")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Punicalagin Inhibits Tert-Butyl Hydroperoxide-Induced Apoptosis and Extracellular Matrix Degradation in Chondrocytes by Activating Autophagy and Ameliorates Murine Osteoarthritis
Authors Kong J, Wang J, Gong X, Zheng X, Chen T
Received 19 September 2020
Accepted for publication 1 December 2020
Published 15 December 2020 Volume 2020:14 Pages 5521—5533
DOI https://doi.org/10.2147/DDDT.S282932
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Georgios Panos
Jinsong Kong, Jiacheng Wang, Xiaokang Gong, Xin Zheng, Tao Chen
Department of Orthopaedics, Taizhou Municipal Hospital, Taizhou, Zhejiang, People’s Republic of China
Correspondence: Tao Chen
Department of Orthopaedics, Taizhou Municipal Hospital, Taizhou, Zhejiang, People’s Republic of China
Email [email protected]
Background: Osteoarthritis (OA) is a prevalent articular disorder and has no entirely satisfactory treatment. Punicalagin (PUG) is a polyphenol which has shown multiple pharmacological effects on various diseases. However, the role of PUG in the treatment of OA has not been well defined.
Methods: The effects of PUG on anti-oxidative stress, anti-apoptosis, extracellular matrix (ECM) degradation and autophagy were evaluated in chondrocytes through Western blot and immunofluorescence (IF) staining. Meanwhile, the effects of PUG on destabilization of the medial meniscus (DMM) model were also assessed in vivo by performing histopathologic analysis and IF staining.
Results: In vitro, PUG treatment not only increased the level of HO-1 and SOD1 against oxidative stress but also suppressed the expression of apoptotic proteins and inhibited ECM degradation. Meanwhile, PUG treatment activated autophagy and restores autophagic flux in chondrocytes after tert-butyl hydroperoxide (TBHP) insult, inhibition of autophagy by 3-methyladenine (3-MA) partly abrogated the protective effects of PUG on chondrocytes. In vivo, degeneration of the articular cartilage following DMM was also ameliorated by PUG treatment.
Conclusion: PUG prevents the progression of OA through inhibition of apoptosis, oxidative stress and ECM degradation in chondrocytes, which mediated by the activation of autophagy.
Keywords: punicalagin, autophagy, osteoarthritis, apoptosis, extracellular matrix
Introduction
Osteoarthritis (OA) is the most prevalent degenerative joint disease and is typically characterized by cell quantity reduction, cartilage damage, extracellular matrix (ECM) degradation; these pathological changes cause persistent chronic pain and functional disability in elderly individuals.1–3
Various pathological factors are involved in the development of OA. The excessive accumulation of reactive oxygen species (ROS) is a common pathological mechanism in degenerative diseases.4,5 Studies have indicated that aggressive oxidative stress can damage molecules and organelles in chondrocytes, thus contributing to cellular apoptosis and senescence and impacting chondrocytes favoring catabolism over anabolism.6,7 As reported previously, chondrocytes are the only resident cells in articular cartilage and play important roles in regulating the synthesis and turnover of ECM and maintaining the structure of cartilage.8 Intriguingly, it has been shown that attenuating oxidative stress and promoting chondrocyte homeostasis by pharmacological treatment is a potential strategy for deferring the progression of OA.6,9,10 However, the drug management of OA is unsatisfactory at present.
As a cellular homeostatic mechanism, autophagy plays a role in degrading dysfunctional intracellular components to maintain cell survival and homoeostasis.11 In actuality, accumulating evidence has suggested that autophagy is not only involved in chondrocyte and cartilage morphogenesis, but also required for the clearance of ROS by removing damaged or dysfunctional mitochondria, maintaining redox homeostasis in the progression of OA.12–14 It has been reported that OA is associated with dysfunctional autophagy, and ATG5 deficiency aggravates chondrocyte apoptosis and the degeneration of cartilage in mice joints.15 Importantly, numerous studies have implicated that activating autophagy by injection with rapamycin or other molecules attenuated adverse molecule accumulation and reduced cartilage degeneration in a mouse OA model.9,16,17 Thus, the modulation of autophagy by several molecules in chondrocytes is an available method for limiting the development of cartilage degeneration.
Pomegranate, the fruit of Punica granatum, has been applied in medical treatment for neural degenerative diseases and cancer.18,19 Punicalagin (PUG) is a hydrolyzable polyphenol, exerts extensive biological activities and exhibits a cell protective effect under pathologic conditions.20,21 Meanwhile, recent studies have revealed that PUG is a critical autophagy regulator, and PUG-mediated autophagy is beneficial against macrophage inflammation and hepatic injury.22,23 Yaidikar et al24 have reported that PUG provides neuroprotection against cerebral ischemia reperfusion injury and improves functional recovery through its antioxidant activity. Similarly, increasing evidences have indicated that PUG pretreatment alleviates myocardial ischemia reperfusion injury, and the underlying mechanism is related to the activation of AMPK phosphorylation and the regulation of the Sirt1-Nrf-2-HO-1 signaling pathway.25,26 Recently, PUG has been reported to promote a switch in mitochondrial biogenesis for reducing endothelial dysfunction in cardiovascular diseases27 and to clear homocysteine, amyloid-β and TNF-α, which is beneficial for the improvement of Alzheimer’s disease and Parkinson’s disease.28 Interestingly, several studies have also suggested that Punica granatum is beneficial for the treatment of OA.29,30 However, the therapeutic effect and underlying mechanism of PUG on chondrocytes and OA has not been well defined.
As PUG exhibited such biological functions above, we evaluated the effects of PUG on chondrocytes under ROS donor treatment and the underlying molecular mechanism, and hypothesized that PUG may provide protection for TBHP treated chondrocytes via the activation of autophagy. Meanwhile, the potential therapeutic effect of PUG was also assessed in a mouse OA model. Collectively, using conventional molecular methods, our results suggested that PUG effectively prevents the progression of OA, and PUG-activated autophagy contributes to maintaining chondrocyte survival and reducing ECM degradation. Therefore, our results provide convincing evidence that PUG could be a potential candidate for the clinical treatment of OA.
Materials and Methods
Animals and Ethical Statement
A total of sixty adult male C57BL/6 mice (7–8 weeks old, 20–25 g) were purchased from Taizhou College and housed in standard conditions (temperature: 23 ± 2 °C, humidity: 50 ± 5%) under a 12-hour light/dark cycle. All experimental operations, reagent administration procedures and mice care protocols were approved (No. 2017042) and strictly conducted according to the guidance of the Animal Care and Use Committee of Taizhou College.
