Back to Journals » Journal of Inflammation Research » Volume 16
Puerarin Attenuates High-Glucose and High-Lipid-Induced Inflammatory Injury in H9c2 Cardiomyocytes via CAV3 Protein Upregulation
Authors Tian Y, Zhou C, Bu X, Lv Q, Huang Q
Received 15 February 2023
Accepted for publication 11 June 2023
Published 29 June 2023 Volume 2023:16 Pages 2707—2718
DOI https://doi.org/10.2147/JIR.S408681
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Ning Quan
YiFu Tian,1 CaiXia Zhou,1 XiaoYang Bu,1 Qian Lv,1 Qin Huang1,2
1Department of Physiology of Basic Medical Sciences, Guangxi Medical University, Nanning, People’s Republic of China; 2Department of Key Laboratory of Longevity and Aging-Related Diseases of Chinese Ministry of Education & School of Basic Medical Sciences, Guangxi Medical University, Nanning, People’s Republic of China
Correspondence: Qin Huang, Department of Physiology, Guangxi Medical University, Building 102, 22 Shuangyong Road, Nanning, Guangxi, 530021, People’s Republic of China, Email [email protected]
Background: Inflammation plays a crucial role in the development of diabetic cardiomyopathy (DCM), including inflammation caused by high-glucose and high-lipid (HGHL). Targeting inflammation may provide a useful strategy for preventing and treating DCM. Puerarin has been shown to reduce the inflammation, apoptosis and hypertrophy of cardiomyocytes induced by HGHL, in which this study aims to investigate the underlying mechanisms.
Methods: H9c2 cardiomyocytes cultured with HGHL were used to establish a cell model of DCM. Puerarin was then placed to these cells for 24 hours. The effects of HGHL and puerarin on cell viability and apoptosis were investigated by the Cell Proliferation, Toxicity Assay Kit (CCK-8) and flow cytometry. Morphological changes of cardiomyocytes was observed by HE staining. CAV3 proteins in H9c2 cardiomyocytes were altered by transient transfection of CAV3 siRNA. IL-6 was detected by ELISA. The Western blot was performed to determine the CAV3, Bcl-2, Bax, pro-Caspase-3, cleaved-Caspase-3, NF-κB (p65) and p38MAPK proteins.
Results: Puerarin treatment reversed the cells viability, hypertrophy in morphology, inflammation (showed by p-p38 and p-p65 and IL-6) and apoptosis-related damage (showed by cleaved-Caspase-3/pro-Caspase-3/Bax, Bcl-2 and flow cytometry) of the H9c2 cardiomyocyte caused by HGHL. Puerarin treatment also restored the decrease of CAV3 proteins of the H9c2 cardiomyocyte caused by HGHL. When silenced the expression of CAV3 proteins with SiRNA, puerarin failed to decreased p-p38 and p-p65 and IL-6, and could not reversed cell viability and morphological damage. In contrast to the simple CAV3 silenced group, the CAV3 silenced with NF-κB pathway or p38MAPK pathway inhibitors, significantly downregulated the p-p38, p-p65 and IL-6.
Conclusion: Puerarin upregulated CAV3 protein expression in H9c2 cardiomyocytes and inhibited the NF-κB and p38MAPK pathways, thereby reducing HGHL-induced inflammation and may related to the apoptosis and hypertrophy of cardiomyocytes.
Keywords: puerarin, cardiomyocyte, inflammation, caveolin-3, siRNA, NF-κB, p38MAPK
Introduction
Patients with diabetes are at an increased risk of cardiovascular complications, including heart failure and death, making it the leading cause of diabetes-related mortality.1 The present research on diabetic cardiomyopathy is still advancing, and some new hypoglycemic drugs like GLP-1 receptor agonists and SGLT-2 inhibitors have shown potential for prevention and treatment of diabetic cardiomyopathy. T cell immunotherapy and anti-inflammatory drugs have also been applied in diabetic cardiomyopathy treatment, and gene therapy was also an important method for diabetic cardiomyopathy treatment. Although these research advances are promising, there is still no effective strategy to prevent or halt the development of DCM. Glycemic control alleviates heart failure but rarely reverses it and has numerous side effects.2 Many natural compounds and their anti-inflammatory effects in DCM are receiving significant attention in preclinical and clinical research. Flavonoids with free radical scavenging, antioxidant and anti-inflammatory properties have been identified as candidates for mediating protection.3 Isoflavones demonstrated cardioprotective effects on mitochondrial Ca2+ intake4 and permeability transition.5
Stimulation of inflammatory cytokine secretion profoundly affects the myocardium, causing at least four changes in cardiac cells that contribute to the reprogramming or remodeling of phenotype, including progressive apoptosis, myocyte hypertrophy, contractility defects, and inflammatory signal transduction.6 These events occur as a result of proinflammatory cascades occurring in different types of cardiac cells. Subsequently, mediators produced by this inflammatory cascade regulate specific intracellular signaling mechanisms in cardiomyocytes, leading to hypertrophy and apoptosis.7 There is evidence to suggest that the induction of myocardial cell inflammation by saturated fatty acids contributes to the occurrence and development of cardiomyopathy.8 Saturated fatty acids were reported to be responsible for systemic and local inflammatory activation, as well as initiation of immune responses in circulating immune cells and cardiomyocytes.9 Furthermore, inflammatory factors triggered by saturated fatty acids worsen cardiac damage through various pathways.10 By activating the intracellular NF-κB pathway, hyperglycemia leads to the production of proinflammatory cytokines contributing to myocardial inflammation, hypertrophy and apoptosis.11 Sustained activation of TNF signaling led to apoptosis and cardiomyocyte remodeling by the activation of intrinsic and extrinsic pathways of cell death, and resulted in elevated cytosolic levels of activated cytochrome c, caspase 3, and 8.12
The enhanced inflammatory response negatively affects cardiac function and triggers cardiomyocyte damage.13 Inflammation and oxidative stress contribute significantly to the apoptosis of cardiomyocytes by elevating inflammatory cytokine.14 The activation of NF-κB and MAPK pathways causes the expression of proinflammatory cytokines, ultimately leading to heart failure and DCM.15–17 Palmitic acid exposure for 24 h resulted in cellular apoptosis, accompanied by an upregulation of Bax and Cleaved-caspase 3, and a downregulation of Bcl-2.8 Targeted inhibition of NF-κB and c-Jun N-terminal kinase/p38MAPK can combat inflammation, oxidative stress, and apoptosis, thus effectively treating DCM.17
Gegen (Radix Puerariae Lobatae) is a widely used herb in Traditional Chinese Medicine (TCM) for treating diabetes and its associated complications.18 Puerarin (daidzein-8-C-glucoside), which is a chemical taxonomic marker of the Pueraria genus, possesses abundant isoflavones.19 Extracted active components of puerarin inhibited the production of IL-6, IL-1β, PGE2, and NO, with anti-inflammatory effects, and regulated transcription levels by suppressing the MAPK signaling pathway.20 Initial observations suggest that puerarin may protect against diabetes and its associated cardiovascular complications, as well as inhibit cellular inflammation, oxidative stress, and apoptosis.1 However, the efficacy of puerarin in DCM and its myocardial protective mechanism remain unclear.
