 (Figure 5B).
 |
Figure 5 Protein-ligand docking and macromolecular docking of Bcl2. (A) Pyrazoline B (green) and Paclitaxel (Orange) binding to Bcl2. (B) Ascorbic acid (blue) and Paclitaxel (Orange) binding to Bcl2. (C) Pyrazoline B interaction with residues on Bcl2. (D) Ascorbic acid interaction with residues on Bcl2. (E) Paclitaxel interaction with residues on Bcl2. (F) Bcl2 without ligand. (G) Bcl2-Paclitaxel. (H) Bcl2-pyrazoline B. (I) Bcl2-Paclitaxel-pyrazoline B. (J) Bcl2-ascorbic acid. (K) Bcl2-Paclitaxel-ascorbic acid. |
Simulation of protein–protein docking revealed that pyrazoline B seems to show a synergistic effect with paclitaxel in inhibiting the binding of Bcl2 (anti-apoptotic protein) to Bax (proapoptotic protein) (Table 12). Without a ligand, Bcl2 has a binding score of ‒510.10 against the Bax fragment (Figure 5F). In the presence of paclitaxel, the binding score was reduced by more than 50% (‒216.61) (Figure 5G), indicating that, in line with previous findings, paclitaxel has an additional molecular action by targeting Bcl2 to prevent this anti-apoptotic protein from binding with Bax. In the presence of pyrazoline B, the binding score was reduced by more than 50% (‒203.93) (Figure 5H). Interestingly, the combination of paclitaxel and pyrazoline B resulted in a lower binding score (‒204.17) (Figure 5I). Bcl2 acts as an anti-apoptotic protein by binding to the Bax monomer and preventing oligomerization of Bax; therefore, pore formation in the mitochondria does not occur, leading to inhibition of the intrinsic apoptosis pathway.40 The synergistic binding of paclitaxel and Pyrazoline B to Bcl2 seem to prevents the binding of Bcl2 to Bax. This may lead to oligomerization of Bax, which causes permeabilization of the mitochondrial outer membrane and promotes cell death by releasing effector apoptosis molecules such as cytochrome c, apoptosis inducing factor (AIF), or SMAC/Diablo.40–42 A comparison with ascorbic acid-paclitaxel showed that ascorbic acid itself slightly reduced the binding between Bcl2-Bax (‒454.09) because the binding site of ascorbic acid is not located in the binding site of Bax. However, binding of the Bcl2-ascorbic acid complex to Bax was similar to binding of Bcl2-ascorbic acid-paclitaxel complex to Bax (−216.61 and ‒216.72, respectively) (Figure 5J and K). This shows that the main molecular mechanism of ascorbic acid in cancer cells might not induce apoptosis by preventing the Bcl2-Bax complex but by inhibiting cell division through stabilization of the α-β-tubulin complex.
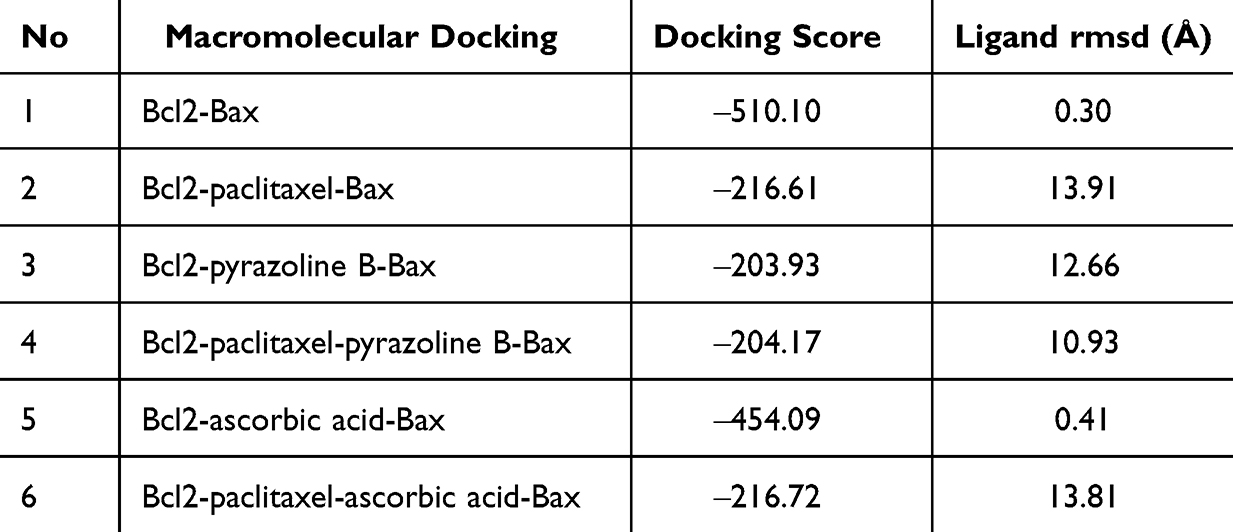 |
Table 12 Macromolecules Docking of Bcl2 and Bax |
Conclusion