Reagents and Antibodies
Punicalagin (MB6504) was purchased from Meilun Bio. (Dalian, China). Tert-butyl hydroperoxide solution (TBHP, 416665), 3-methyladenine (3-MA, M9281), and type II collagenases (C2-28) were purchased from Sigma-Aldrich (WI,USA). Primary antibodies against heme oxygenase 1 (HO-1, 10701–1), superoxide dismutase 1 (SOD1, 10269–1), B-cell lymphoma-2 (Bcl-2, 26593–1), Beclin 1 (11306–1), matrix metalloprotein (MMP13, 18165–1), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH, 10494–1) were purchased from Proteintech (IL, USA), and Bax (ab182733), type II collagen (ab185430), a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS5, ab41037), NAD(P)H quinone dehydrogenase 1 (NQO1, ab80588), p62/SQSTM1 (p62, ab56416), matrix metalloprotein 3 (MMP3, ab52915) and lysosomal Associated Membrane Protein 2 (Lamp2, ab13524) were purchased from Abcam (MA, USA); cleaved caspase 3 was obtained from (AF7022) from Affinity Biosciences (OH, USA); light chain 3B (LC3B, NB600-1384) was obtained from NOVUS Biologicals (CO, USA); and autophagy-related protein 12 (Atg12, 4180T) was purchased from CST (MA, USA). Secondary antibodies against mouse, rat and rabbit were purchased from Proteintech. Alexa Fluor FITC (488) or Cy5 (647) donkey anti-rabbit/mouse/rat secondary antibodies were purchased from Abcam. Reagents, including DMEM/F12 medium (10565018), penicillin/streptomycin solution (15140122), and EDTA (12605028), were purchased from Gibco (CA, USA), and fetal bovine serum (FBS) was purchased from PAN-Biotech GmbH (p30–3301, Baghlia,Germany).The 4,6-Diami-dino-2-phenylindole (DAPI, P0131) was purchased from Beyotime (Shanghai, China).
Primary Chondrocyte Culture
Primary chondrocytes were obtained from postnatal mouse pups (10 days) by dissecting the knee cartilages as described previously.6 In brief, eight immature C57BL/6 mice were sacrificed with an overdose of sodium pentobarbital (8%, 40 mg/kg). Knee joint cartilages were dissected into pieces and digested by collagenase II (2 mg/mL) diluted in DMEM/F12 medium in an incubator at 37°C for 4 hours. Then, chondrocytes were separated, collected and incubated in 12-well culture plates in DMEM/F12 medium with 10% FBS and antibiotics (100 μg/mL streptomycin and 100 U/mL penicillin) in 5% CO2 at 37°C. When up to 80% confluency was reached, chondrocytes were suspended by 0.25% EDTA and reseeded in 12-well culture plates. Passage-1 or passage-2 were used in the following experiments.
Cell Viability Assay
A cell counting kit-8 (CCK-8) assay (CK04, Dojindo Co., Kumamoto, Japan) was used to assess the cytotoxicity of PUG on chondrocytes with the guidance of the manufacturer’s protocol. Chondrocytes were seeded in 96-well plates (1x104 cells per well) in 5% CO2 at 37°C with complete medium for 24 hours. After incubation, cells were administered reagents as experimentally designed and given 10 μL of CCK-8 solution for 2 hours. Then, absorbance was measured at 450 nm using a microplate reader.
Destabilization of the Medial Meniscus (DMM) Mouse Model and Group Setting
The osteoarthritis model was performed by surgical destabilization of the medial meniscus (DMM) as previously described.6 Mice were anesthetized with 2% (w/v) sodium pentobarbital (40 mg/kg, i.p.), and the joint capsule and medial meniscotibial ligament of the right knee were removed and housed for 8 weeks for OA establishment. The mice in the sham group received an arthrotomy without the transaction of the medial meniscotibial ligament. The mice received PUG (20 mg/kg) each day by oral gavage for 8 weeks according to a previous study25 and were divided into the following groups: sham group, DMM group, and DMM + PUG group. The mice in the sham group were injected with an equal dose of saline.
Tissue Preparation
The knee joints of mice were harvested after using an overdose of 8% pentobarbital (40 mg/kg) in each group and were then fixed by 4% (v/v) paraformaldehyde (PFA) for 24 hours and decalcified using 10% (v/v) EDTA for 4 weeks. After that, the joints were embedded in paraffin and cut into 5 μm sagittal slides for histopathologic and staining analysis.
Histopathologic Analysis
For hematoxylin and eosin (H&E) staining, the slides of each groups were dyed with hematoxylin and eosin to evaluate the severity of synovitis, which was graded using a scoring system and evaluated by another group of experienced histology researchers in a blinded manner.31 Safranin O (S-O) fast green staining was also applied according to the manufacturer’s protocol to evaluate the change in cartilage morphology, which was quantified by the Osteoarthritis Research Society International (OARSI) scoring system.32
Western Blot Analysis
The total protein was obtained from chondrocytes by RIPA lysis buffer and was quantified by BCA reagents (23225, Thermo Fisher, MA,USA). Equivalent amounts (40μg) of protein were separated by SDS-PAGE gels and transferred to polyvinylidene fluoride (PVDF) membranes (1620256, Bio-Rad, CA, USA). Using 5% (w/v) nonfat milk, the membranes were blocked for 90 minutes at room temperature and incubated with primary antibodies overnight at 4°C. The primary antibodies included were as follows: HO-1 (1:500), SOD1 (1:300), NQO1 (1:1000), Bcl-2 (1:300), cleaved caspase 3 (1:1000), ADAMTS5 (1:500), MMP3 (1:1000), Atg12 (1:1000), LC3 (1:1000), Beclin-1 (1:1000), p62 (1:1000), and GAPDH (1:1000). Then, the membranes were washed with TBST and incubated with the secondary antibodies (1:5000) for 90 minutes at room temperature. Protein bands were visualized by the ChemiDoc XRS+ Imaging System (Bio-Rad, CA, USA) and quantified by Quantity-One software (Version 4.6.9).
TUNEL Staining
The DNA damage level was evaluated by TUNEL staining with the guidance of the manufacturer’s protocol. Cartilage slides were deparaffinized, rehydrated and stained with 50 μL of solution from the In Situ Cell Death Detection Kit (40307ES60, Yeasen Biochemical, Shanghai, China) for 30 minutes in an incubator and then treated with DAPI for 5 min. The TUNEL results were captured by a Nikon ECLIPSE Ti microscope (Nikon, Tokyo, Japan).
Immunofluorescence
For immunofluorescence (IF) staining, the slides were deparaffinized, rehydrated, and treated with EDTA for antigen restoration, and chondrocytes were washed in PBS and fixed in 4% PFA. After preparation, the samples were treated with 5% BSA at 37°C for 60 minutes and treated with the following primary antibodies overnight at 4°C: cleaved caspase 3 (1:1000), type II collagen (1:1000), MMP13 (1:1000), LC3 (1:1000), Lamp2 (1:1000) and p62 (1:1000). The next day, the samples were treated with Alexa Fluor 488 or 647conjugated secondary antibodies (1:1000) at 37°C for 60 minutes and then treated with DAPI for 5 min. The results of staining were measured by a Nikon ECLIPSE Ti microscopes.