Research reports indicated that CAV3 might have a cardioprotective effect in DCM,21,22 and an inverse relationship between CAV3 gene expression and the expression of NF-κB in BSM cells.23 CAV3 knockout mice exhibited progressive cardiomyopathy, with hyperactivation of the MAPK cascade of p42/44.24 Dimisartan reduced MAPK activity p38 in the absence of angiotensin II in cardiomyocytes, leading to a decrease in expression of the CAV3 related T-type Ca2+channels.25 Nitric oxide ameliorated diabetic cardiomyopathy by improving the Caveolin-3/eNOS complex.22 CAV3 deficiency increased susceptibility to overall palmitic acid-induced insulin resistance, causing cardiomyopathy.26 Propofol offered cardioprotection by suppressing protease degradation of CAV3 in ischemia-reperfusion rat hearts.27 Therefore, these data suggest that CAV3 protects the heart and its downregulation indicates myocardial injury.28
In this study, we use H9c2 cells treated with HGHL to simulate myocardial inflammatory damage to simulate myocardial injury of DCM. Our research endeavored to evaluate the effects and mechanism of Puerarin at both the cellular and molecular levels, with a particular emphasis on investigating if it relates to the protective mechanism of CAV3.
Materials and Methods
Cell Culture and Treatment
Wuhan Punosai Life Sciences Company provided H9c2 cells derived from rat embryonic adult cardiomyocyte-derived cells. The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 µg/mL streptomycin, and incubated in a humidified incubator at 37 °C in 95% air/5% CO2. To test the cytotoxic effects of HGHL, we treated H9c2 cardiomyocytes with a 33mM glucose solution29 and a 100µM palmitate solution30 for 24 hours, based on prior literature protocols. We used various concentrations of puerarin (10−6, 10−5, 10−4, 10−3 M) to assess the cardioprotective effect of puerarin on HGHL-induced injury.1 Treatment with puerarin resulted in maximal cell viability and proliferation at 10−4 M, which we selected as the subsequent experimental concentration. Puerarin was dissolved in dimethyl sulfoxide as a stock solution and diluted with dimethyl sulfoxide at the time of use. The control group did not contain dimethyl sulfoxide, and 0.1% of dimethyl sulfoxide was present in all other groups. To investigate the relationship between CAV3 and the p38MAPK-NF-κB pathway, we employed pathway inhibitors of either p38MAPK (SB203580 10 µM) or NF-κB (BAY11-7082 10 µM).
Hematoxylin and Eosin (HE) Staining
H9c2 cardiomyocyte slides were fixed in 4% paraformaldehyde buffer for 30 min, rinsed three times with PBS, and then stained with HE (Sigma, MHS1, China). The cells were stained with hematoxylin for 8 minutes, followed by running water for 3 minutes, eosin for 8 minutes, and running water for another 3 minutes. At least ten hematoxylin-eosin stained sections were randomly selected from within each group for structural observation. Next, the sections were viewed at 200x using a microscope (Olympus, Japan).
Cell Counting Kit-8 (CCK-8) Assay
H9c2 cardiomyocytes (5×103 cells/well) were seeded in 96-well plates, and each well was supplemented with 100 µL medium. After the cells adhered to the wall, they were treated according to the experimental requirements. After 24 hours, fresh medium was replaced, and 10 µL Cell Counting Kit-8 (CCK8) reagent (Meilunbio, Dalian, China) was added to each well. At the same time, blank control wells (100 µL cell medium, 10 µL CCK8 solution, no cells) were set up and incubated at 37 °C for 2 hours. The absorbance value of each well was measured by a microplate reader (El × 800, BioTek, USA) at OD450 nm. Cell viability % was calculated as (OD of cells in experimental wells -OD of blank wells)/(OD of control wells -OD of blank wells) × 100%. Results were repeated six times for each experiment.
Flow Cytometry
Cells were divided into control group, HGHL group and HGHL+Puerarin group. One million cells from each group were taken into PBS, mixed, centrifuged at 1200 rpm, and the supernatant was removed. 100 µL of 1×Binding Buffer was added to each tube, resuspended, and the solution was gently shaken by adding 5 µL of Annexin V-PE and 5 µL of 7-AAD (Bioswamp, Wuhan, China) and incubated in the dark for 15 min at room temperature (25 °C). 200 µL of 1× Binding Buffer was added to each, and the machine was loaded within 1 hour. Samples were tested by flow cytometry (Beckman Coulter, Fullerton, CA) in three replicates per set. FlowJo software was used to analyze the results.
Transfection
The CAV3 siRNA product was synthesized by Suzhou Jima Gene Co: CAV3 siRNA: sense 5’-GCGAUCACAUGUACUGUAAtt-3’ and antisense 5’-UUACAGUACAUGUGAUCGCtt-3’. Cell transfection was performed following the Lipofectamine 3000 instructions from Invitrogen. Cells were seeded into 6-well plates at a density of 0.25×106, and the groups requiring transfection were cultured in cell medium without penicylyl streptomycin for 24 hours before transfection. After 6 hours, cells were treated with high glucose and high lipid, puerarin, and pathway inhibitors according to the pre-grouping. After co-culture for 48 hours, transfected cells were analyzed and processed. Results were repeated six times for each experiment.
ELISA
Cells were seeded into 6-well plates at a density of 2×105 and cultured, and the supernatants of each cell group were collected after different experimental treatments according to the pre-grouping. The same volume of DMEM was used as a blank control. This experiment was performed using an IL-6 (EK306D) ELISA kit (MULTI SCIENCES, China), and dual-wavelength detection with a microplate reader (El×800, BioTek, USA) according to the manufacturing instructions to determine the OD at 450 nm maximum absorption wavelength and 570 nm or 630 nm reference wavelength. Standard curves were generated by regression fitting using computer software with standard concentration as the abscissa and OD value as the ordinate. Results of each set of experiments were repeated six times.
