Back to Journals » Infection and Drug Resistance » Volume 13
Proteomic Applications in Antimicrobial Resistance and Clinical Microbiology Studies
Authors Khodadadi E, Zeinalzadeh E, Taghizadeh S, Mehramouz B ![]() , Kamounah FS
, Kamounah FS ![]() , Khodadadi E, Ganbarov K
, Khodadadi E, Ganbarov K ![]() , Yousefi B
, Yousefi B ![]() , Bastami M
, Bastami M ![]() , Kafil HS
, Kafil HS ![]()
Received 13 November 2019
Accepted for publication 23 May 2020
Published 16 June 2020 Volume 2020:13 Pages 1785—1806
DOI https://doi.org/10.2147/IDR.S238446
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Eric Nulens
Ehsaneh Khodadadi,1 Elham Zeinalzadeh,2,3 Sepehr Taghizadeh,1 Bahareh Mehramouz,3 Fadhil S Kamounah,4 Ehsan Khodadadi,5 Khudaverdi Ganbarov,6 Bahman Yousefi,7 Milad Bastami,7 Hossein Samadi Kafil1
1Drug Applied Research Center, Faculty of Medicine, Tabriz University of Medical Sciences, Tabriz, Iran; 2Student Research Committee, Tabriz University of Medical Sciences, Tabriz, Iran; 3Hematology and Oncology Research Center, Tabriz University of Medical Sciences, Tabriz, Iran; 4Department of Chemistry, University of Copenhagen, Copenhagen, DK 2100, Denmark; 5Department of Biology, Tabriz Branch, Islamic Azad University, Tabriz, Iran; 6Department of Microbiology, Baku State University, Baku, Azerbaijan; 7Immunology Research Center, Tabriz University of Medical Sciences, Tabriz, Iran
Correspondence: Hossein Samadi Kafil
Drug Applied Research Center, Department of Microbiology, Faculty of Medicine, Tabriz University of Medical Sciences, Golgasht Ave, Tabriz 5166614711, Iran
Tel +98-9127184735
Fax +98-4133364661
Email [email protected]
Abstract: Sequences of the genomes of all-important bacterial pathogens of man, plants, and animals have been completed. Still, it is not enough to achieve complete information of all the mechanisms controlling the biological processes of an organism. Along with all advances in different proteomics technologies, proteomics has completed our knowledge of biological processes all around the world. Proteomics is a valuable technique to explain the complement of proteins in any organism. One of the fields that has been notably benefited from other systems approaches is bacterial pathogenesis. An emerging field is to use proteomics to examine the infectious agents in terms of, among many, the response the host and pathogen to the infection process, which leads to a deeper knowledge of the mechanisms of bacterial virulence. This trend also enables us to identify quantitative measurements for proteins extracted from microorganisms. The present review study is an attempt to summarize a variety of different proteomic techniques and advances. The significant applications in bacterial pathogenesis studies are also covered. Moreover, the areas where proteomics may lead the future studies are introduced.
Keywords: bacterial pathogenesis studies, drug resistance, virulence, pathogen, proteomics
Introduction
Proteins are responsible for the biological functions that are dictated by genes in most cases.1 The vast protein interaction networks control the strange cellular functions mainly. It is not possible to elaborate on these networks by merely relying on a single protein or a few proteins.2 One of the ways to explain the biological systems of microorganisms in a large scale is proteomics. This technique provides us with information as to abundances, post-translational modifications, localization, interactions, and changes.3 The sustained development of different proteomic technologies determines the capacity of proteomics to deal with major issues in the microbial field. There is a need for qualitative and quantitative studies in this field.4 Other systems approaches have also notable benefits for microbial pathogenesis. There is an emerging trend of using proteomics to study infectious agents.5 Using proteomic analysis to study protein profiles of bacterial pathogenesis is one of the main approaches to study proteins and interactions of the host-pathogen to find a deeper knowledge of dysregulations in infection disorders,6 reveal bacterial resistance and virulence mechanisms,7 and significant new targets for future drug discovery.8 The immense potential of proteomic technologies to achieve a deeper insight into pathogenesis and develop therapeutic techniques is undeniable.
Pathogenic microorganisms like viruses, bacteria, or fungi9 are responsible for infectious diseases and represent serious health risks for man, animals, and plants.10 In spite of great works to develop new strategies to fight and prevent infections, the risk of newly emerging infectious diseases is undeniable.11 The key point of infectious disease researches is a deeper insight into the functional interface between pathogenic microbes and their host cells.12 Still, our knowledge of exact molecular adhesion, invasion, and replication is quite limited.13 This lack of knowledge is an obstacle to develop new diagnostic and therapeutic strategies.14 Additionally, the complicated interaction between host and pathogens is controlled by hundreds to thousands of proteins from both sides.15 Most of the research work in this field has concentrated on determining the characterization of individual bacterial virulence factors and their interacting host targets using traditional genetic and biochemical approaches.16 However, these studies fail to elaborate on the complicated multifactorial nature of host-pathogen interactions.17 On the other hand, systems-level analyses give us a panoramic perspective of the functional host-pathogen interplay, which is significant improvement progress from the traditional reductionism-dominant research.18 Therefore, transcriptomic studies have been around for several years and still, there is a great desire for measuring the final gene products, proteins. This is because of the poor correlation between mRNA and protein levels due to extensive post-transcriptional regulations.
One of the most important and interesting aspects of life is the ongoing interaction between hosts and pathogens.19 These interactions take place throughout the long years of evolution; so that the hosts create defense mechanisms to handle pathogenic invasions and pathogens circumvent these new defense mechanisms.20 Thanks to adaptation processes, some hosts can co-exist with or even have the benefit of pathogens. However, many pathogens still function as etiological agents for many life-threatening human diseases.21 Therefore, having a clear understanding of host-pathogen interactions has led to the introduction of different means to prevent and treat infection-induced diseases. This study discusses the advantages and drawbacks of a gel-free/label-free proteomic technique along with introducing the potential application of proteomics in bacterial pathogen studies. In addition, the availability of proteomics approaches to uncover host-pathogen protein interaction networks, changes in the composition, and the organization of the host cell proteome are explained.
Applications of Proteomic Techniques in Bacteria
The metabolic aspects of an organism on a global scale are the subject matter of proteomic studies. Through this, large-scale proteomic technologies are developed prosperously.5 Proteomic studies enable us to identify genome or/and measure proteins from microorganisms in a quantitative manner.22 Researcher keeps developing proteomic techniques so that there are wide range methods and applications available.23 Needless to say, proteomic technologies provide great potential to shed light on pathogenesis and develop new therapeutic techniques based on these insights. The latest studies have conducted reference proteomes for different bacterial pathogens and direct the future studies that need baseline proteomes for performing comparison.24 Valuable information is provided by this technological platform as to signal transduction, adherence, and microbial-host interactions pertinent to bacterial pathogenesis.
Protein Identification
Measuring protein using the 2D gel electrophoresis method is the standard way for proteomic analysis.25 The original separation technology (2-DE) can separate proteins based on their isoelectric point and molecular weight using SDS-polyacrylamide gel electrophoresis in the first and second dimensions, respectively.26 In addition, to have sensitivity, covalent labeling of proteins with fluorescent Cy-dyes is used before separation. This technique is known as 2D difference gel electrophoresis (DIGE) can achieve higher quality and number of protein spots and gives more reliable gel matching.27,28 On the other hand, these gel-based techniques are not sensitive enough to small quantities of proteins and they have limited proteome resolution.29 Another disadvantage of these approaches is their poor performance in detecting different types of post-translational modifications of a single protein that causes crosstalk among signal pathways.30 While one of the disadvantages of membrane proteomics based on the gel-free approach is the solubilization of membranous proteins, which is because of different optimum condition,31 the volume of data available for membrane protein repertoire is growing.32 Several bacterial studies including Mycobacterium tuberculosis,33 Scheffersomyces stipitis,34 and Staphylococcus aureus35 have used a gel-free technique, which further indicates the potential of this method by the identification of a far larger number of proteins. Gel-based and gel-free protein quantification, which are used as complementary approaches, are effective techniques to analyze the regulatory mechanisms utilized by bacteria. In Klebsiella pneumoniae as a successful example, the P13K-mediated vesicular transport was identified by the combination of both approaches.36 Thus, it is essential for studies on plant stress responses to carefully select the proteomic approaches and cellular events that should be resolved by the approach.
Quantitative Proteomics
Both relative and absolute protein quantification are supported by mass spectrometry (MS)-based quantification strategy.37 Metabolic in vivo labeling techniques like SILAC (stable isotope labeling with amino acids in cell culture) and15 N labeling makes it possible to measure smaller measurement bias.38 A chemical in vitro labeling methods like the ICAT (isotope-coded affinity tag),18 O labeling, TMT (tandem mass tags) and iTRAQ (isobaric tags for relative and absolute quantification) can be used for static samples such as clinical samples.39,40 Another identical strategy called isotope-coded protein label (ICPL) labels both N-termini and lysine side chains and is used at the protein level.41 Currently, TMT and iTRAQ are the most commonly used techniques for labeling as it can be used for differential quantification of different protein post-translational modifications.42 The iTRAQ-based differential proteomics of total proteins using a Rhodococcus sp. BAP-1induced by fluoranthene showed a decrease in the abundance of cytochrome ubiquinol oxidase subunit, NAD(P) transhydrogenase subunit alpha, 5-methyltetrahydropteroyltriglutamate-homocysteine methyltransferase; still, there was an increase in the abundance of NADPH-dependent FMN reductase, 30S ribosomal protein S2, and S-ribosylhomocysteinase.43–45 A technique to find differential bacterial proteomic profiling of Staphylococcus aureus46 is the iTRAQ-based strategy; still, limitations of label-based techniques create problems in experimental design to compare samples so that only a few studies have used iTRAQ-based strategy in bacteria. These studies have been mainly on the stress response, which needs comparison among multiple conditions.47 The use of the iTRAQ-based technique to subcellular compartments has the limitation of expensive reagents and the complicated process of preparing samples.48 On the other hand, label-free quantitation is free of any limitations as to the number of samples for analysis.49,50 With label-free quantitation based on MS/MS, liquid chromatography (LC) is used to separate the digested peptides and transferred to a first mass spectrometer (MS1) where the chromatograms depicting signal intensities are obtained to measure the abundance of each peptide.51 The peptide ions are adopted for deeper fragmentation in MS2 and determine the parent ion.52 Label-free LC-MS/MS gives us the chance for wide quantification of proteins.53 Thanks to advantages like easy sample preparation that is done faster in gel-free, label-free quantification allows accumulation of large volume of data in S. aureus proteomics, revealing central responses of S. aureus exposure to cold stress. In the case of subcellular proteomics in S. aureus, changes in the specific factors indicate the importance of citric acid-related signal transduction,54 which controls the early stage of the bacteria’s response to stress. Still, the unsolved problem in this method is how to optimize LC-MS chromatogram alignment for accurate quantification.55 Many platforms use MS/MS scan times or base peak information to align chromatograms.56 The merit of gel-free, label-free proteomics is in the ease of sample preparation and the acceptance for data production.57 That to the large-scale data analysis of accumulated data on protein abundance58 it is possible to elucidate biological processes that are missed in small-scale experiments (Table 1).