Statistical Analysis
The results are displayed as the mean ± SD or median (min - max) from at least three independent experiments. Data normality were evaluated using the method of Shapiro–Wilk test. Statistical analyses were performed by GraphPad Prism (Version 8.0.2). One-way analysis of variance (ANOVA) followed by Tukey’s test for between two-group comparisons were used for parametric analysis. Kruskal–Wallis ANOVA based on ranks followed by Dunn’s post hoc test were used for data that failed the equal variances assumption. Student’s t-test was used for comparisons of two groups. A p value < 0.05 indicated significance.
Results
The Effect of PUG on Chondrocyte Viability
The cytotoxic effects of PUG on chondrocytes were evaluated by the CCK-8 assay. PUG treatment presented no significant cytotoxicity at concentrations up to 50 μg/mL at 24 h (Figure 1A). Meanwhile, PUG treatment at concentration of 50 μg/mL also presented no significant cytotoxicity from 0 to 48 h (Figure 1B). To assess the effect of PUG on the viability of TBHP-treated chondrocytes, we first detected the effect of TBHP on cell viability. The CCK-8 assay showed that TBHP treatment significantly decreased the cell viability in a time-dependent manner at the concentration of 100 μM (Figure 1C). In addition, PUG remarkably reversed the decreased the viability of chondrocytes when subjected to TBHP (Figure 1D).
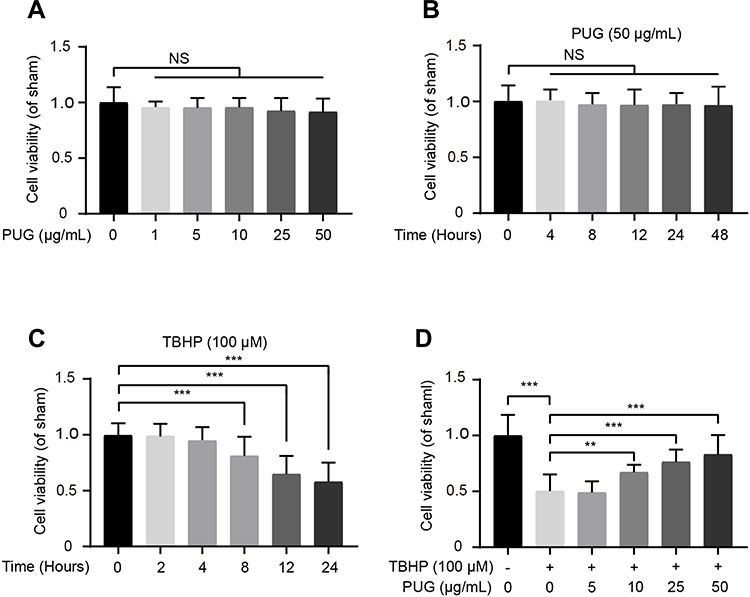 |
Figure 1 Effect of PUG on chondrocyte viability. (A) The cytotoxic effect of PUG on chondrocytes at various doses for 24 hours using a CCK-8 assay. (B) The cytotoxic effect of PUG on chondrocytes from 0 to 48 hours at the dose of 50μg/mL. (C) The cytotoxic effect of TBHP on chondrocytes from 0 to 24 hours. (D) The cell viability of chondrocytes with the treatment of TBHP and different doses of PUG for 24 hours. Data are shown as the mean ± SD, n = 8. Compared with the sham group, significant differences are indicated as **P<0.01, ***P<0.001. Abbreviation: NS, not significant. |
PUG Attenuates TBHP-Induced Oxidative Stress and Apoptosis
Oxidative stress is an important pathological factor in the progression of OA.33 Therefore, the effect of PUG on cellular oxidative stress was evaluated by Western blotting. As shown in Figure 2A–D, TBHP obviously decreased the level of HO-1, SOD1 and NQO1 all of which are common and critical enzymes against oxidative stress. PUG treatment significantly increased the expression of the above proteins. Excessive oxidative stress without intervention can cause cell death. Sequentially, the effect of PUG on chondrocyte apoptosis was also detected. As shown in Figure 2E–H, increased levels of Bcl2 and decreased levels of Bax and cleaved caspase 3 were observed in chondrocytes treated with PUG. Similarly, the results of IF stained with cleaved caspase 3 further showed that PUG decreases the IF intensity of cleaved caspase 3 (Figure 2I and J). Collectively, these data suggested that PUG exerts anti-oxidative stress and anti-apoptosis effects in mouse chondrocytes.
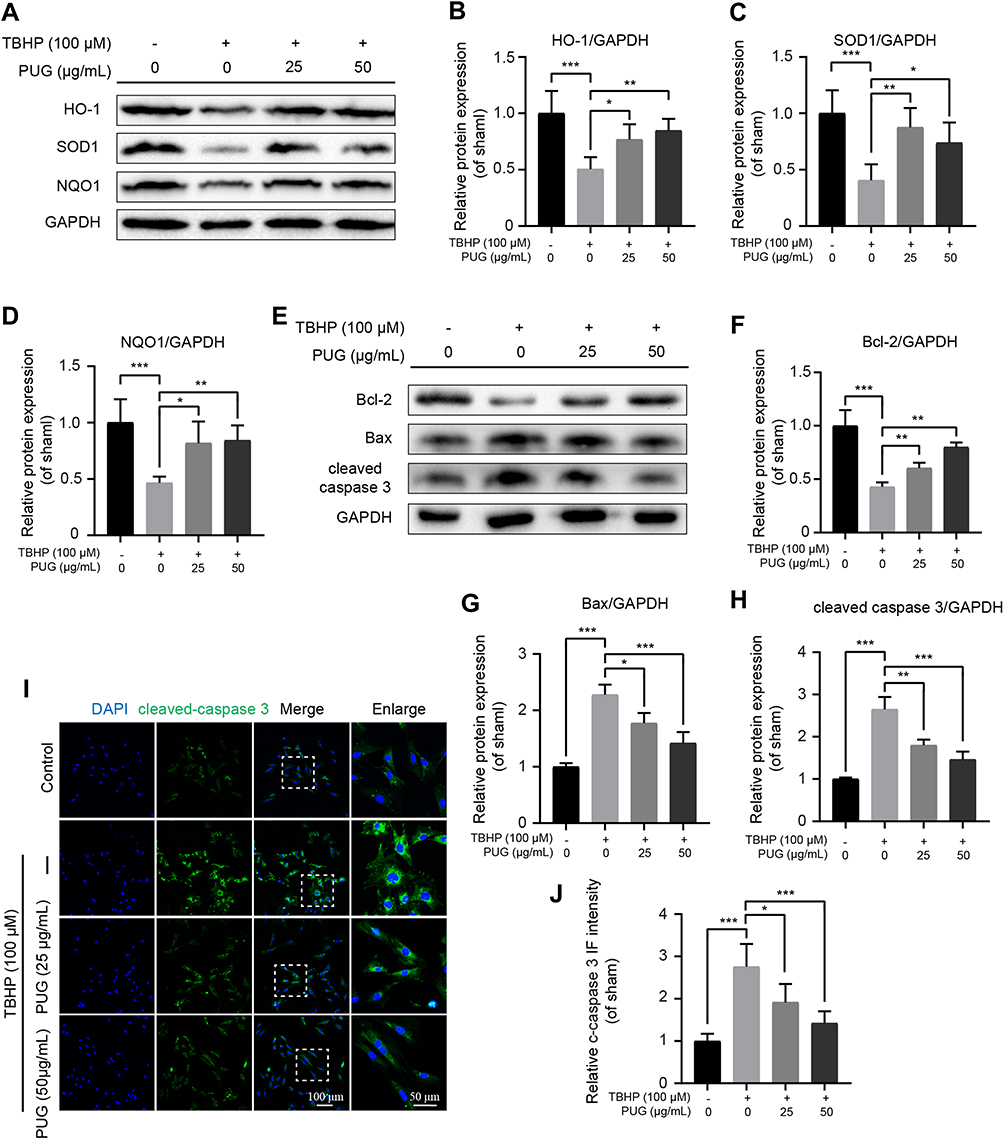 |
Figure 2 PUG inhibits oxidative stress and apoptosis. (A–D) Representative and quantitative analysis by Western blotting of HO-1, SOD1 and NQO1 in chondrocytes treated as above for 6 hours. (E–H) Western blotting and quantification of apoptotic markers in each group as above. (I and J) IF staining and quantitative intensity of cleaved caspase 3 in each chondrocyte group as above. Scale bar = 100 μm; scale bar (enlarged) = 50 μm; n = 5; GAPDH was the loading control; significance: *P<0.05, **P<0.01, ***P<0.001. |
