Back to Journals » Journal of Hepatocellular Carcinoma » Volume 13
Putative Imprinting Control Regions with Aberrant Blood-Based DNA Methylation are Associated with Hepatocellular Carcinoma Risk
Authors DiFrank AJ ![]() , Corliss BA, Park JJH
, Corliss BA, Park JJH ![]() , Rogers M, Skaar DA, Jima DD, Cowley M, Murphy SK
, Rogers M, Skaar DA, Jima DD, Cowley M, Murphy SK ![]() , Liu A, Moylan CA, Hoyo C
, Liu A, Moylan CA, Hoyo C
Received 25 February 2026
Accepted for publication 27 May 2026
Published 18 June 2026 Volume 2026:13 605131
DOI https://doi.org/10.2147/JHC.S605131
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 7
Editor who approved publication: Dr David Gerber
Annie J DiFrank,1 Bruce A Corliss,1,2,* James JH Park,2,* Molly Rogers,1,2,* David A Skaar,1 Dereje D Jima,2 Michael Cowley,1 Susan K Murphy,3 Andrew Liu,4 Cynthia A Moylan,5 Cathrine Hoyo1
1Department of Biological Sciences, North Carolina State University, Raleigh, NC, USA; 2College of Sciences, North Carolina State University, Raleigh, NC, USA; 3Department of Obstetrics and Gynecology, Duke University Medical Center, Durham, NC, USA; 4Department of Neurology, Duke University Health System, Durham, NC, USA; 5Department of Medicine, Duke University School of Medicine, Durham, NC, USA
*These authors contributed equally to this work
Correspondence: Annie J DiFrank, North Carolina State University, 850 Main Campus Dr, Raleigh, NC, 27606, USA, Tel +1 814 969 9246, Email [email protected]
Introduction: Site-specific 5-methylcytosine levels measured in blood can be suitable surrogate markers for epigenetic alterations in inaccessible tissues and are associated with hepatocellular carcinoma (HCC) risk. However, replication has been limited due to prior low genomic coverage and the tissue-specific nature of methylation at many CpG sites. One exception is imprinting control regions (ICRs), which use stable, early-onset methylation to regulate monoallelic expression across somatic tissues, implying aberrant methylation at ICRs detected in blood may be relevant to liver disease.
Purpose: Leveraging primary HCC samples, we aimed to identify putative ICRs with aberrant DNA methylation associated with HCC to inform development of a blood-based risk stratification panel for earlier detection and clinical triage.
Participants and Methods: Whole genome bisulfite sequencing (WGBS) was performed on leukocyte-derived DNA of 10 primary HCC cases and 51 controls to identify differential methylation. An independent comparison of 29 primary HCC cases and 36 controls was used to validate differential methylation measured in leukocytes using the novel Human Imprintome Methylation Array.
Results: We identified 1,519 differentially methylated regions associated with HCC by WGBS (FDR p < 0.05), which mapped to 81 putative ICRs, the majority of which had not previously been linked to HCC. Comparison with published studies further established 16 putative ICRs that have previously been associated with HCC or precancerous chronic liver disease states for a total of 97 loci. Validation using the Human Imprintome Methylation Array replicated 46 (57%) of these regions (FDR p < 0.05). Nearest genes were enriched for pathways related to liver disease including liver hyperplasia, hepatitis, steatosis, and HCC.
Conclusion: Our comprehensive genome profiling identified aberrant methylation at putative ICRs in blood that were associated with HCC. These findings support the hypothesis that ICRs, which exhibit relatively stable methylation across tissues, warrant further investigation as early blood-based HCC biomarkers.
Keywords: DNA methylation, epigenetic markers, risk stratification, epigenetic profiling
Introduction
Recent projections suggest hepatocellular carcinoma (HCC) will be the fifth leading cancer-related cause of death in the US in 2026, accounting for ~31,000 deaths.1 Despite advances in cancer prevention and treatment, HCC remains associated with poor prognosis, and 5-year survival remains low (22%).1,2 The growing population burden of metabolic dysfunction, including obesity and type 2 diabetes mellitus, contributes to HCC occurrence, with epidemiological studies consistently showing obesity as an independent risk factor, even after accounting for viral hepatitis and alcohol use.3–7 The increase of harmful environmental exposures like viruses, mycotoxins, hepatocarcinogens, tobacco smoke, and workplace carcinogens also contribute to HCC incidence.4
In the United States, high mortality is due in part to HCC is typically being undiagnosed until advanced stages, when treatment options are few, with limited efficacy. Notably, HCC arises almost exclusively in the context of chronic liver disease (CLD), most commonly following a prolonged, clinically silent course of hepatic injury that culminates in cirrhosis, the end stage of CLD. This extended and often asymptomatic phase represents a substantial and underutilized opportunity for early intervention. While early-stage HCC is potentially curable, with 5-year survival exceeding 70%,5,8 and surveillance is associated with earlier detection and improved outcomes,6,9,10 existing screening strategies lack sufficient uptake, underscoring the urgent need for novel, reliable approaches to detect HCC earlier along the disease continuum.
Current HCC surveillance guidelines rely heavily on semiannual or annual ultrasound, often combined with serum alpha-fetoprotein (AFP) levels.8 However, ultrasound and AFP serum levels have suboptimal sensitivity and specificity for early-stage disease, limiting their utility as stand-alone risk stratifying markers.11,12 Many risk stratification tools with a variety of approaches have been proposed to make up for this.
For example, etiology-based risk stratification has its shortcomings, as not all HCC patients present with a recognized risk factor, like cirrhosis, and reliance on clinical factors can lead to both overdiagnosis and missed cases.13 Moreover, many models were developed in cohorts dominated by chronic viral hepatitis infection (ie., hepatitis B virus (HBV), hepatitis C virus (HCV)).14–16 With effective antiviral therapies reducing the burden of HBV and HCV, and the increasing prevalence of other CLD etiologies, especially metabolic dysfunction-associated steatotic liver disease (MASLD), these models may be less relevant or accurate; the epidemiology of HCC is changing.
Multi-omics approaches have begun to address this gap. For instance, proteomic approaches, including a measure of circulating biomarker aberration (Hepatoscore-14) and phosphoproteomic subclassifications, have identified molecular alteration subgroups linked to prognosis.17–22 Additionally, polygenic risk scores (PRS) such as PRS-HPC and NAFLD-PRS may be useful as predictors of progressive hepatic damage, including fibrosis, cirrhosis and HCC.23(p90),24–27 Composite models that integrate tumor-associated proteins with demographic factors offer improvements, but remain imperfect.16–18,28–32 For example, the GALAD score (age + sex + AFP ng/mL + AFP-L3% + DCP ng/mL) or newer Oncoguard® Liver test (sex + AFP ng/mL + 3 DNA methylation markers in HOXA1, TSPYL5, and B3GALT6), now in post-market surveillance, improve diagnostic accuracy compared to ultrasound alone. However, given their inclusion of tumor-derived signals (AFP in both), these approaches provide a snapshot of the molecular landscape of HCC, but their utility for early risk stratification remains uncertain, as abnormal AFP levels are not present consistently leading up to tumorigenesis. Additionally, few models incorporate environmental factors, and performance may vary across etiologies and racial/ethnic groups.29
Mounting evidence of the last two decades supports epigenetic modifications as substantial contributors to tumor initiation and progression, making these modifications particularly promising biomarkers for early detection.30–35 DNA methylation changes at CpG dinucleotides, which contribute to regulation of chromatin structure and gene expression, are among the earliest alterations in carcinogenesis and are readily measured in blood.36,37 Profiling liver disease in tissue- or leukocyte-derived DNA using Illumina Infinium Beadchip arrays has revealed differential methylation in CpG sites located near or within genes involved in oxidative stress and CLD, including MASLD, metabolic dysfunction associated steatohepatitis (MASH), and cirrhosis.38–46 Methylation-based classifiers for HCC detection, such as HepaQ and HelioLiver, have demonstrated higher sensitivity and specificity than AFP or GALAD.47,48 Yet, these assays have inherent limitations: both were designed with and for Asian populations and HelioLiver was designed with a control sample of liver disease patients, thus performance can vary based on underlying disease etiology, and limited validation in diverse populations restricts generalizability. Additionally, Illumina Infinium Bead Chip arrays, commonly used for these classifiers, cover <5% of CpG sites, leaving the majority of the genome unexplored.
However, the broad challenge for all of these tools is that although DNA methylation changes regularly arise in cancer, they vary widely across tissues and individuals, including race- and ethnicity-specific differences, and are often consequences of deregulation in tumors, rather than promoters of tumorigenesis. Moreover, deregulation of DNA methylation often occurs due to comorbid conditions such as various neurological, autoimmune, metabolic, infectious and cardiovascular diseases, confounding the identification of specific HCC associations. To effectively utilize methylation as a clinically reliable risk signal, markers must be both consistent across individuals and mechanistically linked to disease predisposition, regardless of time before disease onset.
Imprinting control regions (ICRs) are uniquely suited to meet these criteria for effective prognostic markers. Defined by parent-of-origin–dependent DNA methylation, ICR methylation is established in the gametes and pre-implantation zygote, resisting postfertilization epigenetic reprogramming. Therefore, these methylation profiles are perpetuated through germ layer specification and tissue differentiation and normally maintained throughout the life course. ICRs are also sensitive to environmental exposures during their window of establishment in early development, such as parental smoking, alcohol use, endocrine-disrupting chemicals, and non-chemical stressors (eg. physical, social). Such disruption of ICR methylation can then propagate through the germline, transferring altered gene expression to the next generation, and influence disease susceptibility over the life course,4,49,50 making them robust biomarkers that can reveal mitotically inherited epigenetic disruptions existing prior to tumorigenesis. Because imprinted genes regulate growth, metabolism, and cellular homeostasis, their dysregulation due to aberrant ICR methylation plausibly contributes to cancer initiation.30–32,51,52 By evaluating methylation at ICRs in blood, we are targeting loci that are both biologically constrained and clinically accessible; loci for which deviation from the expected pattern may serve as a strong indicator of mitotically inherited susceptibility, applicable for risk stratification.
Previously, aberrant ICR methylation or loss of imprinting (LOI) has been implicated in HCC and other cancers. Well-characterized loci including the ICRs of growth inhibitory long non-coding RNAs (lncRNA) H19, KCNQ1OT1, and MEG3 and imprinted growth promoters IGF2 and DLK1 have key roles in tumorigenesis and metabolic disfunction, with LOI of these loci reported in several cancers, including HCC, demonstrating key roles in tumorigenesis and metabolic dysfunction.33,34,53–56 More recently, methods such as quantitative chromogenic imprinted gene in-situ hybridization (QCIGISH) have demonstrated the feasibility of measuring imprinting dysregulation in cancer.35,52,57,58 Despite their importance, until the recent characterization of ~1,488 putative ICRs, only a small fraction of this set has been confirmed and thoroughly studied in human diseases.59
Herein, we used whole genome bisulfite sequencing (WGBS) to comprehensively profile the 5-methylcytosine landscape in DNA derived from peripheral leukocytes to identify previously uncharacterized HCC-associated DMRs. We augmented these with published methylation array data, and through cross referencing with previously reported putative ICRs, we identified 97 HCC-associated putative ICRs exhibiting differential methylation between HCC cases and otherwise healthy individuals.
Materials and Methods
Participants and Samples
The characteristics of 39 primary HCC cases and 87 otherwise healthy controls are summarized in Supplementary Table 1. CpG methylation data used in these analyses were derived from two case-control comparisons: 1) WGBS of leukocyte-derived DNA from n = 10 primary HCC cases (mean age 66 ± 6 years) consented at the Lineberger Comprehensive Cancer Center at UNC Chapel Hill who underwent HCC resection surgery, and n = 51 otherwise healthy controls (mean age 63 ± 10 years) consented in primary care or accompanying neurology patients at Duke Health, 2) DNA methylation data using a custom Illumina methylation array (10,000 CpG sites at ICRs) from 29 incident HCC cases and 36 otherwise healthy controls identified using a combination of the rapid case ascertainment system of the North Carolina Cancer Registry, Duke Gastroenterology and the Duke Cancer Institute. The age of the latter participants ranged from 63 to 68 years. Data collection and analyses were approved by Institutional Review Boards at UNC Chapel Hill, Duke University School of Medicine and North Carolina State University.