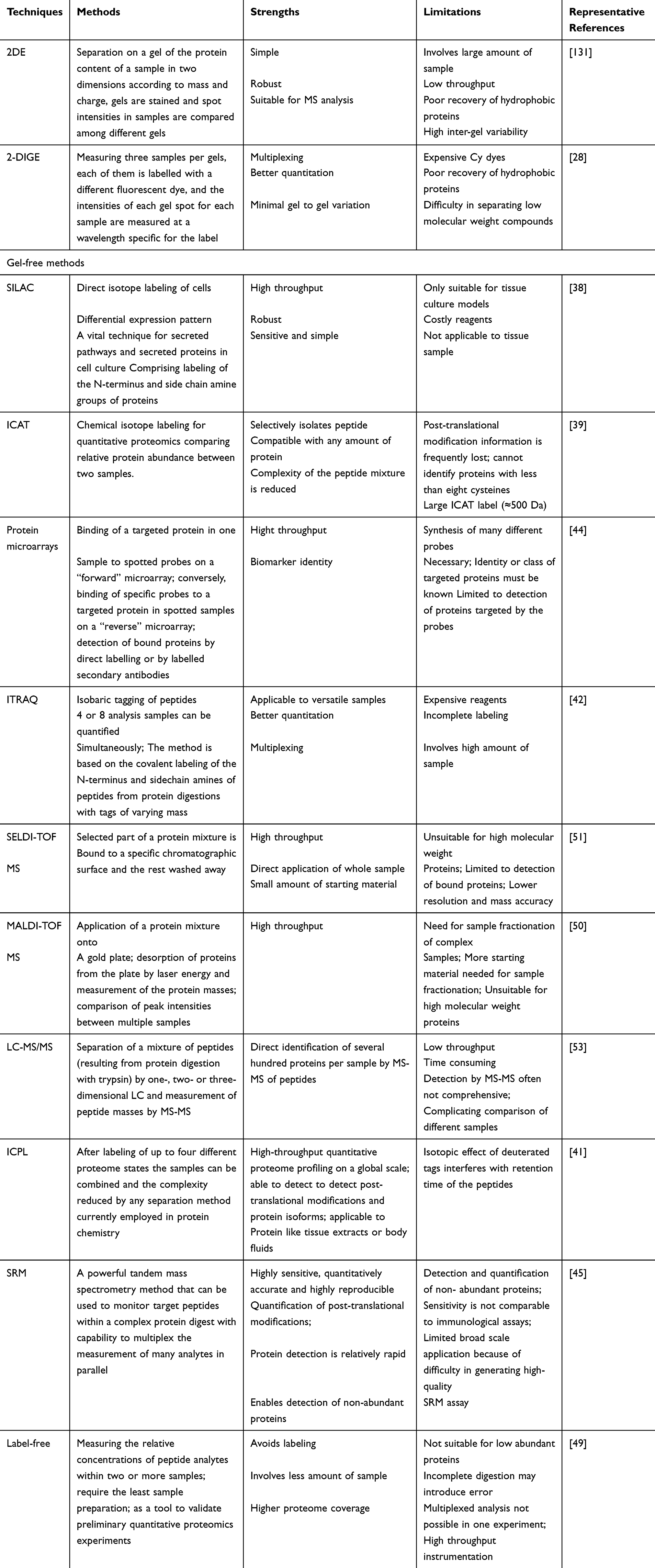 |
Table 1 Different Techniques in Quantitative Proteomic with the Associated Strengths and Limitations |
Proteomics Methods to Provide Mechanistic Insights in Infectious Diseases
Infection by different pathogens that are intrinsic to our ecosystem is the main reason for human disease and death worldwide.59 An interesting aspect of life is the ongoing interaction between hosts and pathogens.20 Researchers have concentrated on creating a molecular picture of pathogen infection and spread in an attempt to control the prevalence of infectious disease and develop better treatments for diseases.60 Therefore, to find more about pathogen-host interactions is a driving force for the event of suggests that to stop and treat infection-induced diseases.61 Over the past years, omic approaches have been introduced as effective tools in basic, translational, and clinical analysis to examine biological pathways effective in pathogen replication, host response, and disease progression. Proteomics tries to study the protein complement of biological systems and it has managed to show the discovery and understanding of pathogen-host interactions.6 This is the outcome of every improved proteomic technology that gives us sensitive protein detection and quantification tools. In addition, it increases awareness inside the biological science community and promotes using these approaches in innovative ways.
Intracellular Host-Pathogen Protein–Protein Interactions
The past ten years have witnessed a great contribution to comprehending host-pathogen interaction in the cellular life cycle of a pathogen by proteomic techniques.62 Notably, the hyphenation of traditional analytical and biochemical techniques based on mass spectrometry has led to proteomic approaches that examine different aspects of the host-pathogen relationship.11 Given that before reproducing to propagate, intracellular pathogens should pass through the host defenses, pathogen proteins interact with host proteins to either suppress or hijack the normal host protein functions.6 Identification of those protein–protein interactions (PPIs) is essential, among many, for understanding the biology of infection; in addition, it can be used for new targets in treatments against human pathogens. In this study, the proteomics strategies that can be used to discover pathogen-host interaction networks, intact protein complexes, or direct interactions are reviewed. Furthermore, their strengths, limitations, and future promising directions in the context of finding out infectious diseases are discussed.
Building Host-Pathogen Protein Interaction Networks
Immunoaffinity purification along with mass spectrometry (IP-MS) are of the methods that have received the widest attention in pathogen-host interaction studies.6 To isolate a protein in IP-MS, an antibody raised against the endogenous protein or epitope-tagging the protein of interest and using an antibody against that epitope are the options.63 Therefore, the protein of interest and co-isolated interacting proteins are identified using MS. As to host-pathogen associations, the main advantages of IP-MS are the fact that experiments can be done in pertinent cellular model systems and the context of viral infection so that unbiased detection of PPIs is possible.64 In the case of bacteria, IP-MS is utilized to detect interactions between effector proteins secreted by intracellular Salmonella and host proteins. Also, SILAC quantification is used to examine the specificity of interactions.65 The multiplexing capability of TMT is not used in host-pathogen PPI studies yet; still, it allows for the simultaneous measurement of different infection time points along with negative controls to examine the specificity of the interactions detected.20 Specific interactions of histone deacetylases by label-free methods and the relative stability of these interactions by SILAC were both determined using the combined analysis.66 Thus, these approaches can be expanded to find valuable information as to dynamic host-pathogen interactions.
The fact that infections can cause significant changes in protein abundances in a cell and that the background of non-specific associations can differ completely from the one observed in an uninfected cell are key issues in pathogen-host interaction studies.67 Thus, controlling isolations should be done in the same biological context under study. There are many computer algorithms available that utilize the data provided by control and experimental isolations to filter false-positive PPIs.68 One of them is the significance analysis of interactome (SAINT).69 This algorithm allocates interaction specificity scores to filter low-confidence interactions. Informatics approaches can also be employed to achieve a more refine identified interactions. For example, by creating extra controls for non-specific associations, like the contaminant repository for affinity purification (CRAPome).70 A recently developed database for HSV-1 interactions, HVint, creates an integrated resource of HSV-1 protein interactions. It uses using evolutionary conservation of herpesvirus proteins to further predict additional interactions.71 Thus, once a list of interactions is ready, these PPIs can be visualized within a functional network. This facilitates identifying the underlying biology in host-pathogen interactions. These results are indicative of the fact that further studies can improve the use of quantitative proteomics for comprehending infectious diseases.
Analysis of Intact Protein Complexes
To perform fully different functions, proteins usually exist simultaneously in distinct protein complexes. Though IP-MS gives us inventories of protein interactions, it averages together several protein complexes that host the same protein of interest.72 Moreover, information about the ratio of associations in a complex is lost in the absence of fractionation and analysis. Top-down MS analyses where proteolytic digestion is not needed for analyzing proteins, can facilitate obtaining information about an intact macromolecule or multiprotein advanced.73 Additionally, it protects each of the non-covalent interactions and consequently the post-translational state of the proteins inside the complex. Moreover, the technique is mostly used to individual infective agent proteins, like the hepatitis c virus pore protein p7,74 and pathogenic complexes reconstituted in vitro (eg the Norwalk virus-like particles).75 Still, top-down MS is not used to study host-pathogen complexes. Moreover, Top-down MS was combined with ion mobility separation to find more about different forms of a multiprotein complex.76 Therefore, top-down MS appears to be a reliable tool for studying host-pathogen protein complexes.
Detecting Direct Interactions
The yeast two-hybrid (Y2H) assay is one of the classic techniques for detecting direct PPIs.77 The Enterohemorrhagic E. coli (EHEC) is not an intracellular pathogen; however, it has a close intracellular interaction with the host, as it injects 39 proteins into the host cytosol at least. The Y2H was also used to explain direct PPIs between EHEC and thus the human host cells.78 A drawback of Y2H is that it has a relatively high false-positive rate, which is due to the non- physiological expression of proteins in cellular compartments where they are not commonly expressed. Moreover, because pathogen proteins are expressed beyond the context of an infection, many potentially relevant interactions might be missed. Along with MS, Hydrogen/deuterium exchange is another in vitro method to find the interacting regions of two proteins.79 Besides, progresses made in search algorithms designed for cross-linking MS studies have added to their simple use.80 Along with the identification of direct PPIs, crosslinkers are capable of stabilizing weaker or transient interactions and improving their identification; still, this increases non-specific associations. A study used those cross-linking tools and computational development to create a large dataset of direct interactions between human lung cells and Acinetobacter baumannii. Results have shown that a subset of that was useful for bacterial invasion.81 Thereby, the examination of RNA-protein interactions by MS can improve our knowledge of post-transcriptional regulation processes that may have an important pathogenic infection.
Pathogen-Induced Proteome Alterations in Time and Space
A central role is played by the production, degradation, and spatial reorganization of proteins for the replication of pathogens.82 Usually, the pathogen causes changes in the levels of specific host proteins required for replication. By global alterations in the proteome organization, the host also reacts to the pathogen invasion, which is critical for mounting effective defenses.83 Thereby, these studies give us a deeper insight into the control of specific time points of infection and the required subcellular compartment reorganization.
Temporal Analysis of the Infected Cellular Proteome
Thanks to the provision of perfectly established protocols and the latest MS instrumentation, temporal proteome alterations are now accepted approaches.84,85 A reliable was to characterize pathways controlled by the infectious agent and key protein effective in pathogenicity is temporal protein analyses.85 Depended on cellular metabolism, viruses have attained several mechanisms such as controlling energy production and lipid synthesis.86 Several studies have been performed on broad alterations in proteins metabolism regulation of human-relevant viruses, like the recently re-emerged Chikungunya virus,87 human cytomegalovirus (HCMV),88 flaviviruses,89 and hepatitis C virus (HCV).90 Additionally, some of these changes are temporally controlled; for instance, HCV regulation of glycolysis proteins happened only early in infection, whereas proteins used in lipid metabolism were increased continuously.90 These proteome alterations are also capable of correlating with pathogenicity as it was reported by temporal proteomic studies on different influenza strains.91 Notably, there is a relationship between regulation of specific proteins by the emerging and extremely virulent H7N9 influenza virus and its increased cytopathic effects.92 Because infections cause a wide range of proteome alterations, further studies have focused on individual pathogenic proteins.
According to proteomic studies that introduce the RTA protein coded by Kaposi’s sarcoma-associated virus (KHSV), which triggers lytic reactivation, known ARID3B as a number protein vital to initiate lytic replication.93 Based on this knowledge, which was used by cell culture systems, temporal proteomic analyses of the infection process have been successfully used for in vivo studies in animal models challenged with viruses and bacterium.94 Based on these findings, this technology makes it possible to carry out the in-depth characterization of specific organelles when infection appears so that there would be no need to eliminate the necessity of doing organelle enrichment and fractionation.
Spatial Cellular Proteome Organization During Infection
It is possible to determine infection-induced changes in protein abundances using proteome analyses on entire cells; however, the spatial information needed to understand proteome organization and characterize molecular mechanisms of pathogen infection is not provided.95,96 To measure protein abundances in different parts of infected and clean cells we can tag cells by SILAC and fractionated technique, which minimizes technical variability in the fractionation steps.97 Another option is to keep the uninfected and infected samples separate throughout fractionation so that quantification can be done through label-free approaches or isobaric tags.98 These alternatives bring the advantage of less limitation in the variety of samples so that analyzing multiple fractions and infection time points becomes possible. Changes that are induced by infection on the cell surface proteome prove the dynamic role of the plasma membrane proteome in the transport of metabolites with the extracellular space,100 intracellular and living thing signaling,99 and cell attachment during infection.101 According to proteomic studies, viral-induced alterations play a role in the mitochondria biogenesis, oxidative phosphorylation, and the electron transport chain in return.102 By integrating quantitative proteomics and live-cell microscopy, the present study introduces a wide range of alterations in organelle composition and form and distinct protein translocations between secretory organelles needed for the production of infectious particles are mentioned.88 Moreover, the integration of strategies to follow the dynamic localization of proteins inside the cell103 gives us more information about the spatial reorganization of the cell proteome when an infection takes place.
