")
Back to Journals » The Application of Clinical Genetics » Volume 14
Pseudo-Meigs’ Syndrome in Tunisian H Syndrome Female Patient: First Case Reported
Authors Zaimi Y , Ayari M , Mensi A, Bel Hadj Kacem L, Achouri L, Bouzrara M, Said Y, Mouelhi L, Debbeche R
Received 11 February 2021
Accepted for publication 23 March 2021
Published 15 April 2021 Volume 2021:14 Pages 235—239
DOI https://doi.org/10.2147/TACG.S306298
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Yosra Zaimi,1 Myriam Ayari,1 Asma Mensi,1 Linda Bel Hadj Kacem,2 Leila Achouri,3 Meriem Bouzrara,4 Yosra Said,1 Leila Mouelhi,1 Radhouane Debbeche1
1Department of Gastroenterology, Charles Nicolle Hospital, Tunis, Tunisia; 2Department of Pathology, Charles Nicolle Hospital, Tunis, Tunisia; 3Department of Oncologic Surgery, University of Tunis El Manar, Tunis, Tunisia; 4Department of Radiology, Charles Nicolle Hospital, Tunis, Tunisia
Correspondence: Myriam Ayari
Department of Gastroenterology, Charles Nicolle Hospital, Boulevard 9 Avril 1938 Bab Saâdoun, Tunis, 1006, Tunisia
Email [email protected]
Abstract: H syndrome is an extremely rare autosomal recessive affection caused by biallelic mutations in the SLC29A3 gene encoding the human equilibrative nucleoside transporter hENT3. The hallmark signs are cutaneous consisting of hyperpigmentation and hypertrichosis patches. Besides, associated systemic manifestations are highly various reflecting phenotypic pleiotropism. Herein, we report a first case of pseudo-Meigs’ syndrome occurring in a young Tunisian H syndrome diagnosed patient with a novel homozygous frameshift mutation in exon 2 of the SLC29A3 gene: p.S15Pfs*86 inducing a premature stop codon. The patient developed ascites associated with left ovarian mass and she underwent surgery. After tumor resection, ascites disappeared rapidly. Histological examination showed serous cystadenoma of the ovary orienting the diagnosis towards pseudo-Meigs’ syndrome.
Keywords: H syndrome, SLC29A3 gene, rare disease, pseudo-Meigs’ syndrome, Tunisian patient
Introduction
H Syndrome is a multifaceted rare autosomal recessive disease first described by Molho-Pessach in 2008, caused by mutations in the SLC29A3 gene encoding the human equilibrative nucleoside transporter 3 (hENT3).1,2 In the literature, nearly 100 cases have been reported worldwide, mostly patients being of Arab origin.3
It’s classified as a genodermatosis4 with multisystemic involvement. Clinical manifestations are highly various and heterogeneous reflecting a phenotypic pleiotropism.5 Indeed, H syndrome is characterized by hyperpigmented cutaneous patches with hypertrichosis most often involving the lower limbs, especially the inner thighs and typically sparing the knees. In addition, systemic manifestations may include hepatosplenomegaly, hearing loss, hypogonadism, short stature, hyperglycemia (insulin-dependent diabetes), hallux valgus, flexion contractures, hematological and heart abnormalities.1 In men, scrotal infiltration is characteristic. As H syndrome is a new and rare disease, clinical spectrum may not have revealed all its manifestations. Herein, we report a case of pseudo-Meigs’ syndrome occurring in a female patient presenting with H syndrome, not described previously.
Case Presentation
A 25 year-old-female Tunisian patient born to a first degree consanguineous marriage was followed for H syndrome. There was no family history, or similar cases. Her past medical history included type 1 diabetes since the age of two years, hyperpigmentation and hypertrichosis discovered at the age of five years on the inner thighs with extension of lesions to the lower limbs, back, abdomen and arms, right-side hypoacousia, delayed puberty occurring at the age of 17 years, hepatosplenomegaly, mild anemia, bilateral camptodactyly of both fifth fingers. Transthoracic ultrasound of the chest performed at the age of 12 years showed cardiomegaly with pericardial effusion. Mutation screening in the SLC29A3 gene was performed using direct analysis of DNA sequence extracted from the samples according to standard techniques. All coding exons of the SLC29A3 gene were amplified using polymerase chain reaction (PCR). Direct sequencing of PCR products was performed with the ABI prism 3500 DNA Genetic Analyzer (Applied Biosystems, Foster City, CA, USA), using the ABI Prism Big Dye Terminator v3.1 Cycle Sequencing Ready Reaction Kit (Applied Biosystems).