PUG Inhibits ECM Degradation
As PUG exhibited regulation of oxidative stress, the effect of PUG on ECM metabolism was assessed in chondrocytes. Using Western blot analysis, the results showed that TBHP treatment obviously increases ADAMTS5 and MMP3 expression. In contrast, PUG pretreatment reversed the above adverse effects caused by TBHP (Figure 3A–C). In addition, the results of IF staining with type II collagen also suggested that PUG enhanced the intensity of type II collagen (Figure 3D and E). Similarly, chondrocytes labeled with MMP13 also indicated that PUG decreased the expression of MMP13 (Figure 3F and G). Consistent with above results, S-O fast green staining also revealed that the cartilage surface of the OA group presented erosion and hypocellularity and obtained a higher OARSI score, whereas the PUG treatment group exhibited a smooth, richer proteoglycan and hypercellularity cartilage surface and obtained a lower OARSI score (Figure 3H and I). Therefore, these data indicated that PUG exhibits protective effects for inhibiting ECM degradation.
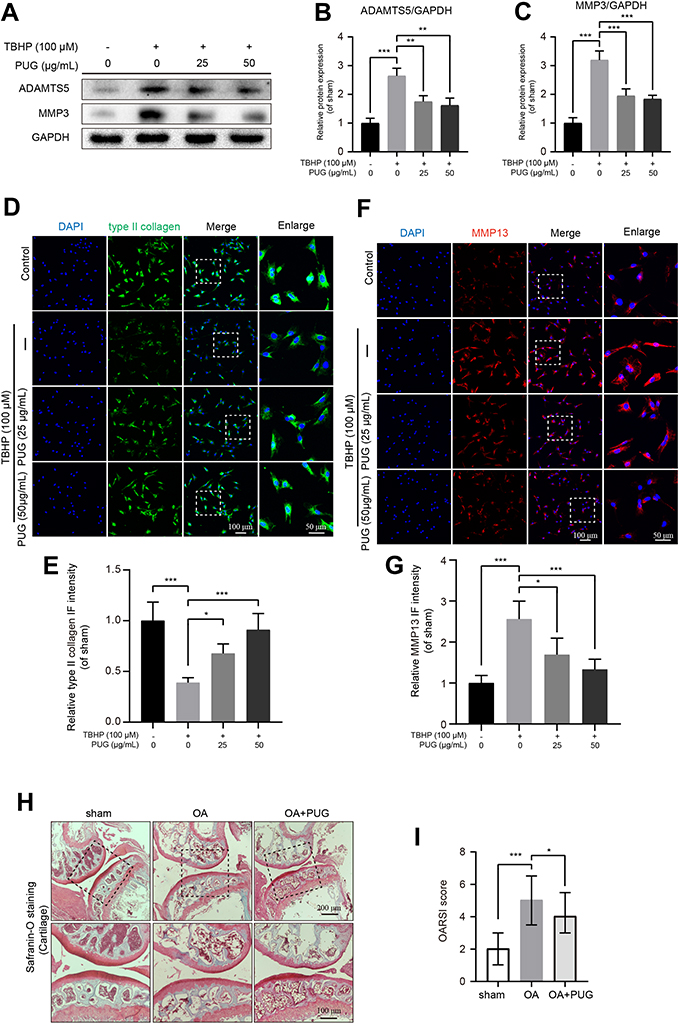 |
Figure 3 PUG reduces ECM degradation. (A–C) Representative and quantitative analysis by Western blotting of ADAMTS5 and MMP3 in chondrocytes treated as above for 6 hours. (D and E) IF staining and quantitative intensity of type II collagen in each chondrocyte group as above; scale bar = 100 μm; scale bar (enlarged) = 50 μm. (F and G) IF staining and quantitative intensity of MMP13 in each chondrocyte group as above; scale bar = 100 μm; scale bar (enlarged) = 50 μm; (H) Safranin-O staining of the cartilage and subchondral cortical bone in each group of mice; scale bar = 200 μm; scale bar (enlarged) = 100 μm. (I) The OARSI scores of each group as above, data were displayed as the median (min - max), n=8. GAPDH was the loading control; significance: *P<0.05, **P<0.01,***P<0.001. |
PUG Activates Autophagy in Chondrocytes
Given PUG’s regulation of oxidative stress, apoptosis and ECM metabolism, we reasoned that PUG’s effects may be due to the regulation of autophagy owing to its function in the clearance of excess or damaged molecules. First, we evaluated the effects of PUG on the modulation of autophagy-related proteins. As results of Figure 4A and B show, PUG obviously enhanced the level of Atg12-5, LC3 II/I, and Beclin1 and the phosphorylation of ULK1, which are essential proteins for autophagosome formation. In addition, we also investigated the function of PUG on autophagy in TBHP-treated chondrocytes. The results showed that chondrocytes present significantly increased p62 expression and a slightly increased LC3 II/I ratio, which indicated that TBHP causes impeditive autophagic flux in cells. Importantly, PUG treatment decreased p62 expression and increased the LC3 II/I ratio (Figure 4C–E). Furthermore, IF for chondrocytes stained with LC3 and Lamp2 also indicated that TBHP treatment reduces the fusion dots of LC3 and Lamp2, whereas PUG remarkably enhances the fusion of LC3 and Lamp2 (Figure 4F and G). Similar to the results in vitro, cartilage stained with LC3 or p62 showed that PUG treatment enhanced LC3 intensity (Figure 4H and I) and decreased p62 expression (Figure 4J and K), respectively, in OA mice. Overall, these data confirmed that PUG enhances the state of autophagy and ameliorates the dysfunctional autophagic flux in chondrocytes.
