Back to Journals » Pediatric Health, Medicine and Therapeutics » Volume 10
Prune belly syndrome: current perspectives
Authors Arlen AM, Nawaf C, Kirsch AJ
Received 14 May 2019
Accepted for publication 13 June 2019
Published 6 August 2019 Volume 2019:10 Pages 75—81
DOI https://doi.org/10.2147/PHMT.S188014
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Roosy Aulakh
Angela M Arlen,1 Cayce Nawaf,1 Andrew J Kirsch2
1Yale University School of Medicine, Department of Urology, New Haven, CT 06520, USA; 2Emory University, Children’s Healthcare of Atlanta, Atlanta, GA 30328, USA
Abstract: Prune belly syndrome (PBS) is a rare but morbid congenital disease, classically defined by a triad of cardinal features that includes cryptorchidism, urinary tract dilation and laxity of the abdominal wall musculature. Children often require numerous surgical interventions including bilateral orchidopexy as well as individually tailored urinary tract and abdominal wall reconstruction. Along with the classic features, patients with PBS often experience gastrointestinal, orthopedic, and cardiopulmonary comorbidities.
Keywords: prune-belly syndrome, cryptorchidism, urinary tract dilation, abdominal wall laxity, abdominoplasty
Introduction
Prune belly syndrome (PBS), also eponymously referred to as Eagle-Barrett syndrome, is a rare multisystem condition typically characterized by deficient or absent abdominal wall musculature, bilateral intra-abdominal cryptorchidism and urinary tract anomalies including megalourethra, megacystis, hydroureteronephrosis and renal dysplasia. Disease severity (ie, degree of renal dysplasia and pulmonary compromise) exists along a broad continuum, and many PBS patients also experience numerous concomitant non-genitourinary associations including cardiopulmonary, gastrointestinal and musculoskeletal anomalies.1–4 No best practice guidelines exist for management of PBS, owing to the scarcity and complex nature of the disease, along with diverse multisystem co-morbidities. The current PBS literature is predominantly comprised of case reports and small, single institution case series, further impeding efforts to reach evidence-based conclusions and standards of care. Herein, we review recent evidence in the pathogenesis, diagnosis and management of children with PBS.
Epidemiology
By strict definition which includes cryptorchidism as one of the three key features, PBS affects boys, with a contemporary incidence of 3.8 per 100,000 live male births in the United States.5 Females represent less than 5% of all PBS cases, and they present with abdominal wall deficiency and a dilated, dysmorphic urinary tract without any associated gonadal anomaly.6 Utilizing the 2000, 2003 and 2006 Kids’ Inpatient Databases, Routh et al reported an increased proportion of black newborns with PBS compared to that of the general population, along with a decreased incidence in Hispanic patients.5
While a clear cause of PBS remains evasive, there is increasing evidence to support a genetic component. Although PBS often presents as a sporadic event, the high concordance rate in twins (12.2 per 100,000 live births), case reports of monozygotic male twins, familial case reports and a higher incidence in males, all suggest a genetic contribution.7–10 However, in cases of monozygotic twins, both discordance and concordance for PBS have been reported, implying that inherited genetic mutations alone cannot explain the pathogenesis of the syndrome.7,11,12
PBS is known to be associated with a genomic HNF1β (hepatocyte nuclear factor) mutation in 3% of cases.13–15 HNF1β is a transcription factor that regulates gene expression necessary for mesodermal and endodermal development, and is expressed in a wide host of tissues. Granberg et al studied 32 DNA samples from PBS patients (30 males and 2 females). In their analysis, one heterozygous mutation was identified in the HNF1β gene, resulting in a missense mutation that was observed to be functionally normal.15 Recently, Boghossian et al systematically screened the genome of 34 PBS patients. They identified numerous copy number variants in genes involved in mesodermal, muscle and urinary tract development (including a duplication that overlaps the BMPR1B gene – the BMP signaling pathway is known to contribute to mesodermal differentiation), further supporting a genetic contribution to the etiology of PBS.16
Pathogenesis
Two predominant theories have been proposed regarding the embryogenesis of PBS. Mesenchymal developmental defects have been suggested as the underlying defect. In this theory, an unknown primary defect in the lateral plate mesoderm occurs between the 6th and 10th weeks of gestation, resulting in maldevelopment of the abdominal wall musculature as well as the urinary tract.17 In the second theory, involving in utero bladder obstruction, a hypoplastic/dysplastic prostate or abnormal urethra prevents the outflow of urine. This obstruction results in proximal bladder, ureteral and renal dilation with secondary poor development of the abdominal wall and urinary muscle.18–20 The exact mechanism remains elusive, as does the molecular basis for PBS.