WGBS Identification of DMRs Associated with Primary HCC
WGBS was performed in peripheral blood of HCC patients (n = 10) and otherwise healthy individuals (n = 51). Libraries for the Illumina NovaSeq 6000 Sequencing System were prepared from bisulfite converted DNA derived from blood using previously described methods.59 To generate long, high-quality reads from both ends of DNA fragments, sequencing was performed using S2 flow cells with 150bp paired-end (PE) reads. The reads were aligned to the hg38 reference genome using BSMAP60 as implemented in the MOABS framework.61 The average number of reads mapped was 132.7 million (75.6 M–258.1 M) and the average alignment percentage was 74.3% (65.1%–90.0%). The average percent of paired reads excluded as duplicates was 4.1% (0.20%–5.6%). Quality control metrics at the BAM level were obtained with samtools flagstat (v1.15.1). Both the control and HCC samples showed mapping rate ~100% and a high proportion of properly paired reads (97.0% and 95.2%, respectively), with very few singleton reads (≤4%) (Supplementary Table 2). The resulting data were processed, and MOABS software was used to call methylation sites using the “MCALL” function.61 Differentially methylated regions (DMRs) were defined using a 300 bp window, requiring at least 4 CpGs per window to all have a minimum credible methylation difference of 10%, in the same direction, between groups. CpG sites were included only if they had a minimum coverage depth of three reads (--minDepthForComp = 3). Approximately 24 million CpG sites were analyzed. Sequencing depth and average methylation levels were consistent across coverage bins, suggesting that both case and control datasets are of similar and sufficient quality for downstream methylation analysis (Supplementary Figures 1 and 2). The WGBS data has been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE302608 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE302608).
We additionally transitioned from the unadjusted and pooled MOABS DMR analysis to a multifactor generalized linear model using the DSS (Dispersion Shrinkage for Sequencing) R package to perform a stratified analysis with age as a covariate. DNA Methylation counts at each locus were obtained using MOABS mcall (the MOABS function was preferred for methylation calling as the initial read alignment performed with BSMAP was not suitable for Bismark pipeline). Spatial smoothing was applied to recover the statistical power at WGBS locus level with bsseq package BSmooth. The generalized linear model was fit using the formula ~ HCC_Status + Age. We then tested for differential methylation against the HCC status coefficients by performing the Wald test on each locus using DMLtest.multifactor to investigate the disease-specific association. The local inclusion parameter for DMR calling was p < 0.05, and false discovery rate was controlled by spatial constraints of minCG = 4, dis.merge = 100bp, and pct.sig = 0.05.
To confirm that methylation changes were not driven by genomic alterations, we performed copy number analysis on the WGBS data using CNVkit (v0.9.10).62 A pooled reference was constructed from control samples. Analysis was conducted using a moving average window of 100 kb to mitigate the inherent variance in bisulfite sequencing coverage.
To post-hoc estimate the power to identify significant DMRs, 500 simulations were run using base R software. Within each, 500 regions were simulated, with 25% randomly selected to be true DMRs. Within each region, a varying number of CpG sites was simulated using a beta-binomial approach to stay methodologically in line with MOABS software, and account for both variation due to coverage and between participants. Replicating our WGBS DMR calling requirements, DMRs were defined as requiring at least 4 CpGs per simulated region window each with a minimum methylation difference of 10%. Comparing the proportion of DMRs declared to the preset DMRs, this simulation gave a power estimation of ~72%.
Gene Annotation
Genomic coordinates were annotated with gene information using the RefSeq gene annotation track for the human genome (GRCh38/hg38). Refseq data were downloaded from the UCSC Genome Browser database (https://hgdownload.soe.ucsc.edu/goldenPath/hg38/database/).
Literature Review of Published HCC-Associated Loci
To comprehensively identify publications reporting altered methylation in association with liver damage, we used the National Library of Medicine’s PubMed database, with search terms including HCC and pre-HCC conditions – liver cirrhosis, non-alcoholic fatty liver disease (NAFLD), MASLD, fibrosis, MASH, steatosis, hepatic fat levels, HBV, HCV, and alcoholic liver disease – and “methylation”, “differential methylation” or “DMR[s]”, and “biomarkers”. With narrowing hits based on methods measuring differential methylation levels between etiologies using Illumina Infinium BeadChip arrays (27k, 450k, EPIC, EPIC v2.0), WGBS, or pyrosequencing, this search yielded n = 66 articles; n = 64 were primary research articles, including meta-analyses, and n = 2 were reviews. From the 64 primary research articles, 19 articles were excluded for missing chromosomal coordinate information. We identified regions reported as significantly differentially methylated between the cases of liver disease (HCC or other degenerative liver disease conditions that can lead to HCC, as listed above in the search terms) and non-cases. All chromosomal positions were converted to the hg38 build. A total of 45 publications were included and are summarized in Supplementary Table 3.38–41,43–46,55,63–97
Determining HCC-Associated Putative ICRs
Among the 1,519 DMRs associated with HCC in mixed leukocytes, we sought to identify overlaps with putative ICRs to facilitate early detection for interventions or triage. We used two sources of published putative ICRs. Briefly, as described in Jima et al (2022), 1,488 putative ICRs were identified in humans by performing WGBS of DNA derived from tissues representing the three germ layers. Additionally, Akbari et al (2022) determined candidate ICRs using publicly available Oxford Nanopore sequencing datasets. Using the NanoMethPhase package, that study identified 143 putative ICRs.98,99
To determine previously reported DMRs associated with HCC risk that are potential biomarkers prior to HCC onset, we determined direct overlaps between the 1,519 HCC-associated DMRs and putative ICRs from Jima et al and Akbari et al, resulting in 81 HCC-associated putative ICRs.
To ensure a comprehensive list of HCC-associated putative ICRs, we additionally intersected previously published loci with the compendium of putative ICRs reported by Jima et al and Akbari et al Analysis of the 45 published studies revealed 3,275 unique regions associated with liver pathology, 673 of which are single dinucleotide CpG sites. Of the 3,275 DMRs/CpG sites previously reported, 59 were identified using WGBS blood-derived DMRs and the remainder from methylation arrays (Supplementary Table 3). The overlap of these regions with the putative ICRs identified 16 additional putative ICRs that contain loci previously implicated in HCC development via published literature, totaling to n = 97 HCC-associated putative ICRs.
Replication via Human Imprintome Methylation Array Platform
Methylation levels in mixed leukocyte-derived DNA isolated from the whole blood of 29 HCC cases and 36 otherwise healthy controls were measured on the Human Imprintome Methylation array. The array, an Illumina Infinium HTS iSelect Custom BeadChip, is a targeted DNA methylation platform designed using the repertoire of 1,488 putative ICRs across the human genome characterized by Jima et al as target regions.59,100 This array contains 22,819 probes (704 control probes and 22,115 CpG probes, of which 10,438 CpG probes target unique CpG sites). Among the CpG probes, 9,757 probes passed design criteria for 1,088 of the 1,488 candidate ICRs; see Carreras-Gallo et al for additional details.100 Additional cgBackground probes enabled background-normalization. The IDAT files containing the raw intensity data for each probe on the microarray chip have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE303108 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE303108).
The raw IDAT files produced from the array were processed using the openSesame function in the Sesame R package.101 The Sesame preprocessing code “0CDB” applies a non-linear dye bias correction and background subtraction using the noob method (Norm-Exp out of band signal), which deconvolves out-of-band fluorescent signal from true signal. The beta values are calculated by the ratio of the methylated signal to the total signal (methylated + unmethylated) for each probe, representing the methylation fraction as a value between 0 and 1. These processing steps produced a beta measurement for each probe, along with a corresponding Sesame signal detection p-value quality score. Signal detection p-values were assigned using the pOOBAH method (P-value with out-of-band (OOB) array hybridization).101
Probes that did not map to a CpG site in the Imprintome were discarded, and any remaining probes were also discarded if their signal detection p-value exceeded 0.2 for more than 10% of samples measured. For cases where multiple probes targeted the same CpG site, averaged beta measurements were used.
A series of Student’s t-tests were performed to test for significant differential methylation of beta values between cases and controls (null hypothesis of µ = 0, two-tailed, α = 0.05, unpaired, unequal variance assumed). Resulting p-values were adjusted based on false discovery rate for the number of ICRs tested. CpG sites were reported as significant based on an adjusted p-value < 0.05. Additionally, CpG beta values were aggregated if within the same ICR, producing beta measurements for each ICR site of each participant for ICR-level case-control comparison. See Supplementary Table 4 for all specialized R packages used.61,101–109
To estimate the power of this study post-hoc, we first estimated the true standard deviation of the data by calculating the median pooled standard deviation of methylation beta values for the subset of CpGs with coverage over the 70 ICRs. Standard deviations were calculated for each group before pooling (σpooled = 0.081). We set the minimum effect size to methylation beta = 0.06 (Cohen’s d = 0.73), marginally below the minimum of 0.1 to define a DMR from WGBS analysis. Statistical power for multiple comparisons within the 70 ICRs was estimated using the Sample Size Calculations for Two-Sample Microarray Experiments from the ssize.fdr package, with parameters (FDR = 0.05, power = 0.8, m = 70, pi0 = 0.5).110 The proportion of nulls (pi0) was set to 50% because this subset of ICRs was already found differentially methylated in the WGBS/previously reported DMR analysis. This test assumes a constant sample size, so we conservatively specified the smaller sample size between groups (n = 29) to calculate the minimum estimated power across all comparisons (βall > 0.73, function:ssize.twoSamp). This analysis confirmed that we had sufficient sample size for statistical power of >73%.
Results
WGBS Identification of DMRs Associated with HCC in Peripheral Blood Leukocytes
Using WGBS on mixed leukocytes from 10 HCC cases and 51 controls, 1,519 DMRs were significantly associated with HCC (FDR q-value range 0–0.000486) (Supplementary Figure 3). Of the 1,519 blood-derived DMRs, the majority (73%; 1103/1519) displayed hypomethylation, and 27% (416/1519) exhibited hypermethylation associated with HCC (Figure 1). The range of the absolute values of the methylation difference (regardless of direction) is 0.100–0.454.
 |
Figure 1 Distribution of hyper- and hypomethylation among the blood-derived DMRs. In peripheral blood between HCC cases and controls, the majority of DMRs (1103/1519) exhibited hypomethylation associated with HCC. |
To assess functional significance, the identified DMR positions were overlapped with publicly available ENCODE chromatin immunoprecipitation sequencing (ChIP-seq) peak datasets for H3K4me3 and H3K27ac – regions enriched for active promoter and enhancer marks – in a B-lymphoblastoid cell line (GM12878) and liver tissue (ENCODE Experiment IDs ENCFF416SIL and ENCFF805YRQ). Overlap with these peak regions indicates localization of DMRs to putative regulatory elements. Additionally, CpG island enrichment in the DMRs was assessed using the UCSC Genome Browser CpG Islands track (hg38) (https://genome.ucsc.edu/cgi-bin/hgTrackUi?hgsid=3564790247_NWaSAOctGsVQ4vD5dafUzrKX1tk4&db=hg38andc=chr7andg=cpgIslandSuper) (Supplementary Table 5). In the liver tissue line, 17.2% (261/1519) of the HCC-associated DMRs were enriched for H3K4Me3 marks, and 16.0% (240/1519) were enriched in the B-lymphoblastoid cell line. Approximately the same proportion of DMRs were enriched for H3K27ac in the liver tissue and B-lymphoblastoid cell line (14.4% (218/1519) and 12.8% (195/1519), respectively). Of these, 72 DMRs were enriched in both liver and B-lymphoblastoid, suggestive of conservation of essential epigenetic regulation and that blood methylation captures HCC-relevant enhancers. Additionally, 26% (401/1519) of the DMRs overlapped with at least one CpG island.
The age-stratified generalized linear model analysis of differential methylation identified 955 DMRs associated with HCC status, accounting for approximately two-thirds of the 1,519 pooled DMRs. The substantial overlap with the 1,519 DMRs identified in the primary analysis suggests that the modest (~3 year) age difference between groups does not meaningfully confound observed associations. Due to potential deflation of significance in stratified/covariate-adjusted analyses, given the small sample size, further analyses were performed on the entire set of 1,519 HCC-associated DMRs to maximize sensitivity.
CNVkit was used to examine copy number variation in each of the ten HCC cases for which WGBS was performed (Supplementary Figure 4A–J). Analysis of log2 copy ratios across all autosomes demonstrated a stable diploid baseline centered at 0.0, indicating the observed depth at the sites in the HCC samples matches the baseline read depth calculated from the control samples. No large-scale aneuploidy or consistently amplified/deleted segments were detected.