Pathogen-Induced Regulation of Protein Post-Translational Modifications
By altering protein interactions, stability, activity, and subcellular localization, post-translational modifications (PTMs) controls protein functions. Thereby, PTM regulation has a key role in the progression and results of infection on either host or pathogen proteins.104 Cellular landscape studies on PTMs and their pathogen-induced regulation have yielded valuable insights into host-pathogen interactions.
Diverse Forms of Post-Translational Modifications are Relevant in the Context of Infection
Different PTMs are efficient means of controlling signal transduction, virulence and regulatory processes on bacterial proteins like phosphorylation, acetylation, methylation, and deamidation.105 The PTMs are a key process in the life cycle of bacteria so that they can modulate main virulence factors and they are attractive targets for novel therapies.106 Finding these PTMs in bacteria is a technical challenge as they are not easy to discover given that the modifications usually exist at low levels of abundance.107 To compensate this, specific enrichment strategies that target certain PTMs are used to lower peptide complexity and increase the chance of finding and characterizing; for instance, immunoaffinity enrichment is a standard way for lysine-acetylated peptides.108 In addition, identical enrichment strategies are used to find phosphorylation events on serine, threonine, and tyrosine (S/T/Y) amino acid residues.109
Novel lysine-acetylation events in virulence factors help host immune response evasions like chitin-binding protein, a serine protease, exotoxin A, and hemolysin. This means that lysine acetylation events in Pseudomonas aeruginosa affect the mechanisms pertinent to virulence.110 Results have shown that cysteine phosphorylation in S. aureus help in controlling bacterial virulence and vancomycin resistance.111 The authors used high-resolution MS to explain in a site-specific fashion, that cysteine phosphorylation events took place in different proteins so that many of them are global regulators that control important biological processes.
MS as a Tool to Study Host and Pathogen Protein PTMs
Post-translational modifications can be observed in cells, and many of them are dynamically regulated when an infection occurs. Therefore, global PTM analyses can be done using proteomic methods.112 Selected global PTM mapping is concentrated on specific types of modifications and it has been done for various pathogenic agents such as bacteria,112 fungi,113 protozoa,114 and viruses115 to detect and measure SUMOylations, phosphorylations, acetylations, and histone modifications.116 The main tool for PTM discovery experiments is the selective enrichment of specific proteins or PTMs and then identifying the modified peptides.117 Normally, this enrichment is done by antibodies against the PTM or protein or by a resin that can enrich a class of PTMs using the chemical properties.118 Along with these discovery-driven experiments, targeted MS/MS methods including selected reaction monitoring (SRM) or parallel reaction monitoring (PRM) are tools for sensitive monitoring of PTMs on proteins of interest.119 Despite their well-recognized value that makes accurate quantification of low abundance PTMs possible,120 pathogen infection studies have not used these approaches frequently; still, they can be used more commonly in the future to widen our knowledge of proteome regulation during infection. Moreover, there is a lack of systematic examination of different types of PTMs and regulation of them as to time and space in infectious contexts.121 This is true for PTMs that are critical regulators of protein functions like phosphorylation, ubiquitination, and acetylation along with emerging PTMs of which our knowledge about their impact on protein functions like malonylation, succinylation, and lipoylation is limited.122 Moreover, for the identified PTMs, the detailed effect of many of these modifications either in uninfected or infected cells is unclear.
Multi-Omics Integration for the Study of Host-Pathogen Interactions
There are several uses for Multi-omic approaches like determining the coding capability of pathogens, identifying key virulence factors, and outlining the responsibilities of the host to pathogenic infection.123 Proteomics is also added to transcriptomic analyses to have a better annotation of infectious agent genomes,124 provide experimental proof for genes, delineate intergenic events, and purify the limits of available gene models of pathogens.125 Although, it is not easy to analyze the data of these varieties of experiments, there are procedural platforms to facilitate future proteogenomic analysis in pathogens.126 Proteomics, glycopeptidomics, and glycomics were used to find glycosylation sites and glycoform distribution in different influenza strains.127 This approach has enabled us to determine the glycosylation patterns of selective pressure obligatory by host immune factors, that influence the strain antigenicity and virulence.
While new omics methods are being introduced every day, it is important to integrate them with alternative omics approaches to achieve higher levels of data that might improve pathogenic research, like as integrating host and infectious agent PTMs128 or subcellular location data.88 A key point in multi-omics studies is that access to informatics platforms that may be accustomed to access and visualize the data such as Immunet.129 Thereby, providing these resources is essential for generating data-driven hypotheses for future pathogenic. While IP-MS is designed for studying protein complexes within bacteria,130 its use has remained limited to study in vivo pathogen-host cell protein interactions and their dynamic regulation throughout infection.131 Proteomic approaches that support protein microarrays, complement IP-MS approaches and demonstrate interesting opportunities for high-throughput screening of infectious agent interactions. By recognizing protein-encoding plasmid DNA and then translating it into exploitation noncellular expression systems merely before using the sample, the nucleic acid Programmable Protein Array (NAPPA) technology outperforms the common pitfalls that influence microarrays imprinted with purified proteins.132 When used together, there would be no need to use antibodies or generate recombinant pathogenic strains. This can be specifically advantageous for basic analysis investigation into the molecular networks of infectious agent interactions.
Proteomics Methods to Provide Mechanistic Insights in Bacterial Antibiotic Resistance
Human health is growingly threatened by bacterial pathogens as the number and distribution of antibiotic-resistant bacterium and the rate of discovery of recent antimicrobials dwindles is increasing.133 Since using antibiotics to fight infectious diseases, microorganisms have started to fight back. Using resistance mechanisms microbes can by-pass and survive the action of antibiotic drugs.134 There are several strategies to find these mechanisms and associated in-progress efforts to lower the steady increase in the number of treatment failures due to multi-drug-resistant microbes.135 Proteomics is one of the key tools in this area of research. They have key roles in realizing the molecular mechanisms of bacterial pathogenesis and in distinctive disease outcome determinants.136 The physical associations find by proteomics lead to tools to develop pathogen-specific treatment strategies that lower the spread of antibiotic resistance.137 After the recent fast advances in whole-genome sequencing, proteomic technologies are used extensively to examine microbial gene expression.138 Therefore, proteomics has emerged as a reliable tool to review bacteria. There are many comparative proteomic studies on bacteria-resistant to develop different antibiotics and some are mentioned in the following sections (Figure 1; Table 2).
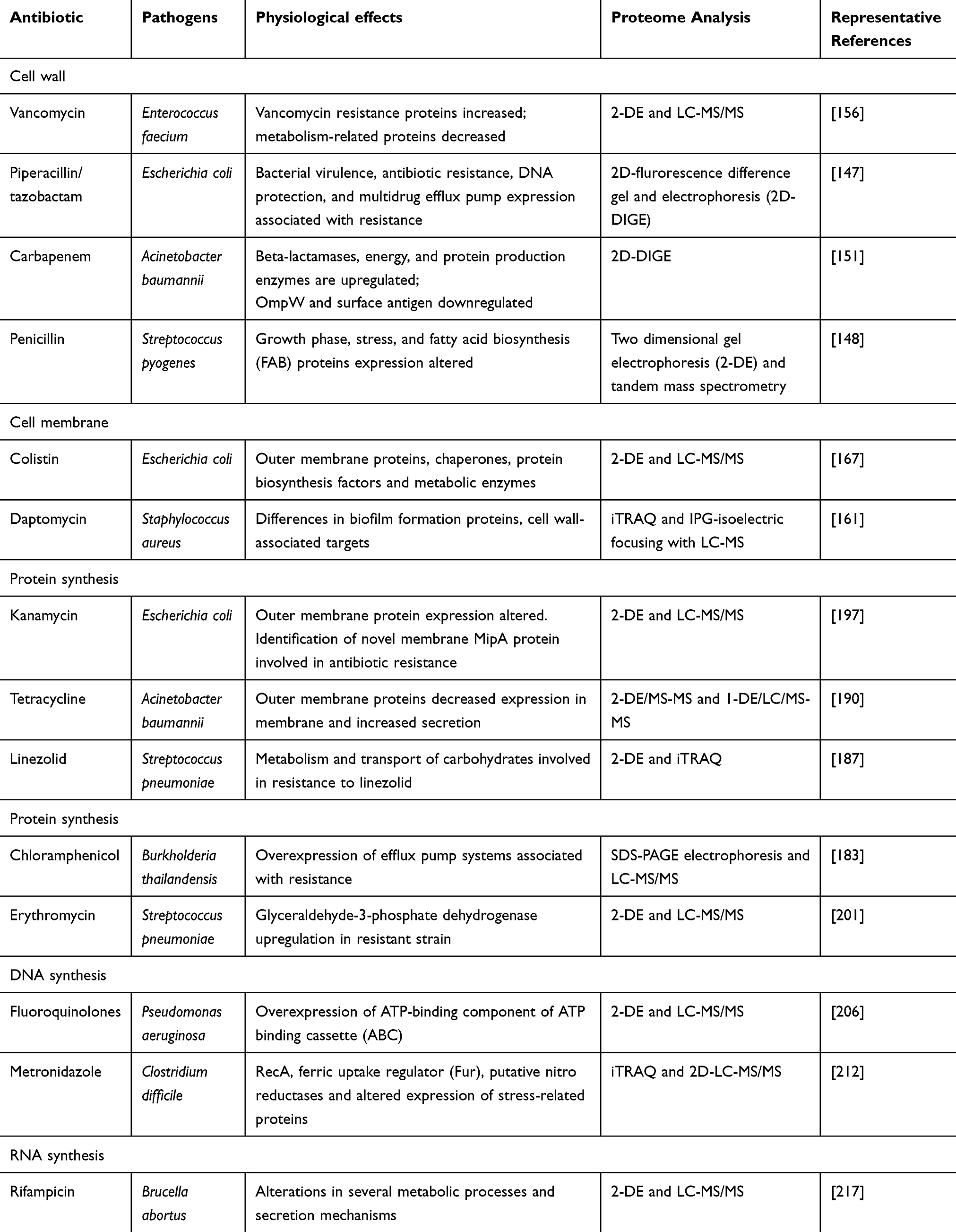 |
Table 2 Proteomic Studies of Bacterial Antibiotic Resistance Mechanisms |
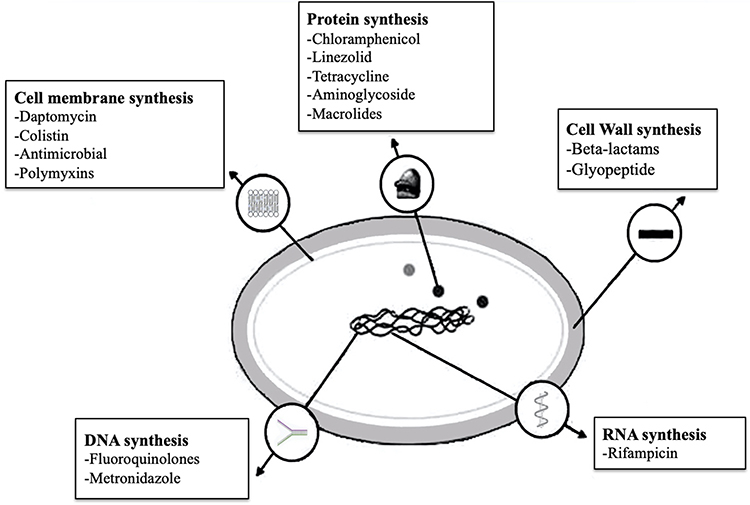 |
Figure 1 Overview of bacterial antibiotic resistance mechanisms. Antibiotics target essential bacterial processes and structures to inhibit cell growth and/or causing cell death. The major cellular targets for antibiotics include DNA replication (eg, fluoroquinolones), protein synthesis (eg aminoglycosides), cell wall integrity (eg, penicillins) and folic acid metabolism (eg, sulfonamides). |
Cell Wall-Acting Antibiotics
Beta-Lactams
Beta-lactams antibiotics are generally categorized as penicillin, cephalosporin, carbapenems, monobactam, beta-lactamase inhibitors, and other minor categories.139 The beta-lactams halt the synthesis and/or stability of the cell envelope, which results in the biogenesis of cell-wall and loss of selective permeability and osmotic integrity in return and finally bacterial cell death.140 Beta-lactam antibiotics resistance is one of the commonly studied resistance based on proteomics methods.141 Antibiotic hydrolyzing proteins is the main resistance mechanisms to beta-lactam antibiotics, which is also known as beta-lactamases.142 There are other major mechanisms like imbalance in transport proteins such as efflux pumps and porins and alteration in the penicillin-binding protein targets.143 The growing trend of using antibiotics has resulted in the rate of some key resistance strains like methicillin-resistant Staphylococcus aureus, penicillin-resistant Streptococcus pneumoniae, and extended-spectrum beta-lactamase (ESBL),144 as well as carbapenemase-producing Enterobacteriaceae, Pseudomonas aeruginosa, and Acinetobacter baummanni.145 The findings by proteomic researchers give us deep insights into ampicillin-resistant Pseudomonas aeruginosa, where novel porins are involved in resistance.146 Studies on the resistance to piperacillin/tazobactam in Escherichia coli have shown that the expression of porin OmpX was lowered and the expression of TolC increased.147 Wither regard to the penicillin-tolerant Gram-positive Streptococcus pyogenes, overexpression of murein metabolism proteins and general alteration of bacterial physiology are reported.148 Studies on methicillin-resistant S. aureus have revealed changes in cell physiology and overexpression of catalase and superoxide dismutase.149 Alanine dehydrogenase has been found effective in antibiotic resistance.150 Studies on inner membrane fraction of carbapenem-resistant A. baumanni have revealed a relationship with beta-lactamase AmpC and OXA-51 production along with metabolic enzymes, elongation factor Tu, and ribosomal proteins.151
Glycopeptide
Glycopeptide vancomycin functions through stopping peptidoglycan synthesis. It binds to the DAla-DAla terminus of the nascent peptidoglycan and therefore blocks the correct synthesis.152 Substitution of the DAla residue from peptidoglycan termini by D-lactose or D-Serine, in Enterococcus spp., was found to be the key mechanism of resistance to vancomycin.153 In addition, in S. aureus, a more complicated scenario was proposed with diverse enzymes and gene clusters implicated in vancomycin-resistance. Resistant strains like vancomycin-resistant Staphylococcus aureus (VRSA) and vancomycin-resistant enterococci (VRE) are of main clinical concern.154 Wang et al155 studied vancomycin-resistant Enterococcus faecalis and investigated a reference strain (V583) and a clinical isolate (V309) with and without vancomycin. The results supported the regulation of the proteins involved in vancomycin resistance functions, virulence factors, stress, metabolism, translation, and conjunction. Ramos et al156 determined the proteomic profiles of vancomycin-resistant E. faecium SU18 strain treated and not treated with vancomycin. 14 proteins are differentially expressed in SU18. Proteins that played a role in the vancomycin resistance mechanisms demonstrated an increase in the presence of vancomycin; while there was a decrease in metabolism-related proteins, which results in compensatory effects. Notably, the proteomic profile of a group of heterogeneous vancomycin-intermediate Staphylococcus aureus (hVISA) vancomycin susceptible S. aureus has been compared.157 At first, five upregulated proteins in hVISA were detected and only one of them supported by real-time quantitative reverse transcription PCR (qRT-PCR) – ie the protein encoded by the isaA gene involved in cell wall biogenesis.