Western Blot
We performed assays for Western blot of CAV3, p65, p-p65, p38, p-38, cleaved-caspase-3, pro-caspases-3, Bax and Bcl-2 protein levels. Extraction of total protein was performed by RIPA lysis (Solarbio, China) and protein concentration was measured by BCA assay (Bioswamp, China). Each lane was supplemented with protein samples (30 μg); proteins were separated by electrophoresis using sodium dodecyl sulfate-polyacrylamide gels (SDS-PAGE, China) at 80 V, 120 g/L for 90 min. Membrane transfer was performed at 220 mA for 60 minutes using a wet transfer system, followed by skimmed milk powder at 25°C for 1 h. The concentrations of antibodies were as follows: CAV3 (1:1000), p-p65 (1:1000), p65 (1:1000), p-p38 (1:1000), p38 (1:1000), Bax (1:500), Bcl-2 (1:500), pro-Caspases-3 (1:500), cleaved-Caspase-3 (1:500), GAPDH (1:1000) and β-tublin (1:1000), respectively. The primary antibodies (Cell Signalling Technology, Inc. USA) were overnight incubated at 4°C before the secondary antibody (1:1000) at 25°C for 2 hours. Finally, the films were measured using the Odyssey LI-COR infrared imaging system (LICOR Biosciences Inc., USA). The combined IR signal intensity at 800 nm was calculated for each band using Li-Cor Odyssey Infrared Imaging System analysis software.
Statistical Analysis
Data were presented as mean ± standard deviation (SD). The SPSS 23.0 software (SPSS, Chicago, USA) was used for statistical analysis. Comparisons between the two samples were performed using the LSD t-test, and that between groups was analyzed using one-way analysis of variance (ANOVA). All the reported P values were two-tailed, and P<0.05 was considered to indicate statistical significance.
Results
Expression Downregulation of CAV3 Protein and Cardiomyocyte Morphology Damage by HGHL
CAV3 protein expression was downregulated in a time-dependent manner in cardiomyocytes treated with HGHL, as confirmed by Western blot analysis (Figure 1A and B). A remarkable downregulation was observed at 24 hours after treatment. Microscopic analysis of cardiac myocyte morphology revealed that HGHL treatment significantly increased damage to these cells, as indicated by the presence of round or oval-shaped cardiomyocytes with damaged nuclear membranes, and enlarged cells with vacuolation after 36–48 hours of treatment. Conversely, no evidence of cardiomyocyte morphological changes was noted at 0–12 hours of treatment, during which time cells were normal in shape and dense. Based on these findings and further analysis (Figure 1C), a duration of 24 hours of HGHL treatment was selected for subsequent experiments.
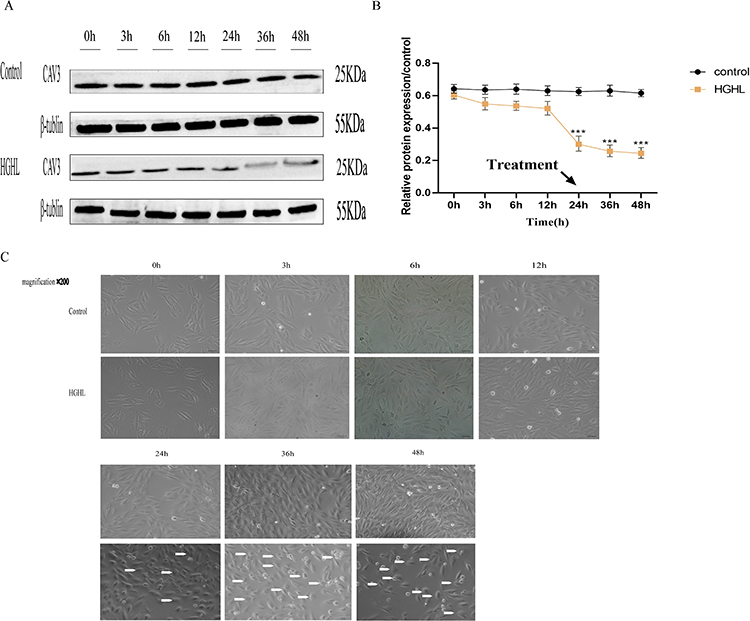 |
Figure 1 HGHL-Induced CAV3 Protein Expression Downregulation and Morphological Injury in H9c2 Cells. HGHL treatment downregulated CAV3 protein expression in a time-dependent manner, with a significant decrease at 24 hours observed using Western blot analysis (A and B). The data were presented as mean ± SD, with n = 6 per group. Microscopic observation (200× amplification) revealed HGHL treatment resulted in increased morphological damage to H9c2 cardiomyocytes (C). The scale bars in the panel were 50 μm. The statistical significance of the observed effects was indicated by ***P<0.001. |
Myocardial Cell was Injuried by HGHL in Inflammatory Conditions
Cell Counting Kit-8 (CCK-8) and flow cytometry results confirmed that HGHL treatment significantly inhibited H9c2 cell activity (Figure 2A) and enhanced apoptosis (Figure 2B). HGHL treatment also caused massive morphological damage to cardiomyocytes, as evidenced by H&E staining (Figure 2C). Western blot analysis revealed significantly increased expression levels of cleaved-caspase-3/pro-Caspase-3 and Bax, whereas expression of Bcl-2 was significantly downregulated (Figure 2D-G). Moreover, HGHL treatment resulted in a significant upregulation of IL-6 expression in H9c2 cells (Figure 2H).
