Back to Journals » International Journal of General Medicine » Volume 7
Pseudomyxoma peritonei: a clinical case of this poorly understood condition
Authors de Oliveira AM, Rodrigues C, Borges A, Martins A, dos Santos S, Pires F, Araújo J, de Deus J
Received 13 July 2013
Accepted for publication 6 August 2013
Published 3 March 2014 Volume 2014:7 Pages 137—141
DOI https://doi.org/10.2147/IJGM.S51504
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Ana Maria de Oliveira,1 Catarina Graça Rodrigues,1 Alexys Borges,2 Alexandra Martins,1 Sofia Loureiro dos Santos,3 Francisco Rocha Pires,4 João Mascarenhas Araújo,2 João Ramos de Deus1
1Department of Gastroenterology, 2Department of Internal Medicine, 3Department of Pathology and Anatomical Sciences, 4Department of Surgery, Pathology and Anatomical Sciences, Prof. Doutor Fernando Fonseca Hospital, Amadora, Portugal
Background: Pseudomyxoma peritonei is an uncommon condition with an estimated incidence of one to two per million (worldwide) per year. It is characterized by the peritoneal deposition of mucinous tumors, most commonly of the appendix, and occasionally from the ovary, coupled by mucinous ascites.
Case presentation: We report the case of a 76-year-old woman who presented with increased abdominal girth and dyspnea for 2 weeks. She was diagnosed as a case of pseudomyxoma peritonei. She was submitted to right oophorectomy, omentectomy, and pseudomyxoma debulking. The histology was compatible with a mucinous tumor of colorectal/appendicular origin. Chemotherapy was not administered because of her functional status. Two years and 8 months later, she refers with postprandial fullness and has moderate ascites.
Keywords: pseudomyxoma peritonei, mucinous tumor, pseudomyxoma debulking
A Letter to the Editor has been published for this article
Introduction
Pseudomyxoma peritonei (PMP) is a tumor with an estimated incidence of one to two million per year.1 It is a poorly understood disease that is known for its production of mucin in the abdominal cavity and mucinous implants, diffusely involving the peritoneal surfaces.2
The term PMP has been applied broadly and includes a heterogeneous group of pathological lesions from the benign to the borderline to the frankly malignant lesion.3,4 Ronnett et al have suggested a classification based on tumor pathology in which they place all PMP in three groups in order of decreasing prognosis: disseminated peritoneal adenomucinosis, peritoneal mucinous carcinoma with intermediate or discordant features, and peritoneal mucinous carcinoma.4
A definitive diagnosis of PMP requires the presence both of mucinous neoplastic cells/epithelium and of mucinous ascites and diffuse intra-abdominal mucin. Some specialists also require the presence of diffuse mucinous implants for this diagnosis. Viable epithelial glandular cells must be identified within the mucin pools by histological analysis to diagnose PMP. Cases without epithelium are regarded as mucinous ascites.5
Case report
A 76-year-old woman presented to our emergency department (Hospital Professor Doutor Fernando Fonseca, Amadora, Portugal) with dyspnea, increased abdominal girth, and peripheral edema for 2 weeks. Her medical history included diabetes mellitus type 2, hypertension, and dyslipidemia. She was medicated with ramipril, furosemide, aspirin, simvastatin, rabeprazole, and lorazepam.
The physical examination revealed tachypnea, a blood pressure of 144/82 mmHg, a pulse rate of 83 beats per minute (bpm), and apyrexia. Expiratory time was increased, and there were rales at the base of the lungs. The cardiovascular system did not reveal any remarkable changes. There was collateral venous circulation on the abdomen wall, the wall was slightly depressible, and percussion revealed tympanic resonance in the center of the abdomen and dullness in both lumbar regions. There was no palpable organomegaly or lymphadenopathy. Peripheral edema was present.
Laboratory investigations showed an absence of leukocytosis (8,400 leukocytes/μL) with neutrophilia (6,100 neutrophils/μL) and a C-reactive protein value of 6.21 mg/dL. The patient’s erythrocyte sedimentation rate was 88 mm, her carcinoembryonic antigen level was 177 ng/mL (0–3 ng/mL), her cancer antigen 15–3 level was 55 U/mL (0–30 U/mL), her cancer antigen 125 level was 57.5 U/mL (0–35 U/mL), and her cancer antigen 19.9 level was lower than 2.5 U/mL (0–37 U/mL).
Paracentesis was attempted but was unsuccessful. Abdomen ultrasonography and computed tomography showed images that were suggestive of pseudomyxoma and moderate, multiseptate ascites (Figure 1).