Cell Membrane-Acting Antibiotics
Daptomycin
A new mechanism of action that is demonstrated by daptomycin is a cyclic lipopeptide antibiotic.158 This agent functions on the cell wall membrane structure and it is synthesized through binding to the cell membrane using a calcium-dependent mechanism. This results in the efflux of potassium ions of the bacterial cells.159 This process results in bacterial cell death in.160 Daptomycin is active against Gram-positive bacteria and it is clinically used to treat intense infections by these organisms (MRSA bacteremia, skin and soft tissue infections, endocarditis, and VRE infections,).7 Based on comparative proteomics profiles in the daptomycin-susceptible S. aureus strain and the daptomycin-resistant S. aureus strain 701, there is a differential abundance of proteins in different functional categories, such as cell wall-associated targets and biofilm formation proteins.161 In addition, LiaI and LiaH proteins caused (429-fold) by daptomycin, using the proteomic approach of a daptomycin-susceptible B. subtilis strain W168 in presence of daptomycin treatment of sublethal amount (1 μg/mL).162 The removal of the response regulator LiaR controls the expression of liaIH in daptomycin-resistant E. faecalis and reversed resistance to daptomycin. This leads to hypersusceptibility to daptomycin.163 Thereby, it can be concluded that LiaR is the main regulator that protects cell membranes against diverse antimicrobial agents, by regulating the expression of different genes like liaH gene. Thus, the study showed that several proteins of different functional categories, including cell wall-associated targets, had different expressions.
Colistin
As an antimicrobial peptide, Colistin interacts with the bacterial outer membrane, by replacing bacterial counter ions in the lipopolysaccharide (LPS).164 Hydrophobic and hydrophilic regions interact with the cytoplasmic membrane as a detergent and make the membrane solubilized.165 The main common mechanisms of resistance to colistin are modifications to LPS.166 Li et al167 studied proteins in mcr-1-mediated colistin-resistant and -susceptible Escherichia coli to achieve a deeper insight into the colistin resistance mechanism. They showed that the substrate phosphoethanolamine (PEA) for mcr-1 that mediated colistin resistance was accumulated in colistin-resistant E. coli. It is notable that along with PEA modification of the bacterial cell membrane lipid A, mcr-1 has an effect on the biosynthesis and transport of lipoprotein in colistin resistance through disrupting the expression of efflux pump proteins that play a role in the resistance pathway of cationic antimicrobial peptide (CAMP). There is an association between the low intracellular c-di-GMP level in dispersed cells of a P. aeruginosa strain and a higher abundance of proteins required by the virulence and development of antimicrobial peptide resistance in P. aeruginosa.168 Therefore, P. aeruginosa cells with low c-di-GMP levels act as an extra immunity to colistin than P. aeruginosa cells with high c-di-GMP levels.
Antimicrobial
A polypeptide known as antimicrobial peptides (AMPs) is generated endogenously to defend the host against microbial invasion. Also, they function actively against a wide range of microorganisms such as MDR bacteria.169 The bacterium, in Vibrio parahaemolyticus, reacts to AMPs by up-regulating the efflux channel, increasing the energy consumption performance, repairing damaged membranes effectively, and down-regulating of carbohydrate and nucleotide metabolism to preserve energy.170 In the case of Mycoplasma pulmonis, we know that the activation of the stress response, which also triggers mutations in the hrcA gene, can improve the development of resistance to AMPs like melittin or gramicidin D.171 Furthermore, 2-DE analyses, in M. pulmonis, indicated the up-regulation of enzymes playing a role in energy metabolism as a feasible outcome of the increased energy demand of the resistant strains.172 Proteins that are effective in Vibrio parahaemolyticus AMP resistance were indicated by Shen et al.173 In addition, subculture of V.parahaemolyticus strains exposed to four different AMPs demonstrated resistant strains. Additionally, two OMPs (TolC, flagellin) and five IMPs (transcription termination factor NusA, EF-Tu, ATP synthase α subunit, dihydrolipoamide dehydrogenase, long-chain FA transport protein, FadL) were spotted by analyses, which had changed the expression between the WT and AMP-resistant strain significantly. Moreover, it is believed that up-regulation of the energy-dependent MDR efflux transporter (TolC and F1-ATPa), repair of damaged membranes effectively (DLD) and AMPs cellular penetration (down-regulation of FadL)174 mediate AMP resistance. These findings showed that the upregulation of the TolC pump is a form of probable resistance mechanism described with different antibiotics.
Polymyxins
As well-established antibiotics, Polymyxins have lately drawn a great deal of attention as a result of the growing incidence of infections caused by multidrug-resistant Gram-negative bacteria.175 The polymyxins that are produced by Bacillus polymyxa are a set of cyclic polypeptides that altering the permeability of the cytoplasmic membrane176 to induce their effect. Based on MALDI-TOF analysis of the lipid A extracted from RamA-overexpressing strains of K. pneumoniae, RamA increases colistin/polymyxin resistance levels.177 This increase was done by RamA that is directly bound to lipid A biosynthesis genes like lpxC that modifies the structure of lipid A. A study showed that overexpression of a pagL-specific sRNA, Sr006 increased pagL mRNA, lipid A deacetylation, and polymyxin B resistance in P. aeruginosa. It also revealed that a pagL knockout led to a decrease in polymyxin B resistance.178 Thus, the fact that PagL is upregulated in chlorhexidine-resistant P. aeruginosa means that the resistance action mechanism to chlorhexidine might be the same, partially, as of polymyxins.
Protein Synthesis-Acting Antibiotics
Chloramphenicol
Chloramphenicol is a broad-spectrum antibiotic that plays a role in the synthesis of mitochondrial protein.179 The chloramphenicol functions through creating bounds to the 50S bacterial ribosomal subunit and inhibiting the synthesis of protein.142 Studies have explained resistance to chloramphenicol as part of the presence of the chloramphenicol acetyltransferase (CAT), which is an enzyme that inactivates the drug.180 Li et al181 found six outer membrane proteins and one protein of the location was unknown and in charge of chloramphenicol (CAP)-resistant Escherichia coli and for survival in medium with suddenly strong CAP treatment. The study argued that 4 out of the 7 proteins, including OmpC, TolC, OmpT, and OmpW, were notably changed and they could be considered as potential targets for developing new medicines against CAP-resistant E. coli. Therefore, antibodies that acted against the known OM proteins were utilized to show antibody-combating bacterial growth.182 As the results showed anti-TolC had highly significant inhibition on bacterial growth in medium with CAP. This highlights a potential novel method to treat infection by antibiotic-resistant bacteria. Antibiotic resistance mechanisms Burkholderia thailandensis were used to examine SDS-PAGE coupled with LC nanoelectrospray MS/MS.183 The resistance induced by the chloramphenicol was effective with structurally unrelated antibiotics such as quinolones and tetracyclines.184 In general, the results showed that there was an association between the multidrug resistance phenotype, found in chloramphenicol-resistant variants and the over-expression of two different efflux pumps, which were able to expel antibiotics from several families.
Linezolid
One of the oxazolidinone antibiotics for clinical treatment of severe infections with resistance against Gram-positive bacteria is Linezolid.185 Linezolid an oxazolidinone that binds to the 23S rRNA (Ribosomal ribonucleic acid) and it demonstrates different resistance mechanisms such as a higher expression of ABC transporters, mutations in 23S rRNA, mutations in ribosomal proteins L3 and L4, and mutations in an RNA methyltransferase.7 Voigt et al186 studied expressions of the protein in S. aureus after a short exposure to MCB3681, a new quinolonyl-oxazolidinone antibacterial. They tried to answer the question if MCB3681 can influence the expression of proteins different from those influenced by ciprofloxacin or linezolid. Their findings indicated that the effect of MCB3681 on the proteome signature of treated S. aureus cells was not the same as ciprofloxacin or linezolid. Proteomic and transcriptomic screening of linezolid indicated that it is feasible to increase the metabolism and transport of carbohydrates in like linezolid-resistant S. pneumoniae mutants.187 That is, resistant strains overexpressed several glycolytic proteins, enzymes, and transporters involved in sugar metabolism.
Tetracycline
Aaminoacyl tRNA binding to the mRNA-ribosome complex can be inhibited by tetracycline.188 There are at least three mechanisms that create cell resistance to tetracycline including enzymatic inactivation of tetracycline, efflux, and ribosomal protection.189 Yun et al190 utilized proteomic techniques to examine the surface proteome of A. baumannii DU202 outer membrane vesicles (OMV). This surface is notable resistant to tetracycline, after imipenem treatment. They reported a higher OMV secretion after exposure to imipenem treatment and an increase cytotoxicity towards A549 human lung carcinoma cells. The differential proteome of E. coli K12 BW25113 exposed to chlortetracycline stress was labeled using isobaric tags and quantitative proteomics technology for relative and absolute quantitation of the labeling (Lin et al191). The role of ribosome protein complexes in the translation process was improved in general in the presence of chlortetracycline stress, which is a compensatory mechanism created by the chlortetracycline effect on the ribosome. Therefore, these findings give us deeper insights to hypothesize the role of energy to guarantees cell survival. It appears that they change their metabolism to achieve a basic level of energy production and ensure their survival in the presence of the stress caused by a harmful antibiotic agent. This hypothesis can be the subject to future studies on proteomics.