Diagnosis
The routine implementation of maternal sonography has rendered prenatal diagnosis of PBS the most common presentation.21–23 PBS presents prenatally with many sonogram findings comparable to that of lower urinary tract obstruction (LUTO). Although accurate diagnosis has been reported as early as 11–12 weeks of gestation, the classical findings of hydroureteronephrosis, megacystis, irregular abdominal wall circumference and/or oligohydramnios may not be consistently identified until later in pregnancy.24 Differential diagnosis includes causes of LUTO including posterior urethral valves, ureterocele and urethral atresia as well mimics such as megacystis-microcolon-intestinal-hypoperistalsis syndrome (MMIHS).23 Prenatal diagnosis of PBS should be considered whenever the following ultrasound anomalies are clearly identified: oligohydramnios, urinary abnormalities (dilatation of the urinary tract, megacystis, bilateral hydroureteronephrosis), and the absence of abdominal musculature. Early, accurate diagnosis allows not only for prompt multidisciplinary management of newborns in a tertiary center at birth, resulting in improved survival, but also allows for the option of voluntary termination if desired.
Case reports of decompression of the urinary tract in utero with vesicoamniotic shunting in PBS fetuses exist.25–28 Criteria for intrauterine intervention includes second or third trimester gestation, oligohydramnios, megacystis, advanced hydronephrosis, normal karyotype, and an encouraging urinary index. Complications include dislodgement of the shunt, urinary ascites, premature labor, and chorioamnionitis. Although shunting may be effective in correcting oligohydramnios, there remains a lack of standardization and its ability to achieve adequate renal function is variable while pulmonary function cannot be assured despite restoration of normal amniotic fluid levels.29
Neonatal presentation
Abdominal wall appearance of the neonate immediately suggests PBS (Figure 1), regardless of whether the diagnosis was known prenatally. Children with PBS present a challenge and require prompt specialized care with a multidisciplinary care team, including a neonatologist, urologist, and nephrologist as well as cardiology and orthopedics when indicated. As is the case with LUTO, the initial postnatal course is dictated by the severity of co-morbidities such as pulmonary hypoplasia. Upwards of three-fourths of children with PBS have concomitant diagnoses with prematurity being the most common.1,5,30,31 Despite marked advances in neonatal care, perinatal mortality rates remain high, ranging between 10% and 25%. Early demise is largely attributed to the degree of prematurity and pulmonary hypoplasia.
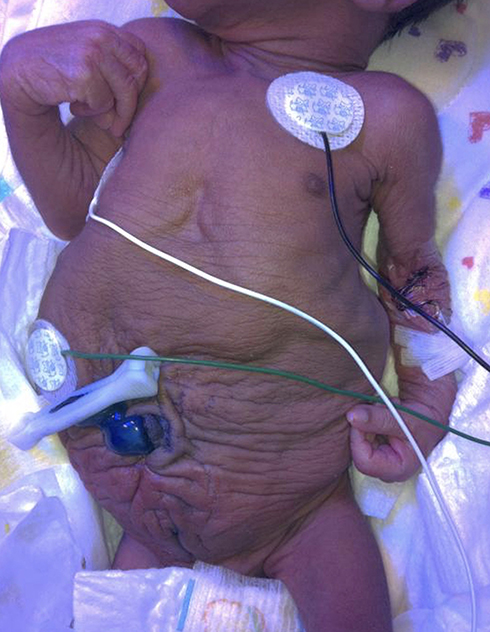 |
Figure 1 Appearance of a newborn with PBS: wrinkled, redundant skin with bulging at the flanks due to deficient of abdominal wall musculature. |
There are three major categories of neonatal presentation as described by Woodard (Table 1).32 Category I consists of patients with pronounced oligohydramnios secondary to renal dysplasia and/or outlet obstruction resulting in severe pulmonary hypoplasia and skeletal abnormalities; these neonates often expire within a few days and interventions are limited. Potter’s facies may be present and is secondary to oligohydramnios. Features include micrognathia, wide set eyes, flattened palpebral fissures, prominent epicanthus, flattened nasal bridge, low-set ears lacking cartilage and skeletal deformities.33 Cases of urethral atresia are typically in this most severe category. Boys in category II experience moderate renal insufficiency and moderate-severe hydroureteronephrosis, though pulmonary hypoplasia is not a prominent feature. Category III consists of patients with mild triad features or incomplete forms; renal function is typically normal or mildly impaired and there is no pulmonary insufficiency.