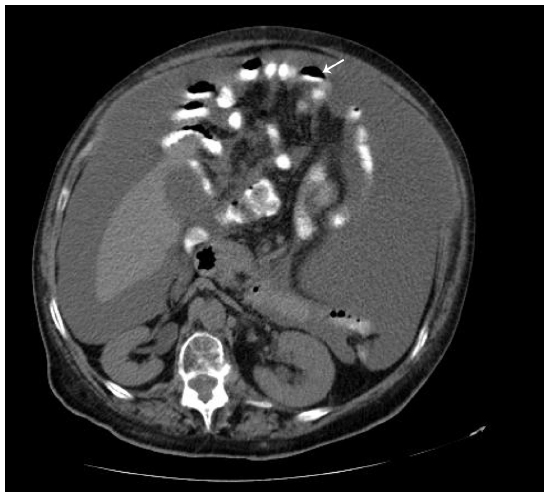 | Figure 1 Computed tomographic scan of abdomen showing pseudomyxoma peritonei with multiple peritoneal masses (arrow) with “scalloping effect.” |
The patient was submitted to laparotomy with right oophorectomy, omentectomy, and pseudomyxoma debulking (Figure 2). At surgery, 6,200 g yellow-orange gelatinous material (Figure 3) was removed. The histological examination of biopsies (from small intestine, ovary, peritoneum, and epiploon) revealed mucinous neoplastic cells and diffuse intra-abdominal mucin, characteristics that allowed the diagnosis of PMP.
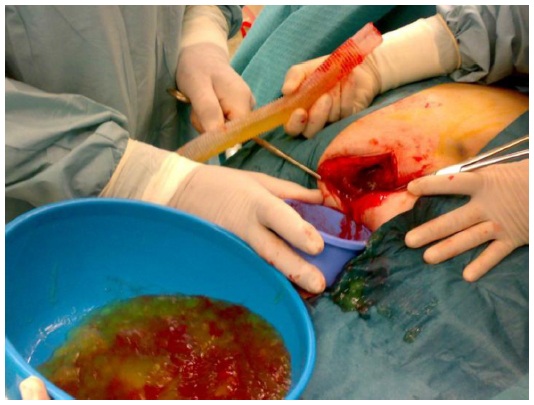 | Figure 2 Laparotomy with right oophorectomy, omentectomy, and pseudomyxoma debulking. |
 | Figure 3 Yellow-orange gelatinous material. |
The morphological (Figure 4) and immunophenotypic (cytokeratin 7 focally positive [Figure 5], cytokeratin 20 diffusedly positive [Figure 6], and p16 negative [Figure 7]) patterns were compatible with colorectal/appendicular origin.
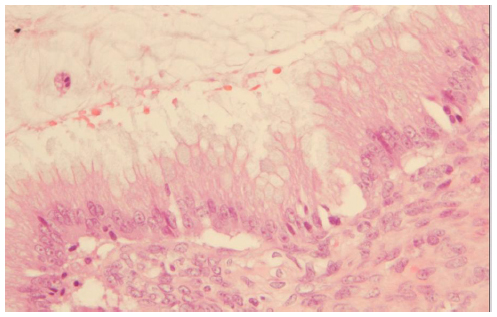 | Figure 4 Hematoxylin and eosin staining (×20). |
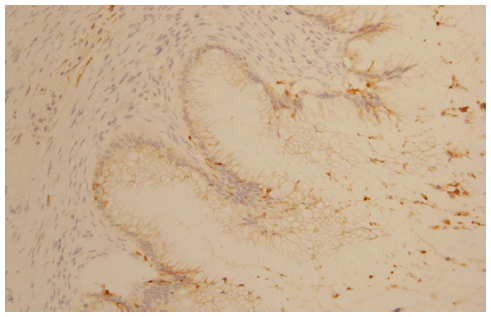 | Figure 5 Cytokeratin 7 focally positive staining (×10). |
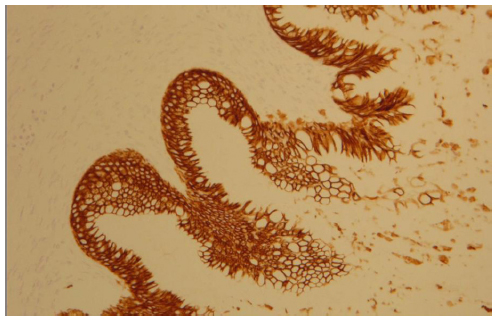 | Figure 6 Cytokeratin 20 diffusely positive staining (×10). |
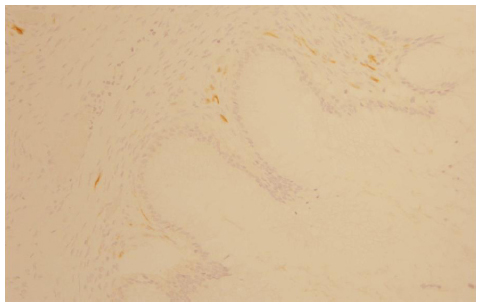 | Figure 7 p16-negative staining (×10). |
In the postoperative course, the patient had hemodynamic instability that reverted with fluid replacement and amines for a short period.