The nearest genes of the 1,519 blood-derived DMRs were used in Qiagen Ingenuity Pathway Analysis (IPA) to determine if there was over-representation of HCC-related pathways in the set of DMR-associated genes. Using bedtools suite and RefSeq hg38 gene annotations, 1,321 unique nearest genes mapped to the DMRs, with several DMRs mapping to the same gene. After removing four genes not annotated in IPA (“LOC730100”, “LOC124903068”, “LOC102724219”, “LOC124905221”), 1,317 genes were processed for pathway enrichment. The top five “Diseases and Disorders” pathways are cancer (n = 838 genes, FDR adjusted p-value range 3.82E-03 – 1.05E-42), organismal injury and abnormalities (n = 878 genes, 3.82E-03 – 1.05E-42), endocrine system disorders (n = 748 genes, 3.82E-03 – 1.06E-39), hereditary disorder (n = 317 genes, 3.82E-03 – 4.1E-37), and dermatological diseases and conditions (n = 638 genes, 3.82E-03 – 2.94E-33). The top five hepatotoxicity pathways are liver hyperplasia/hyperproliferation (n = 412 genes, FDR adjusted p-value range 1.00E00 – 5.51E-11), liver inflammation/hepatitis (n = 39 genes, 1.00E00–4.3E-03), hepatocellular carcinoma (n = 147 genes, 1.00E00 – 7.2E-03), liver steatosis (n = 40 genes, 1.00E00 – 1.81E-02), and liver regeneration (n = 8 genes, 2.80E-01 – 3.46E-01). The top 5 significant canonical pathways are Protein Kinase A Signaling (n = 31/396 genes in the pathway, FDR adjusted p-value 1.49E-05), Hepatic Fibrosis Signaling Pathway (n = 30/416, 2.18E-04), 3-phosphoinositide Biosynthesis (n = 19/215, 2.18E-04), mRNA 3 Prime End Processing Signaling Pathway (n = 12/102, 2.18E-04), and Superpathway of Inositol Phosphate Compounds (n = 20/241, 2.18E-04). All canonical pathways are available in Supplementary Table 6.
Defining a Comprehensive List of HCC-Associated Putative ICRs
For comparison of our findings with results from published data, we created a compendium of previously reported DMRs or differentially methylated CpG sites found in multiple source tissues, including circulating cell-free DNA (ccfDNA) from whole blood or plasma and liver tissue biopsies. The overlap of these regions with the putative ICRs identified 16 additional putative ICRs that overlap differential methylation previously implicated in HCC development via published literature. Of the 16 ICR/DMR regions, 15 of the ICRs were first identified in liver tissue, and one was first identified in peripheral blood associated with alanine aminotransferase (ALT) levels.45,55,68–70
With 81 HCC-associated blood-derived DMRs from our WGBS and 16 DMRs previously published as associated with HCC or pre-HCC risk, we compiled a set of 97 HCC-associated DMRs that overlap with putative ICRs, of which 92 are unique Imprintome regions (Supplementary Table 7). This includes 21 regions co-located with previously identified ICRs (see Supplementary Table 8 for the list of previously identified ICRs). All but five ICRs from Akbari et al that overlapped with the DMR compendium were also reported in the 1,488 Imprintome ICRs (Figure 2).
 |
Figure 2 Intersection of HCC-associated DMRs with Putative ICR targets. Our compendium of HCC-associated regions, containing the 1,519 blood-derived DMRs from WGBS and from our literature analysis, contained 4,794 DMRs. The 143 Nanopore-detected ICRs were reported by Akbari et al,98 and the 1,488 putative Imprintome ICRs were reported by Jima et al.59 For some ICRs, there were multiple overlapping DMRs, thus we note the number of unique ICRs included. |
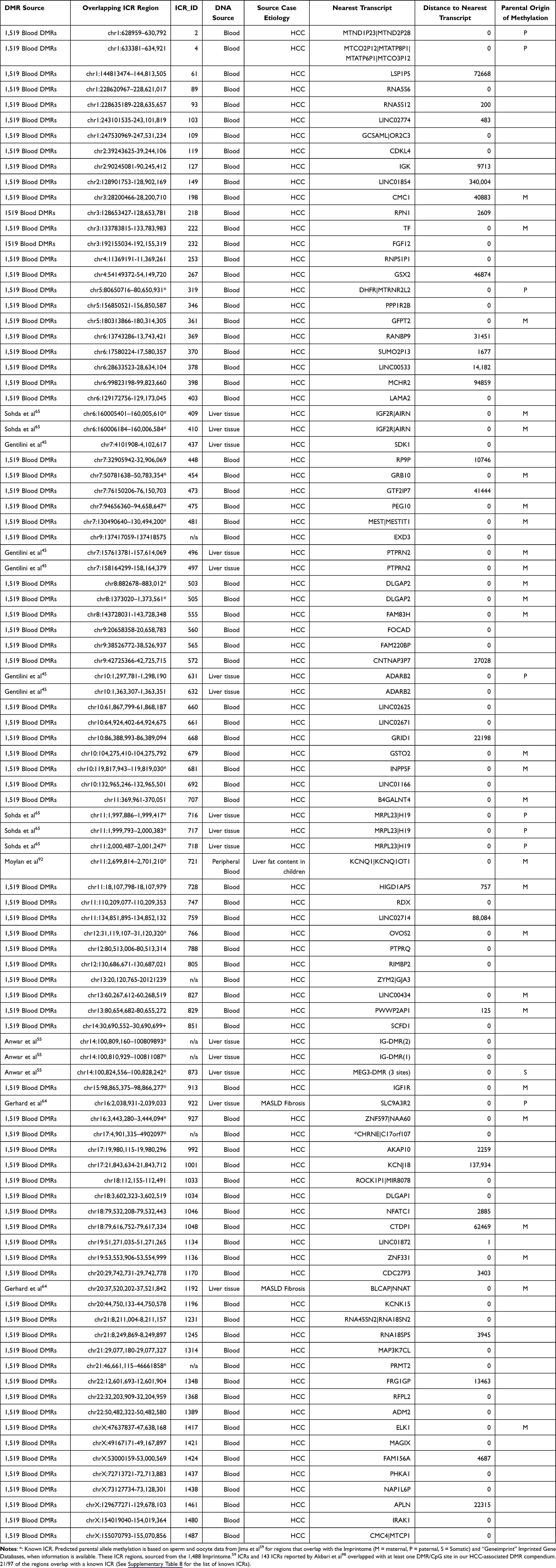 |
Table 1 HCC/Liver Disease-Associated Differentially Methylated Regions Detectable in Mixed Leukocytes, cfDNA and Liver Tissue |
See Table 1 for further details of the 97 regions and Figure 3 for a schematic workflow for identifying putative ICR targets. These 97 regions represent a subset of putative ICRs that have exhibited differential methylation between HCC cases and controls in an accessible surrogate tissue and are therefore potential biomarkers for early detection prior to HCC onset due to the establishment of imprinting regulation in utero.
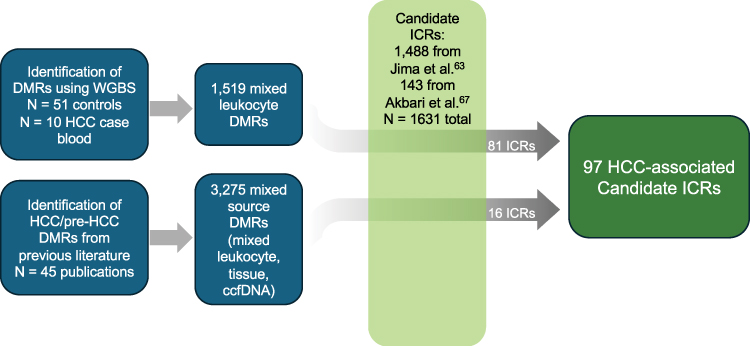 |
Figure 3 Schematic workflow for identifying putative ICR targets. We combined the 1,519 blood DMRs with the DMRs reported in previous literature from mixed blood and tissue sources. Using a Perl script, we intersected those with the 1,488 putative ICRs previously described111 as well as 143 putative ICRs reported by Akbari et al98 to find 97 direct overlaps, of which 81 DMRs came from our WGBS of HCC case/control blood methylation levels. |
Determining Molecular Pathways of Liver Disease-Associated Targets
We used Qiagen IPA to analyze the genes overlapping or closest to the 97 putative ICR regions. The two most significant metabolic gene pathways are associated with HCC risk: PTEN signaling (FDR adjusted p = 9.12E-03) and IGF-1 signaling (p = 1.28E-02). Additional significant (p<0.05) HCC risk-associated pathways include growth hormone signaling, signaling by MET, toll-like receptor signaling, p38 MAPK signaling, PDGF signaling, signaling by IGF1R, acute phase response signaling, and the folate signaling pathway (Supplementary Table 9). Dysregulation of these pathways is associated with cellular proliferation and several cancers. The 10 most significant canonical pathways are shown in Table 2.
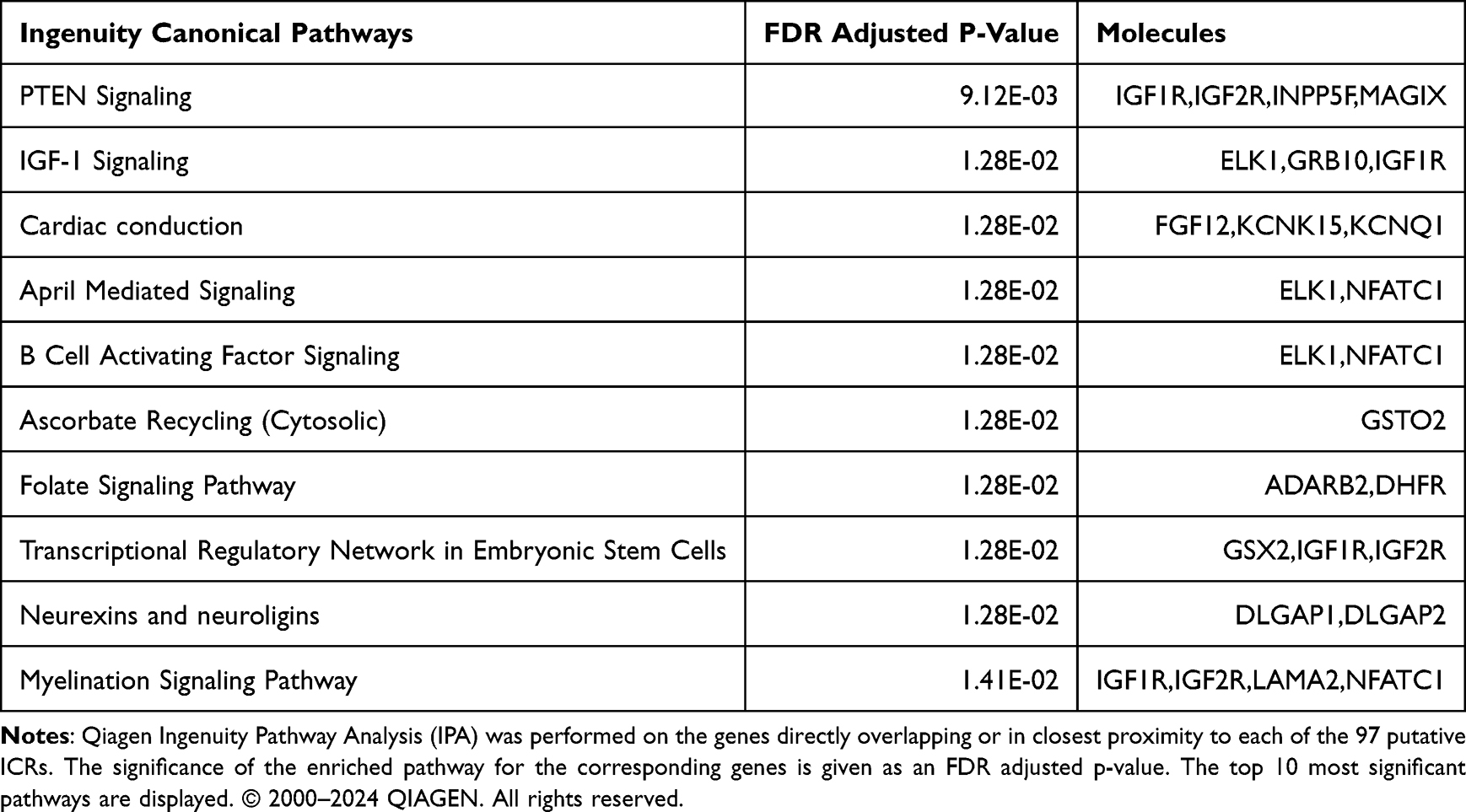 |
Table 2 Top Pathways Associated with HCC-Associated Genes by Ingenuity Pathway Analysis |
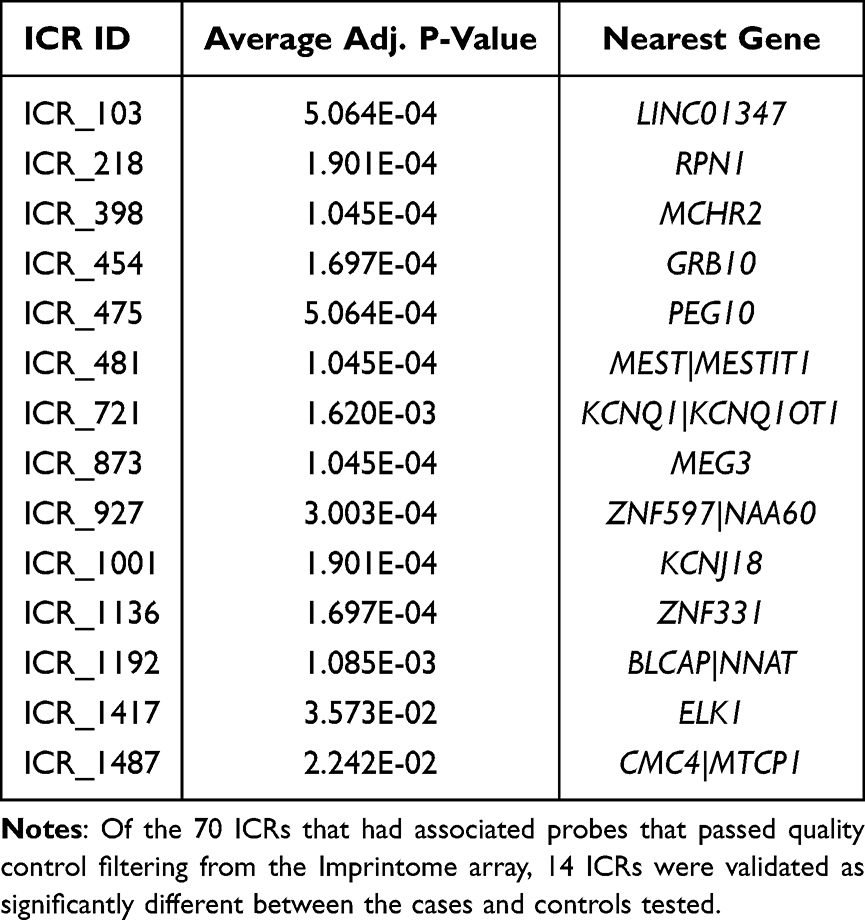 |
Table 3 Independent Validation of Differentially Methylated ICRs Using the Imprintome Array |
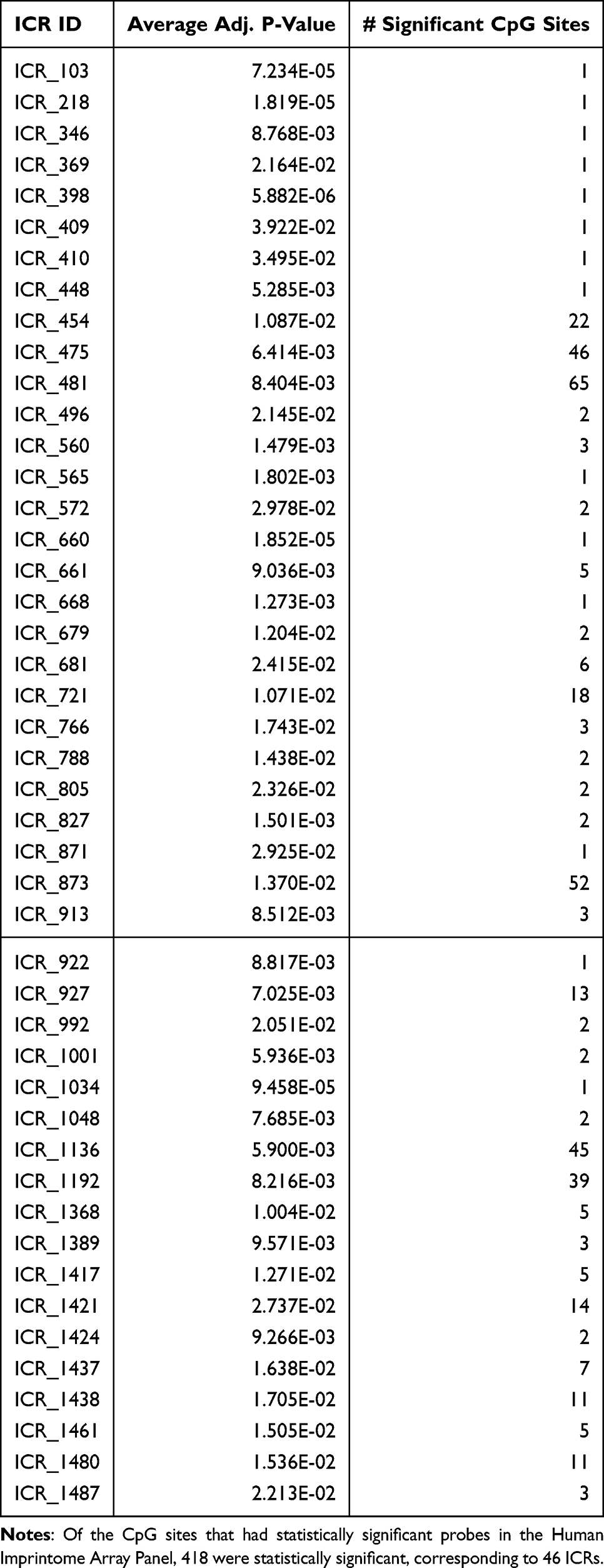 |
Table 4 Significantly Differentially Methylated ICRs at the CpG Level in the Human Imprintome Array |
Replication of the Putative ICRs in an External HCC Case-Control Analysis