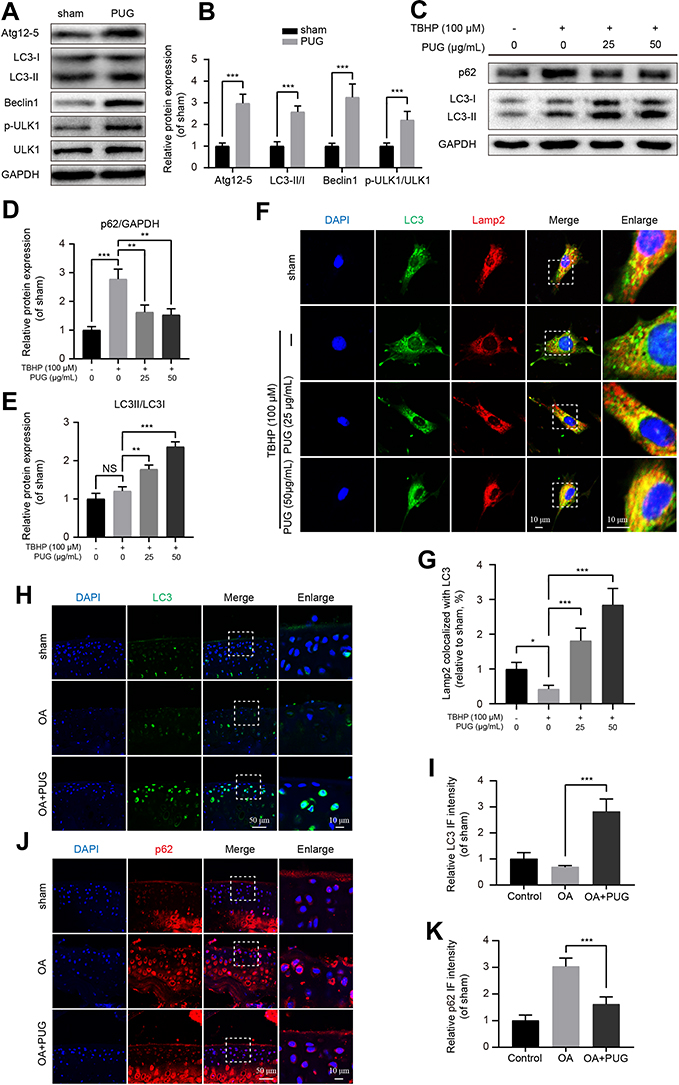 |
Figure 4 PUG activates autophagy and restores autophagic flux. (A and B) Representative and quantitative analysis by Western blotting of autophagy-related markers in chondrocytes treated as above for 6 hours. (C–E) Representative and quantitative analysis by Western blotting of LC3 and p62 in chondrocytes treated as above for 6 hours. (F) Co-IF staining for LC3 (green) and Lamp2 (red) in chondrocytes treated with TBHP and PUG for 6 hours in each group; scale bar = 10 μm. (G) Quantitative analysis of the fusion of LC3 and Lamp2 in each group as above. (H and I) IF staining and quantitative intensity of LC3 in each mouse group as above; scale bar = 50 μm; scale bar (enlarged) = 10 μm. (J and K) IF staining and quantitative intensity of p62 in each mouse group as above; scale bar = 50 μm; scale bar (enlarged) = 10 μm; n = 5; GAPDH was the loading control; significance: *P<0.05, **P<0.01, ***P<0.001. |
3-MA Abolishes the Effects of PUG on Oxidative Stress, Apoptosis and ECM Degradation
The activation of autophagy has been reported to be beneficial for cell homeostasis and survival under stress stimulation.12 To confirm whether PUG-activated autophagy is responsible for the effect against oxidative stress, apoptosis and ECM degradation, a well-established autophagy inhibitor, 3-MA, was used to suppress autophagy in chondrocytes. Here, the results of Western blotting showed that when compared with the PUG treatment group, antioxidant-related enzyme expression was remarkably reduced after cotreatment with 3-MA, which indicated that autophagy promoted antioxidants in chondrocytes (Figure 5A–D). In addition, the inhibition of autophagy by 3-MA also enhanced the expression of apoptosis-related proteins, including Bax and cleaved caspase 3, but inhibited Bcl-2 expression, suggesting that autophagy repressed apoptosis in PUG-treated chondrocytes (Figure 5A and E–G). In addition, by staining with type II collagen and MMP13 in IF analysis, cotreatment of PUG and 3-MA significantly reduced the level of collagen type II (Figure 5H and I) and increased the intensity of MMP13 (Figure 5J and K). These data suggested that PUG inhibiting oxidative stress, apoptosis and ECM degradation is required for the activation of autophagy.
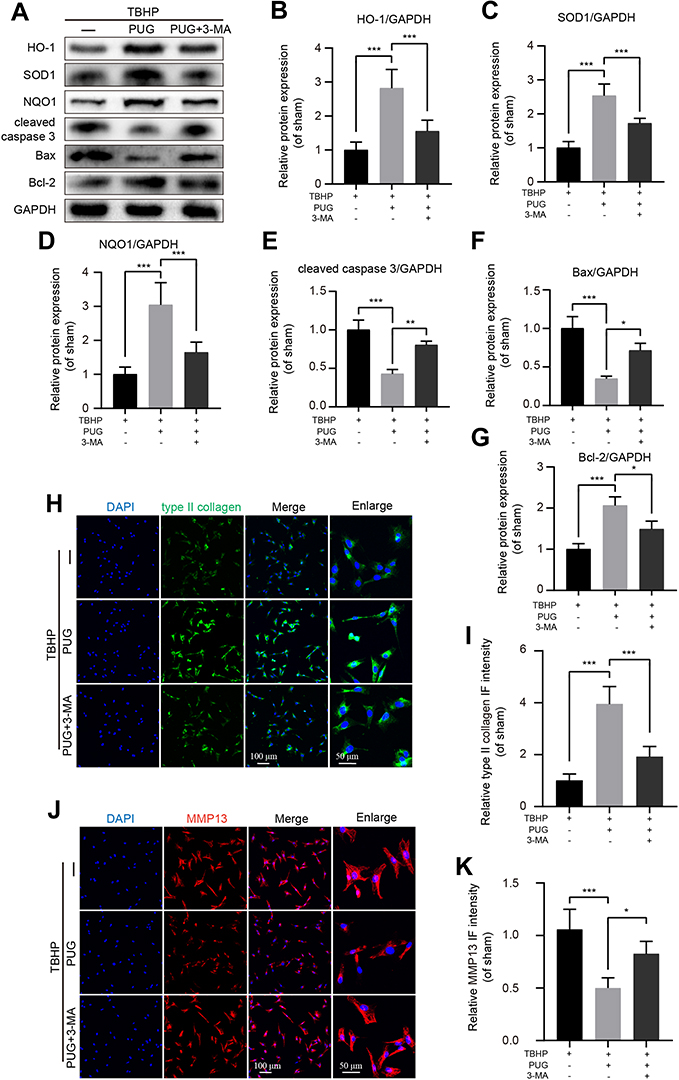 |
Figure 5 Treatment with 3-MA reverses the beneficial effects of PUG in chondrocytes. (A–G) Representative and quantitative analysis by Western blotting of antioxidant-related enzymes and apoptotic proteins in chondrocytes treated as above for 6 hours. (H and I) IF staining and quantitative intensity of type II collagen in each chondrocyte group as above; scale bar = 100 μm; scale bar (enlarged) = 50 μm. (J and K) IF staining and quantitative intensity of MMP13 in each chondrocyte group as above; scale bar = 100 μm; scale bar (enlarged) = 50 μm; n = 5; GAPDH was the loading control; significance: *P<0.05, **P<0.01, ***P<0.001. |
PUG Alleviates OA in a DMM Mouse Model
As PUG exhibited the above biological activities in vitro, we further evaluated the underlying effects of PUG on OA in vivo. After mice were fed PUG, IF, TUNEL, HE and Safranin-O staining were performed. First, cartilages labeled with cleaved caspase 3 revealed that the intensity of cleaved caspase3 was significantly decreased after treatment with PUG (Figure 6A and B). Similarly, TUNEL staining also suggested that PUG treatment reduces the higher proportion of cellular apoptosis induced by OA (Figure 6C and D). Furthermore, HE staining was performed to assess the morphology of the synovium, and results indicated that PUG ameliorates the thickening and hypercellularity of the synovium caused by OA (Figure 6E). PUG also reduced the synovitis score (Figure 6F), which suggested that PUG improves the synovitis of OA. As the above results revealed, PUG provided beneficial effects for preventing OA development in a DMM mouse model.