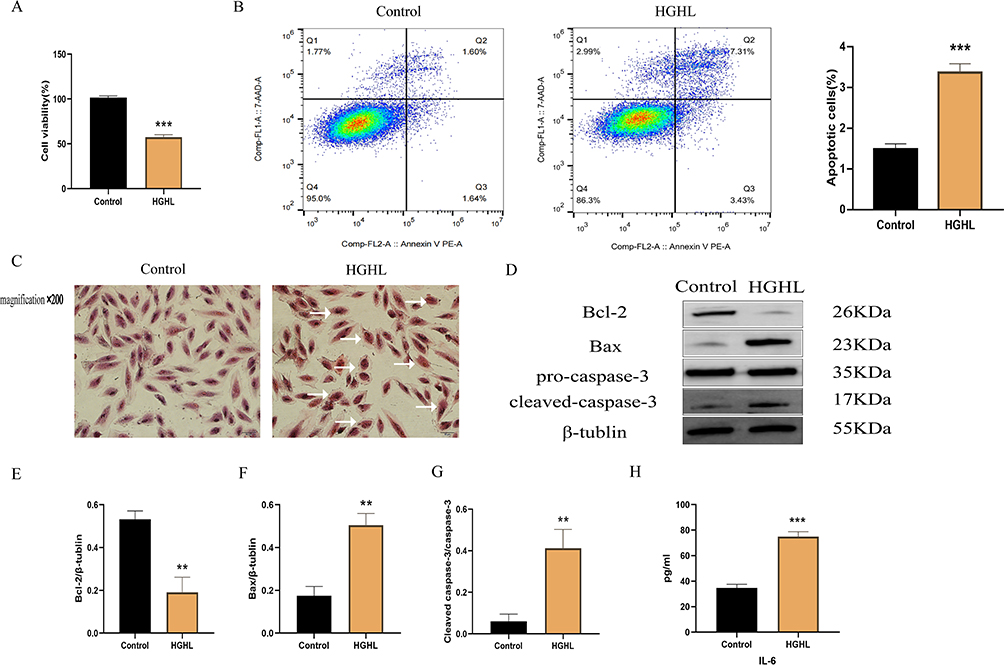 |
Figure 2 HGHL Provoked Inflammatory Injury in H9c2 Cells. Results showed that treatment with HGHL significantly decreased cell viability and increased cell apoptosis (A and B). Additionally, HGHL treatment resulted in significant morphological changes to cardiomyocytes when compared to the normal group, as indicated by hematoxylin and eosin (H&E) staining (C). Western blot analysis indicated significant upregulation of cleaved-Caspase-3/pro-Caspase-3, and Bax expression, while Bcl-2 showed significant downregulation following HGHL treatment (D-G). Moreover, HGHL treatment resulted in a significant upregulation of IL-6 expression in H9c2 cells (H). The IL-6 release and cell viability assays were performed with n = 6 per group and Western blot analysis with n = 3 per group. Flow cytometry assays were conducted with n = 3 per group. All HE staining images were obtained using microscopy with 200× magnification, and the scale bars in the panel were 50 µm. The statistical significance of the observed effects was indicated by **p<0.01 and ***p<0.001. |
Puerarin Attenuated HGHL-Induced Inflammatory Injury in Myocardial Cells
Puerarin is a flavonoid glycoside, derived from wild legume puerarin, and its chemical structure is shown in Figure 3A. Cell viability remained unchanged upon incubating cardiomyocytes with different concentrations of puerarin (Figure 3B). However, a significant dose-dependent increase in cell viability was observed in HGHL-treated cardiomyocytes upon the addition of puerarin at different concentrations (Figure 3C). The optimum concentration of puerarin was determined to be 10−4 M, based on its maximum protective effect on cell viability. H&E staining revealed reduced morphological damage in cardiomyocytes after treatment with puerarin compared to the HGHL-treated group (Figure 3D). Furthermore, flow cytometry analysis indicated that puerarin treatment significantly inhibited HGHL-induced apoptosis (Figure 3E). Western blot analysis revealed significantly upregulated expression levels of Bcl-2, while expression of cleaved-Caspase-3/pro-Caspase-3, and Bax was downregulated following puerarin treatment of HGHL-induced cardiomyocytes (Figure 3F-I). Moreover, puerarin treatment resulted in significant downregulation of IL-6 expression levels (Figure 3J).
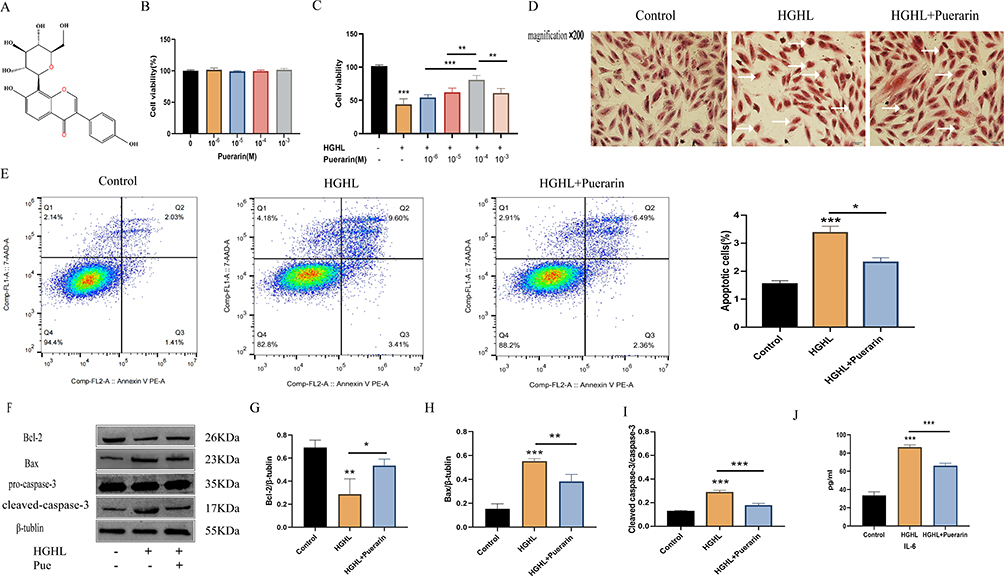 |
Figure 3 Puerarin alleviated HGHL-induced inflammatory injury in H9c2 cells. (A) Chemical Structure of Puerarin. (B) No obvious changes in cell viability with puerarin (0,10−6,10−5,10−4,10−3M) treatment. (C) When cells were treated with similar puerarin concentrations in HGHL-induced cells, cell viability was significantly improved with the increased concentration of puerarin and results show a dose-dependent way. (D) Puerarin groups reduced myocardial cell morphology injury. (E) Puerarin significantly suppressed the HGHL-induced apoptosis. (F-I) Puerarin noticeably inhibited HGHL-induced levels of cleaved-Caspase-3/pro-Caspase-3 and Bax and enhanced the level of Bcl-2. (J) Puerarin markedly suppressed the production of IL-6. This experiment was conducted with six replicates in the IL-6 release and cell viability assays, four replicates in the Western blot, and three replicates in the Flow cytometry assay. The HE staining images were obtained via microscope with 200× amplification and the scale bars in the panel were 50 µm. The statistical significance of the observed effects was indicated by *p <0 0.05, **p<0.01 and ***p<0.001. |
Puerarin Restrained the NF-κB and p38MAPK Pathways in Myocardial Cells
Phosphorylation levels of p/t-p38MAPK (Figure 4A and B) and p/t-p65 (Figure 4C and D) were significantly increased after HGHL treatment. Nevertheless, these upregulated expressions were significantly downregulated following puerarin treatment (Figure 4A-D). These observations suggest that puerarin suppressed NF-κB and p38MAPK signalling pathways.