To externally replicate the identified ICR targets, an independent population of n = 29 cases and n = 36 otherwise healthy controls was used to measure DNA methylation on the Human Imprintome Methylation Array, which contains probes for CpGs within 1,088 of the 1,488 putative ICRs.100 Because this array was designed to the ICRs reported by Jima et al, five of the 97 regions that were reported by Akbari et al are not included.98 In addition, 12 ICRs lacked associated probes after quality control filtering, limiting this replication to 70 ICR sites overlapping HCC-associated DMRs (containing a total of 1,049 CpG sites). We observed significant differential methylation associated with HCC at both the CpG site-level and ICR-level. At the ICR level, 14 ICRs were significant after adjustment for false discovery rate (Table 3). These regions are in proximity to the transcripts LINC02774, RPN1, MCHR2, GRB10, PEG10, MEST, KCNQ1, MEG3, ZNF597, KCNJ18, ZNF331, BLCAP, ELK1, and CMC4. Nine of the 14 genes are known imprinted genes.
When considering CpGs individually, 46 ICRs had at least one significantly differentially methylated CpG site between the cases and controls (Table 4). Together, these results provide supporting evidence for the association of these ICRs with HCC risk.
CpG methylation summary plots were created for each ICR to visualize the difference between case and control methylation levels. Figure 4 displays the mean methylation of CpG sites in cases and controls for ICR 454 (GRB10, previously identified ICR), ICR 1417 (ELK1, putative ICR), ICR 1192 (BLCAP|NNAT, previously identified ICR), and ICR 1438 (NAP1L6P, putative ICR), as examples. Each ICR displays methylation levels consistent with hypomethylation in HCC cases. Full plots and CpG methylation summaries can be found at: https://mollyrbio.shinyapps.io/HCCIMPlots/.
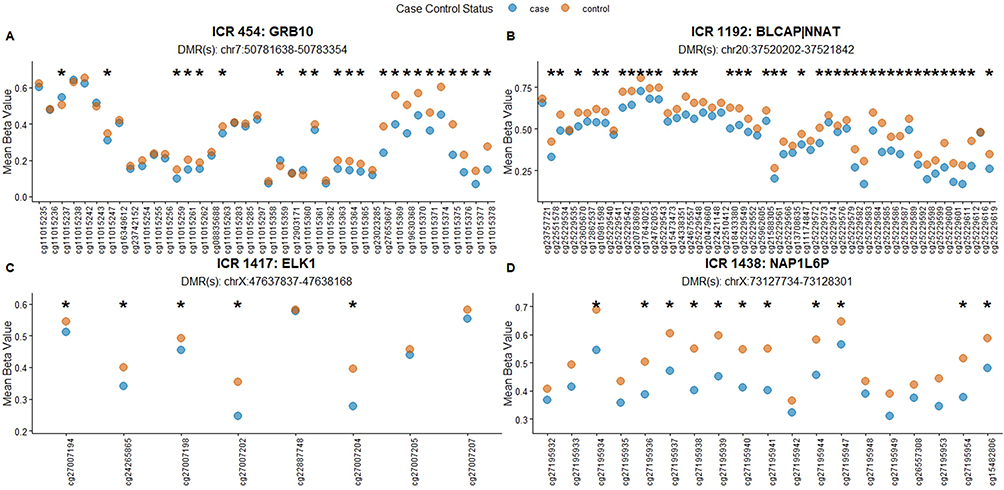 |
Figure 4 Differential CpG methylation for four ICRs. Visualization of differential methylation between HCC cases and controls for two previously identified ICRs (A and B) and two putative ICRs (C and D). Mean methylation beta values for each CpG site across cases and controls were estimated as detailed in methods and plotted for each ICR (https://mollyrbio.shinyapps.io/HCCIMPlots/). X-axis: Mean methylation beta value, a continuous measure (0–1) representing the proportion of methylated CpGs at the site. Y-axis: Illumina array CG identifier number. * Adjusted p-value <0.05. |
Discussion
We conducted WGBS in mixed leukocytes to characterize HCC-associated alterations in the 5mC landscape. This analysis was complemented by a comprehensive literature review of previously reported liver disease-associated DMRs to provide broader biological context. We focused specifically on loci overlapping putative ICRs. Given their established methylation stability across tissues and cell types and persistence throughout the life course, diseased-associated aberrant methylation at these regulatory regions has potential to serve as early-risk biomarkers.59,98 Findings were subsequently evaluated in an additional case-control study using leukocytes, accessible cells that could serve as surrogates for inaccessible liver cells, given the conservation of ICR methylation. Ultimately, these efforts were designed to identify epigenetic alterations detectable in leukocytes that may inform early risk assessment for HCC, by identification of susceptible individuals otherwise appearing healthy.
The WGBS of leukocytes derived from a cohort of HCC cases and otherwise healthy controls identified 1,519 blood-derived DMRs, 81 of which coincide with ICRs, both demonstrated and putative.
Functional enrichment analysis of these regions with histone marks suggests essential epigenetic regulatory function and that methylation analysis of blood-derived DNA captures HCC-relevant enhancers. Enrichment for H3K4me3, which is associated with active promoters, indicates potential transcriptional effects from methylation changes on active genes, with these effects conserved across tissues. Similarly, DMR overlap with H3K27ac suggests involvement of active enhancers with cell type-specific regulatory roles. Enrichment at CpG islands – regions typically exhibiting tightly regulated and stable methylation in blood – further supports the interpretation that these DMRs reflect biologically meaningful regulatory variation rather than shifts in cell composition.112
IPA analysis of genes nearest to the 1,519 HCC DMRs revealed enrichment across three major functional themes: (i) fibrosis and cirrhosis signaling, (ii) metabolic and obesity-related pathways, and (iii) canonical oncogenic signaling cascades implicated in HCC progression. Notably, several of these pathways include known or putative imprinted genes, reinforcing the biological relevance of our ICR-focused approach.
Among oncogenic signaling networks, Protein kinase A (PKA) signaling emerged as significant. Multiple PTPN family members were represented in this pathway. PTPN members regulate signal transduction and metabolic processes, have been implicated in obesity-related phenotypes, and are generally considered tumor suppressors in HCC, where reduced expression is associated with poor prognosis.113–118
Growth factor signaling genes were also prominent, including IGF2 and IGF2R. IGF2, a well-established imprinted gene, is frequently overexpressed in HCC and functions as a key mitogenic driver of tumor growth, while IGF2R, which modulates IGF2 availability, is often downregulated, further contributing to cancer progression.119–121 The representation of these genes within enriched pathways supports a model in which altered methylation near imprinted growth regulators may contribute to dysregulated proliferative signaling. Also, notably, emerging evidence suggests that epigenetic dysregulation in HCC intersects with metabolic reprogramming to promote tumor progression, including mechanisms such as lactylation- and acetylation-mediated activation of glycolytic and angiogenic pathways.122,123 In this context, hypermethylation at the IGF2R|AIRN ICR may have implications beyond growth factor signaling, as IGF2R has been linked to lysosomal function and iron homeostasis, raising the possibility that its dysregulation contributes to ferroptosis resistance. Future studies integrating iron-related biomarkers (eg. ferritin, hepcidin) and ferroptosis regulators such as GPX4 could help determine whether altered imprinting at this locus promotes metabolic vulnerabilities that support therapeutic evasion.