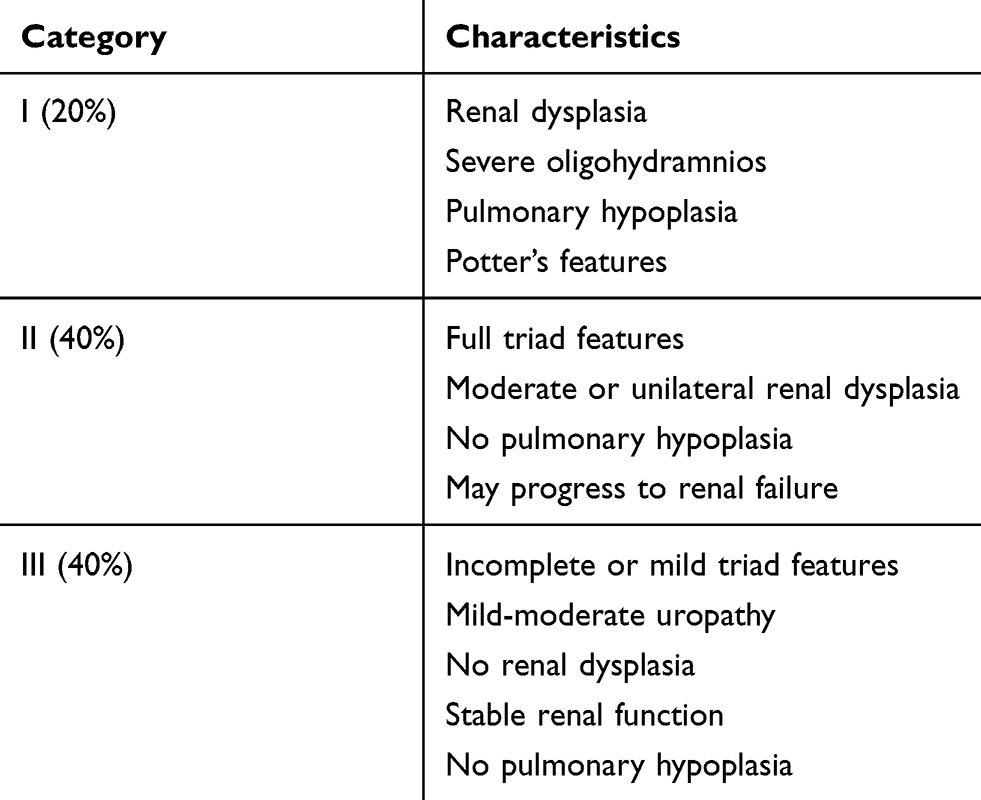 |
Table 1 Spectrum of prune belly syndrome |
Management issues in PBS
There are no guidelines nor consensus regarding management of children with PBS, and standardized treatment is further confounded by the rarity and broad spectrum of severity of the syndrome. Eventual repair of abdominal wall flaccidity and various urinary tract anomalies must be considered, along with the mandatory correction of cryptorchidism. Timing, and whether to stage surgical interventions or perform a more comprehensive approach, is also a factor.34
Urinary tract
One consistent hallmark of PBS is low-pressure dilation of the urinary tract, extending from the renal pelvis proximally to the urethra distally. The bladder is typically enlarged and hypotonic, with elevated compliance and low-pressure vesicoureteral reflux (VUR) present in approximately 75% of patients.35 While these capacious bladders are competent at storage, they often demonstrate incomplete emptying secondary to reduced detrusor contractility.36 Therefore, initial urologic intervention is directed at bladder drainage in order to preserve renal function. Retroperitoneal ultrasound is necessary to assess the renal parenchyma for dysplasia and degree of dilation. While instrumentation is to be avoided in general, a voiding cystourethrogram (VCUG) should be obtained to determine emptying and assess the bladder outlet.23 Avoidance of urinary tract infection (UTI) is crucial in the child with PBS as dilation, stasis, urinary reflux and compromised baseline kidney function are frequently present. As such, circumcision and prophylactic antibiotics are often implemented to decrease the incidence of UTI and protect the upper tracts from further insult.