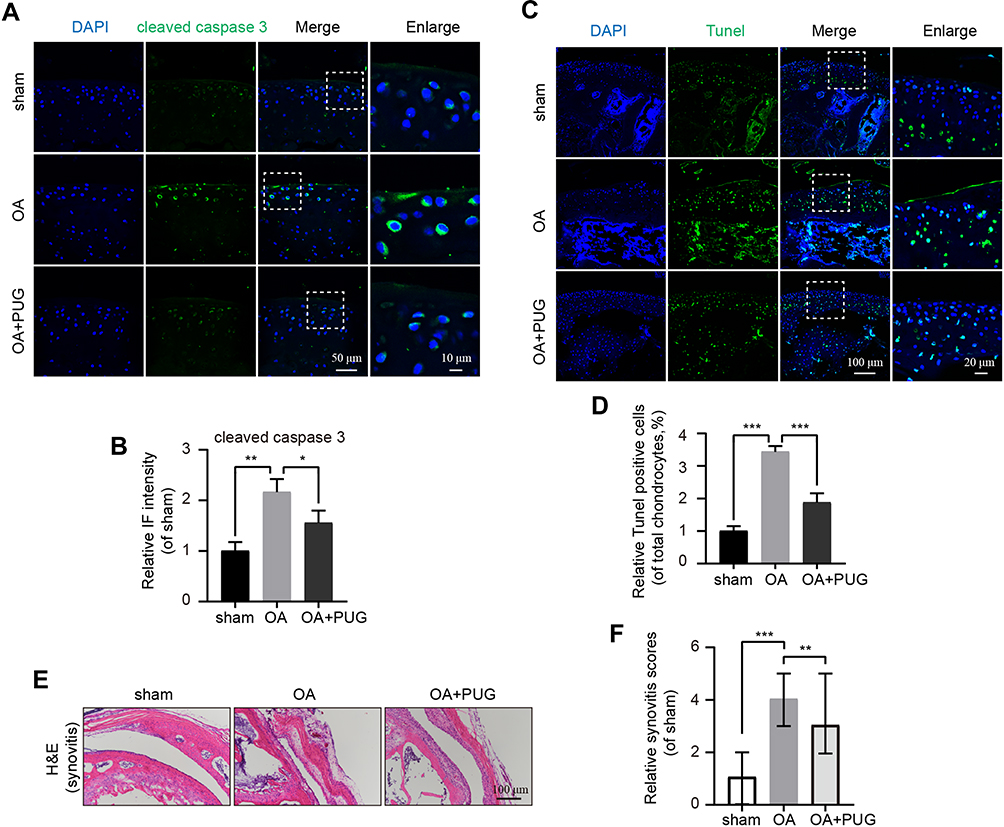 |
Figure 6 PUG inhibits apoptosis and ameliorates OA in a mouse DMM model. (A and B) IF staining and quantitative intensity of cleaved caspase 3 in each mouse group as above; scale bar = 50 μm; scale bar (enlarged) = 10 μm. (C) TUNEL staining in each group of mouse cartilage; scale bar: 100 μm; scale bar (enlarged) = 20 μm. (D) Assessment of relative TUNEL-positive cells in each group. (E) H&E staining of synovitis in each group at 8 weeks after surgery; scale bar: 100 μm. (F) The scores of synovitis in each group as above, data were displayed as the median (min - max). n = 8; significance: *P<0.05, **P<0.01, ***P<0.001. |
Discussion
OA is the most common age-related joint disease and is involved in a series of pathophysiological processes; pharmacological administration has been reported to exert beneficial effects for preventing OA development.6,13,34 Currently, we found that PUG treatment ameliorates OA progression in vivo and inhibits oxidative stress, apoptosis and ECM degradation in TBHP-treated chondrocytes. In addition, we also revealed that PUG-mediated protection is partly due to the activation of autophagy and restoration of autophagic flux (Figure 7).
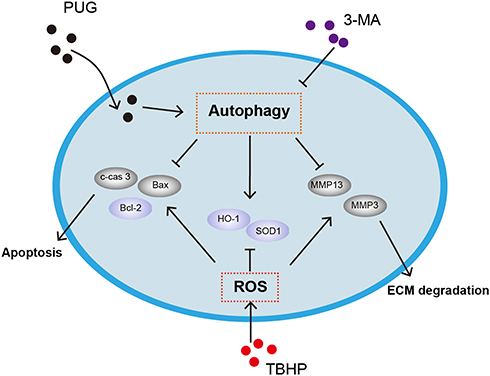 |
Figure 7 Schematic graph of the potential protective effects of PUG on osteoarthritis treatment. |
It is well-established that articular cartilage is an avascularity tissue with a low rate of cell turnover, chondrocytes trend to quiescence to adapt to the low oxygen supply, and mechanical and metabolic stressors cannot be effectively cleared in the development of OA. Particularly, oxidative stress is a result of ROS overproduction and the imbalance between oxidant and antioxidant enzymes. Various studies have showed that ROS are excessively accumulated and contribute to the destruction of chondrocyte homeostasis, initiating and aggravating the progression of OA.5,33 Correspondingly, targeting the clearance of ROS and inhibiting oxidative stress is a useful strategy for the treatment of OA.13 As an antioxidant, the effects of PUG on oxidative stress in OA have currently been detected. Here, we selected TBHP as a ROS donor to simulate chondrocytes in vitro, and the results showed that TBHP intervention successfully reduced the level of HO-1, SOD1 and NQO1, which indicated that chondrocytes were subjected to oxidative stress. Similar to the results of previous studies, PUG treatment successfully enhanced the expression of the above antioxidant enzymes, which suggests that PUG is a potent promoter against oxidative stress in chondrocytes.