Pathways related to metabolic dysfunction and fibrosis were additionally represented, including hepatic fibrosis signaling. LEP (leptin), a hormone that regulates body weight, appeared among the significant genes, consistent with crucial role of leptin in obesity, cirrhosis, and MASLD – conditions that elevate HCC risk.124–126 Several imprinted genes are known regulators of leptin production or signaling, further linking imprinting biology to metabolic and inflammatory pathways implicated in hepatocarcinogenesis.127–131 High leptin levels are also linked to increased HCC risk due to its ability to promote tumor cell proliferation, invasion, and angiogenesis through various signaling pathways including JAK2/STAT3 (both of which were enriched pathways) and PIK3/AKT (a gene among the 1,519 DMRs).132,133
Collectively, these findings indicate that genes proximal to the HCC-associated DMRs converge on interconnected signaling networks governing growth regulation, metabolic homeostasis, and fibrotic progression – core biological processes underlying HCC development. Incorporating previously reported liver-disease associated DMRs, we identified 16 additional regions overlapping putative ICRs, bringing the total to 97 HCC-associated ICR-linked loci. Of these, 21 overlap established imprinted domains, including regions within or proximal to IGF2R|AIRN, MEST|MESTIT1, H19, KCNQ1|KCNQ1OT1, MEG3|DLK1 (including the intragenic IG-DMR and the MEG3-DMR), GRB10, PEG10, CHRNE, PRMT2, DHFR, DLGAP2, INPP5F, OVOS2, and ZNF597. These loci broadly cluster into growth factor signaling, metabolic regulation, and canonical tumor suppressor/oncogene networks – processes central to hepatocarcinogenesis.
Several of the identified imprinted regions converge on the IGF signaling axis. As discussed above, dysregulation of IGF2 and its receptors is a recurrent feature of HCC, with aberrant activation of the axis reported in a subset of early tumors.121,134,135 The presence of IGF1R and IGF2R within these putative ICR regions reinforces the relevance of altered imprinting and growth factor regulation in HCC development. Other loci, including MEST, H19, MEG3, DLK1, PEG10, and GRB10, have established roles in tumor growth, metabolic regulation, or both, further supporting the biological plausibility of imprinting-linked epigenetic disruption in liver disease and cancer progression.136–142 Indeed, single ICRs can regulate multiple tumor-associated genes; for example, the IG-DMR of MEG3 regulates not only MEG3 and DLK1, but also is responsible for establishing the imprinting status of >80 genes in the 14q32.2 imprinted domain.143 PRMT2, INPP5F, and OVOS2 are also novel oncogenes.142,144–146
Additional genes proximal to putative ICRs, such as KCNK15, have not been established as imprinted, but have been associated with HCC prognosis in prior transcriptomic analyses.147 We identified a hypomethylated DMR with KCNK15, and hypothesize that the methylation at this locus may regulate gene expression and alterations can serve as a marker of disease progression.
IPA of genes nearest to these 97 DMRs revealed significant enrichment of pathways previously implicated in HCC, notably PTEN signaling and IGF-1 signaling. These findings mirror the broader IPA results from the 1,519 DMRs, again highlighting convergence on growth factor signaling and PI3K/AKT pathway regulation. PTEN signaling, a critical negative regulator of PI3K/AKT activity and cellular metabolism, has also been shown to influence the expression of multiple imprinted genes.148,149 A 2021 study in mice showed that when Pten is absent, the expression of several imprinted genes, including Igf2, Plagl1 (Zac1), Cdkn1c, Dlk1, Mkrn1, Magel2, and Dlx5, changes in mouse embryonic stem cells, embryoid bodies, and cardiomyocytes.150 The second top pathway, IGF-1 signaling, mediates cell proliferation, survival, migration, and blocks apoptosis in HCC, as detailed above.151,152
This may provide a potential mechanistic bridge between imprinting dysregulation and oncogenic metabolic reprogramming.
Despite the small sample size (N = 10 cases), we determined the reproducibility of our findings in mixed leukocyte-derived DNA of an additional HCC case-control study using a novel methylation array targeting a majority of ICRs comprising the Human Imprintome.59,100 Of the 97 HCC-associated putative ICRs, 46 (47%) had significantly associated CpGs, and further 14 of these ICRs, several of which had not previously been linked to HCC, were significant across their spans. While we do not report the direction of methylation change between cases and controls for these ICRs, both hyper- and hypomethylation could coincide with imprinting dysregulation. Whether this dysregulation happens via hypo- or hypermethylation is dependent on the method of imprinting regulation at the locus (ie. parent-of-origin specific methylation at the gene promoter, long-range chromatin mediated interactions such as enhancer blocking, insulator function, or lncRNA-mediated silencing). Future studies are needed to determine which mechanisms are in operation at each candidate ICR.
Additional imprinted genes implicated in HCC and liver disease were not represented among the 97 regions, but warrant consideration in future biomarker refinement. For example, PLAGL1 (ICR-404), involved in cell-proliferation control, was not included in our DMR compendium. However, this gene has been shown to have abnormal transcription in cell lines derived from HCC patients with HCC, and its murine ortholog Zac1 has been demonstrated to directly coordinate imprinted gene networks involved in MASLD pathophysiology.153–155 While genes such as PLAGL1 may play mechanistic roles in hepatocarcinogenesis, our biomarker strategy prioritizes loci demonstrating stable germline imprinting detectable in blood, as these are more suitable for non-invasive risk stratification. These excluded loci may show variable methylation, partial imprinting, or tissue specificity, hence not meeting our stringent criteria.
Other studies have investigated blood-based DNA methylation marks in HCC. Hernandez-Meza et al67 used Illumina HumanMethylation450K arrays (485,000 CpGs, <2% of known CpG islands) to profile a cohort of normal liver tissues, cirrhotic tissues, dysplastic nodules, and HCC tissues. They found an increased proportion of hypermethylated samples in the progression from cirrhotic tissue (<1%) to dysplastic nodules (≥25%) to HCC (>50%). The study also observed an inverse correlation between DNA methylation and gene expression for TSPYL5, KCNA3, LDBH, and SPINT2 (all p < 0.001).67 Interestingly, KCNA3 encodes a potassium voltage-gated channel, similar to KCNK15, which was identified in our study. Lubecka et al found hypermethylation at LSP1 in HCC relative to cirrhosis controls, a gene we have identified in our list of putative ICRs.91 While each of these panels show promise for differential methylation at CpG sites as biomarkers, they are not selective for ICRs, so aberrant methylation identified may be consequential to tumorigenesis and progression, and thus useful in early risk stratification. Thus, while multiple studies have identified epigenetic marks with high sensitivity and specificity to detect HCC in blood-derived DNA, such marks are difficult to interpret.15,16 Additionally, many previous studies were limited by small sample sizes lacking population diversity, inferred methylation levels using novel techniques, and/or lack of target validation.
Our findings identify epigenetic biomarkers that are present across the life course and independent of cirrhosis status, which can be incorporated into existing risk models, such as GALAD. The biomarker panel used as part of GALAD has recently shown 82% specificity and 62% sensitivity when retrospectively evaluated for cirrhosis patients within 12 months before clinical diagnosis of HCC.156 Merging epigenetic risk markers with protein markers of progressive liver disease can be evaluated for their incremental predictive performance.
Like other HCC studies discussed, we are limited by HCC-patient and control sample sizes (WGBS: n = 10 cases, 51 controls, replication case control: n = 29 cases, 36 controls). Because HCC remains a rare—though highly lethal—cancer despite rising incidence, assembling larger well-characterized cohorts remains inherently challenging. However, post-hoc power analyses show sufficient power to detect meaningful differences in both the WGBS discovery of DMRs and the Human Imprintome Methylation Array to replicate these DMRs (72% and 73% at our given sample sizes, respectively). Also, although several of the 97 putative ICRs did not replicate in the external validation, despite being previously implicated in HCC risk, likely due to population heterogeneity, these findings pave the way for experimental validation of the 97 regions and identifying potential mechanisms that could be targeted for dietary or pharmacological interventions. Additionally, our studies were performed in case-control settings with methylation measurements made in samples obtained at diagnosis. Although ICR disruption occurs early in development, methylation changes could still arise concurrently with HCC onset. From contemporary cohort studies being assembled such as All of Us or Cohorts for Environmental Exposure and Cancer Risk (CEECR), it will soon be possible to determine if methylation differences identified here precede cancer onset or arise secondarily from tumorigenesis.
Nonetheless, these results provide comprehensive evidence for association of a set of ICRs with HCC risk. These regions are thus strong candidates for further validation in diverse cohorts, to be developed as early-risk stratifying HCC biomarkers. Given the stability of ICRs, temporally and spatially, such biomarkers would be applicable to DNA from accessible cell types, such as mixed leukocytes, and at any time during the life course, providing efficient, non-invasive, early detection.
Conclusions
Here, we address the critical need to identify early-detection epigenetic markers of HCC risk that may allow providers to facilitate interventions while the liver has the capacity to regenerate and, thus, reduce overall HCC incidence. For public health screening, it is critically important that DNA methylation marks be consistent across individuals, and within individuals, both across age and cell types, so that accessible DNA sources, such as blood and saliva, are informative as proxies for affected tissues.
We have achieved a pivotal step towards this goal of creating a clinically applicable screening panel, with our key finding of 97 putative ICRs with demonstrated differential methylation between patients with HCC and otherwise healthy individuals in DNA from a clinically accessible source, blood. When replicated in larger prospective studies, these marks could be developed into a biomarker panel for early detection in any setting, including primary care.
Abbreviations
HCC, Hepatocellular carcinoma; ICRs, imprinting control regions; DMRs, differentially methylated regions; WGBS, whole genome bisulfite sequencing; CLD, chronic liver disease; NAFLD, nonalcoholic fatty live disease; MASLD, metabolic associated steatotic liver disease; NASH, nonalcoholic steatohepatitis; AFP, alpha fetoprotein; NH, Non-Hispanic; PRS, polygenic risk score; MASH, metabolic dysfunction associated steatohepatitis; ccfDNA, circulating cell free DNA; IPA, Ingenuity Pathway Analysis; ALT, alanine Aminotransferase.
Data Sharing Statement
All sequencing and metadata is available as of July 31, 2026 at the following links: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE302608. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE303108.
Ethics Approval and Informed Consent
Primary data included here were collected with ethics oversight from NC State University (IRB#16558) and Duke University School of Medicine (IRB # Pro00102567). All participants signed a consent form. This study complies with the Declaration of Helsinki.
Acknowledgments
We thank the participants from the Lineberger Comprehensive Cancer Center at UNC Chapel Hill, Duke Health, Duke Gastroenterology, and Duke Cancer Institute. We thank Dr. Adriana Vidal for her contributions drafting the manuscript and Dr. Andrew Moon for cataloging Lineberger Comprehensive Cancer Center patient samples.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
Research reported in this publication was supported in part by National Institute of Health grants UH3CA265842 (C.H.), R01HD098857 (D.A.S., C.H.) and R21CA227023 (C.H., C.A.M., D.A.S., C.H., R.L.J.), and R01MD011746, R01MD017696, R01ES032462, P30ES025128 (C.H., D.A.S., D.D.J., R.L.J.) and Office of Extramural Research, National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosure
Dr David Skaar reports grants from National Institutes of Health, during the conduct of the study; personal fees from TruDiagnostics, outside the submitted work; In addition, Dr David Skaar has a patent U.S. Patent Application Serial No. 17/372,113 pending to TruDiagnostic Inc. The authors declare no competing interest.
References
1. Cancer. Cancer Facts & Figures 2026. Available from: https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/2026-cancer-facts-figures.html.
2. SEER. Cancer of the Liver and Intrahepatic Bile Duct - Cancer Stat Facts. Available from: https://seer.cancer.gov/statfacts/html/livibd.html.