Extent and timing of urinary tract reconstruction remains a source of debate and is best tailored to a given child’s bladder dynamics while also taking into consideration respiratory status. Early simultaneous correction of PBS anomalies with individualized urinary tract intervention, bilateral orchidopexy and abdominal wall reconstruction has been described in the literature.3,34,37 Preservation of renal function is of utmost importance. Children with incomplete bladder emptying and/or recurrent UTIs with VUR may require appendicovesicostomy to facilitate clean intermittent catheterization and/or anti-reflux surgery when conservative management such as prophylactic antibiotics and timed/double voiding fails to prevent infection and achieve adequate emptying. In the setting of complex urinary tract anatomy, magnetic resonance imaging (MRU) has proven useful as it provides both functional and anatomical detail. The utility of MRU in boys with PBS has been reported; it allows for detailed characterization of upper tract abnormalities such as calyceal diverticula and renal dysplasia, which may also help with operative planning.38
Abdominal wall
Abdominal wall reconstruction is an important consideration in the surgical management of PBS patients. Histologic analysis of muscle in these patients has revealed fatty infiltration and fibrous replacement, along with normal nerve distribution and the presence of striated muscle cells consistent with a developmental anomaly rather than muscular atrophy.39 The basis of abdominoplasty is to advance the more normal peripheral abdominal wall to support the abnormal, deficient central portions.40,41 Traditionally, three surgical procedures have been reported in the literature for correction of the abdominal wall defects associated with PBS. The Randolph procedure describes excision of a portion of the lower abdominal wall to correct vertical fascial redundancy. The Monfort and Ehrlich procedures both describe correction of the lateral redundancy along with strengthening of the abdominal wall by vertical overlapping of the fascia.42–44 A laparoscopic approach to abdominoplasty has also been reported, proving beneficial in both assisting with the reconstruction itself as well as delineating the varying degrees of muscular deficiency.45
The severity of muscle/fascia deficiency is not consistent across the abdomen – often there is absence of the muscle in the medial and inferior portions of the abdominal wall while the upper rectus and external oblique muscles are present but hypoplastic. As a consequence of the laxity and elongation of the inferior fascia, the umbilicus tends to be displaced superiorly. Furthermore, PBS patients may each display unique features with regard to the variation of severity and pattern of abdominal musculature deficiency, such as a particular area with a complete paucity of muscle. In order to provide more individualized correction of abdominal wall laxity and allow for positioning the umbilicus to a more anatomically correct location, Smith and colleagues recently reported their adaptations to the Monfort procedure.46 These modifications are detailed in Figure 2. The addition of rectus femoris muscle transposition has also been reported to supplement traditional abdominal wall reconstruction.47
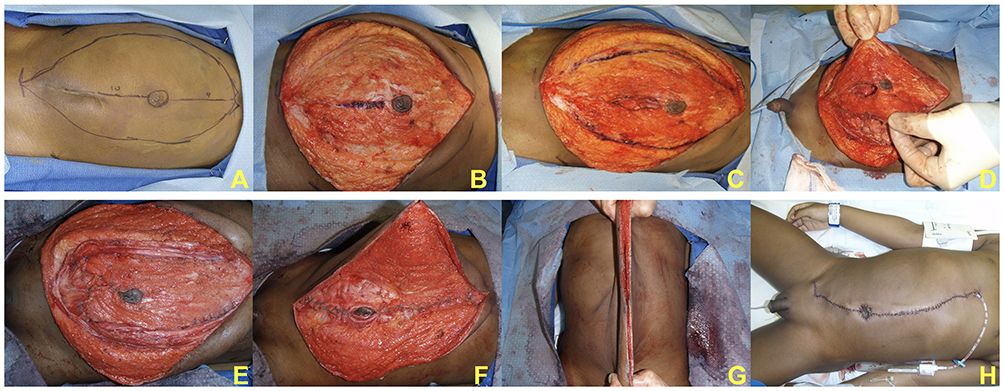 |
Figure 2 Monfort modifications. Preoperative asymmetric laxity with midline incision and extent of subcutaneous dissection marked out; note that the right (upper in image) laxity is more significant than left (A). After subcutaneous dissection (B), the lateral fascial incisions are marked as well as the central fascial bridge with the umbilical island (C). Lateral incisions provide exposure to the peritoneal cavity for urinary reconstruction when necessary (D). Closure of infraumbilical incision shifts umbilicus inferiorly to a more anatomic position (E). Lateral abdominal walls are brought over the midline fascial plate, plicating the abdominal wall in a double-breasted fashion (F). Excess skin removed (G) and brought back together in midline completing the procedure (H). |
In addition to cosmesis, abdominoplasty has also been shown to improve functional bladder dynamics independent of genitourinary reconstruction.48 Urodynamic testing is often performed preoperatively to determine the need for any concomitant bladder procedures such as appendicovesicostomy in cases of incomplete emptying and/or ureteral reimplantation when VUR is present. Durable, satisfactory cosmetic results following abdominal wall reconstruction have been reported with long-term follow-up of up to two decades.49
Cryptorchidism