Furthermore, it should be specified that excessive oxidative stress is detrimental for chondrocyte survival.2,33 Previous studies suggested that attenuating oxidative stress is beneficial for the inhibition of chondrocyte apoptosis, which is responsible for the improvement of OA.6,7 PUG has been reported to play a protective role in repressing apoptosis.21,25 Similar to that in previous studies, our IF, TUNEL staining and Western blotting results indicated that PUG effectively reduces DNA damage and apoptotic protein expression. Moreover, previous investigations demonstrated that oxidative stress serves as an adverse contributor to chondrocyte ECM metabolism, and increasing ECM degradation is a critical factor promoting cell senescence and cartilage degeneration.6 In line with the above evidence, ECM-related markers were detected, and the results led to the finding that PUG markedly suppresses ECM degradation after TBHP treatment.
Previous literature has elucidated that autophagy plays a critical role in regulating cellular physiological processes and maintaining intracellular homeostasis during OA progression.12,35 As previous studies have described, the status of autophagy is decreased and insufficient for clearing the toxins and waste released by damaged intracellular organelles.14 In our current work, the administration of TBHP slightly increased the LC3-II/I ratio in vitro, while decreased LC3 expression was observed in the cartilage of OA mice. We speculated that this discrepancy was owed to long-term durative ROS invasion, which aggravates irreparable autophagy dysfunction damage, whereas short-term ROS stimulation activates the compensatory mechanism. In addition, previous studies revealed that the autophagic flux is damaged due to the impaired lysosomal function induced by ROS.6,7 In accordance with the finding of a previous study, increased p62 levels were also observed in the current study, which implicated damaged autophagic flux. Intriguingly, PUG has been reported to regulate autophagy in several studies.22,23 Consistent with the data of previous studies, our results indicated that PUG could activate autophagy in chondrocytes. Notably, through IF staining analysis, PUG treatment successfully improved the formation of autophagosomes and lysosomes. In addition, we applied an autophagic inhibitor to treat chondrocytes, and the results indicated that PUG-activated autophagy is responsible for resistance to oxidative stress, apoptosis and ECM degradation.
Certainly, there are several limitations to the use of PUG as a therapy for OA, which still require further investigation. For example, the ideal dose and optimized treatment time investigated by further studies would provide a better understanding of its therapeutic value. In addition, considering its complex chemical structure, PUG may be associated with different effects on chondrocytes, and the treatment effect on OA by PUG, which was entirely attributed to its effect on autophagy, may be one-sided. Furthermore, we admit that using gene knockdown instead of molecular inhibitors would be more persuasive. Nevertheless, the protective effect of PUG was confirmed to be induced by autophagy, at least in part.
In conclusion, the current study indicated that the administration of PUG prevents cartilage damage and ameliorated osteoarthritis in a mouse DMM model. Moreover, PUG treatment increases antioxidant enzymes and reduces apoptotic proteins and ECM degradation, which are mediated by the activation of autophagy. Therefore, our findings provide insight into the regulatory molecular mechanisms accounting for the benefits of PUG in TBHP-treated chondrocytes, suggesting that PUG has therapeutic value for the treatment of OA.
Abbreviations
OA, osteoarthritis; PUG, punicalagin; ECM, extracellular matrix; IF, immunofluorescence; DMM, destabilization of the medial meniscus; TBHP, tert-butyl hydroperoxide; 3-MA, 3-methyladenine; ROS, reactive oxygen species; FBS, foetal bovine serum; DAPI, 4,6-Diami-dino-2-phenylindole; CCK-8, cell counting kit-8; PFA, paraformaldehyde; PVDF, polyvinylidene fluoride; OARSI, Osteoarthritis Research Society International; ANOVA, One-way analysis of variance.
Data Sharing Statement
The datasets used and analyzed during the current study are available from the corresponding authors on reasonable request.
Consent for Publication
We declare that our institutes are aware of the work and declare consent for publication of the manuscript. We received consent to publish the animal experimental data from the animal ethical committee.
Acknowledgments
This study was partially supported by a research grant from Medical and Health Foundation of Zhejiang Province (2020KY367, 2020PY091) and Taizhou science project (XM20190640).
Disclosure
The authors declare that they have no conflicts of interest.
References
1. Hunter D, Bierma-Zeinstra S. Osteoarthritis. Lancet. 2019;393(10182):1745–1759. doi:10.1016/S0140-6736(19)30417-9
2. Kloppenburg M, Berenbaum F. Osteoarthritis year in review 2019: epidemiology and therapy. Osteoarthr Cartil. 2020;28(3):242–248. doi:10.1016/j.joca.2020.01.002
3. Maly M, Marriott K, Chopp-Hurley J. Osteoarthritis year in review 2019: rehabilitation and outcomes. Osteoarthr Cartil. 2020;28(3):249–266. doi:10.1016/j.joca.2019.11.008