3. Dhanasekaran R, Suzuki H, Lemaitre L, Kubota N, Hoshida Y. Molecular and immune landscape of hepatocellular carcinoma to guide therapeutic decision-making. Hepatology. 2025;81(3):1038. doi:10.1097/HEP.0000000000000513
4. Klibaner-Schiff E, Simonin EM, Akdis CA, et al. Environmental exposures influence multigenerational epigenetic transmission. Clin Clin Epigenet. 2024;16(1):145. doi:10.1186/s13148-024-01762-3
5. Galle PR, Forner A, Llovet JM, et al. EASL Clinical Practice Guidelines: management of hepatocellular carcinoma. J Hepatol. 2018;69(1):182–24. doi:10.1016/j.jhep.2018.03.019
6. Shiha G, Hassan A, Mousa N, et al. Individualized HCC surveillance using risk stratification scores in advanced fibrosis and cirrhotic HCV patients who achieved SVR: prospective study. Aliment Pharmacol Ther. 2025;61(1):99–108. doi:10.1111/apt.18291
7. Sun B, Karin M. Obesity, Inflammation and Liver Cancer. J Hepatol. 2012;56(3):704–713. doi:10.1016/j.jhep.2011.09.020
8. Heimbach JK, Kulik LM, Finn RS, et al. AASLD guidelines for the treatment of hepatocellular carcinoma. Hepatology. 2018;67(1):358–380. doi:10.1002/hep.29086
9. Manea I, Iacob R, Iacob S, et al. Liquid biopsy for early detection of hepatocellular carcinoma. Front Med. 2023;10:1218705. doi:10.3389/fmed.2023.1218705
10. Phan TH, Chi Nguyen VT, Thi Pham TT, et al. Circulating DNA Methylation Profile Improves the Accuracy of Serum Biomarkers for the Detection Of Nonmetastatic Hepatocellular Carcinoma. Future Oncol. 2022;18(39):4399–4413. doi:10.2217/fon-2022-1218
11. Gastrojournal. Surveillance Imaging and Alpha Fetoprotein for Early Detection of Hepatocellular Carcinoma in Patients With Cirrhosis: a Meta-analysis - Gastroenterology. Available from: https://www.gastrojournal.org/article/s0016-5085(18)30155-0/fulltext.
12. Del Poggio P, Olmi S, Ciccarese F, et al. Factors That Affect Efficacy of Ultrasound Surveillance for Early Stage Hepatocellular Carcinoma in Patients With Cirrhosis - Clinical Gastroenterology and Hepatology. Clin Gastroenterol Hepatol. 2014;12(11):1927–1933.e2. doi:10.1016/j.cgh.2014.02.025
13. Petrasek J, Singal AG, Rich NE. Harms of hepatocellular carcinoma surveillance. Curr Hepatol Rep. 2019;18(4):383. doi:10.1007/s11901-019-00488-8
14. Fan R, Papatheodoridis G, Sun J, et al. aMAP risk score predicts hepatocellular carcinoma development in patients with chronic hepatitis. J Hepatol. 2020;73(6):1368–1378. doi:10.1016/j.jhep.2020.07.025
15. Kim H, Yu X, Kramer J, et al. Comparative performance of risk prediction models for hepatitis B-related hepatocellular carcinoma in the United States. J Hepatol. 2022;76(2):294–301. doi:10.1016/j.jhep.2021.09.009
16. Lok AS, Seeff LB, Morgan TR, et al. Incidence of hepatocellular carcinoma and associated risk factors in hepatitis c-related advanced liver disease. Gastroenterology. 2009;136(1):138–148. doi:10.1053/j.gastro.2008.09.014
17. Morris JS, Hassan MM, Zohner YE, et al. HepatoScore-14: measures of Biological Heterogeneity Significantly Improve Prediction of Hepatocellular Carcinoma Risk. Hepatology. 2021;73(6):2278–2292. doi:10.1002/hep.31555
18. Jiang Y, Sun A, Zhao Y, et al. Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature. 2019;567(7747):257–261. doi:10.1038/s41586-019-0987-8
19. Gao Q, Zhu H, Dong L, et al. Integrated Proteogenomic Characterization of HBV-Related Hepatocellular Carcinoma. Cell. 2019;179(5):1240. doi:10.1016/j.cell.2019.10.038
20. Zhang Q, Lou Y, Yang J, et al. Integrated multiomic analysis reveals comprehensive tumour heterogeneity and novel immunophenotypic classification in hepatocellular carcinomas. Gut. 2019;68(11):2019–2031. doi:10.1136/gutjnl-2019-318912
21. Cky N, Dazert E, Boldanova T, et al. Integrative proteogenomic characterization of hepatocellular carcinoma across etiologies and stages. Nat Commun. 2022;13(1):2436. doi:10.1038/s41467-022-29960-8
22. Zhang Z, Zhang Z, Zhang Y, et al. Phosphoproteomics delineates hepatocellular carcinoma subtypes and pinpoints therapeutic targets. Hepatology. 2025;82(6):1432–1449. doi:10.1097/HEP.0000000000001250
23. Miao Z, Garske KM, Pan DZ, et al. Identification of 90 NAFLD GWAS loci and establishment of NAFLD PRS and causal role of NAFLD in coronary artery disease. HGG Adv. 2022;3(1):100056. doi:10.1016/j.xhgg.2021.100056
24. De Vincentis A, Tavaglione F, Jamialahmadi O, et al. A Polygenic Risk Score to Refine Risk Stratification and Prediction for Severe Liver Disease by Clinical Fibrosis Scores. Clin Gastroenterol Hepatol. 2022;20(3):658–673. doi:10.1016/j.cgh.2021.05.056
25. Zou Y, Zhu J, Song C, et al. A polygenetic risk score combined with environmental factors better predict susceptibility to hepatocellular carcinoma in Chinese population. Cancer Med. 2024;13(9):e7230. doi:10.1002/cam4.7230
26. Bianco C, Jamialahmadi O, Pelusi S, et al. Non-invasive stratification of hepatocellular carcinoma risk in non-alcoholic fatty liver using polygenic risk scores. J Hepatol. 2021;74(4):775–782. doi:10.1016/j.jhep.2020.11.024
27. Thomas CE, Diergaarde B, Kuipers AL, et al. NAFLD polygenic risk score and risk of hepatocellular carcinoma in an East Asian population. Hepatol Commun. 2022;6(9):2310–2321. doi:10.1002/hep4.1976
28. Fujiwara N, Lopez C, Marsh TL, et al. Phase 3 Validation of PAaM for Hepatocellular Carcinoma Risk Stratification in Cirrhosis. Gastroenterology. 2025;168(3):556–567.e7. doi:10.1053/j.gastro.2024.10.035
29. Rich NE, Hester C, Odewole M, et al. Racial and Ethnic Differences in Presentation and Outcomes of Hepatocellular Carcinoma. Clin Gastroenterol Hepatol. 2019;17(3):551–559.e1. doi:10.1016/j.cgh.2018.05.039
30. Dolinoy DC, Weidman JR, Jirtle RL. Epigenetic gene regulation: linking early developmental environment to adult disease. Reprod Toxicol. 2007;23(3):297–307. doi:10.1016/j.reprotox.2006.08.012
31. Schofield PN, Joyce JA, Lam WK, et al. Genomic imprinting and cancer; new paradigms in the genetics of neoplasia. Toxicol Lett. 2001;120(1):151–160. doi:10.1016/S0378-4274(01)00294-6
32. Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet. 2007;8(4):253–262. doi:10.1038/nrg2045
33. Xie G, Si Q, Zhang G, et al. The role of imprinting genes’ loss of imprints in cancers and their clinical implications. Front Oncol. 2024;14:1365474. doi:10.3389/fonc.2024.1365474
34. Zhou J, Cheng T, Li X, et al. Epigenetic imprinting alterations as effective diagnostic biomarkers for early-stage lung cancer and small pulmonary nodules. Clin Clin Epigenet. 2021;13(1):220. doi:10.1186/s13148-021-01203-5
35. CareAcross Lung Cancer service. Lung Cancer Diagnosis by Detecting Epigenetic Imprinting Alterations in Bronchoalveolar Lavage (NCT05707585). Available from: https://www.careacross.com/clinical-trials/trial/NCT05707585.
36. Toh TB, Lim JJ, Chow EKH. Epigenetics of hepatocellular carcinoma. Clin Transl Med. 2019;8(1):13. doi:10.1186/s40169-019-0230-0
37. Parikh ND, Tayob N, Singal AG. Blood-based biomarkers for hepatocellular carcinoma screening: approaching the end of the ultrasound era? J Hepatol. 2023;78(1):207–216. doi:10.1016/j.jhep.2022.08.036
38. Panagopoulou M, Fanidis D, Aidinis V, Chatzaki E. ENPP2 Methylation in Health and Cancer. Int J Mol Sci. 2021;22(21):11958. doi:10.3390/ijms222111958
39. Revill K, Wang T, Lachenmayer A, et al. Genome-Wide Methylation Analysis and Epigenetic Unmasking Identify Tumor Suppressor Genes in Hepatocellular Carcinoma. Gastroenterology. 2013;145(6):1424–1435.e25. doi:10.1053/j.gastro.2013.08.055
40. Tao R, Li J, Xin J, et al. Methylation Profile of Single Hepatocytes Derived from Hepatitis B Virus-Related Hepatocellular Carcinoma. PLoS One. 2011;6(5):e19862. doi:10.1371/journal.pone.0019862
41. Shen J, Wang S, Zhang YJ, et al. Genome-wide DNA methylation profiles in hepatocellular carcinoma. Hepatology. 2012;55(6):1799–1808. doi:10.1002/hep.25569
42. Urbańska K, Orzechowski A. Unappreciated Role of LDHA and LDHB to Control Apoptosis and Autophagy in Tumor Cells. Int J Mol Sci. 2019;20(9):2085. doi:10.3390/ijms20092085
43. Ammerpohl O, Pratschke J, Schafmayer C, et al. Distinct DNA methylation patterns in cirrhotic liver and hepatocellular carcinoma. Int J Cancer. 2012;130(6):1319–1328. doi:10.1002/ijc.26136
44. Slowly M, Domingo-Relloso A, Santella RM, et al. Blood DNA methylation and liver cancer in American Indians: evidence from the Strong Heart Study. Cancer Causes Control. 2023;35(4):661–669. doi:10.1007/s10552-023-01822-8
45. Gentilini D, Scala S, Gaudenzi G, et al. Epigenome-wide association study in hepatocellular carcinoma: identification of stochastic epigenetic mutations through an innovative statistical approach. Oncotarget. 2017;8(26):41890–41902. doi:10.18632/oncotarget.17462
46. Gonçalves E, Gonçalves-Reis M, Pereira-Leal JB, Cardoso J. DNA methylation fingerprint of hepatocellular carcinoma from tissue and liquid biopsies. Sci Rep. 2022;12(1):11512. doi:10.1038/s41598-022-15058-0
47. Yang XR, Liu R, Zhou J, et al. Discovery and clinical validation of cost-effective noninvasive early detection of hepatocellular carcinoma (HCC) through circulating tumor DNA (ctDNA) methylation signature. J Clin Oncol. 2022;40(16_suppl):4103. doi:10.1200/JCO.2022.40.16_suppl.4103
48. Lin N, Lin Y, Xu J, et al. A multi-analyte cell-free DNA-based blood test for early detection of hepatocellular carcinoma. Hepatol Commun. 2022;6(7):1753–1763. doi:10.1002/hep4.1918
49. Breton-Larrivée M, Elder E, McGraw S. DNA methylation, environmental exposures and early embryo development. Anim Reprod. 2019;16(3):465–474. doi:10.21451/1984-3143-AR2019-0062
50. Peral-Sanchez I, Hojeij B, Ojeda DA, Steegers-Theunissen RPM, Willaime-Morawek S. Epigenetics in the Uterine Environment: how Maternal Diet and ART May Influence the Epigenome in the Offspring with Long-Term Health Consequences. Genes. 2021;13(1):31. doi:10.3390/genes13010031
51. Hoyo C, Murphy SK, Jirtle RL. Imprint regulatory elements as epigenetic biosensors of exposure in epidemiological studies. J Epidemiol Community Health. 2009;63(9):683–684. doi:10.1136/jech.2009.090803
52. Clinical Epigenetics. Epigenetic imprinting alterations as effective diagnostic biomarkers for early-stage lung cancer and small pulmonary nodules. doi:10.1186/s13148-021-01203-5.