Aminoglycoside
Through blocking the small 16S subunit of the bacterial ribosome, aminoglycoside antibiotic family can stop protein synthesis.192 We know three aminoglycoside resistance mechanisms including lowered uptake or decreased cell permeability, modification at the ribosomal binding sites, and generation of aminoglycoside modifying enzymes.193 Low levels of NarG and NarH and two elements of respiratory nitrate reductase (Nar) were found in streptomycin, gentamicin, ceftazidime, tetracycline, and nalidixic acid-resistant E. coli strains in a proteomic study based on native/SDS-PAGE.194 The protein expression profiles of a high-level spectinomycin-resistant (clinical isolate) and a susceptible (reference strain) Neisseria gonorrhoeae treated by sub minimal inhibitory concentrations (subMICs) of spectinomycin were compared by Nabu et al.195 Both strains demonstrated overexpression of 50S ribosomal protein L7/L12 which is a key element for ribosomal translocation. This means that compensatory mechanisms function might be in response to antibiotics that inhibit protein synthesis. To create the effects of gentamicin on the proteomes of aerobic and oxygen-limited E. coli, Proteomics techniques are an options.196 In addition, protein involvement in kanamycin resistance was reported in a proteomic and Western blotting study of the E. coli K-12 outer membrane (OM). Zhang et al197 reported an increase of some OM proteins like Tolc, TsX, and OstA, and a decrease of MipA, OmpA, FadL, and OmpW OM proteins in the kanamycin-resistant E. coli K-12 strain. They argued that MipA is a new OM protein implicated in antibiotic resistance.
Macrolides
Macrolide antibiotics function through creating a reversible bound to the P site on the subunit 23 S of the bacterial ribosome.198 The main tool of bacterial to resist against macrolides is through post-transcriptional methylation of the 23S bacterial ribosomal RNA.199 Among experimental types of acquired resistance are a generation of drug-inactivating enzymes (esterases or kinases) and generation of active ATP (Adenosine triphosphate)-dependent efflux proteins that transport the drug outside of the cell.200 Cash et al201 studies the proteins synthesized by erythromycin-susceptible and erythromycin-resistant S. pneumoniae using peptide mass mapping to find a 38500 Dalton protein upregulated in resistant strains as glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Assuming that this a probable reason for the resistance against erythromycin, the authors maintained that was an increase in energy production for the efflux system. Smiley et al202 conducted a proteomic study on isolated sarcosine- insoluble outer membrane protein (OMP) fractions obtained from clarithromycin-susceptible and resistant Helicobacter pylori strains. They demonstrated a decrease in iron-regulated membrane protein, UreaseB, EF-Tu, and putative OMP; and an increase in the HopT (BabB) transmembrane protein, HofC, and OMP31 in clarithromycin-resistant H. pylori. These findings indicate changing the outer membrane protein profile can be considered as a new mechanism effective in clarithromycin resistance in H. pylori.
DNA Synthesis-Acting Antibiotics
Fluoroquinolones
A commonly used family of quinolones in clinical settings is Fluoroquinolones.203 Quinolones inhibit the essential bacterial enzymes DNA gyrase and DNA topoisomerase IV.204 There are three quinolones resistance mechanism namely mutations that change the drug targets, mutations that lower drug accumulation and plasmids that defend cells against the lethal effects of quinolones.205 Proteomic studies on protein expression levels have found 43 proteins with higher expression in Salmonella enterica serovar Typhimurium strains when a fluoroquinolone is added to the bacterial culture.206 This means that the majority of these proteins were only a physiological reaction to fluoroquinolone; still, there was an association between the identified over-expressed AcrAB/TolC efflux pump and resistance. Proteomic analyses are conducted to examine the mechanisms at the protein level that confer resistance to fluoroquinolones. A comparison between the proteomes of fluoroquinolone-susceptible Coxiella burnetii and fluoroquinolone-resistant samples of the bacterium was done by Vranakis et al.207 They showed diverse expressions of 15 bacterial proteins that had a role in different cellular processes, which indicate the multifaceted feature of the antibiotic resistance mechanism in the bacterium. Additionally, Lin et al208 showed an increase in the OM proteins TolC, OmpT, OmpC, and OmpW and a decrease in FadL in the nalidixic acid-resistant E. coli strains. Generally, TolC and OmpC can have a stronger role in controlling nalidixic acid resistance comparing with the other identified outer membrane proteins.
Metronidazole
To inhibit nucleic acid synthesis, Metronidazole, as an antibiotic of the nitroimidazole class, disrupts the DNA of microbial cells.209 A study on the protein profiles of a derivative of Helicobacter pylori strain 26695, featured with resistance to moderate levels of metronidazole, showed that the mutant strain improved the production of the resistant phenotype of different isoforms of alkyl hydroperoxide reductase when exposed to metronidazole.210 A study on a metronidazole-resistant strain derived from B. fragilis ATCC 25285 indicated that the proteomic changes influenced a wide range of metabolic proteins such as lactate dehydrogenase and flavodoxin.147 Changes in the metabolic pathway effective in pyruvate-ferredoxin oxidoreductase has been also reported by a multidisciplinary analysis of a non-toxigenic Clostridium difficile strain that was resistive to metronidazole.211 Moreover, according to proteomic analysis, DNA repair proteins, putative nitroreductases and the ferric uptake regulator are regulated in a NAP1 C. difficile clinical isolate that is resistive to metronidazole.212 The results mean that there can be an association between a multi-factorial response and high-level metronidazole-resistance in C. difficile, such as the probable roles of altered iron metabolism and/or DNA repair.
RNA Synthesis-Acting Antibiotics
Rifampicin
By inhibiting bacterial DNA-dependent RNA polymerase, rifampicin can inhibit bacterial DNA-dependent RNA synthesis.213 Rifampicin resistance is rooted in mutations that change the residues of the rifampicin binding site on RNA polymerase, which also leads to a lower affinity for rifampicin.214 The possible to map resistant mutations to the rpoB gene, encoding RNA polymerase beta subunit.215 Neri et al216 reported different expressions of 23 proteins in two rifampicin-resistant and one susceptible meningococcus. Moreover, they report an increase in the proteins involved in the major metabolic pathways such as pyruvate catabolism and the tricarboxylic acid cycle; still, they showed a decrease in the proteins related to gene regulation in polypeptide folding. Rifampicin-resistant in a rifampicin resistant strain of Brucella abortus 2308 developed in vitro was analyzed by Sandalakis et al.217 The resistant strain indicated the described mutation V154F, in the rpoB gene. Among 456 proteins found by MS/MS, the resistant strain had 39 differentially affected proteins that play a role in different metabolic pathways. Moreover, rifampicin resistance in Brucella is mostly effective in the excitation of many metabolic processes and possible use of the secretion mechanisms that exist at a more efficient level.218 In general, these results indicate that rather than an outcome of changes in single proteins, resistance is the outcome of a complicated cellular processes network.
Proteomics Methods to Provide Mechanistic Insights in Bacterial Virulence
Growingly, proteomic techniques are attracting attention as key tools for studying bacterial pathogenesis.134 Uses of these tools are finding of virulence factors and examining the response of both host and pathogen to infection. Provenzano et al219 studied the metaproteome of microbial communities caused by endodontic infections featured with severe apical abscesses and asymptomatic apical periodontal lesions. They argued that many of the detected human proteins had a role in cellular processes and metabolism and immune defense. Wang et al220 compared the proteome profile of the S. enterica subsp. enterica serovar Typhimurium and S. typhi. These profiles are in charge of gastroenteritis and typhoid fever types. The authors first found a set of proteins with the serovar-specific expression as a novel biomarkers for finding clinical serotypes. They also reported that compared with S. typhimurium, the expression of flagella and chemotaxis proteins was lower in S. typhi. Mirrashidi et al131 employed affinities purification-mass spectroscopy to find Inc-human interactions for 38/58 Incs that plays a role in intracellular life cycles of the host, including retromer components as sorting nexin. Observation of inc targets and overlapping of viral proteins indicates common pathogenic mechanisms among obligate intracellular microbes. In general, the findings mean that a better understanding of virulence factors and resistance mechanisms to antibiotics is achievable through realizing the functionalities of the involved proteins.
Conclusion
Using proteomic analysis gives us a valuable systematic approach to study the protein complement of bacterial pathogenesis. However, studies on using proteomic analysis to examine the interactions between bacterial pathogenesis and host are at early stages. That is, the new frontline of studies on pathogens is at the interface between the pathogen and host and examining the interaction of virulence proteins with cognate host entities, coordination of their actions, and finally subverting the host cell function as part of the disease process. However, we can use systems-level proteomic analyses to examine the intrinsically delicate balance of host-pathogen interactions. In addition, the host cells possess many defense strategies to defend against and kill invading pathogens. These key aspects of host-pathogen interactions are visible in proteomic differences. Research works on human infectious diseases have been extended notably thanks to proteomic approaches to pathogenic research. Proteomic tools are becoming promising ways for clinical studies and diagnosis. In another word, these proteomic studies have led to discoveries about different pathogenic infections by studying pathogenic factors, host anti-pathogen proteins, and protein complexes and profiling host and pathogen PTM sites during infection. The convergence of proteomics and omic technologies provides chances to have a clearer picture of the dynamics of diseases and find therapeutic targets. There is an immense potential for proteomic studies on PTMs to uncover mechanisms that mediate the progression, spread, and pathogenicity of infection.
Ethics Committee Approval
The present study was approved in National ethic committee with registration number IR.TBZMED.REC.1397.514.
Acknowledgments
We thank Drug Applied Research Center, Tabriz University of Medical Sciences for all their supports.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Branden CI, Tooze J. Introduction to Protein Structure. Garland Science; 2012.
2. Aebersold R, Mann M. Mass-spectrometric exploration of proteome structure and function. Nature. 2016;537(7620):347. doi:10.1038/nature19949
3. Jung RH, Kim M, Bhatt B, Choi JM, Roh JH. Identification of pathogenic bacteria from public libraries via proteomics analysis. Int J Environ Res Public Health. 2019;16(6):912. doi:10.3390/ijerph16060912
4. Han MJ, Lee JW, Lee SY. Understanding and engineering of microbial cells based on proteomics and its conjunction with other omics studies. Proteomics. 2011;11(4):721–743. doi:10.1002/pmic.201000411
5. He F, Wu F, Zhong F, He F. Microbial proteomics: approaches, advances, and applications. J Bioinfo Proteom Imag Anal. 2016;2(1).
6. Lum KK, Cristea IM. Proteomic approaches to uncovering virus–host protein interactions during the progression of viral infection. Expert Rev Proteomics. 2016;13(3):325–340. doi:10.1586/14789450.2016.1147353
7. Lee C-R, Lee JH, Park KS, Jeong BC, Lee SH. Quantitative proteomic view associated with resistance to clinically important antibiotics in Gram-positive bacteria: a systematic review. Front Microbiol. 2015;6:828. doi:10.3389/fmicb.2015.00828
8. Stekhoven DJ, Omasits U, Quebatte M, Dehio C, Ahrens CH. Proteome-wide identification of predominant subcellular protein localizations in a bacterial model organism. J Proteomics. 2014;99:123–137. doi:10.1016/j.jprot.2014.01.015
9. Morens DM, Fauci AS. Emerging infectious diseases: threats to human health and global stability. PLoS Pathog. 2013;9(7):e1003467. doi:10.1371/journal.ppat.1003467
10. Beceiro A, Tomás M, Bou G. Antimicrobial resistance and virulence: a successful or deleterious association in the bacterial world? Clin Microbiol Rev. 2013;26(2):185–230.
11. Yang Y, Hu M, Yu K, Zeng X, Liu X. Mass spectrometry-based proteomic approaches to study pathogenic bacteria-host interactions. Protein Cell. 2015;6(4):265–274. doi:10.1007/s13238-015-0136-6
12. Sampson SL. Mycobacterial PE/PPE proteins at the host-pathogen interface. Clin Dev Immunol. 2011;2011:1–11. doi:10.1155/2011/497203
13. Senthilkumar B, Senbagam D, Prahalathan C, Anbarasu K. Gateways of pathogenic bacterial entry into host cells—salmonella. In: Garrigues P, editor. Pocket Guide to Bacterial Infections. CRC Press; 2019:59–78.