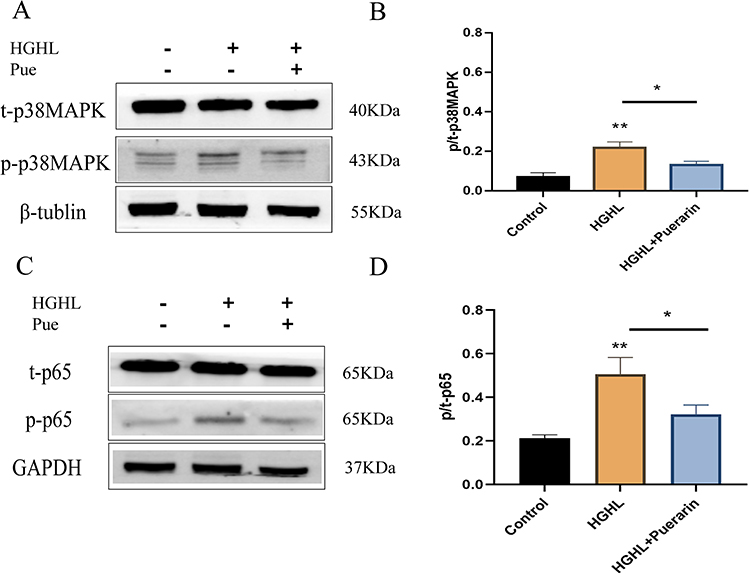 |
Figure 4 Puerarin inhibited the NF-κB and p38MAPK pathways in H9c2 cells. (A and B) Puerarin inhibited the p38MAPK pathway. (C and D) Puerarin inhibited the NF-κB pathway. n = 4 per group for the Western blot. *p < 0.05; **p <0 0.01. |
Puerarin-Mediated the Upregulation of CAV3 in Myocardial Cells
Western blot results showed significant downregulation of protein CAV3 expression levels after treatment with HGHL. However, when puerarin was added to HGHL-induced cells, CAV3 protein expression was significantly upregulated (Figure 5A and B). Therefore, this implies that puerarin may positively regulate CAV3 protein expression.
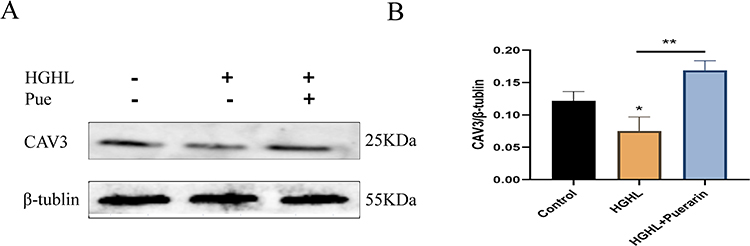 |
Figure 5 Puerarin positively regulates the expression level of CAV3. (A and B) The protein expression level of CAV3 was significantly downregulated after HGHL treatment. n = 4 per group for the Western blot. *p < 0.05; **p < 0.01. |
Puerarin Inhibited NF-κB and p38MAPK Pathways via CAV3 Upregulation in Myocardial Cells
After transfecting with FAM NC, green fluorescence was observed under a fluorescent inverted microscope, indicating successful transfection of CAV3 siRNA in the H9c2 cardiomyocytes (Figure 6A). Western blot confirmed that transfection with CAV3 siRNA markedly downregulated the CAV3 protein expression. This indicates that we successfully silenced the CAV3 protein expression of H9c2 cardiomyocytes (Figure 6B). The classic bands of Western blot for CAV3 siRNA before and after transfection were observed (Figure 6C). On this basis, SB203580 significantly decreased the level of p/t-p38MAPK phosphorylation induced via CAV3 siRNA transfection (Figure 6D); besides, BAY11-7082 significantly decreased the level of p/t-p65 phosphorylation induced by CAV3 siRNA transfection (Figure 6E). Moreover, CAV3 siRNA transfection significantly upregulated the IL-6 expression level after puerarin treatment (Figure 6F). After CAV3 siRNA transfection, H&E staining showed apparent morphological changes in hypertrophic cardiomyocytes and an increased number of apoptotic cells (Figure 6G). Therefore, puerarin may inhibit NF-κB and p38MAPK signaling pathways by upregulating the CAV3 protein.
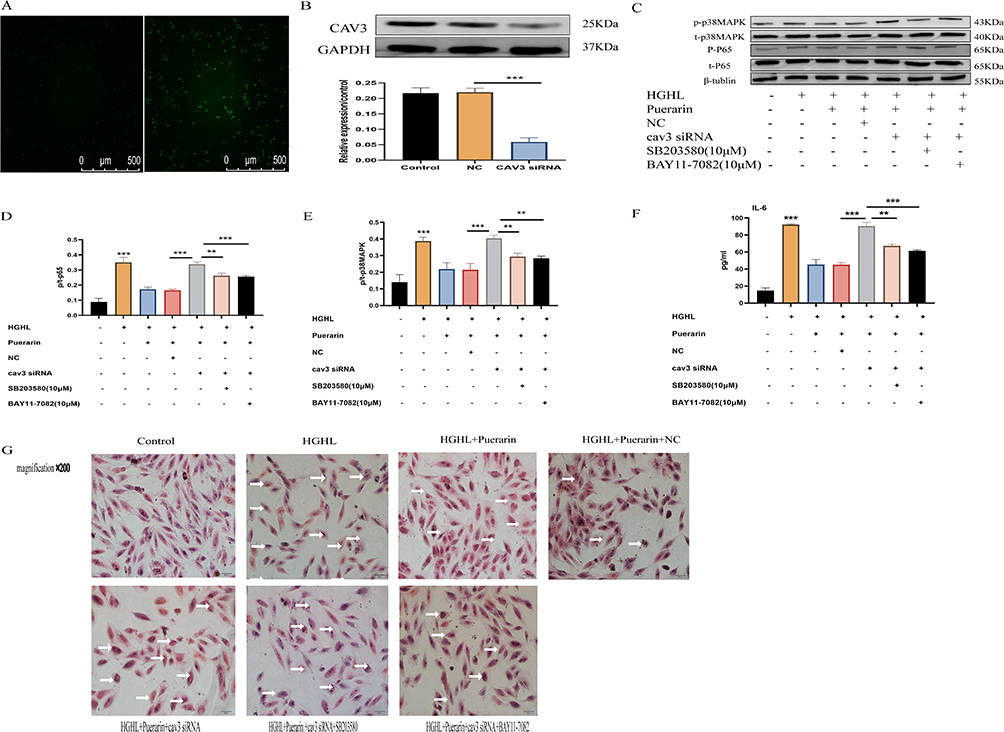 |
Figure 6 Puerarin inhibited HGHL-induced inflammatory damage via up-regulated CAV3 expression. (A and B) CAV3 expression was significantly downregulated upon transfection with CAV3siRNA. (C-E) SB203580 significantly decreased phosphorylation levels of p/t-p38MAPK induced by CAV3 siRNA transfection; BAY11-7082 significantly reduced phosphorylation levels of p/t-p38MAPK induced by CAV3 siRNA transfection. (F) CAV3 siRNA significantly improved puerarin-treated IL-6 expression level. (G) The myocardial cells had significant morphological changes after the transfection of CAV3 siRNA. The Western blot analysis was conducted with 6 replicates for each group, while the IL-6 release assay was conducted with 6 replicates per group. All HE staining images were obtained via a microscope with 200× amplification and the scale bars in the panel were 50 µm. The statistical significance of the observed effects was indicated by **p<0.01 and ***p<0.001. |
Discussion
Cardiac inflammation in the pathophysiology of cardiac disease has received increasing research attention. The underlying mechanisms of diabetic cardiomyopathy are unclear, however, many studies have shown that inflammation contributes to the pathogenesis of DCM.31 Inflammation caused by HGHL plays an important role in cardiovascular diseases.32 Effectively therapy targeting myocardial inflammation is importance in moderating both the onset and progression in DCM of diabetes patients.