Preliminary analysis using the Way2Drug web platform revealed that the combination of doxorubicin-cyclophosphamide, doxorubicin-pyrazoline B, paclitaxel-ascorbic acid, and paclitaxel-pyrazoline B was safe because it was estimated to have no contraindications or side-effects. Ligand-protein docking demonstrated that doxorubicin-pyrazoline B and doxorubicin-cyclophosphamide exhibited high affinity for the DNA-binding site of topoisomerase I in a non-competitive manner. Paclitaxel-pyrazoline B also exhibited high affinity for the Bax fragment binding site of Bcl2 in a non-competitive manner. Paclitaxel-pyrazoline B also showed high affinity for the binding site between α- and β-tubulin. Furthermore, simulation of macromolecular docking revealed that doxorubicin-pyrazoline B and doxorubicin-cyclophosphamide synergistically inhibited topoisomerase I to bind with the DNA chain, that will leading to the inhibition of cancer cell proliferation by inhibiting DNA replication, inducing DNA damage, and stimulating cell cycle arrest. Pyrazoline B-paclitaxel has synergistic activity, causing apoptosis by inhibiting Bcl2 binding to the Bax fragment or inhibiting cell division by inhibiting α-β-tubulin disintegration. Paclitaxel-ascorbic acid had a synergistic effect in inhibiting α-β-tubulin disintegration. All molecular simulation data indicated that the combination could be applied for further in vitro or in vivo research to observe the inhibition of cell proliferation or apoptosis in cancer cells or animal model.
Acknowledgments
This study was supported by the Universitas Padjadjaran Research Grant “Riset Kolaborasi, Indonesia (RKI)”. We thank the Directorate of Research and Community Service and Innovation of the Universitas Padjadjaran.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Kars MD, Iseri OD, Gündüz U, Ural AU, Arpaci F, Molnár J. Development of rational in vitro models for drug resistance in breast cancer and modulation of MDR by selected compounds. Anticancer Res. 2006;26(6b):4559.
2. Xu F, Wang F, Yang T, Sheng Y, Zhong T, Chen Y. Differential drug resistance acquisition to doxorubicin and paclitaxel in breast cancer cells. Can Cell Inter. 2014;14(1):538. doi:10.1186/s12935-014-0142-4
3. Celebi R, Bear Don’t Walk O, Movva R, Alpsoy S, Dumontier M. In-silico prediction of synergistic anti-cancer drug combinations using multi-omics data. Sci Rep. 2019;9(1):8949. doi:10.1038/s41598-019-45236-6
4. Jeon M, Kim S, Park S, Lee H, Kang J. In silico drug combination discovery for personalized cancer therapy. BMC Syst Biol. 2018;12(Suppl 2):16. doi:10.1186/s12918-018-0546-1
5. Meng F, Li F, Liu J-X, et al. NEXGB: a network embedding framework for anticancer drug combination prediction. Int J Mol Sci. 2022;23(17):9838. doi:10.3390/ijms23179838
6. Gupta A, Chauhan SS, Gaur AS, Parthasarathi R. Computational screening for investigating the synergistic regulatory potential of drugs and phytochemicals in combination with 2-deoxy-D-glucose against SARS-CoV-2. Struct Chem. 2022;33(6):2179–2193. doi:10.1007/s11224-022-02049-0