14. Alston CL, Rocha MC, Lax NZ, Turnbull DM, Taylor RW. The genetics and pathology of mitochondrial disease. J Pathol. 2017;241(2):236–250. doi:10.1002/path.4809
15. Hartlova A, Krocova Z, Cerveny L, Stulik J. A proteomic view of the host–pathogen interaction: the host perspective. Proteomics. 2011;11(15):3212–3220. doi:10.1002/pmic.201000767
16. Nicod C, Banaei-Esfahani A, Collins BC. Elucidation of host–pathogen protein–protein interactions to uncover mechanisms of host cell rewiring. Curr Opin Microbiol. 2017;39:7–15. doi:10.1016/j.mib.2017.07.005
17. Ravinder K. Gill RK, Hecht GA. Host-Pathogen Interactions in Pathophysiology of Diarrheal Disorders.In: Said HM, editor. Physiology of the Gastrointestinal Tract (Sixth Edition). Academic Press; 2018:1547–1577.
18. Inal JM, Ansa-Addo EA, Lange S. Interplay of Host–Pathogen Microvesicles and Their Role in Infectious Disease. Portland Press Limited; 2013.
19. Solanki V, Tiwari M, Tiwari V. Host-bacteria interaction and adhesin study for development of therapeutics. Int J Biol Macromol. 2018;112:54–64. doi:10.1016/j.ijbiomac.2018.01.151
20. Beltran PMJ, Federspiel JD, Sheng X, Cristea IM. Proteomics and integrative omic approaches for understanding host–pathogen interactions and infectious diseases. Mol Syst Biol. 2017;13(3).
21. Cook HV, Jensen LJ. An integrative approach to virus–host protein–protein interactions. Methods Mol Biol. 2018;1819:175–196.
22. Van Els CA, Corbière V, Smits K, et al. Toward understanding the essence of post-translational modifications for the mycobacterium tuberculosis immunoproteome. Front Immunol. 2014;5:361. doi:10.3389/fimmu.2014.00361
23. Du C, van Wezel GP. Mining for microbial gems: integrating proteomics in the postgenomic natural product discovery pipeline. Proteomics. 2018;18(18):1700332. doi:10.1002/pmic.201700332
24. Patel H, Whitehouse DB. 4 Microbial Proteomics. In: Rapley R, editor. Genomics and Clinical Diagnostics. 2019:103.
25. Blundon M, Ganesan V, Redler B, Van PT, Minden JS. Two–dimensional differencegel electrophoresis. Methods Mol Biol. 2019;1855:229–247.
26. Magdeldin S, Enany S, Yoshida Y, et al. Basics and recent advances of two dimensional-polyacrylamide gel electrophoresis. Clin Proteomics. 2014;11(1):16. doi:10.1186/1559-0275-11-16
27. Drabik A, Bodzoń-Kułakowska A, Silberring J. Gel electrophoresis. In: Silberring PC, editor. Proteomic Profiling and Analytical Chemistry. Elsevier; 2016:115–143.
28. May C, Brosseron F, Chartowski P, Meyer HE, Marcus K. Differential proteome analysis using 2D-DIGE. Methods Mol Biol. 2012;893:75–82.
29. Novotny MV, Alley WR, Mann BF. Analytical glycobiology at high sensitivity: current approaches and directions. Glycoconj J. 2013;30(2):89–117. doi:10.1007/s10719-012-9444-8
30. Venne AS, Kollipara L, Zahedi RP. The next level of complexity: crosstalk of posttranslational modifications. Proteomics. 2014;14(4–5):513–524. doi:10.1002/pmic.201300344
31. Di Venere M, Viglio S, Sassera D, et al. Do the complementarities of electrokinetic and chromatographic procedures represent the “Swiss knife” in proteomic investigation? An overview of the literature in the past decade. Electrophoresis. 2017;38(12):1538–1550. doi:10.1002/elps.201600504
32. Johnson N, Březinová J, Stephens E, et al. Quantitative proteomics screen identifies a substrate repertoire of rhomboid protease RHBDL2 in human cells and implicates it in epithelial homeostasis. Sci Rep. 2017;7(1):7283. doi:10.1038/s41598-017-07556-3
33. Bespyatykh J, Smolyakov A, Guliaev A, et al. Proteogenomic analysis of Mycobacterium tuberculosis Beijing B0/W148 cluster strains. J Proteomics. 2019;192:18–26. doi:10.1016/j.jprot.2018.07.002
34. Huang EL, Lefsrud MG. Temporal analysis of xylose fermentation by Scheffersomyces stipitis using shotgun proteomics. J Ind Microbiol Biotechnol. 2012;39(10):1507–1514. doi:10.1007/s10295-012-1147-4
35. Liu X, Hu Y, Pai P-J, Chen D, Lam H. Label-free quantitative proteomics analysis of antibiotic response in Staphylococcus aureus to oxacillin. J Proteome Res. 2014;13(3):1223–1233. doi:10.1021/pr400669d
36. Kamaladevi A, Balamurugan K. Global proteomics revealed Klebsiella pneumoniae induced autophagy and oxidative stress in caenorhabditis elegans by inhibiting PI3K/AKT/mTOR pathway during infection. Front Cell Infect Microbiol. 2017;7:393. doi:10.3389/fcimb.2017.00393
37. Dayon L, Sanchez J-C. Relative protein quantification by MS/MS using the tandem mass tag technology. Methods Mol Biol. 2012;893:115–127.
38. Han J, Yi S, Zhao X, et al. Improved SILAC method for double labeling of bacterial proteome. J Proteomics. 2019;194:89–98. doi:10.1016/j.jprot.2018.12.011
39. Wang B, Hom G, Zhou S, et al. The oxidized thiol proteome in aging and cataractous mouse and human lens revealed by ICAT labeling. Aging Cell. 2017;16(2):244–261.
40. Rauniyar N, Yates III JR. Isobaric labeling-based relative quantification in shotgun proteomics. J Proteome Res. 2014;13(12):5293–5309. doi:10.1021/pr500880b
41. Lottspeich F, Kellermann J. ICPL labeling strategies for proteome research. Methods Mol Biol. 2011;753:55–64.
42. Chahrour O, Cobice D, Malone J. Stable isotope labelling methods in mass spectrometry-based quantitative proteomics. J Pharm Biomed Anal. 2015;113:2–20. doi:10.1016/j.jpba.2015.04.013
43. Xu J, Wang H, Kong D 2-DE Compared with iTRAQ-based proteomic analysis of the functional regulation of proteins in Rhodococcus sp. BAP-1 response to fluoranthene.
44. Syahir A, Usui K, Tomizaki K-Y, Kajikawa K, Mihara H. Label and label-free detection techniques for protein microarrays. Microarrays. 2015;4(2):228–244. doi:10.3390/microarrays4020228
45. Krizman DB, Hembrough T, Thyparambil S, Liao W-L. SRM/MRM Assay for the GTPase KRas Protein (Kras). Google Patents; 2017.
46. Zahedi Bialvaei A, Rahbar M, Yousefi M, Asgharzadeh M, Samadi Kafil H. Linezolid: a promising option in the treatment of Gram-positives. J Antimicrob Chemother. 2017;72(2):354–364. doi:10.1093/jac/dkw450
47. Bonar E, Wójcik I, Wladyka B. Proteomics in studies of Staphylococcus aureus virulence. Acta Biochim Pol. 2015;62(3):367–381. doi:10.18388/abp.2015_1083
48. Neilson KA, Ali NA, Muralidharan S, et al. Less label, more free: approaches in label free quantitative mass spectrometry. Proteomics. 2011;11(4):535–553. doi:10.1002/pmic.201000553
49. Zhang Y, Wen Z, Washburn MP, Florens L. Improving label-free quantitative proteomics strategies by distributing shared peptides and stabilizing variance. Anal Chem. 2015;87(9):4749–4756. doi:10.1021/ac504740p
50. Greco V, Piras C, Pieroni L, et al. Applications of MALDI-TOF mass spectrometry in clinical proteomics. Expert Rev Proteomics. 2018;15(8):683–696. doi:10.1080/14789450.2018.1505510
51. Schlichtemeier SM, Nahm CB, Xue A, Gill AJ, Smith RC, Hugh TJ. SELDI-TOF MS analysis of hepatocellular carcinoma in an Australian cohort. J Surg Res. 2019;238:127–136. doi:10.1016/j.jss.2019.01.008
52. Anand S, Samuel M, Ang C-S, Keerthikumar S, Mathivanan S. Label-Based and Label-free strategies for protein quantitation. Methods Mol Biol. 2017;1549:31–43.
53. Bereman MS. Tools for monitoring system suitability in LC MS/MS centric proteomic experiments. Proteomics. 2015;15(5–6):891–902. doi:10.1002/pmic.201400373
54. Alreshidi MM, Dunstan RH, Macdonald MM, Smith ND, Gottfries J, Roberts TK. Metabolomic and proteomic responses of Staphylococcus aureus to prolonged cold stress. J Proteomics. 2015;121:44–55. doi:10.1016/j.jprot.2015.03.010
55. Tsou -C-C, Tsai C-F, Tsui Y-H, et al. IDEAL-Q, an automated tool for label-free quantitation analysis using an efficient peptide alignment approach and spectral data validation. Mol Cell Proteomics. 2010;9(1):131–144. doi:10.1074/mcp.M900177-MCP200
56. Kind T, Tsugawa H, Cajka T, et al. Identification of small molecules using accurate mass MS/MS search. Mass Spectrom Rev. 2018;37(4):513–532. doi:10.1002/mas.21535
57. Paul D, Kumar A, Gajbhiye A, Santra MK, Srikanth R. Mass spectrometry-based proteomics in molecular diagnostics: discovery of cancer biomarkers using tissue culture. Biomed Res Int. 2013;2013:1–16. doi:10.1155/2013/783131
58. Matsumoto M, Matsuzaki F, Oshikawa K, et al. A large-scale targeted proteomics assay resource based on an in vitro human proteome. Nat Methods. 2017;14(3):251. doi:10.1038/nmeth.4116
59. Lee M-Y, Huang C-H, Kuo C-J, Lin C-LS, Lai W-T, Chiou S-H. Clinical proteomics identifies urinary CD14 as a potential biomarker for diagnosis of stable coronary artery disease. PLoS One. 2015;10(2):e0117169. doi:10.1371/journal.pone.0117169
60. Greco TM, Cristea IM. Proteomics tracing the footsteps of infectious disease. Mol Cell Proteomics. 2017;16(4 suppl 1):S5–S14. doi:10.1074/mcp.O116.066001
61. Ayres JS. Cooperative microbial tolerance behaviors in host-microbiota mutualism. Cell. 2016;165(6):1323–1331. doi:10.1016/j.cell.2016.05.049
62. Goodman AG, Rasmussen AL. Host-pathogen interactions during arboviral infections. Front Cell Infect Microbiol. 2019;9:77. doi:10.3389/fcimb.2019.00077
63. Federspiel JD, Cristea IM. Considerations for identifying endogenous protein complexes from tissue via immunoaffinity purification and quantitative mass Spectrometry. Methods Mol Biol. 2019;1977:115–143.