4. Alcaraz MJ, Ferrándiz ML. Relevance of Nrf2 and heme oxygenase-1 in articular diseases. Free Radic Biol Med. 2019.
5. Drevet S, Gavazzi G, Grange L, Dupuy C, Lardy B. Reactive oxygen species and NADPH oxidase 4 involvement in osteoarthritis. Exp Gerontol. 2018;111:107–117. doi:10.1016/j.exger.2018.07.007
6. Li Y, Wu Y, Jiang K, et al. Mangiferin prevents TBHP-induced apoptosis and ECM degradation in mouse osteoarthritic chondrocytes via restoring autophagy and ameliorates murine osteoarthritis. Oxid Med Cell Longev. 2019;2019:17. doi:10.1155/2019/8783197
7. Zheng G, Zhan Y, Li X, et al. TFEB, a potential therapeutic target for osteoarthritis via autophagy regulation. Cell Death Dis. 2018;9(9):858. doi:10.1038/s41419-018-0909-y
8. Varela-Eirin M, Loureiro J, Fonseca E, et al. Cartilage regeneration and ageing: targeting cellular plasticity in osteoarthritis. Ageing Res Rev. 2018;42:56–71. doi:10.1016/j.arr.2017.12.006
9. D’Adamo S, Cetrullo S, Guidotti S, et al. Spermidine rescues the deregulated autophagic response to oxidative stress of osteoarthritic chondrocytes. Free Radic Biol Med. 2020;153:159–172. doi:10.1016/j.freeradbiomed.2020.03.029
10. Feng K, Ge Y, Chen Z, et al. αCurcumin Inhibits the PERK-eIF2-CHOP pathway through promoting SIRT1 expression in oxidative stress-induced rat chondrocytes and ameliorates osteoarthritis progression in a rat model. Oxid Med Cell Longev. 2019;2019:8574386. doi:10.1155/2019/8574386
11. Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal. 2014;20(3):460–473. doi:10.1089/ars.2013.5371
12. Duarte JH. Osteoarthritis: autophagy prevents age-related OA. Nat Rev Rheumatol. 2015;11(12):683. doi:10.1038/nrrheum.2015.145
13. Tang Q, Zheng G, Feng Z, et al. Trehalose ameliorates oxidative stress-mediated mitochondrial dysfunction and ER stress via selective autophagy stimulation and autophagic flux restoration in osteoarthritis development. Cell Death Dis. 2017;8(10):e3081. doi:10.1038/cddis.2017.453
14. Goutas A, Syrrou C, Papathanasiou I, Tsezou A, Trachana V. The autophagic response to oxidative stress in osteoarthritic chondrocytes is deregulated. Free Radic Biol Med. 2018;126:122–132. doi:10.1016/j.freeradbiomed.2018.08.003
15. Sasaki H, Takayama K, Matsushita T, et al. Autophagy modulates osteoarthritis-related gene expression in human chondrocytes. Arthritis Rheum. 2012;64(6):1920–1928. doi:10.1002/art.34323
16. Bao J, Chen Z, Xu L, Wu L, Xiong Y. Rapamycin protects chondrocytes against IL-18-induced apoptosis and ameliorates rat osteoarthritis. Aging. 2020;12(6):5152–5167. doi:10.18632/aging.102937
17. Meng Z, Shen B, Gu Y, et al. Diazoxide ameliorates severity of experimental osteoarthritis by activating autophagy via modulation of the osteoarthritis-related biomarkers. J Cell Biochem. 2018;119(11):8922–8936. doi:10.1002/jcb.27145
18. Panth N, Manandhar B, Paudel K. Anticancer activity of Punica granatum (pomegranate): a review. Phytother Res. 2017;31(4):568–578. doi:10.1002/ptr.5784
19. Yuan T, Ma H, Liu W, et al. Pomegranate’s neuroprotective effects against Alzheimer’s disease are mediated by urolithins, its ellagitannin-gut microbial derived metabolites. ACS Chem Neurosci. 2016;7(1):26–33. doi:10.1021/acschemneuro.5b00260
20. Yan C, Sun W, Wang X, et al. Punicalagin attenuates palmitate-induced lipotoxicity in HepG2 cells by activating the Keap1-Nrf2 antioxidant defense system. Mol Nutr Food Res. 2016;60(5):1139–1149. doi:10.1002/mnfr.201500490
21. Foroutanfar A, Mehri S, Kamyar M, Tandisehpanah Z, Hosseinzadeh H. Protective effect of punicalagin, the main polyphenol compound of pomegranate, against acrylamide-induced neurotoxicity and hepatotoxicity in rats. Phytother Res. 2020. doi:10.1002/ptr.6774
22. Zhang Y, Cao Y, Chen J, Qin H, Yang L, New Possible A. Mechanism by which punicalagin protects against liver injury induced by type 2 diabetes mellitus: upregulation of autophagy via the Akt/FoxO3a signaling pathway. J Agric Food Chem. 2019;67(50):13948–13959. doi:10.1021/acs.jafc.9b05910
23. Cao Y, Chen J, Ren G, Zhang Y, Tan X, Yang L. Punicalagin prevents inflammation in LPS-induced RAW264.7 macrophages by inhibiting FoxO3a/autophagy signaling pathway. Nutrients. 2019;11(11):2794. doi:10.3390/nu11112794
24. Yaidikar L, Byna B, Thakur S. Neuroprotective effect of punicalagin against cerebral ischemia reperfusion-induced oxidative brain injury in rats. J Stroke Cerebrovasc Dis. 2014;23(10):2869–2878. doi:10.1016/j.jstrokecerebrovasdis.2014.07.020
25. Yu L, Dong X, Xue X, et al. Protection of the myocardium against ischemia/reperfusion injury by punicalagin through an SIRT1-NRF-2-HO-1-dependent mechanism. Chem Biol Interact. 2019;306:152–162. doi:10.1016/j.cbi.2019.05.003
26. Ding M, Wang Y, Sun D, et al. Punicalagin pretreatment attenuates myocardial ischemia-reperfusion injury via activation of AMPK. Am J Chin Med. 2017;45(1):53–66. doi:10.1142/S0192415X17500057
27. Liu X, Cao K, Lv W, et al. Punicalagin attenuates endothelial dysfunction by activating FoxO1, a pivotal regulating switch of mitochondrial biogenesis. Free Radic Biol Med. 2019;135:251–260. doi:10.1016/j.freeradbiomed.2019.03.011
28. El-Missiry M, ElKomy M, Othman A, AbouEl-Ezz A. Punicalagin ameliorates the elevation of plasma homocysteine, amyloid-β, TNF-α and apoptosis by advocating antioxidants and modulating apoptotic mediator proteins in brain. Biomed Pharmacother. 2018;102:472–480. doi:10.1016/j.biopha.2018.03.096
29. Lee C, Chen L, Wang C. Anti-inflammatory effects of Punica granatum Linne in LPS-induced primary human chondrocytes. Planta Med. 2010;76(12):1376. doi:10.1055/s-0030-1265835
30. Lee CJ, Chen LG, Liang WL, Hsieh MS, Wang CC. Inhibitory effects of punicalagin from Punica granatum against type II collagenase-induced osteoarthritis. J Funct Food. 2018;41:216–222. doi:10.1016/j.jff.2017.12.026
31. Lewis J, Hembree W, Furman B, et al. Acute joint pathology and synovial inflammation is associated with increased intra-articular fracture severity in the mouse knee. Osteoarthr Cartil. 2011;19(7):864–873. doi:10.1016/j.joca.2011.04.011
32. Kraus V, Huebner J, DeGroot J, Bendele A. The OARSI histopathology initiative - recommendations for histological assessments of osteoarthritis in the guinea pig. Osteoarthr Cartil. 2010;18:S35–52. doi:10.1016/j.joca.2010.04.015
33. Bolduc J, Collins J, Loeser R. Reactive oxygen species, aging and articular cartilage homeostasis. Free Radic Biol Med. 2019;132:73–82. doi:10.1016/j.freeradbiomed.2018.08.038
34. Zhu Z, Li J, Ruan G, Wang G, Huang C, Ding C. Investigational drugs for the treatment of osteoarthritis, an update on recent developments. Expert Opin Investig Drugs. 2018;27(11):881–900. doi:10.1080/13543784.2018.1539075
35. Lotz M, Caramés B. Autophagy and cartilage homeostasis mechanisms in joint health, aging and OA. Nat Rev Rheumatol. 2011;7(10):579–587. doi:10.1038/nrrheum.2011.109
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.