This molecular analysis revealed a novel homozygous frameshift mutation in exon 2 of the SLC29A3 gene: p.S15Pfs*86 (c.42delC) inducing a premature stop codon (Figure 1). Genetic analysis was also performed for the parents and both were found heterozygous for the mutation (Figure 1). At the age of 22 years, the patient was referred to our department to investigate newly diagnosed ascites. The patient reported progressive abdominal distension since one month and worsening abdominal discomfort. There was no fever, no change in bowel habits, no intestinal obstruction signs, no weight loss neither asthenia. Vital signs were stable Physical examination revealed distended abdomen with shifting dullness. There were no dilated veins, no evidence of pleural effusion or cardiac dysfunction signs nor oedema. Abdominal ultrasound showed high abundance ascites, known hepatomegaly without portal hypertension signs and 6 cm left latero-uterine mass. Analysis of the ascetic fluid showed a white blood cell count of 200/µL with 80% lymphocytes and fluid protein level of 3.1 g/dl. Gram staining, bacterial culture, Mycobacterium tuberculosis screening and cytology were negative. While investigating cardiac origin of ascites, new transthoracic ultrasound was performed showing non-compressive circumferential pericardial effusion without deep deterioration of ventricular function (59%). Upper endoscopy and colonoscopy were normal. Seric tumor markers showed high CA-125 concentration of 59 U/mL (normal value 0–35 U/mL), with normal blood levels of alpha-fetoprotein and carcinoembryonic antigen. Magnetic resonance imaging (MRI) confirmed the presence of left ovarian cystic mass of 6*3*3 cm with thin regular wall and enhanced septa after Gadolinium injection (Figure 2). As the combination of ascites with negative work-up and ovarian mass was suggestive of Meigs’ syndrome, she was referred to the department of gynecology and underwent surgery. Perioperative exploration showed a benign-looking left ovarian tumor and a massive amount of ascites (3.5 L). Ovarian cystectomy was successfully performed. Histopathological examination of the resected ovarian mass revealed serous cystadenoma (Figure 3), orienting the diagnosis towards pseudo-Meigs’ syndrome. After tumor removal, ascites decreased rapidly then subsequently resolved postoperatively and the patient become asymptomatic. During 3 years follow-up there was no relapse of ascites.
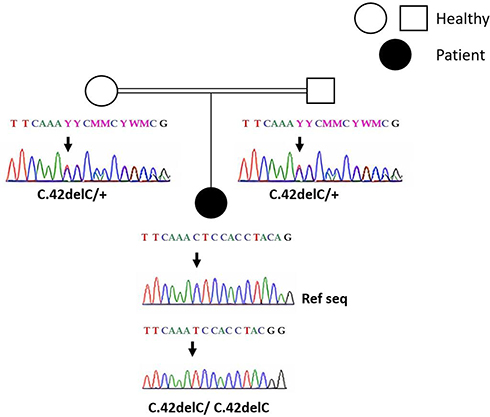 |
Figure 1 Sanger sequencing results: electropherograms of the affected patient and her parents. (+) indicated the wild type allele. The position of the mutation is indicated by the arrow. |
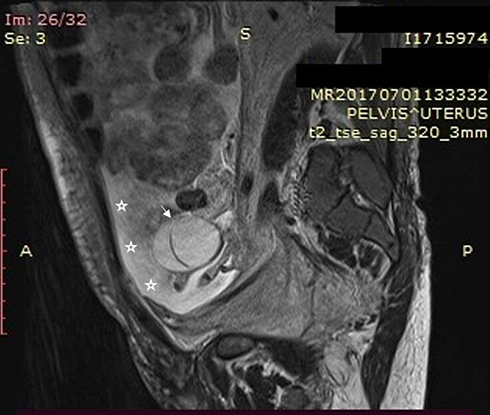 |