She was discharged in a satisfactory condition with follow-up at the oncology division. Because of her functional status, the patient has not received chemotherapy. The treatment is being based on symptomatic relief.
Two years and 8 months later, she has referred occasionally with postprandial fullness and moderate ascites. Her carcinoembryonic antigen and cancer antigen 125 levels are 133 ng/mL and 139 U/mL, respectively. The last image exam (abdomen ultrasonography) showed a small volume of perihepatic, perisplenic, and interloop fluid with peritoneal leaflets thickening. It was possible to identify a mass with 58 × 30 mm in the left parietocolic groove, indicative of peritoneal carcinomatosis (Figure 8). Enlarged ganglia were also seen, the biggest of which was at the hepatic hilum, at 32 mm (Figure 9).
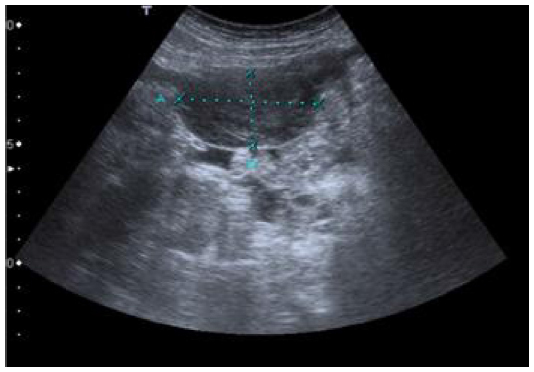 | Figure 8 Abdomen ultrasonography showing a mass with 58 × 30 mm in the left parietocolic groove. |
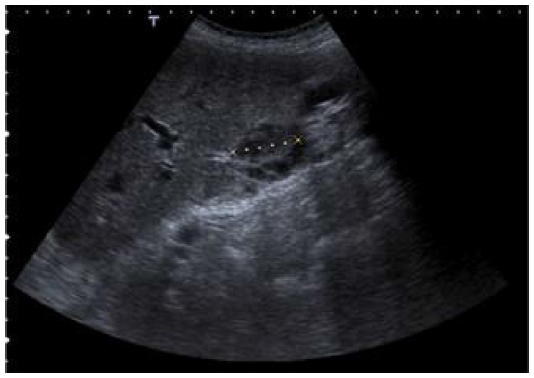 | Figure 9 Abdomen ultrasonography showing a ganglion with 32 mm at hepatic hilum. |
Discussion
PMP was first described in 1842 by Karl F Rokitansky,6 but the term was used in 1884 in association with a mucinous carcinoma of the ovary.7 Later, in 1901, Franckel described PMP in association with an appendiceal cystic tumor.8
PMP is a disease of the MUC (a gel-forming mucin that forms strong bonds with the surrounding stroma and is also believed to have tumor-suppressor activities)9 expressing goblet cells that are specific for mucin production. Extracellular mucin accumulates dramatically and has no place to drain.4 A copious amount of mucinous fluid gradually fills the peritoneal cavity, resulting in the so-called “jelly belly” abdomen.5
The origin of PMP is a controversial subject.10 Most acknowledge that PMP predominantly originates in the appendix in men, and increasingly, evidence suggests a similar origin in women. In women, synchronous ovarian and appendiceal disease is common. However, immunohistochemistry and molecular genetic techniques support the hypothesis that in the majority of women, the ovarian tumor is metastatic from a perforated appendiceal mucinous tumor.1 This hypothesis is illustrated by the presented case, as histological analysis of the oophorectomy piece revealed a pattern compatible with colorectal/appendicular origin.