64. Vinayagam A, Gibson TE, Lee H-J, et al. Controllability analysis of the directed human protein interaction network identifies disease genes and drug targets. Proc Natl Acad Sci. 2016;113(18):4976–4981. doi:10.1073/pnas.1603992113
65. Auweter SD, Bhavsar AP, de Hoog CL, et al. Quantitative mass spectrometry catalogues Salmonella pathogenicity island-2 effectors and identifies their cognate host binding partners. J Biol Chem. 2011;286(27):24023–24035. doi:10.1074/jbc.M111.224600
66. Joshi P, Greco TM, Guise AJ, et al. The functional interactome landscape of the human histone deacetylase family. Mol Syst Biol. 2013;9(1). doi:10.1038/msb.2013.26
67. Fels U, Gevaert K, Van Damme P. Proteogenomics in aid of host–pathogen interaction studies: a bacterial perspective. Proteomes. 2017;5(4):26. doi:10.3390/proteomes5040026
68. Armean IM, Lilley KS, Trotter MW. Popular computational methods to assess multiprotein complexes derived from label-free affinity purification and mass spectrometry (AP-MS) experiments. Mol Cell Proteomics. 2013;12(1):1–13. doi:10.1074/mcp.R112.019554
69. Choi H, Larsen B, Lin Z-Y, et al. SAINT: probabilistic scoring of affinity purification–mass spectrometry data. Nat Methods. 2011;8(1):70. doi:10.1038/nmeth.1541
70. Mellacheruvu D, Wright Z, Couzens AL, et al. The CRAPome: a contaminant repository for affinity purification–mass spectrometry data. Nat Methods. 2013;10(8):730. doi:10.1038/nmeth.2557
71. Ashford P, Hernandez A, Greco TM, et al. HVint: a strategy for identifying novel protein-protein interactions in herpes simplex virus type 1. Mol Cell Proteomics. 2016;15(9):2939–2953. doi:10.1074/mcp.M116.058552
72. Miteva YV, Budayeva HG, Cristea IM. Proteomics-based methods for discovery, quantification, and validation of protein–protein interactions. Anal Chem. 2013;85(2):749–768. doi:10.1021/ac3033257
73. Toby TK, Fornelli L, Kelleher NL. Progress in top-down proteomics and the analysis of proteoforms. Annu Rev Anal Chem. 2016;9:499–519. doi:10.1146/annurev-anchem-071015-041550
74. Konijnenberg A, Bannwarth L, Yilmaz D, Koçer A, Venien Bryan C, Sobott F. Top down mass spectrometry of intact membrane protein complexes reveals oligomeric state and sequence information in a single experiment. Protein Sci. 2015;24(8):1292–1300. doi:10.1002/pro.2703
75. Shoemaker GK, van Duijn E, Crawford SE, et al. Norwalk virus assembly and stability monitored by mass spectrometry. Mol Cell Proteomics. 2010;9(8):1742–1751.
76. Uetrecht C, Barbu IM, Shoemaker GK, Van Duijn E, Heck AJ. Interrogating viral capsid assembly with ion mobility–mass spectrometry. Nat Chem. 2011;3(2):126. doi:10.1038/nchem.947
77. Calderwood MA, Venkatesan K, Xing L, et al. Epstein–Barr virus and virus human protein interaction maps. Proc Natl Acad Sci. 2007;104(18):7606–7611. doi:10.1073/pnas.0702332104
78. Blasche S, Arens S, Ceol A, et al. The EHEC-host interactome reveals novel targets for the translocated intimin receptor. Sci Rep. 2014;4:7531. doi:10.1038/srep07531
79. Ciferri C, Chandramouli S, Leitner A, et al. Antigenic characterization of the HCMV gH/gL/gO and pentamer cell entry complexes reveals binding sites for potently neutralizing human antibodies. PLoS Pathog. 2015;11(10):e1005230. doi:10.1371/journal.ppat.1005230
80. Leitner A, Faini M, Stengel F, Aebersold R. Crosslinking and mass spectrometry: an integrated technology to understand the structure and function of molecular machines. Trends Biochem Sci. 2016;41(1):20–32. doi:10.1016/j.tibs.2015.10.008
81. Schweppe DK, Harding C, Chavez JD, et al. Host-microbe protein interactions during bacterial infection. Chem Biol. 2015;22(11):1521–1530. doi:10.1016/j.chembiol.2015.09.015
82. Tandon R, Mocarski ES. Viral and host control of cytomegalovirus maturation. Trends Microbiol. 2012;20(8):392–401. doi:10.1016/j.tim.2012.04.008
83. Janssens S, Pulendran B, Lambrecht BN. Emerging functions of the unfolded protein response in immunity. Nat Immunol. 2014;15(10):910. doi:10.1038/ni.2991
84. Weekes MP, Tomasec P, Huttlin EL, et al. Quantitative temporal viromics: an approach to investigate host-pathogen interaction. Cell. 2014;157(6):1460–1472. doi:10.1016/j.cell.2014.04.028
85. Fraisier C, Koraka P, Belghazi M, et al. Kinetic analysis of mouse brain proteome alterations following Chikungunya virus infection before and after appearance of clinical symptoms. PLoS One. 2014;9(3):e91397. doi:10.1371/journal.pone.0091397
86. Lopez V, Villar M, Queiros J, et al. Comparative proteomics identifies host immune system proteins affected by infection with Mycobacterium bovis. PLoS Negl Trop Dis. 2016;10(3):e0004541. doi:10.1371/journal.pntd.0004541
87. Abere B, Wikan N, Ubol S, et al. Proteomic analysis of chikungunya virus infected microgial cells. PLoS One. 2012;7(4):e34800. doi:10.1371/journal.pone.0034800
88. Beltran PMJ, Mathias RA, Cristea IM. A portrait of the human organelle proteome in space and time during cytomegalovirus infection. Cell Syst. 2016;3(4):361–373 e366. doi:10.1016/j.cels.2016.08.012
89. Grabowski JM, Perera R, Roumani AM, et al. Changes in the proteome of Langat-infected Ixodes scapularis ISE6 cells: metabolic pathways associated with flavivirus infection. PLoS Negl Trop Dis. 2016;10(2):e0004180. doi:10.1371/journal.pntd.0004180
90. Diamond DL, Syder AJ, Jacobs JM, et al. Temporal proteome and lipidome profiles reveal hepatitis C virus-associated reprogramming of hepatocellular metabolism and bioenergetics. PLoS Pathog. 2010;6(1):e1000719.
91. PFo S, McCorrister S, Hu P, et al. Highly pathogenic H5N1 and novel H7N9 influenza A viruses induce more profound proteomic host responses than seasonal and pandemic H1N1 strains. J Proteome Res. 2015;14(11):4511–4523. doi:10.1021/acs.jproteome.5b00196
92. Ding X, Lu J, Yu R, et al. Preliminary proteomic analysis of A549 cells infected with avian influenza virus H7N9 and influenza A virus H1N1. PLoS One. 2016;11(5):e0156017. doi:10.1371/journal.pone.0156017
93. Wood JJ, Boyne JR, Paulus C, et al. ARID3B: a novel regulator of the Kaposi’s sarcoma-associated herpesvirus lytic cycle. J Virol. 2016;90(20):9543–9555. doi:10.1128/JVI.03262-15
94. Shen S, Li J, Hilchey S, et al. Ion-current-based temporal proteomic profiling of influenza-a-virus-infected mouse lungs revealed underlying mechanisms of altered integrity of the lung microvascular barrier. J Proteome Res. 2016;15(2):540–553. doi:10.1021/acs.jproteome.5b00927
95. Vogels MW, van Balkom BW, Heck AJ, et al. Quantitative proteomic identification of host factors involved in the Salmonella typhimurium infection cycle. Proteomics. 2011;11(23):4477–4491. doi:10.1002/pmic.201100224
96. Kaloyanova D, Vogels M, van Balkom BW, Helms JB. Quantitative proteomic identification of host factors involved in the Salmonella typhimurium infection cycle. Methods Mol Biol. 2015;1225:29–45.
97. Villeneuve LM, Purnell PR, Stauch KL, Callen SE, Buch SJ, Fox HS. HIV-1 transgenic rats display mitochondrial abnormalities consistent with abnormal energy generation and distribution. J Neurovirol. 2016;22(5):564–574. doi:10.1007/s13365-016-0424-9
98. Wu X, Wang H, Bai L, et al. Mitochondrial proteomic analysis of human host cells infected with H3N2 swine influenza virus. J Proteomics. 2013;91:136–150. doi:10.1016/j.jprot.2013.06.037
99. Gudleski-O’Regan N, Greco TM, Cristea IM, Shenk T. Increased expression of LDL receptor-related protein 1 during human cytomegalovirus infection reduces virion cholesterol and infectivity. Cell Host Microbe. 2012;12(1):86–96. doi:10.1016/j.chom.2012.05.012
100. Hsu J-L, van den Boomen DJ, Tomasec P, et al. Plasma membrane profiling defines an expanded class of cell surface proteins selectively targeted for degradation by HCMV US2 in cooperation with UL141. PLoS Pathog. 2015;11(4):e1004811. doi:10.1371/journal.ppat.1004811
101. Matheson NJ, Sumner J, Wals K, et al. Cell surface proteomic map of HIV infection reveals antagonism of amino acid metabolism by Vpu and Nef. Cell Host Microbe. 2015;18(4):409–423. doi:10.1016/j.chom.2015.09.003
102. Karniely S, Weekes MP, Antrobus R, et al. Human cytomegalovirus infection upregulates the mitochondrial transcription and translation machineries. MBio. 2016;7(2):e00029–00016. doi:10.1128/mBio.00029-16
103. Itzhak DN, Tyanova S, Cox J, Borner GH. Global, quantitative and dynamic mapping of protein subcellular localization. Elife. 2016;5:e16950. doi:10.7554/eLife.16950
104. Ribet D, Cossart P. Pathogen-mediated posttranslational modifications: a re-emerging field. Cell. 2010;143(5):694–702. doi:10.1016/j.cell.2010.11.019
105. Liao G, Xie L, Li X, Cheng Z, Xie J. Unexpected extensive lysine acetylation in the trump-card antibiotic producer Streptomyces roseosporus revealed by proteome-wide profiling. J Proteomics. 2014;106:260–269. doi:10.1016/j.jprot.2014.04.017
106. Zhang K, Zheng S, Yang JS, Chen Y, Cheng Z. Comprehensive profiling of protein lysine acetylation in Escherichia coli. J Proteome Res. 2013;12(2):844–851. doi:10.1021/pr300912q
107. Kim D, Yu BJ, Kim JA, et al. The acetylproteome of Gram positive model bacterium Bacillus subtilis. Proteomics. 2013;13(10–11):1726–1736. doi:10.1002/pmic.201200001
108. Rardin MJ, Held JM, Gibson BW. Targeted quantitation of acetylated lysine peptides by selected reaction monitoring mass spectrometry. Methods Mol Biol. 2013;1077:121–131.
109. Manteca A, Ye J, Sánchez J, Jensen ON. Phosphoproteome analysis of Streptomyces development reveals extensive protein phosphorylation accompanying bacterial differentiation. J Proteome Res. 2011;10(12):5481–5492. doi:10.1021/pr200762y
110. Ouidir T, Cosette P, Jouenne T, Hardouin J. Proteomic profiling of lysine acetylation in Pseudomonas aeruginosa reveals the diversity of acetylated proteins. Proteomics. 2015;15(13):2152–2157. doi:10.1002/pmic.201500056
111. Sun F, Ding Y, Ji Q, et al. Protein cysteine phosphorylation of SarA/MgrA family transcriptional regulators mediates bacterial virulence and antibiotic resistance. Proc Natl Acad Sci. 2012;109(38):15461–15466. doi:10.1073/pnas.1205952109
112. Liu F, Yang M, Wang X, et al. Acetylome analysis reveals diverse functions of lysine acetylation in Mycobacterium tuberculosis. Mol Cell Proteomics. 2014;13(12):3352–3366. doi:10.1074/mcp.M114.041962
113. Leach MD, Brown AJ. Posttranslational modifications of proteins in the pathobiology of medically relevant fungi. Eukaryot Cell. 2012;11(2):98–108. doi:10.1128/EC.05238-11
114. Croken MM, Nardelli SC, Kim K. Chromatin modifications, epigenetics, and how protozoan parasites regulate their lives. Trends Parasitol. 2012;28(5):202–213. doi:10.1016/j.pt.2012.02.009
115. Bagdonaite I, Nordén R, Joshi HJ, et al. Global mapping of O-glycosylation of varicella zoster virus, human cytomegalovirus, and Epstein-Barr virus. J Biol Chem. 2016;291(23):12014–12028. doi:10.1074/jbc.M116.721746
116. Kulej K, Avgousti DC, Weitzman MD, Garcia BA. Characterization of histone post-translational modifications during virus infection using mass spectrometry-based proteomics. Methods. 2015;90:8–20. doi:10.1016/j.ymeth.2015.06.008
117. Zielinska DF, Gnad F, Schropp K, Wiśniewski JR, Mann M. Mapping N-glycosylation sites across seven evolutionarily distant species reveals a divergent substrate proteome despite a common core machinery. Mol Cell. 2012;46(4):542–548. doi:10.1016/j.molcel.2012.04.031
118. Udeshi ND, Svinkina T, Mertins P, et al. Refined preparation and use of anti-diglycine remnant (K-ε-GG) antibody enables routine quantification of 10,000 s of ubiquitination sites in single proteomics experiments. Mol Cell Proteomics. 2013;12(3):825–831. doi:10.1074/mcp.O112.027094
119. Hendriks IA, D’souza RC, Yang B, Verlaan-de Vries M, Mann M, Vertegaal AC. Uncovering global SUMOylation signaling networks in a site-specific manner. Nat Struct Mol Biol. 2014;21(10):927. doi:10.1038/nsmb.2890
120. Kusebauch U, Campbell DS, Deutsch EW, et al. Human SRMAtlas: a resource of targeted assays to quantify the complete human proteome. Cell. 2016;166(3):766–778. doi:10.1016/j.cell.2016.06.041
121. Champasa K, Longwell SA, Eldridge AM, Stemmler EA, Dube DH. Targeted identification of glycosylated proteins in the gastric pathogen Helicobacter pylori (Hp). Mol Cell Proteomics. 2013;12(9):2568–2586. doi:10.1074/mcp.M113.029561
122. Engholm-Keller K, Larsen MR. Improving the phosphoproteome coverage for limited sample amounts using TiO2-SIMAC-HILIC (TiSH) phosphopeptide enrichment and fractionation. Methods Mol Biol. 2016;1355:161–177.