Figure 2 Sagittal T2-weighted MRI without injection showing bilobular ovarian mass (arrow) and ascites (asterisk). |
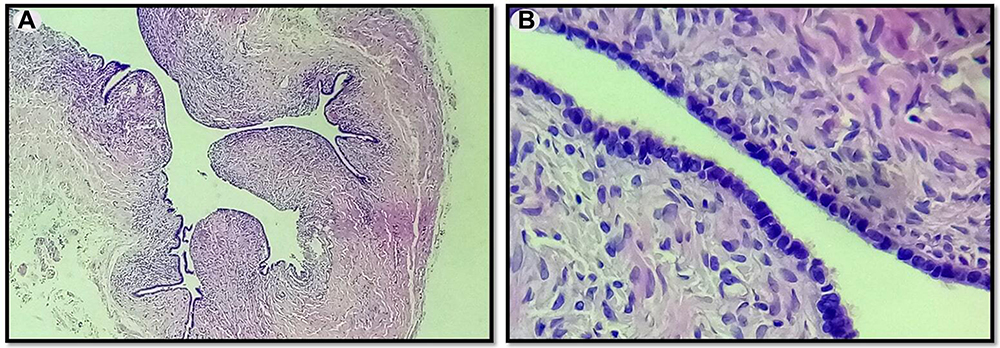 |
Figure 3 Serous cystadenoma of the ovary. (A) Thin-walled unilocular cyst that is lined by cubo-cylindrical monostratified epithelium (Low magnification x 10). (B) Monostratified, focally pseudostratified lining with monotonous, cuboidal or columnar, ciliated cells with round or oval nuclei. The epithelium is supported by variable amounts of spindle cell stroma with no cytologic atypia seen (Medium magnification x 20). |
Discussion
H syndrome (OMIM 612391) is an extremely rare autosomal recessive multisystemic disorder. It’s currently considered as an inherited histiocytic disorder of the R group according to a recent revised classification of histiocytoses and related neoplasms by the Histiocyte Society.6 Clinical findings are widely variable. The main characteristic lesions are cutaneous bilateral hyperpigmentation with indurated patches accompanied by hypertrichosis and sclerodermatous thickening mainly located on the inner thighs and the lower limbs. Typical reported histopathological findings are hyperpigmentation of basal layer, widespread fibrosis, thickened collagen bundles, extensive inflammatory mononuclear infiltrate consisting mainly of monocyte derived cells expressing CD68+ CD34+ and FXIIIa+, plasma cells and perivascular lymphocytic aggregates.7
In addition to cutaneous signs that may sometimes be missing, symptoms may range a constellation of other clinical and laboratory findings including hepatosplenomegaly, hearing loss, heart anomalies, hypogonadism, hyperglycemia, growth delay, musculoskeletal deformities, anemia and red cell aplasia/dyserythropoiesis. H syndrome occurs because of a mutation in the SLC29A3 gene that encodes hENT3 which is a nucleoside transporter protein found in intracellular membranes, especially in lysosomes as well as mitochondria, and had vast biochemical roles. Thus, any disorder could result in various abnormal phenotypes. In addition to well-established symptoms, some new findings occurring in this rare syndrome are being reported recently. El-Bassyouni et al presented a case of H syndrome young patient with areas of hypopigmentation on the abdomen and elevated serum amyloid unreported previously.8 In another study-case, Rezvani et al described the first case of a patient with H syndrome and idiopathic thrombocytopenic purpura, an autoimmune phenomenon.9 Also, glomerular involvement was observed in affected children suggesting that generalized peripheral edema and glomerulopathy may broaden the clinical spectrum of H syndrome.10