Basic and clinical investigations suggest that puerarin is a promising therapeutic candidate for a diverse range of disorders, including diabetes and its complications, osteonecrosis, cancer, Alzheimer’s disease, Parkinson’s disease, and endometriosis. Puerarin improved the heart function of db/db mice by reducing pro-inflammatory cytokine secretion and diminishing NF-κB transcriptional activity.1 Moreover, puerarin exhibits a pivotal role in managing several cardiovascular diseases, such as atherosclerosis, cardiac hypertrophy, heart failure, diabetic cardiovascular complications, myocardial infarction, stroke, and hypertension.33
HGHL induced cardiomyocyte hypertrophy and myocardial damage,30,34 and significantly inhibited viability and promoted apoptosis.35,36 In our study, cardiomyocytes treated with HGHL caused a significant downregulation of CAV3 protein; increased phosphorylation levels of p-p65 protein of the NF-κB pathway as well as the p-p38 protein of the p38MAPK pathway; upregulated the expression and release of IL-6; increased in the expression levels of Bax and cleaved-Caspase-3/pro-Caspase-3, a decreased in the expression level of Bcl-2, and an increased in myocardial hypertrophy.; decreased cell viability associated with inflammatory injury in cardiomyocytes.
Puerarin has been reported to reduce HGHL-induced cellular damage,37 as well as suppress oxidative stress and inflammation,38 it also exerted anti-inflammatory effects by inhibiting NF-κB and p38MAPK pathways.20,39 Our study reveals that puerarin significantly inhibits NF-κB and p38MAPK pathways. Puerarin treatment significantly decreased the phosphorylation of p-p38 and p-p65 proteins, and apoptosis-related protein expression: cleaved -Caspase-3/pro-Caspase-3/Bax.
The NF‐κB‐dependent mechanism plays an important role in hyperglycemia‐induced myocardial injury and inflammation40 and the development of DCM.41 NF‐κB is activated in cardiac myocytes in patients with DCM. NF-κB activation induced a release of downstream pro-inflammatory cytokines, involving IL-6.11 P38MAPK is the most common member of the MAPK family, responsible for regulating inflammation, apoptosis, and other physiological and pathological processes. Activation of p38MAPK further promotes the secretion of IL-6 and MCP-1.42 Erythropoietin can reduce apoptosis and cardiac hypertrophy by inhibiting the activation of NF-κB, phosphorylation of p38-MAPK.43 In terms of proinflammatory molecules, elevated levels of IL-6, TNF-α and matrix metalloproteinase 7 were reported in diabetic patients with diastolic dysfunction44 and predicted the onset of type 2 diabetes.45 IL-6 was sufficient to induce myocardial hypertrophy, inflammation, fibrosis and diastolic dysfunction in rats,46 and deletion of IL-6 can reverse myocardial inflammation and cardiac metabolic abnormalities in diabetes induced by a HL diet.47 In our study, IL-6 was significantly increased and there were significantly damage, apoptosis and hypertrophy of cardiomyocytes after HGHL treatment.
Many studies confirmed the cardiomyocyte protective effect of puerarin.48,49 Nonetheless, the mechanism of its cardioprotective effect on DCM is not fully clarified. Investigations have reported the cardioprotective effect of CAV3 in DCM,21,22,50,51 and CAV3 mediated reduced inflammatory damage.52 In our study, puerarin treated with silencing CAV3 in H9c2 cells enhanced p38MAPK and NF-κB pathways as well as upregulated expression of inflammatory factors IL-6, proved that Puerarin exerted cardioprotective effects via an anti-inflammatory pathway by promoting CAV3, thereby inhibiting the p38MAPK-NF-κB signalling pathway in HGHL-induced inflammatory damage of cardiomyocytes.
There was a very close relationship between CAV3 and the p38MAPK pathway, as well as the NF-κB pathway. Dexamethasone upregulated the transcriptional activity of T-type CAV3 Ca2+channel, inhibiting the activation of NF-κB and the Glucocorticoid receptor.53 The hindlimb suspension reduced CAV3 levels in cod muscle but increases p38 mitogen-activated protein kinase phosphorylation levels.54 Our study also found that CAV3 inhibiting the p38MAPK and NF-κB signaling pathway. All these reports focused on the relationship between CAV3 and p38MAPK or CAV3 and NF-κB. However, we further discover that CAV3 may inhibit inflammation mediated by the p38MAPK-NF-κB, and which had been reported that there were a relationship with apoptosis and hypertrophy injuries.
To sum up, treated with HGHL and puerarin, compared with simple HGHL, we observed a significant upregulation in CAV3, a significant decrease in p-p38 and p-p65 protein phosphorylation, IL-6 expression, as well as depressed inflammation, viability damage, apoptosis and hypertrophy of H9c2 cells. Consequently, our results demonstrate that puerarin can inhibits the NF-κB and p38MAPK pathways after HGHL treatment that facilitate inflammatory lesions in cardiomyocytes, leading to a notable decrease in inflammatory cytokines and suppressed viability damage, apoptosis and hypertrophy of the myocardial cells. Further, Caveolin-3 protein is downregulated using siRNA and added pathway blockers including NF-κB and p38MAPK, respectively. We found that this protective effect of puerarin increased CAV3 proteins, hence inhibited the NF-κB and p38MAPK pathways, upregulated CAV3 proteins and attenuated inflammatory lesions in HGHL-induced cardiomyocytes.
Our results provide a new reference for puerarin applying in clinical DCM and additional investigation into its mechanism. The pharmacological mechanism of Puerarin including the molecular interaction and its key targets still need to be further explored. Since puerarin has no significant toxic side effects in practice and reports, it would be a safe, effective and promising clinical drug for DCM.