Bilateral intra-abdominal testicles are another trademark feature of PBS. Failure of testicular descent is hypothesized to be secondary to mechanical obstruction from megacystis and hydroureteronephrosis. Anatomic changes in the anterior abdominal wall are also thought to play a role by hindering the elevation of intra-abdominal pressure, one of the factors necessary for testicular descent.50 Traditional single-stage orchidopexy and single versus staged Fowler-Stephens orchidopexy are all potential options for managing the testicles, depending on gonadal vessel length, and can be performed via an open or laparoscopic approach depending upon surgeon preference. Philip et al reported on the use of radially expanding trocars and high gas flow rates during laparoscopic orchidopexy to address potential technical issues unique to PBS boys.51
Orchidopexy can be performed at the same time as other surgical procedures, such as appendicovesicostomy and abdominoplasty,50 to limit the number of general anesthetics. This may be of particular benefit in children with compromised pulmonary status. It should be emphasized, however, that in the absence of any overriding cardiopulmonary issue, orchidopexy should be performed as recommended by the current AUA guideline and not delayed until older in childhood.52,53 Rather, since germinative epithelium is significantly reduced in males with PBS, orchidopexy should be performed as early as possible to enhance the chances of fertility.54–57 Division of the gonadal vessels is less likely when orchidopexy is performed early. There are multiple reports of live births resulting from intracytoplasmic sperm injection (ICSI) using spermatozoa from adult patients with PBS, further highlighting the importance of appropriate, timely management of the cryptorchid testes.58
Renal failure
Renal dysplasia and dysfunction is common in the PBS population, with approximately 40–50% of patients ultimately requiring renal replacement therapy.59 Early end-stage renal disease (ESRD) is thought to be secondary to renal dysplasia, whereas kidney failure occurring later is often attributed to parenchymal damage from repeated infections and the increased pressure transmitted to the upper tracts generated from incomplete emptying.60 Presence of one normal kidney on ultrasound and a nadir serum creatinine of <0.7 mg/dL in infancy are predictive of satisfactory long-term renal function.61
Kidney transplantation remains the gold standard for pediatric patients with ESRD regardless of etiology, as it provides a known survival advantage over peritoneal or hemodialysis.62 A recent study by Yalcinkaya et al reported the median age of initial renal replacement therapy for boys with PBS was 7 years, significantly younger than male patients with other types of LUTO or renal dysplasia, with a median age at first transplant at just 9.3 years.63 One and 5 year renal transplant graft survival rates have been reported to be lower for children with lower urinary tract dysfunction including PBS, with a 5 year 67% graft survival rate. This highlights a serious concern over time, as PBS patients are often transplanted at a younger age, and thus are at higher risk for requiring multiple transplants over their lifetime.64,65 Despite the young age at which PBS patients are transplanted, in 2016 Bagga et al reported a trend toward delayed time to first renal transplant in patients with congenital urinary tract anomalies, indicating that improved urological and nephrological care may result in renal preservation.66 These findings stress the importance of early and lifelong nephrology care.
Quality of life
It is well documented that children with chronic illness often experience physical and/or cognitive limitations, which in turn may be associated with fatigue, reduced activity level and emotional distress, all of which impacts well-being.67 Health-related quality of life in children, which strives to understand the impact of a disease and its treatment, is increasingly being recognized as a salient outcome measure along with more traditional health indicators such as survival.68 Not surprisingly, PBS has been shown to profoundly affect quality of life in pediatric patients, negatively impacting their physical, emotional, social and school functioning, highlighting the need to determine which interventions positively influence patient-reported quality of life.69
Conclusions
As with most complex congenital anomalies, the key to management of PBS is a multidisciplinary team-based approach providing individualized care. Long-term surveillance of the urinary tract is crucial as bladder dynamics and renal function can change over time. As technological advances continue to improve the overall survival and life expectancy of PBS patients, the challenge remains to develop best practice standards and provide comprehensive care while mitigating potential negative disease sequelae.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Hassett S, Smith GHH, Holland AJA. Prune belly syndrome. Pediatr Surg Int. 2012;28:219–228. doi:10.1007/s00383-011-3046-6
2. Smolkin T, Soudack M, Goldstein I, Sujov P, Makhoul IR. Prune belly syndrome: expanding the phenotype. Clin Dysmorphol. 2008;17:133–135.