The reported sex distribution is contradictory in the literature: some authors have noted a significant preponderance of women5,8,10 whereas others found no difference.3 However, as some authors highlight, it is improbable that the female preponderance found in some studies has a biological explanation; instead, the effect is more likely the result of some referral bias, possibly resulting from the use of iterative surgery in secondary care hospitals to treat male patients with PMP and not referring them to a tertiary unit. Female patients were more likely to be referred because of presumed ovarian malignancy requiring complex procedures available only in tertiary care units.1,10
The symptoms of this uncommon disease are nonspecific; the most commonly recorded are abdominal pain10 resembling acute appendicitis, distension,10,11 or mass.12 Nausea, vomiting, and asthenia have also been described.13 In rare cases, the mucinous collection can lead to signs of raised intra-abdominal pressure, such as uterine prolapse or abdominal hernia.10
In addition to symptoms being nonspecific, routine laboratory studies also are seldom helpful in making this diagnosis.5
An accurate preoperative diagnosis of PMP can be aided by radiological imaging,5 with a computed tomography scan being the currently optimal imaging modality for the diagnosis and staging of this disease. Computed tomography scan findings may be pathognomonic. Typical appearances include areas of low attenuation, with islands of higher attenuation resulting from solid elements within mucinous material. Classically, “scalloping” of visceral surfaces, particularly of the liver and spleen, distinguishes mucinous from fluid ascites.1
However, diagnosis of PMP is challenging because of the mimicking nature of the disease. In the study conducted by Järvinen and Lepistö, only 28% of patients underwent initial surgery for presumed PMP.10 Preoperative diagnosis is often appendicitis,1 gynecological cancer1/ovarian tumor,10 or peritonitis,1 which did not happen in our case, as imaging suggested from the beginning a diagnosis of PMP.
A treatment strategy for PMP should pursue complete cytoreduction and prevention of recurrence or progression. Combined modality treatment, consisting of cytoreductive surgery with hyperthermic intraperitoneal chemotherapy, seems very efficient in this regard.14–17 This approach is currently carried out in many centers throughout the world, with promising results,18 and seems to win ground as the standard treatment approach.14
However, not all cases are suitable for this approach.10 Patients must be medically fit to safely undergo these treatments, and patients with peritoneal carcinomatosis with an Eastern Cooperative Oncology Group (ECOG) performance score of 2 to 3 have significantly poorer overall survival after the treatment compared with those with an ECOG score of 1.1
In the presented case, the patient had an ECOG performance status of 3; therefore, we considered her to be not medically fit to be submitted to the referred approach.
Unfortunately, the majority of patients develop recurrence after treatment,19 which requires repeated surgical intervention.6 Elevated tumor markers such as carcinoembryonic antigen level, cancer antigen 19-9 level, and cancer antigen 125 level indicate advanced disease and may rise in association with recurrent disease.12,20
Surgical reintervention is usually difficult because adhesions and fibrosis greatly increase the risk of unintentional enterotomies with subsequent leaks and fistulae formation; it is, however, widely accepted that recurrences should be investigated vigorously and treated with further surgical debulking, with or without adjuvant chemotherapy, in the expectation that many patients will enjoy substantial additional symptom-free survival.6
The reported prognostic factors of PMP are age,21 histology,11 residual tumor volume,11 and intraperitoneal chemotherapy.13
An interesting fact is that chemotherapy may improve survival only if administered intraperitoneally,13,19 as reported by the Mayo Clinic, Gough DB.13 Systemic chemotherapy seems not to affect prognosis.19
In addition, survival rates differ greatly according to histology: Ronnett et al observed 5- and 10-year survival rates of 75% and 68%, respectively, for cases of disseminated peritoneal adenomucinosis; 50% and 21%, respectively, for cases of intermediate malignancy; and 14% and 3%, respectively, for peritoneal mucinous carcinoma.22
Conclusion
We described a case of a female patient who developed PMP, a rare and heterogeneous condition for which the diagnosis is often misleading in the preoperative period.
In the light of current knowledge, the optimal treatment involves a combination of surgery and hyperthermic intraperitoneal chemotherapy.
Awareness of this condition, although it is rare, is an important prerequisite for appropriate management, as the long-term outcomes for cytoreductive surgery with hyperthermic intraperitoneal chemotherapy are impressive for patients with low-grade histology susceptible to complete cytoreduction.
Acknowledgments
Carlos Carvalho, Oncology Department, Prof. Doutor Fernando Fonseca Hospital, Amadora, Portugal.
Disclosure
The author reports no conflicts of interest in this work.