Data Avail Ability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Ethics Approval and Consent to Participate
The present study was approved by the Ethics Committee of Guangxi Medical University (Guangxi, China) and followed the Guangxi Medical University guidelines and regulations.
Author Contributions
All authors contributed significantly to the work reported, whether in conception, study design, execution, data acquisition, analysis and interpretation, or in all of these areas. Participate in the drafting, revision or review of the article; Final approval of the version for publication; Journals that agreed to submit the manuscript; And agree to be accountable for all aspects of the work.
Funding
This research was supported by the National Natural Science Foundation of China (No.82260077) and the Guangxi Natural Science Foundation Project (No.2020GXNSFAA238001).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Yin MS, Zhang YC, Xu SH, et al. Puerarin prevents diabetic cardiomyopathy in vivo and in vitro by inhibition of inflammation. J Asian Nat Prod Res. 2019;21(5):476–493.
2. Gilbert RE, Krum H. Heart failure in diabetes: effects of anti-hyperglycaemic drug therapy. Lancet. 2015;385(9982):2107–2117.
3. Li L, Luo W, Qian Y, et al. Luteolin protects against diabetic cardiomyopathy by inhibiting NF-kappaB-mediated inflammation and activating the Nrf2-mediated antioxidant responses. Phytomedicine. 2019;59:152774.
4. Colareda GA, Matera SI, Bayley M, et al. Lepidium meyenii (maca) and soy isoflavones reduce cardiac stunning of ischemia-reperfusion in rats by mitochondrial mechanisms. J Tradit Complement Med. 2021;11(6):471–480.
5. Zhao Y, Guo R, Li L, et al. Tongmai formula improves cardiac function via regulating mitochondrial quality control in the myocardium with ischemia/reperfusion injury. Biomed Pharmacother. 2020;132:110897.
6. Nian M, Lee P, Khaper N, Liu P. Inflammatory cytokines and postmyocardial infarction remodeling. Circ Res. 2004;94(12):1543–1553.
7. Tan Y, Zhang Z, Zheng C, Wintergerst KA, Keller BB, Cai L. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: preclinical and clinical evidence. Nat Rev Cardiol. 2020;17(9):585–607.
8. Lin K, Yang N, Luo W, et al. Direct cardio-protection of Dapagliflozin against obesity-related cardiomyopathy via NHE1/MAPK signaling. Acta Pharmacol Sin. 2022;43(10):2624–2635.
9. Kennedy A, Martinez K, Chuang -C-C, LaPoint K, McIntosh M. Saturated fatty acid-mediated inflammation and insulin resistance in adipose tissue: mechanisms of action and implications. J Nutr. 2009;139(1):1–4.
10. Nakamura M, Sadoshima J. Cardiomyopathy in obesity, insulin resistance and diabetes. J Physiol. 2019;598(14):2977–2993.
11. Frati G, Schirone L, Chimenti I, et al. An overview of the inflammatory signalling mechanisms in the myocardium underlying the development of diabetic cardiomyopathy. Cardiovasc Res. 2017;113(4):378–388.
12. Haudek SB, Taffet GE, Schneider MD, Mann DL. TNF provokes cardiomyocyte apoptosis and cardiac remodeling through activation of multiple cell death pathways. J Clin Invest. 2007;117(9):2692–2701.
13. Kaludercic N, Di Lisa F. Mitochondrial ROS Formation in the Pathogenesis of Diabetic Cardiomyopathy. Front Cardiovasc Med. 2020;7:12.
14. Almourani R, Chinnakotla B, Patel R, Kurukulasuriya LR, Sowers J. Diabetes and cardiovascular disease: an update. Curr Diab Rep. 2019;19(12):161.
15. Tsai KH, Wang WJ, Lin CW, et al. NADPH oxidase-derived superoxide anion-induced apoptosis is mediated via the JNK-dependent activation of NF-kappaB in cardiomyocytes exposed to high glucose. J Cell Physiol. 2012;227(4):1347–1357.
16. Nizamutdinova IT, Guleria RS, Singh AB, Kendall JA, Baker KM, Pan J. Retinoic acid protects cardiomyocytes from high glucose-induced apoptosis through inhibition of NF-kappaB signaling pathway. J Cell Physiol. 2013;228(2):380–392.
17. Zuo G, Ren X, Qian X, et al. Inhibition of JNK and p38 MAPK-mediated inflammation and apoptosis by ivabradine improves cardiac function in streptozotocin-induced diabetic cardiomyopathy. J Cell Physiol. 2019;234(2):1925–1936.
18. Xue B, Wang L, Zhang Z, et al. Puerarin may protect against Schwann cell damage induced by glucose fluctuation. J Nat Med. 2017;71(3):472–481.
19. Andor B, Danciu C, Alexa E, et al. Germinated and Ungerminated Seeds Extract from Two Lupinus Species. Biol Compounds Character In Vitro In Vivo Evaluat Evid Based Complement Alternat Med. 2016;2016:7638542.
20. Wu H, Zhao G, Jiang K, et al. Puerarin Exerts an Antiinflammatory Effect by Inhibiting NF-kB and MAPK Activation in Staphylococcus aureus-Induced Mastitis. Phytother Res. 2016;30(10):1658–1664.
21. Su W, Zhang Y, Zhang Q, et al. N-acetylcysteine attenuates myocardial dysfunction and postischemic injury by restoring caveolin-3/eNOS signaling in diabetic rats. Cardiovasc Diabetol. 2016;15(1):146.
22. Sun HJ, Xiong SP, Wu ZY, et al. Induction of caveolin-3/eNOS complex by nitroxyl (HNO) ameliorates diabetic cardiomyopathy. Redox Biol. 2020;32:101493.
23. Thangavel C, Gomes CM, Zderic SA, et al. NF-kappaB and GATA-Binding Factor 6 Repress Transcription of Caveolins in Bladder Smooth Muscle Hypertrophy. Am J Pathol. 2019;189(4):847–867.
24. Woodman SE, Park DS, Cohen AW, et al. Caveolin-3 knock-out mice develop a progressive cardiomyopathy and show hyperactivation of the p42/44 MAPK cascade. J Biol Chem. 2002;277(41):38988–38997.
25. Hiramoto S, Tsubota M, Yamaguchi K, et al. Cystitis-Related Bladder Pain Involves ATP-Dependent HMGB1 Release from Macrophages and Its Downstream H(2)S/Ca(v)3.2 Signaling in Mice. Cells. 2020;9(8):65.
26. Talukder MA, Preda M, Ryzhova L, Prudovsky I, Pinz IM. Heterozygous caveolin-3 mice show increased susceptibility to palmitate-induced insulin resistance. Physiol Rep. 2016;4(6):3456.