3. Seidel NE, Arlen AM, Smith EA, Kirsch AJ. Clinical manifestations and management of prune-belly syndrome in a large contemporary pediatric population. Urology. 2015;85:211–215.
4. Grimsby GM, Harrison SM, Granberg CF, Berstein IH, Baker LA. Impact and frequency of extra-genitourinary manifestations of prune belly syndrome. J Pediatr Urol. 2015;11:280e1–6.
5. Routh JC, Huang L, Retik AB, Nelson CP. Contemporary epidemiology and characterization of newborn males with prune belly syndrome. Urology. 2010;76:44–48.
6. Lloyd JC, Wiener JS, Gargollo PC, Inman BA, Ross SS, Routh JC. Contemporary epidemiological trends in complex congenital genitourinary anomalies. J Urol. 2013;194:1590–1595.
7. Balaji KC, Patil A, Townes PL, Primack W, Skare J, Hopkins T. Concordant prune belly syndrome in monozygotic twins. Urology. 2000;55:949.
8. Garlinger P, Ott J. Prune belly syndrome: possible genetic implications. Birth Defects Orig Artic Ser. 1974;10:173.
9. Lockhart JL, Reeve HR, Bredael JJ, Krueger RP. Siblings with prune belly syndrome and associated pulmonic stensosis, mental retardation, and deafness. Urology. 1979;14:140.
10. Ramasamy R, Haviland M, Woodard JR, Barone JG. Patterns of inheritance in familial prune belly syndrome. Urology. 2005;65:1227. doi:10.1016/j.urology.2004.09.047
11. Druschel CM. A descriptive study of prune belly in New York State, 1983 to 1989. Arch Pediatr Adolesc Med. 1995;149:70–76.
12. Petersen DS, Fish L, Cass AS. Twins with congenital deficiency of abdominal musculature. J Urol. 1972;107:670–672.
13. Murray PJ, Thomas K, Mulgrew CG, Ellard S, Edgehill EL, Bingham C. Whole gene deletion of the hepatocyte nuclear factor-1beta gene in a patient with the prune-belly syndrome. Nephrol Dial Transplant. 2008;23:2412–2415.
14. Haeri S, Devers PL, Kaiser-Rogers KA, et al. Deletion of hepatocyte nuclear factor-1beta in an infant with prune belly syndrome. Am J Perinatol. 2010;27:559–563.
15. Granberg CF, Harrison SM, Dajusta D, et al. Genetic basis of prune belly syndrome: screening for HNF1B gene. J Urol. 2012;187(1):272–278.
16. Boghossian NS, Sicko RJ, Giannakou A, et al. Rare copy number variants identified in prune belly syndrome. Eur J Med Genet. 2018;61(3):145–151.
17. Ives EJ. The abdominal muscle deficiency triad syndrome – experience with ten cases. Birth Defects Orig Artic Ser. 1974;10:127–135.
18. Manivel JC, Pettinato G, Reinberg Y, Gonzalez R, Burke B, Dehner LP. Prune belly syndrome: clinicopathologic study of 29 cases. Pediatr Pathol. 1989;9:691–711.
19. Gonzalez R, Reinberg Y, Burke B, Wells T, Vernier RL. Early bladder outlet obstruction in fetal lambs induces renal dysplasia and the prune-belly syndrome. J Pediatr Surg. 1990;25:342–345.
20. Hutson JM, Beasley SW. Aetiology of the prune belly syndrome. Aust Paediatr J. 1987;23:309–310.
21. Tonni G, Ida V, Alessandro V, Bonasoni MP. Prune-belly syndrome: case series and review of the literature regarding early prenatal diagnosis, epidemiology, genetic factors, treatment, and prognosis. Fetal Pediatr Pathol. 2013;31(1):13–24.
22. Achour R, Bennour W, Ksibi I, et al. Prune belly syndrome: approaches to its diagnosis and management. Intractable Rare Dis Res. 2018;7(4):271–274.