53. Krushkal J, Jensen TL, Wright G, Zhao Y. Allelic expression patterns of imprinted and non-imprinted genes in cancer cell lines from multiple histologies. Clin Clin Epigenet. 2025;17(1):83. doi:10.1186/s13148-025-01883-3
54. Harrison K, Hoad G, Scott P, et al. Breast cancer risk and imprinting methylation in blood. Clin Clin Epigenet. 2015;7(1):92. doi:10.1186/s13148-015-0125-x
55. Anwar SL, Krech T, Hasemeier B, et al. Loss of Imprinting and Allelic Switching at the DLK1-MEG3 Locus in Human Hepatocellular Carcinoma. PLoS One. 2012;7(11):e49462. doi:10.1371/journal.pone.0049462
56. He Y, Luo Y, Liang B, Ye L, Lu G, He W. Potential applications of MEG3 in cancer diagnosis and prognosis. Oncotarget. 2017;8(42):73282–73295. doi:10.18632/oncotarget.19931
57. Xu H, Zhang Y, Wu H, et al. High Diagnostic Accuracy of Epigenetic Imprinting Biomarkers in Thyroid Nodules. J Clin Oncol. 2023;41(6):1296–1306. doi:10.1200/JCO.22.00232
58. Shen R, Cheng T, Xu C, et al. Novel visualized quantitative epigenetic imprinted gene biomarkers diagnose the malignancy of ten cancer types. Clin Clin Epigenet. 2020;12(1):71. doi:10.1186/s13148-020-00861-1
59. Jima DD, Skaar DA, Planchart A, et al. Genomic map of candidate human imprint control regions: the imprintome. Epigenetics. 2022;17(13):1920–1943. doi:10.1080/15592294.2022.2091815
60. Xi Y, Li W. BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinf. 2009;10(1):232. doi:10.1186/1471-2105-10-232
61. Morgan M, Wang J, Obenchain V, Lang M, Thompson R, Turaga N. BiocParallel: bioconductor facilities for parallel evaluation. 2025. Available from: https://github.com/Bioconductor/BiocParallel.
62. Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLOS Comput Biol. 2016;12(4):e1004873. doi:10.1371/journal.pcbi.1004873
63. Zeybel M, Vatansever S, Hardy T, et al. DNA methylation profiling identifies novel markers of progression in hepatitis B-related chronic liver disease. Clin Clin Epigenet. 2016;8(1):48. doi:10.1186/s13148-016-0218-1
64. Gerhard GS, Malenica I, Llaci L, et al. Differentially methylated loci in NAFLD cirrhosis are associated with key signaling pathways. Clin Clin Epigenet. 2018;10(1):93. doi:10.1186/s13148-018-0525-9
65. Sohda T, Iwata K, Soejima H, Kamimura S, Shijo H, Yun K. In situ detection of insulin-like growth factor II (IGF2) and H19 gene expression in hepatocellular carcinoma. J Hum Genet. 1998;43(1):49–53. doi:10.1007/s100380050036
66. Fu S, Deger T, Boers RG, et al. Hypermethylation of DNA Methylation Markers in Non-Cirrhotic Hepatocellular Carcinoma. Cancers. 2023;15(19):4784. doi:10.3390/cancers15194784
67. Hernandez-Meza G, von Felden J, Gonzalez-Kozlova EE, et al. DNA Methylation Profiling of Human Hepatocarcinogenesis. Hepatology. 2021;74(1):183–199. doi:10.1002/hep.31659
68. Murphy SK, Yang H, Moylan CA, et al. Relationship Between Methylome and Transcriptome in Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology. 2013;145(5):1076–1087. doi:10.1053/j.gastro.2013.07.047
69. Tsuda N, Tian Y, Fujimoto M, et al. DNA methylation status of the SPHK1 and LTB genes underlies the clinicopathological diversity of non-alcoholic steatohepatitis-related hepatocellular carcinomas. J Cancer Res Clin Oncol. 2023;149(8):5109–5125. doi:10.1007/s00432-022-04445-9
70. Johnson ND, Wu X, Still CD, et al. Differential DNA methylation and changing cell-type proportions as fibrotic stage progresses in NAFLD. Clin Clin Epigenet. 2021;13(1):152. doi:10.1186/s13148-021-01129-y
71. Zhang E, Li C, Fang Y, et al. STMN1 as a novel prognostic biomarker in HCC correlating with immune infiltrates and methylation. World J Surg Oncol. 2022;20(1):301. doi:10.1186/s12957-022-02768-y
72. Villanueva A, Portela A, Sayols S, et al. DNA methylation-based prognosis and epidrivers in hepatocellular carcinoma. Hepatology. 2015;61(6):1945–1956. doi:10.1002/hep.27732
73. Bai Y, Tong W, Xie F, et al. DNA methylation biomarkers for diagnosis of primary liver cancer and distinguishing hepatocellular carcinoma from intrahepatic cholangiocarcinoma. Aging. 2021;13(13):17592–17606. doi:10.18632/aging.203249
74. Kuramoto J, Arai E, Fujimoto M, et al. Quantification of DNA methylation for carcinogenic risk estimation in patients with non-alcoholic steatohepatitis. Clin Clin Epigenet. 2022;14(1):1. doi:10.1186/s13148-022-01379-4
75. Ahrens M, Ammerpohl O, von Schönfels W, et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab. 2013;18(2):296–302. doi:10.1016/j.cmet.2013.07.004
76. de Mello VD, Matte A, Perfilyev A, et al. Human liver epigenetic alterations in non-alcoholic steatohepatitis are related to insulin action. Epigenetics. 2017;12(4):287–295. doi:10.1080/15592294.2017.1294305
77. Wegermann K, Henao R, Diehl AM, Murphy SK, Abdelmalek MF, Moylan CA. Branched chain amino acid transaminase 1 (BCAT1) is overexpressed and hypomethylated in patients with non-alcoholic fatty liver disease who experience adverse clinical events: a pilot study. PLoS One. 2018;13(9):e0204308. doi:10.1371/journal.pone.0204308
78. Walle P, Männistö V, de Mello VD, et al. Liver DNA methylation of FADS2 associates with FADS2 genotype. Clin Clin Epigenet. 2019;11(1):10. doi:10.1186/s13148-019-0609-1
79. Kuramoto J, Arai E, Tian Y, et al. Genome-wide DNA methylation analysis during non-alcoholic steatohepatitis-related multistage hepatocarcinogenesis: comparison with hepatitis virus-related carcinogenesis. Carcinogenesis. 2017;38(3):261–270. doi:10.1093/carcin/bgx005
80. Tian Y, Arai E, Makiuchi S, et al. Aberrant DNA methylation results in altered gene expression in non-alcoholic steatohepatitis-related hepatocellular carcinomas. J Cancer Res Clin Oncol. 2020;146(10):2461–2477. doi:10.1007/s00432-020-03298-4
81. Zheng Y, Huang Q, Ding Z, et al. Genome-wide DNA methylation analysis identifies candidate epigenetic markers and drivers of hepatocellular carcinoma. Brief Bioinform. 2018;19(1):101–108. doi:10.1093/bib/bbw094
82. Xu Q, Hu Y, Chen S, et al. Immunological Significance of Prognostic DNA Methylation Sites in Hepatocellular Carcinoma. Front Mol Biosci. 2021;8:683240. doi:10.3389/fmolb.2021.683240
83. Holmila R, Sklias A, Muller DC, et al. Targeted deep sequencing of plasma circulating cell-free DNA reveals Vimentin and Fibulin 1 as potential epigenetic biomarkers for hepatocellular carcinoma. PLoS One. 2017;12(3):e0174265. doi:10.1371/journal.pone.0174265
84. Hardy T, Zeybel M, Day CP, et al. Plasma DNA methylation: a potential biomarker for stratification of liver fibrosis in non-alcoholic fatty liver disease. Gut. 2017;66(7):1321–1328. doi:10.1136/gutjnl-2016-311526
85. Miller BF, Petrykowska HM, Elnitski L. Assessing ZNF154 methylation in patient plasma as a multicancer marker in liquid biopsies from colon, liver, ovarian and pancreatic cancer patients. Sci Rep. 2021;11(1):1. doi:10.1038/s41598-020-80345-7
86. Hlady RA, Zhao X, Pan X, et al. Genome-wide discovery and validation of diagnostic DNA methylation-based biomarkers for hepatocellular cancer detection in circulating cell free DNA. Theranostics. 2019;9(24):7239–7250. doi:10.7150/thno.35573
87. He N, Feng G, Zhang C, Wu F, Zhang T, Yang Y. Plasma levels of methylated septin 9 are capable of detecting hepatocellular carcinoma and hepatic cirrhosis. Mol Med Rep. 2020;22(4):2705–2714. doi:10.3892/mmr.2020.11356
88. Kisiel JB, Dukek BA, Kanipakam R, et al. Hepatocellular Carcinoma Detection by Plasma Methylated DNA: discovery, Phase I Pilot, and Phase II Clinical Validation. Hepatology. 2019;69(3):1180–1192. doi:10.1002/hep.30244
89. Xu RH, Wei W, Krawczyk M, et al. Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma. Nat Mater. 2017;16(11):1155–1161. doi:10.1038/nmat4997
90. Luo B, Ma F, Liu H, et al. Cell-free DNA methylation markers for differential diagnosis of hepatocellular carcinoma. BMC Med. 2022;20(1):8. doi:10.1186/s12916-021-02201-3
91. Lubecka K, Flower K, Beetch M, et al. Loci-specific differences in blood DNA methylation in HBV-negative populations at risk for hepatocellular carcinoma development. Epigenetics. 2018;13(6):605–626. doi:10.1080/15592294.2018.1481706
92. Moylan CA, Mavis AM, Jima D, et al. Alterations in DNA methylation associate with fatty liver and metabolic abnormalities in a multi-ethnic cohort of pre-teenage children. Epigenetics. 2022;17(11):1446–1461. doi:10.1080/15592294.2022.2039850
93. Zhang Y, Petropoulos S, Liu J, et al. The signature of liver cancer in immune cells DNA methylation. Clin Clin Epigenet. 2018;10(1):8. doi:10.1186/s13148-017-0436-1
94. Nano J, Ghanbari M, Wang W, et al. Epigenome-Wide Association Study Identifies Methylation Sites Associated With Liver Enzymes and Hepatic Steatosis. Gastroenterology. 2017;153(4):1096–1106.e2. doi:10.1053/j.gastro.2017.06.003
95. Zhang RN, Pan Q, Zheng RD, et al. Genome-wide analysis of DNA methylation in human peripheral leukocytes identifies potential biomarkers of nonalcoholic fatty liver disease. Int J Mol Med. 2018;42(1):443–452. doi:10.3892/ijmm.2018.3583
96. Wu J, Zhang R, Shen F, et al. Altered DNA Methylation Sites in Peripheral Blood Leukocytes from Patients with Simple Steatosis and Nonalcoholic Steatohepatitis (NASH). Med Sci Monit Int Med J Exp Clin Res. 2018;24:6946–6967. doi:10.12659/MSM.909747
97. Ma J, Nano J, Ding J, et al. A Peripheral Blood DNA Methylation Signature of Hepatic Fat Reveals a Potential Causal Pathway for Nonalcoholic Fatty Liver Disease. Diabetes. 2019;68(5):1073–1083. doi:10.2337/DB18-1193
98. Akbari V, Garant JM, O’Neill K, et al. Genome-wide detection of imprinted differentially methylated regions using nanopore sequencing. eLife. 2022;11:e77898. doi:10.7554/eLife.77898
99. Akbari V, Garant JM, O’Neill K, et al. Megabase-scale methylation phasing using nanopore long reads and NanoMethPhase. Genome Biol. 2021;22(1):68. doi:10.1186/s13059-021-02283-5
100. Carreras-Gallo N, Dwaraka VB, Jima DD, et al. Creation and Validation of the First Infinium DNA Methylation Array for the Human Imprintome. BioRxiv Prepr Serv Biol. 2024;2024:575646. doi:10.1101/2024.01.15.575646
101. Zhou W, Triche Jr TJ, Laird PW, Shen H. SeSAMe: reducing artifactual detection of DNA methylation by Infinium BeadChips in genomic deletions. Nucleic Acids Res. 2018;46(20):e123. doi:10.1093/nar/gky691
102. Gentleman RC, Carey VJ, Bates DM, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5(10):R80. doi:10.1186/gb-2004-5-10-r80
103. R-universe. Cowplot: streamlined Plot Theme and Plot Annotations for “ggplot2.”. Available from: https://wilkelab.r-universe.dev/cowplot.
104. Daniel F, Corporation M, Weston S. doSNOW: foreach Parallel Adaptor for the “snow” Package. 2022. Available from: https://cran.r-project.org/web/packages/doSNOW/index.html.
105. Wickham H, Henry L, Software P, PBC [cph, fnd. magrittr: a Forward-Pipe Operator for R. 2026. https://cran.r-project.org/web/packages/magrittr/index.html.