27. Zhu A, Wei X, Zhang Y, et al. Propofol Provides Cardiac Protection by Suppressing the Proteasome Degradation of Caveolin-3 in Ischemic/Reperfused Rat Hearts. J Cardiovasc Pharmacol. 2017;69(3):170–177.
28. Murfitt L, Whiteley G, Iqbal MM, Kitmitto A. Targeting caveolin-3 for the treatment of diabetic cardiomyopathy. Pharmacol Ther. 2015;151:50–71.
29. Varela R, Rauschert I, Romanelli G, Alberro A, Benech JC. Hyperglycemia and hyperlipidemia can induce morphophysiological changes in rat cardiac cell line. Biochem Biophys Rep. 2021;26:100983.
30. Chang W, Zhang M, Meng Z, et al. Berberine treatment prevents cardiac dysfunction and remodeling through activation of 5’-adenosine monophosphate-activated protein kinase in type 2 diabetic rats and in palmitate-induced hypertrophic H9c2 cells. Eur J Pharmacol. 2015;769:55–63.
31. Varga ZV, Giricz Z, Liaudet L, Hasko G, Ferdinandy P, Pacher P. Interplay of oxidative, nitrosative/nitrative stress, inflammation, cell death and autophagy in diabetic cardiomyopathy. Biochim Biophys Acta. 2015;1852(2):232–242.
32. Lee SH, Kim MK, Rhee EJ. Effects of cardiovascular risk factor variability on health outcomes. Endocrinol Metab. 2020;35(2):217–226.
33. Zhou YX, Zhang H, Peng C. Effects of puerarin on the prevention and treatment of cardiovascular diseases. Front Pharmacol. 2021;12:771793.
34. Wang X, McLennan SV, Allen TJ, Tsoutsman T, Semsarian C, Twigg SM. Adverse effects of high glucose and free fatty acid on cardiomyocytes are mediated by connective tissue growth factor. Am J Physiol Cell Physiol. 2009;297(6):C1490–500.
35. Kui L, Weiwei Z, Ling L, et al. Ghrelin inhibits apoptosis induced by high glucose and sodium palmitate in adult rat cardiomyocytes through the PI3K-Akt signaling pathway. Regul Pept. 2009;155(1–3):62–69.
36. Zhang B, Li X, Liu G, et al. Peroxiredomin-4 ameliorates lipotoxicity-induced oxidative stress and apoptosis in diabetic cardiomyopathy. Biomed Pharmacother. 2021;141:111780.
37. Liu Y, Qiu Y, Chen Q, Han X, Cai M, Hao L. Puerarin suppresses the hepatic gluconeogenesis via activation of PI3K/Akt signaling pathway in diabetic rats and HepG(2) cells. Biomed Pharmacother. 2021;137:111325.
38. Zhang L, Liu L, Wang M. Effects of puerarin on chronic inflammation: focus on the heart, brain, and arteries. Aging Med. 2021;4(4):317–324.
39. Tang W, Xiao L, Ge G, et al. Puerarin inhibits titanium particle-induced osteolysis and RANKL-induced osteoclastogenesis via suppression of the NF-kappaB signaling pathway. J Cell Mol Med. 2020;24(20):11972–11983.
40. Jia G, DeMarco VG, Sowers JR. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat Rev Endocrinol. 2016;12(3):144–153.
41. Ramesh P, Yeo JL, Brady EM, McCann GP. Role of inflammation in diabetic cardiomyopathy. Ther Adv Endocrinol Metab. 2022;13:20420188221083530.
42. Zhang S, Zhang S, Liang X, et al. Guanxinping ameliorates atherosclerosis via MAPK/NF-kappaB signaling pathway in ApoE(-/-) mice. Perfusion. 2023;38(3):557–566.
43. Savira F, Cao L, Wang I, et al. Apoptosis signal-regulating kinase 1 inhibition attenuates cardiac hypertrophy and cardiorenal fibrosis induced by uremic toxins: implications for cardiorenal syndrome. PLoS One. 2017;12(11):e0187459.
44. Shaver A, Nichols A, Thompson E, et al. Role of Serum Biomarkers in Early Detection of Diabetic Cardiomyopathy in the West Virginian Population. Int J Med Sci. 2016;13(3):161–168.
45. Wu S, Zhu J, Wu G, et al. 6-Gingerol Alleviates Ferroptosis and Inflammation of Diabetic Cardiomyopathy via the Nrf2/HO-1 Pathway. Oxid Med Cell Longev. 2022;2022:3027514.
46. Melendez GC, McLarty JL, Levick SP, Du Y, Janicki JS, Brower GL. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension. 2010;56(2):225–231.
47. Ko HJ, Zhang Z, Jung DY, et al. Nutrient Stress Activates Inflammation and Reduces Glucose Metabolism by Suppressing AMP-Activated Protein Kinase in the Heart. Diabetes. 2009;58(11):2536–2546.
48. Li WQ, Wu JY, Xiang DX, et al. Micelles Loaded With Puerarin And Modified With Triphenylphosphonium Cation Possess Mitochondrial Targeting And Demonstrate Enhanced Protective Effect Against Isoprenaline-Induced H9c2 Cells Apoptosis. Int J Nanomedicine. 2019;14:8345–8360.
49. Gao S, Li L, Li L, et al. Effects of the combination of tanshinone IIA and puerarin on cardiac function and inflammatory response in myocardial ischemia mice. J Mol Cell Cardiol. 2019;137:59–70.
50. Li M, Zhang M, Huang L, et al. T-type Ca2+ channels are involved in high glucose-induced rat neonatal cardiomyocyte proliferation. Pediatr Res. 2005;57(4):550–556.
51. Lei S, Li H, Xu J, et al. Hyperglycemia-induced protein kinase C beta 2 activation induces diastolic cardiac dysfunction in diabetic rats by impairing caveolin-3 expression and Akt/eNOS signaling. Diabetes. 2013;62(7):2318–2328.
52. Stemkowski PL, Garcia-Caballero A, Gadotti VM, et al. Identification of interleukin-1 beta as a key mediator in the upregulation of Cav3.2-USP5 interactions in the pain pathway. Mol Pain. 2017;13:1744806917724698.
53. BenMohamed F, Ferron L, Ruchon Y, Gouadon E, Renaud J-F, Capuano V. Regulation of T-type Cav3.1 channels expression by synthetic glucocorticoid dexamethasone in neonatal cardiac myocytes. Mol Cell Biochem. 2008;320:1–2.
54. Ohno Y, Sugiura T, Ohira Y, Yoshioka T, Goto K. Loading-associated expression of TRIM72 and caveolin-3 in antigravitational soleus muscle in mice. Physiol Rep. 2014;2(12):24.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.