7. Jaaks P, Coker EA, Vis DJ, et al. Effective drug combinations in breast, colon and pancreatic cancer cells. Nature. 2022;603(7899):166–173. doi:10.1038/s41586-022-04437-2
8. Karabacak M, Altıntop MD, Ibrahim Çiftçi H, et al. Synthesis and evaluation of new pyrazoline derivatives as potential anticancer agents. Molecules. 2015;20(10):19066–19084. doi:10.3390/molecules201019066
9. Menier -A-A, Melati K, O. Al-Najjar B, et al. Synthesis, anticancer activity and docking studies of pyrazoline and pyrimidine derivatives as potential epidermal growth factor receptor (EGFR) inhibitors. Arabian J Chem. 2022;15(7):103864. doi:10.1016/j.arabjc.2022.103864
10. Mohamad Y, Payal J. Synthetic and biological studies of pyrazolines and related heterocyclic compounds. Arabian J Chem. 2014;7(5):553–596. doi:10.1016/j.arabjc.2011.09.013
11. Rana M, Faizan MI, Dar SH, et al. Design and synthesis of carbothioamide/carboxamide-based pyrazoline analogs as potential anticancer agents: apoptosis, molecular docking, ADME assay, and DNA binding studies. ACS Omega. 2022;7(26):22639–22656. doi:10.1021/acsomega.2c02033
12. Wang H, Zheng J, Xu W, et al. A new series of cytotoxic pyrazoline derivatives as potential anticancer agents that induce cell cycle arrest and apoptosis. Molecules. 2017;22(10). doi:10.3390/molecules22101635
13. Suma A, Wahyuningsih TD, Mustofa M. Synthesis, cytotoxicity evaluation and molecular docking study of N-phenylpyrazoline derivatives. Indonesian J Chem. 2019;19(4):1081. doi:10.22146/ijc.45777
14. Tutik Dwi W, Artania Adnin Tri S, Endang Puji A. Synthesis, anticancer activity, and docking study of N-acetyl pyrazolines from veratraldehyde. J Appl Pharm Sci. 2019;2019:1.
15. Suma A, Wahyuningsih TD, Pranowo D. Synthesis and antibacterial activities of N-phenylpyrazolines from veratraldehyde. Mater Scie Forum. 2017;901:124–132. doi:10.4028/www.scientific.net/MSF.901.124
16. Mustofa S, Satriyo PB, Suma AAT, et al. A potent EGFR inhibitor, N-phenyl pyrazoline derivative suppresses aggressiveness and cancer stem cell-like phenotype of cervical cancer cells. Drug Des Devel Ther. 2022;16:2325–2339. doi:10.2147/DDDT.S350913
17. Matiadis D, Sagnou M. Pyrazoline hybrids as promising anticancer agents: an up-to-date overview. Int J Mol Sci. 2020;21(15):5507. doi:10.3390/ijms21155507
18. Wiraswati HL, Bashari MH, Alfarafisa NM, et al. Virtual screening of anticancer activity of chalcones and pyrazolines as potential EGFR, VEGFR, and cytochrome P450 inhibitors. J Pharm Pharmacogn Res. 2023;11(4):699–713. doi:10.56499/jppres23.1591_11.4.699
19. Wiraswati HL, Satriyo PB. Pyrazoline B Induces Caspase-Dependent Apoptosis, Inhibits the EGFR and VEGFR-2 Signaling Pathways, and Potentiates Paclitaxel in Luminal B Breast Cancer Cells. Manuscript submitted for publication; 2023.