106. R-universe. Snow: simple Network of Workstations. Available from: https://ltierney.r-universe.dev/snow.
107. Zeileis A, Grothendieck G. zoo: S3 Infrastructure for Regular and Irregular Time Series. J Stat Softw. 2005;14:1–27. doi:10.18637/jss.v014.i06
108. Wickham H, Henry L, Software P, PBC [cph, fnd. purrr: functional Programming Tools. 2026. Available from: https://cran.r-project.org/web/packages/purrr/index.html.
109. Wickham H. Reshaping Data with the reshape Package. J Stat Softw. 2007;21(12):1–20. doi:10.18637/jss.v021.i12
110. Orr M, Liu P. ssize.fdr: sample Size Calculations for Microarray Experiments. 2022. Available from: https://cran.r-project.org/web/packages/ssize.fdr/index.html.
111. Jima DD, Skaar DA, Planchart A, et al. Genomic map of candidate human imprint control regions: the imprintome. Epigenetics. 2022;17(13):13. doi:10.1080/15592294.2022.2091815
112. Kim M, Costello J. DNA methylation: an epigenetic mark of cellular memory. Exp Mol Med. 2017;49(4):e322. doi:10.1038/emm.2017.10
113. Requena D, Medico JA, Soto-Ugaldi LF, et al. Liver cancer multiomics reveals diverse protein kinase A disruptions convergently produce fibrolamellar hepatocellular carcinoma. Nat Commun. 2024;15(1):10887. doi:10.1038/s41467-024-55238-2
114. Shirani M, Levin S, Shebl B, et al. Increased Protein Kinase A Activity Induces Fibrolamellar Hepatocellular Carcinoma Features Independent of DNAJB1. Cancer Res. 2024;84(16):2626–2644. doi:10.1158/0008-5472.CAN-23-4110
115. Wang Y, Liu S, Jia T, Feng Y, Xin X, Zhang D. T Cell Protein Tyrosine Phosphatase in Glucose Metabolism. Front Cell Dev Biol. 2021;2021:1. doi:10.3389/fcell.2021.682947
116. Salazar-Tortosa DF, Labayen I, González-Gross M, et al. Association between PTPN1 polymorphisms and obesity-related phenotypes in European adolescents: influence of physical activity. Pediatr Res. 2023;93(7):2036–2044. doi:10.1038/s41390-022-02377-1
117. Xie L, Qi H, Tian W, Bu S, Wu Z, Wang H. High-expressed PTPN1 promotes tumor proliferation signature in human hepatocellular carcinoma. Heliyon. 2023;9(9):e19895. doi:10.1016/j.heliyon.2023.e19895
118. Tang X, Qi C, Zhou H, Liu Y. Critical roles of PTPN family members regulated by non-coding RNAs in tumorigenesis and immunotherapy. Front Oncol. 2022;12:972906. doi:10.3389/fonc.2022.972906
119. Lau MM, Stewart CE, Liu Z, Bhatt H, Rotwein P, Stewart CL. Loss of the imprinted IGF2/cation-independent mannose 6-phosphate receptor results in fetal overgrowth and perinatal lethality. Genes Dev. 1994;8(24):2953–2963. doi:10.1101/gad.8.24.2953
120. Adachi Y, Nojima M, Mori M, et al. Insulin-Like Growth Factor 2 and Incidence of Liver Cancer in a Nested Case–Control Study. Cancer Epidemiol Biomarkers Prev. 2021;30(11):2130–2135. doi:10.1158/1055-9965.EPI-21-0481
121. Zhong Y, Ren X, Cao X, et al. Insulin-like growth factor 2 receptor is a key immune-related gene that is correlated with a poor prognosis in patients with triple-negative breast cancer: a bioinformatics analysis. Front Oncol. 2022;12:871786. doi:10.3389/fonc.2022.871786
122. Hong H, Hexu H, Lei W, et al. ABCF1-K430-Lactylation promotes HCC malignant progression via transcriptional activation of HIF1 signaling pathway - PMC. Nat Cell Death Differ. 2025;32:613–631. doi:10.1038/s41418-024-01436-w
123. Han H, Wang S, Ma L, et al. ASH2L-K312-Lac Stimulates Angiogenesis in Tumors to Expedite the Malignant Progression of Hepatocellular Carcinoma - PMC. Adv Sci. 2025;12(40). doi:10.1002/advs.202509477
124. Košuta I, Mrzljak A, Kolarić B, Vučić Lovrenčić M. Leptin as a Key Player in Insulin Resistance of Liver Cirrhosis? A Cross-Sectional Study in Liver Transplant Candidates. J Clin Med. 2020;9(2):560. doi:10.3390/jcm9020560
125. la Cruz-Color L D, Dominguez-Rosales JA, Maldonado-González M, et al. Evidence That Peripheral Leptin Resistance in Omental Adipose Tissue and Liver Correlates with MASLD in Humans. Int J Mol Sci. 2024;25(12):6420. doi:10.3390/ijms25126420
126. Obradovic M, Sudar-Milovanovic E, Soskic S, et al. Leptin and Obesity: role and Clinical Implication. Front Endocrinol. 2021;12:585887. doi:10.3389/fendo.2021.585887
127. Curley JP, Pinnock SB, Dickson SL, et al. Increased body fat in mice with a targeted mutation of the paternally expressed imprinted gene Peg3. FASEB J. 2005;19(10):1302–1304. doi:10.1096/fj.04-3216fje
128. Nikonova L, Koza RA, Mendoza T, Chao PM, Curley JP, Kozak LP. Mesoderm-specific transcript is associated with fat mass expansion in response to a positive energy balance. FASEB J. 2008;22(11):3925–3937. doi:10.1096/fj.08-108266
129. Cowley M, Garfield AS, Madon-Simon M, et al. Developmental Programming Mediated by Complementary Roles of Imprinted Grb10 in Mother and Pup. PLoS Biol. 2014;12(2):e1001799. doi:10.1371/journal.pbio.1001799
130. Li Y, Zhang Y, Hu Q, et al. Functional significance of gain-of-function H19 lncRNA in skeletal muscle differentiation and anti-obesity effects. Genome Med. 2021;13(1):137. doi:10.1186/s13073-021-00937-4
131. Cleaton MAM, Dent CL, Howard M, et al. Fetus-derived DLK1 is required for maternal metabolic adaptations to pregnancy and is associated with fetal growth restriction. Nat Genet. 2016;48(12):1473–1480. doi:10.1038/ng.3699
132. Stefanou N, Papanikolaou V, Furukawa Y, Nakamura Y, Tsezou A. Leptin as a critical regulator of hepatocellular carcinoma development through modulation of human telomerase reverse transcriptase. BMC Cancer. 2010;10(1):442. doi:10.1186/1471-2407-10-442
133. Chen C, Chang YC, Liu CL, Liu TP, Chang KJ, Guo IC. Leptin Induces Proliferation and Anti-Apoptosis in Human Hepatocarcinoma Cells by up-Regulating Cyclin D1 and Down-Regulating Bax via a Janus Kinase 2-Linked Pathway; 2007. doi:10.1677/ERC-06-0027
134. Boone DN, Lee AV. Targeting the Insulin-like Growth Factor Receptor: developing Biomarkers from Gene Expression Profiling. Crit Rev Oncog. 2012;17(2):161–173. doi:10.1615/critrevoncog.v17.i2.30
135. Tovar V, Alsinet C, Villanueva A, et al. IGF activation in a molecular subclass of hepatocellular carcinoma and pre-clinical efficacy of IGF-1R blockage. J Hepatol. 2010;52(4):550–559. doi:10.1016/j.jhep.2010.01.015
136. Berenguier C, Chen X, Allegrini B, et al. Cancer-associated loss-of-function mutations in KCNQ1 enhance Wnt/β-catenin signalling disrupting epithelial homeostasis. Oncogene. 2025;44(31):2715–2729. doi:10.1038/s41388-025-03447-4
137. Wang Y, Zhang J, Li YJ, et al. MEST promotes lung cancer invasion and metastasis by interacting with VCP to activate NF-κB signaling. J Exp Clin Cancer Res CR. 2021;40:301. doi:10.1186/s13046-021-02107-1
138. Zhang X, Luo M, Zhang J, et al. The role of lncRNA H19 in tumorigenesis and drug resistance of human Cancers. Front Genet. 2022;13:1005522. doi:10.3389/fgene.2022.1005522
139. Xu J, Wang X, Zhu C, Wang K. A review of current evidence about lncRNA MEG3: a tumor suppressor in multiple cancers. Front Cell Dev Biol. 2022;10:997633. doi:10.3389/fcell.2022.997633
140. Chen Y, Tang M, Xiong J, Gao Q, Cao W, Huang J. GRB10 is a novel oncogene associated with cell proliferation and prognosis in glioma. Cancer Cell Int. 2022;22(1):223. doi:10.1186/s12935-022-02636-5
141. Luo L, Jiang W, Liu H, et al. De-silencing Grb10 contributes to acute ER stress-induced steatosis in mouse liver. J Mol Endocrinol. 2018;60(4):285–297. doi:10.1530/JME-18-0018
142. Xie T, Pan S, Zheng H, et al. PEG10 as an oncogene: expression regulatory mechanisms and role in tumor progression. Cancer Cell Int. 2018;18(1):112. doi:10.1186/s12935-018-0610-3
143. Kagami M, O’Sullivan MJ, Green AJ, et al. The IG-DMR and the MEG3-DMR at Human Chromosome 14q32.2: hierarchical Interaction and Distinct Functional Properties as Imprinting Control Centers. PLoS Genet. 2010;6(6):e1000992. doi:10.1371/journal.pgen.1000992
144. Li Z, Chen C, Yong H, et al. PRMT2 promotes RCC tumorigenesis and metastasis via enhancing WNT5A transcriptional expression. Cell Death Dis. 2023;14(5):1–12. doi:10.1038/s41419-023-05837-6
145. Zhou Q, Lin J, Yan Y, et al. INPP5F translocates into cytoplasm and interacts with ASPH to promote tumor growth in hepatocellular carcinoma. J Exp Clin Cancer Res. 2022;41(1):13. doi:10.1186/s13046-021-02216-x
146. Huang YX, Song H, Tao Y, et al. Ovostatin 2 knockdown significantly inhibits the growth, migration, and tumorigenicity of cutaneous malignant melanoma cells. PLoS One. 2018;13(4):e0195610. doi:10.1371/journal.pone.0195610
147. Li WC, Xiong ZY, Huang PZ, et al. KCNK levels are prognostic and diagnostic markers for hepatocellular carcinoma. Aging. 2019;11(19):8169–8182. doi:10.18632/aging.102311
148. Chow JTS, Salmena L. Recent advances in PTEN signalling axes in cancer. Fac Rev. 2020;9:31. doi:10.12703/r/9-31
149. Endersby R, Baker SJ. PTEN signaling in brain: neuropathology and tumorigenesis. Oncogene. 2008;27(41):5416–5430. doi:10.1038/onc.2008.239
150. Wang W, Lu G, Liu HB, et al. Pten Regulates Cardiomyocyte Differentiation by Modulating Non-CG Methylation via Dnmt3. Adv Sci. 2021;8(17):2100849. doi:10.1002/advs.202100849
151. Ngo MHT, Jeng HY, Kuo YC, et al. The Role of IGF/IGF-1R Signaling in Hepatocellular Carcinomas: stemness-Related Properties and Drug Resistance. Int J Mol Sci. 2021;22(4):1931. doi:10.3390/ijms22041931
152. Al-Gayyar MMH, Bagalagel A, Noor AO, Almasri DM, Diri R. The therapeutic effects of nicotinamide in hepatocellular carcinoma through blocking IGF-1 and effecting the balance between Nrf2 and PKB. Biomed Pharmacother. 2019;112:108653. doi:10.1016/j.biopha.2019.108653
153. Baptissart M, Bradish CM, Jones BS, et al. Zac1 and the Imprinted Gene Network program juvenile NAFLD in response to maternal metabolic syndrome. Hepatology. 2022;76(4):1090–1104. doi:10.1002/hep.32363
154. Riegl SD, Starnes C, Jima DD, et al. The imprinted gene Zac1 regulates steatosis in developmental cadmium-induced nonalcoholic fatty liver disease. Toxicol Sci. 2023;191(1):34–46. doi:10.1093/toxsci/kfac106
155. Vega-Benedetti AF, Saucedo CN, Zavattari P, et al. PLAGL1 gene function during hepatoma cells proliferation. Oncotarget. 2018;9(67):32775–32794. doi:10.18632/oncotarget.25996
156. Marsh TL, Parikh ND, Roberts LR, et al. A Phase 3 Biomarker Validation of GALAD for the Detection of Hepatocellular Carcinoma in Cirrhosis. Gastroenterology. 2025;168(2):316–326. doi:10.1053/j.gastro.2024.09.008
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.