Our patient presented a novel frameshift mutation in exon 2, p.S15Pfs*86 reported by Jaouadi et al.11 The pathogenicity of this mutation was assessed by using in silico prediction tools showing a pathogenic influence of the mutation on the hENT3 protein function.11 In this case, the patient demonstrated quite severe presentation with classic findings of H syndrome (Table 1). Also, she is the first H syndrome patient presenting pseudo-Meigs’ syndrome. Indeed, up to our knowledge, there is no reported case of ovarian tumor in H syndrome patient in the literature. Meigs’ syndrome is a rare condition defined by the presence of a benign ovarian fibroma (or a fibroma-like tumor) with ascites and pleural effusion that resolve spontaneously after removal of the tumor.12 When the ovary tumor is other than fibromas, like in our case, it is called pseudo-Meigs’ syndrome. Serous cystadenomas are common ovarian masses, however rarely associated with ascites. Recently, a case of pseudo-Meigs’ syndrome secondary to serous cystadenoma was reported.13 It’s incomplete or partial if just ascites is present. The pathophysiological mechanism underlying the constitution of these effusions is not exactly known. Several theories have been proposed to explain the physiopathology of ascites observed in this syndrome, such as direct irritation of the peritoneum, lymphatic obstruction due to the development of the tumor, or to a liquid flow coming directly from the tumor. Prior to histopathological examination, this syndrome often leads to fears ovarian neoplasia due to the existence of ascites and the commonly observed elevation of CA-125.14 Unlike our case where the patient was 22 years old, pseudo-Meigs’ syndrome is more frequent in postmenopausal women with an average age of about 50 years and is extremely rare in women aged less than 30 years.15 This may be related to a link with H syndrome as its manifestations usually occur in the first or second decade of life.2 In fact, pseudo-Meigs’ syndrome happening in young H syndrome patient contains too much rarity to be a mere coincidence. This suggest that begin ovarian tumor may be among H syndrome features.
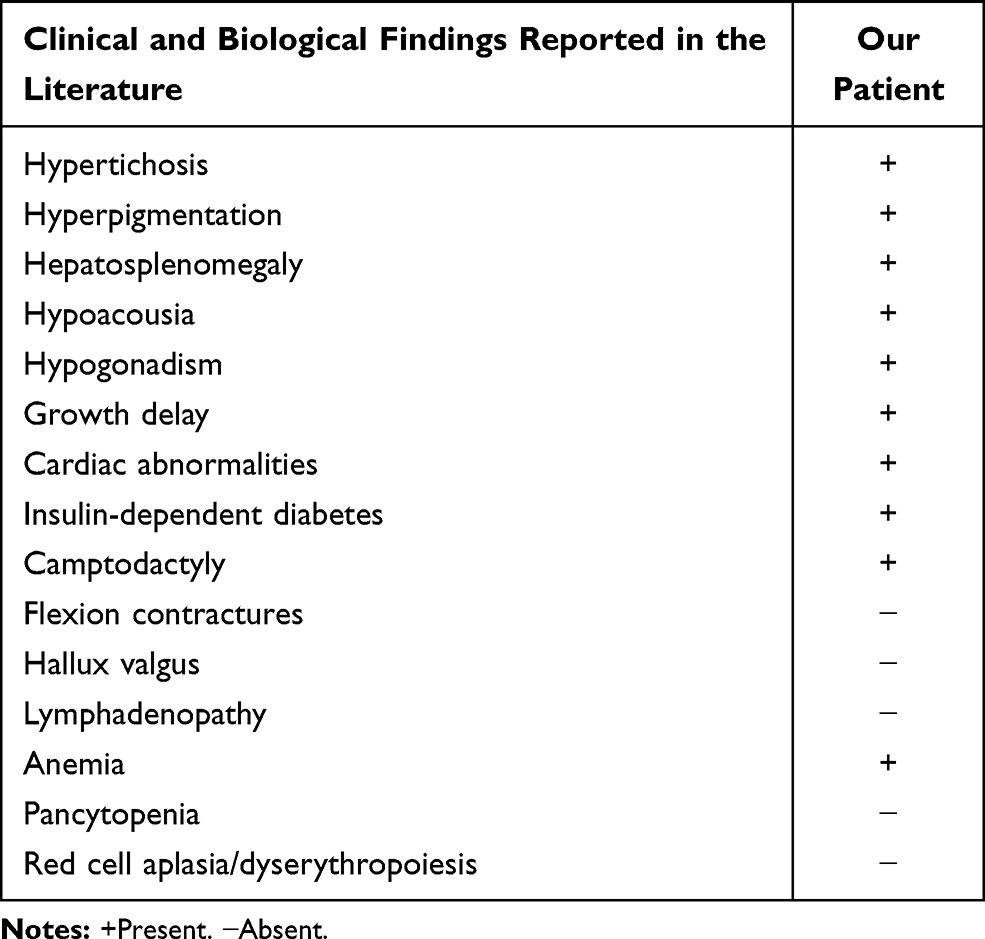 |
Table 1 Reported Clinical and Biological Findings Characterizing H Syndrome in the Literature Compared with Those in Our Patient |
In conclusion, H syndrome is a rare genetic disorder with huge phenotypic spectrum ranging from mild to severe. Due to its rarity it may be misdiagnosed. However, every physician should be aware of this condition and the possible manifestations as it includes multisystem involvement, in order to timely perform genetic testing for its implication in early clinical management. This case may extend the phenotypic spectrum of this disorder by reporting a first description of pseudo-Meigs’ syndrome, an unusual entity, occurring in H syndrome patient presenting with a novel frameshift mutation.