23. Clayton DB, Brock JW
24. Chen L, Cai A, Wang X, Li J. Two-and three-dimenstional prenatal sonographic diagnosis of prune-belly syndrome. J Clin Ultrasound. 2010;38:279–282.
25. Austin JC, Canning DA, Johnson MP, Flake AW, Carr MC. Vesicoamniotic shunt in a female fetus with the prune belly syndrome. J Urol. 2001;166:2382.
26. Galati V, Beeson JH, Confer SD, et al. A favorable outcome following 32 vesicocentesis and amnioinfusion procedures in a fetus with severe prune belly syndrome. J Pediatr Urol. 2008;4:170–172.
27. Muller Brochut AC, Thomann D, Kluwe W, Di Naro E, Kuhn A, Raio L. Fetal megacystis: experience of a single tertiary center in Switzerland over 20 years. Fetal Diagn Ther. 2014;36(3):215–222.
28. White JT, Sheth KR, Bilgutay AE, et al. Vesicoamniotic shunting improves outcomes in a subset of prune belly syndrome patients at a single tertiary center. Front Pediatr. 2018;6:229.
29. McLorie G, Farhat W, Khoury A, Geary D, Ryan G. Outcome analysis of vesicoamniotic shunting in a comprehensive population. J Urol. 2001;166(3):1036–1040.
30. Fallat ME, Skoog SJ, Belman AB, Eng G, Randolph JC. The prune belly syndrome: a comprehensive approach to management. J Urol. 1989;142:802–805.
31. Burbige KA, Amodio J, Berdon WE, Hensle TW, Blanc W, Lattimer JK. Prune belly syndrome: 35 years of experience. J Urol. 1987;137:86–90.
32. Woodard JR. The prune belly syndrome. Urol Clinic North Am. 1978;5:75–93.
33. Woods AG, Brandon DH. Prune belly syndrome: a focused physical assessment. Adv Neonatal Care. 2007;7(3):132–143.
34. Lopes RI, Tavares A, Srougi M, Denes FT. 27 years of experience with the comprehensive surgical treatment of prune belly syndrome. J Pediatr Urol. 2015;11:276e1–7.
35. Bellah RD, States LJ, Duckett JW. Pseudoprune-belly syndrome: imaging findings and clinical outcome. AJR Am J Roentgenol. 1996;167:1389–1393.
36. Kinahan TJ, Churchill BM, McLorie GA, Gilmour RF, Khoury AE. The efficiency of bladder emptying in the prune belly syndrome. J Urol. 1992;148:600–603.
37. Denes FT, Arap MA, AM S, FA A. Comprehensive surgical treatment of prune belly syndrome: 17 years’ experience with 32 patients. Urology. 2004;64:789–793.
38. Garcia-Roig ML, Grattan-Smith JD, Arlen AM, Smith EA, Kirsch AJ. Detailed evaluation of the upper urinary tract in patients with prune belly syndrome using magnetic resonance urography. J Pediatr Urol. 2016;12:122e1–7.
39. Loder RT, Guiboux JP, Bloom DA, Hensinger RN. Musculoskeletal aspects of prune-belly syndrome: description and pathogenesis. Am J Dis Child. 1992;146:1224–1229.
40. Mininberg DT, Montoya F, Okada K, Galioto F, Presutti R. Subcellular muscle studies in the prune belly syndrome. J Urol. 1973;109:524–526.
41. Afifi AK, Rebeiz J, Mire J, Andonian J, Kaloustian V. The myopathology of the prune belly syndrome. J Neurol Sci. 1972;15:153–165.
42. Randolph J, Eng G. Abdominal wall reconstruction in the prune belly syndrome. J Pediatr Surg. 1981;16(6):960–964.
43. Monfort G, Guys JM, Bocciardi A, Coquet M, Chevallier D. A novel technique for reconstruction of the abdominal wall in the prune belly syndrome. J Urol. 1991;146(2):639–640.
44. RM E, MA L, RN F. Total abdominal wall reconstruction in the prune belly syndrome. J Urol. 1986;136:282–285.
45. Franco I. Laparoscopic assisted modification of the Firlit abdominal wall plication. J Urol. 2005;174:280–283.
46. Smith EA, Srinivasan A, Scherz HC, Tracey AJ, Broecker B, Kirsch AJ. Abdominoplasty in prune belly syndrome: modifications in monfort technique to address variable patterns of abdominal wall weakness. J Pediatr Urol. 2017;13:502e1–6.