References
Bevan KE, Mohamed F, Moran BJ. Pseudomyxoma peritonei. World J Gastrointest Oncol. 2010;2(1):44–50. | |
Gupta S, Singh G, Gupta A, et al. Pseudomyxoma peritonei: An uncommon tumor. Indian J Med Paediatr Oncol. 2010;31(2):58–61. | |
Bryant J, Clegg AJ, Sidhu MK, Brodin H, Royle P, Davidson P. Systematic review of the Sugarbaker procedure for pseudomyxoma peritonei. Br J Surg. 2005;92(2):153–158. | |
Ronnett BM, Zahn CM, Kurman RJ, Kass ME, Sugarbaker PH, Shmookler BM. Disseminated peritoneal adenomucinosis and peritoneal mucinous carcinomatosis. A clinicopathologic analysis of 109 cases with emphasis on distinguishing pathologic features, site of origin, prognosis, and relationship to “pseudomyxoma peritonei.” Am J Surg Pathol. 1995;19(12):1390–1408. | |
Li C, Kanthan R, Kanthan SC. Pseudomyxoma peritonei – a revisit: report of 2 cases and literature review. World J Surg Oncol. 2006;4:60. | |
Al-Buainain H, Al-Jehani Y, Moghazy K, Al-Quorain A. Psuedomyxoma peritonei secondary to adenocarcinoma of the cecum. J Family Community Med. 2010;17(2):103–106. | |
Werth R. Klinische und anatomische Untersuchungen zur Lehre von den Bauchgeschwullsten und der Laparotomie. [Clinical and anatomical studies in teaching abdominal tumors and laparotomy] Arch Gynecol Obstet. 1884;24:100–118. German. | |
Franckel E. Uber das sogennate pseudomyxome peritonei. [About the so-called pseudomyxoma peritonei] Med Wochenschr. 1901;48:965–970. German. | |
O’Connell JT, Tomlinson JS, Roberts AA, McGonigle KF, Barsky SH. Pseudomyxoma peritonei is a disease of MUC2-expressing goblet cells. Am J Pathol. 2002;161(2):551–564. | |
Järvinen P, Lepistö A. Clinical presentation of pseudomyxoma peritonei. Scand J Surg. 2019;9(4):213–216. | |
Miner TJ, Shia J, Jaques DP, Klimstra DS, Brennan MF, Coit DG. Long-term survival following treatment of pseudomyxoma peritonei: an analysis of surgical therapy. Ann Surg. 2005;241(2):300–308. | |
Meshikhes AW, Al-Abkari HA, Al-Momen SA, Saad FE. Pseudomyxoma peritonei. Saudi Med J. 2006;27(3):389–391. | |
Gough DB, Donohue JH, Schutt AJ, et al. Pseudomyxoma peritonei. Long-term patient survival with an aggressive regional approach. Ann Surg. 1994;219(2):112–119. | |
Smeenk RM, Verwaal VJ, Zoetmulder FA. Pseudomyxoma peritonei. Cancer Treat Rev. 2007;33(2):138–145. | |
Arjona-Sanchez A, Muñoz-Casares FC, Casado-Adam A, et al. Outcome of patients with aggressive pseudomyxoma peritonei treated by cytoreductive surgery and intraperitoneal chemotherapy. World J Surg. 2013;37(6):1263–1270. | |
Buell-Gutbrod R, Gwin K. Pathologic diagnosis, origin, and natural history of pseudomyxoma peritonei. Am Soc Clin Oncol Educ Book. 2013;2013:221–225. | |
McBride K, McFadden D, Osler T. Improved survival of patients with pseudomyxoma peritonei receiving intraperitoneal chemotherapy with cytoreductive surgery: a systematic review and meta-analysis. J Surg Res. 2013;183(1):246–252. | |
Virzì S, Iusco D, Bonomi S, Grassi A. Pseudomyxoma peritonei treated with cytoreductive surgery and hyperthermic chemotherapy: a 7-year single-center experience. Tumori. 2012;98(5):588–593. | |
Kojimahara T, Nakahara K, Shoji T, et al. Identifying prognostic factors in Japanese women with pseudomyxoma peritonei: a retrospective clinico-pathological study of the Tohoku Gynecologic Cancer Unit. Tohoku J Exp Med. 2011;223(2):91–96. | |
Kusamura S, Hutanu I, Baratti D, Deraco M. Circulating tumor markers: predictors of incomplete cytoreduction and powerful determinants of outcome in pseudomyxoma peritonei. J Surg Oncol. 2013;108(1):1–8. | |
Lee JK, Song SH, Kim I, et al. Retrospective multicenter study of a clinicopathologic analysis of pseudomyxoma peritonei associated with ovarian tumors (KGOG 3005). Int J Gynecol Cancer. 2008;18(5):916–920. | |
Ronnett BM, Yan H, Kurman RJ, Shmookler BM, Wu L, Sugarbaker PH. Patients with pseudomyxoma peritonei associated with disseminated peritoneal adenomucinosis have a significantly more favorable prognosis than patients with peritoneal mucinous carcinomatosis. Cancer. 2001;92(1):85–91. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.