20. Narayan RS, Molenaar P, Teng J, et al. A cancer drug atlas enables synergistic targeting of independent drug vulnerabilities. Nat Commun. 2020;11(1):2935. doi:10.1038/s41467-020-16735-2
21. Fogel DB. Factors associated with clinical trials that fail and opportunities for improving the likelihood of success: a review. Contemp Clin Trials Commun. 2018;11:156–164. doi:10.1016/j.conctc.2018.08.001
22. Dmitriev AV, Filimonov DA, Rudik AV, et al. Drug-drug interaction prediction using PASS. SAR QSAR Environ Res. 2019;30(9):655–664. doi:10.1080/1062936x.2019.1653966
23. Berman HM, Westbrook J, Feng Z, et al. The protein data bank. Nucleic Acids Res. 2000;28(1):235–242. doi:10.1093/nar/28.1.235
24. Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi:10.1002/jcc.20084
25. Trott O, Olson AJ. AutoDock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. doi:10.1002/jcc.21334
26. Laskowski RA, Swindells MB. LigPlot+: multiple ligand–protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51(10):2778–2786. doi:10.1021/ci200227u
27. Pondugula SR, Salamat JM, Abbott KL, et al. A clinically relevant combination treatment with doxorubicin and cyclophosphamide does not induce hepatotoxicity in C57BL/6J mice. Liver Res. 2021;5(4):239–242. doi:10.1016/j.livres.2021.04.002
28. Park JH, Davis KR, Lee G, et al. Ascorbic acid alleviates toxicity of paclitaxel without interfering with the anticancer efficacy in mice. Nutr Res. 2012;32(11):873–883. doi:10.1016/j.nutres.2012.09.011
29. Yan Y, Zhang D, Zhou P, Li B, Huang S-Y. HDOCK: a web server for protein–protein and protein–DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 2017;45(W1):W365–W373. doi:10.1093/nar/gkx407
30. Poroikov VV, Filimonov DA, Ihlenfeldt WD, et al. PASS biological activity spectrum predictions in the enhanced open NCI database browser. J Chem Inf Comput Sci. 2003;43(1):228–236. doi:10.1021/ci020048r
31. Ai D, Cai H, Wei J, Zhao D, Chen Y, Wang L. DEEPCYPs: a deep learning platform for enhanced cytochrome P450 activity prediction. Front Pharmacol. 2023;14:1099093. doi:10.3389/fphar.2023.1099093
32. Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol. 2013;65(2):157–170. doi:10.1111/j.2042-7158.2012.01567.x
33. Zhao H, Yu J, Zhang R, Chen P, Jiang H, Yu W. Doxorubicin prodrug-based nanomedicines for the treatment of cancer. Eur J Med Chem. 2023;258:115612. doi:10.1016/j.ejmech.2023.115612
34. Liu J, Qu L, Meng L, Shou C. Topoisomerase inhibitors promote cancer cell motility via ROS-mediated activation of JAK2-STAT1-CXCL1 pathway. J Exp Clin Cancer Res. 2019;38(1):370. doi:10.1186/s13046-019-1353-2
35. Harris LN, Broadwater G, Abu-Khalaf M, et al. Topoisomerase II{alpha} amplification does not predict benefit from dose-intense cyclophosphamide, doxorubicin, and fluorouracil therapy in HER2-amplified early breast cancer: results of CALGB 8541/150013. J Clin Oncol. 2009;27(21):3430–3436. doi:10.1200/jco.2008.18.4085
36. Kampan NC, Madondo MT, McNally OM, Quinn M, Plebanski M. Paclitaxel and its evolving role in the management of ovarian cancer. Biomed Res Int. 2015;2015:413076. doi:10.1155/2015/413076
37. Srivastava RK, Mi QS, Hardwick JM, Longo DL. Deletion of the loop region of Bcl-2 completely blocks paclitaxel-induced apoptosis. Proc Natl Acad Sci U S A. 1999;96(7):3775–3780. doi:10.1073/pnas.96.7.3775
38. Ferlini C, Cicchillitti L, Raspaglio G, et al. Paclitaxel directly binds to Bcl-2 and functionally mimics activity of Nur77. Cancer Res. 2009;69:6906–6914. doi:10.1158/0008-5472.CAN-09-0540
39. Ben-Hamo R, Zilberberg A, Cohen H, et al. Resistance to paclitaxel is associated with a variant of the gene BCL2 in multiple tumor types. NPJ Precis Oncol. 2019;3(1):12. doi:10.1038/s41698-019-0084-3
40. Dlugosz PJ, Billen LP, Annis MG, et al. Bcl-2 changes conformation to inhibit Bax oligomerization. EMBO J. 2006;25(11):2287–2296. doi:10.1038/sj.emboj.7601126
41. Sharpe JC, Arnoult D, Youle RJ, et al. Control of mitochondrial permeability by Bcl-2 family members. Biochim Biophys Acta. 2004;1644(2–3):107–113. doi:10.1016/j.bbamcr.2003.10.016
42. Lorenzo HK, Susin SA, Penninger J, et al. Apoptosis inducing factor (AIF): a phylogenetically old, caspase-independent effector of cell death. Cell Death Differ. 1999;6(6):516–524. doi:10.1038/sj.cdd.4400527
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.