47. Fearon JA, Varkarakis G. Dynamic abdominoplasty for the treatment of prune belly syndrome. Plast Reconstr Surg. 2012;130(3):648–657.
48. Smith CA, Smith EA, Parrott TS, Broecker BH, Woodard JR. Voiding function in patients with the prune-belly syndrome after monfort abdominoplasty. J Urol. 1998;159:1675–1679.
49. Lesavoy MA, Chang E, Suliman A, Taylor J, Kim SE, Ehrlich RM. Long-term follow-up of total abdominal wall reconstruction for prune belly syndrome. Plast Reconstr Surg. 2012;129:104–109.
50. Zugor V, Schott GE, Labanaris AP. The prune belly syndrome: urological aspects and long-term outcomes of a rare disease. Pediatr Rep. 2012;4:e20.
51. Philip J, Mullassery D, Craigie RJ, Manikandan R, Kenny SE. Laparoscopic orchidopexy in boys with prune belly syndrome–outcome and technical considerations. J Endourol. 2011;25(7):1115–1117.
52. Kolon TF, Herndon CD, Baker LA, et al. Evaluation and treatment of cryptorchidism: AUA guideline. J Urol. 2014;192:337–345.
53. Hutson JM, Balic A, Nation T, Southwell B. Cryptorchidism. Semin Pediatr Surg. 2010;19(3):215–224.
54. Orvis BR, Bottles K, Kogan BA. Testicular histology in fetuses with the prune belly syndrome and posterior urethral valves. J Urol. 1988;139(2):335–337.
55. Massad CA, Cohen MB, Kogan BA, Beckstead JH. Morphology and histochemistry of infant testes in the prune belly syndrome. J Urol. 1991;146(6):1598–1600.
56. Uehling DT, Zadina SP, Gilbert E. Testicular histology in triad syndrome. Urology. 1984;23(4):364–366.
57. Favorito LA, Costa SF, Costa WS, Vieiralves R, Bernado FO, Sampaio FJB. Study of testicular structure in fetuses with prune belly syndrome. Adv Urol. 2017;2017:3254980.
58. Fleming SD, Varughese E, Hua VK, Robertson A, Dalzell F, Boothroyd CV. Normal live births after intracytoplasmic sperm injection in a man with the rare condition of eagle-barrett syndrome (prune-belly syndrome). Fertil Steril. 2013;100(6):1532–1535.
59. Penna FJ, Elder JS. CKD and bladder problems in children. Adv Chronic Kidney Dis. 2011;18:362–369.
60. Noh PH, Cooper CS, Winkler AC, Zderic SA, Snyder HM
61. Reinberg Y, Manivel JC, Pettinato G, Gonzalez R. Development of renal failure in children with the prune belly syndrome. J Urol. 1991;195:1017–1019.
62. Dharnidharka VR, Fiorina P, Harmon WE. Kidney transplantation in children. N Engl J Med. 2014;371:549–558.
63. Yalcinkaya F, Bonthuis M, Erdogan BD, et al. Outcomes of renal replacement therapy in boys with prune belly syndrome: findings from the ESPN/ERA-EDTA registry. Pediatr Neph. 2018;33(1):117–124.
64. Fusaro F, Zanon GF, Ferreli AM, et al. Renal transplantation in prune-belly syndrome. Transpl Int. 2004;17:549–552.
65. Adams J, Mehls O, Wiesel M. Pediatric renal transplantation and the dysfunctional bladder. Transpl Int. 2004;17:596–602.
66. Bagga HS, Lin S, Williams A, et al. Trends in renal transplantation rates in patients with congenital urinary tract disorders. J Urol. 2016;195:1257–1262.
67. Kim J, Chung H, Amtmann D, et al. Symptoms and quality of life indicators among children with chronic medical conditions. Disabil Health J. 2014;7:96–104.
68. Varni JW, Limbers CA, Burwinkle TM. Impaired health-related quality of life in children and adolescents with chronic conditions: a comparative analysis of 10 disease clusters and 33 disease categories/severities utilizing the PedsQL 4.0 generic core scales. Health Qual Life Outcomes. 2007;5:43.
69. Arlen AM, Kirsch SS, Seidel NE, Garcia-Roig M, Smith EA, Kirsch AJ. Health-related quality of life in children with prune belly syndrome and their caregivers. Urology. 2016;87:224–227.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.