Data Sharing Statement
All data underlying the results are available as part of the article and no additional source data are required.
Ethics Statement
Written informed consent was obtained from the patient for publication of this case report and any accompanying images.
Acknowledgment
We express our gratitude to the staff at Charles Nicolle Hospital who were involved in the patient care.
Disclosure
The authors report no conflicts of interest in this work. Institutional approval was not required to publish the case details.
References
1. Molho-Pessach V, Agha Z, Aamar S, et al. The H syndrome: a genodermatosis characterized by indurated, hyperpigmented, and hypertrichotic skin with systemic manifestations. J Am Acad Dermatol. 2008;59(1):
2. Molho-Pessach V, Lerer I, Abeliovich D, et al. The H syndrome is caused by mutations in the nucleoside transporter hENT3. Am J Hum Genet. 2008;83(4):
3. Molho-Pessach V, Ramot Y, Camille F, et al. H syndrome: the first 79 patients. J Am Acad Dermatol. 2014;70(1):80–8. doi:10.1016/j.jaad.2013.09.019
4. Bhatti S, Jamil A, Siddiqui SH, Yaqoob U, Virk LN, Bhatti A. The H Syndrome: a genodermatosis. Cureus. 2018;10(6):e2763.
5. Spiegel R, Cliffe ST, Buckley MF, et al. Expanding the clinical spectrum of SLC29A3 gene defects. Eur J Med Genet. 2010;53(5):
6. Emile J-F, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127(22):
7. Doviner V, Maly A, Ne’eman Z, et al. H syndrome: recently defined genodermatosis with distinct histologic features. A morphological, histochemical, immunohistochemical, and ultrastructural study of 10 cases. Am J Dermatopathol. 2010;32(2):
8. El-Bassyouni HT, Thomas MM, Tosson AMS. Mutation in the SLC29A3 Gene in an Egyptian patient with H syndrome: a case report and review of literature. J Pediatr Genet. 2020;9(2):
9. Rezvani A, Taherifard E, Mokhtari M, Feili A, Movahed H. H syndrome with a possibly new immunological phenotype. Int J Dermatol. 2020;59(7):
10. David O, Geylis M, Kristal E, Ling G, Schreiber R. Glomerular involvement in children with H syndrome. Pediatr Nephrol Berl Ger. 2021;36(3):
11. Jaouadi H, Zaouak A, Sellami K, et al. H syndrome: clinical, histological and genetic investigation in Tunisian patients. J Dermatol. 2018;45(8):
12. Meigs JV. Fibroma of the ovary with ascites and hydrothorax; Meigs’ syndrome. Am J Obstet Gynecol. 1954;67(5):
13. Daoudi M, Herrak L, Ftouh ME, Achachi L. Pseudo-Meigs syndrome secondary to ovarian serous cystadenoma. Pan Afr Med J. 2019;33:11. doi:10.11604/pamj.2019.33.11.18128
14. Benjapibal M, Sangkarat S, Laiwejpithaya S, Viriyapak B, Chaopotong P, Jaishuen A. Meigs’ syndrome with elevated serum CA125: case report and review of the literature. Case Rep Oncol. 2009;2(1):
15. Shiau C-S, Chang M-Y, Hsieh -C-C, Hsieh -T-T, Chiang C-H. Meigs’ syndrome in a young woman with a normal serum CA-125 level. Chang Gung Med J. 2005;28(8):
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.