123. Nesvizhskii AI. Proteogenomics: concepts, applications and computational strategies. Nat Methods. 2014;11(11):1114. doi:10.1038/nmeth.3144
124. Abd-Alla AM, Kariithi HM, Cousserans F, et al. Comprehensive annotation of Glossina pallidipes salivary gland hypertrophy virus from Ethiopian tsetse flies: a proteogenomics approach. J Gen Virol. 2016;97(4):1010. doi:10.1099/jgv.0.000409
125. Miranda-CasoLuengo AA, Staunton PM, Dinan AM, Lohan AJ, Loftus BJ. Functional characterization of the Mycobacterium abscessus genome coupled with condition specific transcriptomics reveals conserved molecular strategies for host adaptation and persistence. BMC Genomics. 2016;17(1):553. doi:10.1186/s12864-016-2868-y
126. Fan J, Saha S, Barker G, et al. Galaxy integrated omics: web-based standards-compliant workflows for proteomics informed by transcriptomics. Mol Cell Proteomics. 2015;14(11):3087–3093. doi:10.1074/mcp.O115.048777
127. Khatri K, Klein JA, White MR, et al. Integrated omics and computational glycobiology reveal structural basis for influenza A virus glycan microheterogeneity and host interactions. Mol Cell Proteomics. 2016;15(6):1895–1912. doi:10.1074/mcp.M116.058016
128. Chen W-H, van Noort V, Lluch-Senar M, et al. Integration of multi-omics data of a genome-reduced bacterium: prevalence of post-transcriptional regulation and its correlation with protein abundances. Nucleic Acids Res. 2016;44(3):1192–1202. doi:10.1093/nar/gkw004
129. Gorenshteyn D, Zaslavsky E, Fribourg M, et al. Interactive big data resource to elucidate human immune pathways and diseases. Immunity. 2015;43(3):605–614. doi:10.1016/j.immuni.2015.08.014
130. Kühner S, van Noort V, Betts MJ, et al. Proteome organization in a genome-reduced bacterium. Science. 2009;326(5957):1235–1240. doi:10.1126/science.1176343
131. Mirrashidi KM, Elwell CA, Verschueren E, et al. Global mapping of the Inc-human interactome reveals that retromer restricts Chlamydia infection. Cell Host Microbe. 2015;18(1):109–121. doi:10.1016/j.chom.2015.06.004
132. Miersch S, LaBaer J. Nucleic acid programmable protein arrays: versatile tools for array‐based functional protein studies. Curr Protoc Protein Sci. 2011;64(1):
133. Gagarinova A, Phanse S, Cygler M, Babu M. Insights from protein-protein interaction studies on bacterial pathogenesis. Expert Rev Proteomics. 2017;14(9):779–797. doi:10.1080/14789450.2017.1365603
134. Vranakis I, Goniotakis I, Psaroulaki A, et al. Proteome studies of bacterial antibiotic resistance mechanisms. J Proteomics. 2014;97:88–99. doi:10.1016/j.jprot.2013.10.027
135. Laxminarayan R, Duse A, Wattal C, et al. Antibiotic resistance—the need for global solutions. Lancet Infect Dis. 2013;13(12):1057–1098. doi:10.1016/S1473-3099(13)70318-9
136. Chen B, Zhang D, Wang X, et al. Proteomics progresses in microbial physiology and clinical antimicrobial therapy. Eur J Clin Microbiol Infect Dis. 2017;36(3):403–413. doi:10.1007/s10096-016-2816-4
137. Karlsson C, Malmström L, Aebersold R, Malmström J. Proteome-wide selected reaction monitoring assays for the human pathogen Streptococcus pyogenes. Nat Commun. 2012;3:1301. doi:10.1038/ncomms2297
138. Schneider T, Riedel K. Environmental proteomics: analysis of structure and function of microbial communities. Proteomics. 2010;10(4):785–798. doi:10.1002/pmic.200900450
139. Bush K, Bradford PA. β-Lactams and β-lactamase inhibitors: an overview. Cold Spring Harb Perspect Med. 2016;6(8):a025247. doi:10.1101/cshperspect.a025247
140. Dam S, Pagès J-M, Masi M. Stress responses, outer membrane permeability control and antimicrobial resistance in Enterobacteriaceae 2 3. Microbiology. 2018;164(3):260–267. doi:10.1099/mic.0.000613
141. Saleh S, Staes A, Deborggraeve S, Gevaert K. Targeted proteomics for studying pathogenic bacteria. Proteomics. 2019;19(16):1800435. doi:10.1002/pmic.201800435
142. Kapoor G, Saigal S, Elongavan A. Action and resistance mechanisms of antibiotics: A guide for clinicians. J Anaesthesiol Clin Pharmacol. 2017;33(3):300. doi:10.4103/joacp.JOACP_349_15
143. Blair JM, Bavro VN, Ricci V, et al. AcrB drug-binding pocket substitution confers clinically relevant resistance and altered substrate specificity. Proc Natl Acad Sci. 2015;112(11):3511–3516. doi:10.1073/pnas.1419939112
144. Pérez-Llarena FJ, Bou G. Proteomics as a tool for studying bacterial virulence and antimicrobial resistance. Front Microbiol. 2016;7:410. doi:10.3389/fmicb.2016.00410
145. Vala MH, Hallajzadeh M, Hashemi A, et al. Detection of Ambler class A, B and D ß-lactamases among Pseudomonas aeruginosa and Acinetobacter baumannii clinical isolates from burn patients. Ann Burns Fire Disasters. 2014;27(1):8.
146. Park AJ, Surette MD, Khursigara CM. Antimicrobial targets localize to the extracellular vesicle-associated proteome of Pseudomonas aeruginosa grown in a biofilm. Front Microbiol. 2014;5:464. doi:10.3389/fmicb.2014.00464
147. Dos Santos KV, Diniz CG, de Castro Veloso L, et al. Proteomic analysis of Escherichia coli with experimentally induced resistance to piperacillin/tazobactam. Res Microbiol. 2010;161(4):268–275. doi:10.1016/j.resmic.2010.03.006
148. Chaussee MA, McDowell EJ, Rieck LD, Callegari EA, Chaussee MS. Proteomic analysis of a penicillin-tolerant rgg mutant strain of Streptococcus pyogenes. J Antimicrob Chemother. 2006;58(4):752–759. doi:10.1093/jac/dkl319
149. Monteiro R, Vitorino R, Domingues P, et al. Proteome of a methicillin-resistant Staphylococcus aureus clinical strain of sequence type ST398. J Proteomics. 2012;75(10):2892–2915. doi:10.1016/j.jprot.2011.12.036
150. Solis N, Parker BL, Kwong SM, Robinson G, Firth N, Cordwell SJ. Staphylococcus aureus surface proteins involved in adaptation to oxacillin identified using a novel cell shaving approach. J Proteome Res. 2014;13(6):2954–2972. doi:10.1021/pr500107p
151. Tiwari V, Tiwari M. Quantitative proteomics to study carbapenem resistance in Acinetobacter baumannii. Front Microbiol. 2014;5:512. doi:10.3389/fmicb.2014.00512
152. Lebreton F, Cattoir V. Resistance to Glycopeptide Antibiotics. In: Bonev BB, Brown NM, editors. Bacterial Resistance to Antibiotics: From Molecules to Man. 2019:8051.
153. Miller WR, Munita JM, Arias CA. Mechanisms of antibiotic resistance in enterococci. Expert Rev Anti Infect Ther. 2014;12(10):1221–1236. doi:10.1586/14787210.2014.956092
154. Lima TB, Pinto MFS, Ribeiro SM, et al. Bacterial resistance mechanism: what proteomics can elucidate. FASEB J. 2013;27(4):1291–1303. doi:10.1096/fj.12-221127
155. Wang X, He X, Jiang Z, et al. Proteomic analysis of the Enterococcus faecalis V583 strain and clinical isolate V309 under vancomycin treatment. J Proteome Res. 2010;9(4):1772–1785. doi:10.1021/pr901216e
156. Ramos S, Chafsey I, Silva N, et al. Effect of vancomycin on the proteome of the multiresistant Enterococcus faecium SU18 strain. J Proteomics. 2015;113:378–387. doi:10.1016/j.jprot.2014.10.012
157. Chen H, Liu Y, Zhao C, et al. Comparative proteomics-based identification of genes associated with glycopeptide resistance in clinically derived heterogeneous vancomycin-intermediate Staphylococcus aureus strains. PLoS One. 2013;8(6):e66880. doi:10.1371/journal.pone.0066880
158. Dabul ANG, Kos VN, Gilmore MS, Camargo IL. Draft genome sequence of methicillin-resistant Staphylococcus aureus strain SA16, representative of an endemic clone from a Brazilian hospital. Genome Announc. 2013;1(5):e00754–00713. doi:10.1128/genomeA.00754-13
159. Müller A, Grein F, Otto A, et al. Differential daptomycin resistance development in Staphylococcus aureus strains with active and mutated gra regulatory systems. Int J Med Microbiol. 2018;308(3):335–348. doi:10.1016/j.ijmm.2017.12.002
160. Yu R, Dale SE, Yamamura D, Stankus V, Lee C. Daptomycin-nonsusceptible, vancomycin-intermediate, methicillin-resistant Staphylococcus aureus endocarditis. Can J Infect Dis Med Microbiol. 2012;23(2):e48–e50. doi:10.1155/2012/138470
161. Fischer A, Yang S-J, Bayer AS, et al. Daptomycin resistance mechanisms in clinically derived Staphylococcus aureus strains assessed by a combined transcriptomics and proteomics approach. J Antimicrob Chemother. 2011;66(8):1696–1711. doi:10.1093/jac/dkr195
162. Wecke T, Zühlke D, Mäder U, et al. Daptomycin versus friulimicin B: in-depth profiling of Bacillus subtilis cell envelope stress responses. Antimicrob Agents Chemother. 2009;53(4):1619–1623. doi:10.1128/AAC.01046-08
163. Reyes J, Panesso D, Tran TT, et al. A liaR deletion restores susceptibility to daptomycin and antimicrobial peptides in multidrug-resistant Enterococcus faecalis. J Infect Dis. 2015;211(8):1317–1325. doi:10.1093/infdis/jiu602
164. Yahav D, Farbman L, Leibovici L, Paul M. Colistin: new lessons on an old antibiotic. Clin Microbiol Infect. 2012;18(1):18–29. doi:10.1111/j.1469-0691.2011.03734.x
165. Blair JM, Webber MA, Baylay AJ, Ogbolu DO, Piddock LJ. Molecular mechanisms of antibiotic resistance. Nat Rev Microbiol. 2015;13(1):42. doi:10.1038/nrmicro3380
166. Aghapour Z, Gholizadeh P, Ganbarov K, et al. Molecular mechanisms related to colistin resistance in Enterobacteriaceae. Infect Drug Resist. 2019;12:965. doi:10.2147/IDR.S199844
167. Li H, Wang Y, Meng Q, et al. Comprehensive proteomic and metabolomic profiling of mcr-1-mediated colistin resistance in Escherichia coli. Int J Antimicrob Agents. 2019;53(6):795–804. doi:10.1016/j.ijantimicag.2019.02.014
168. Chua SL, Tan S-Y-Y, Rybtke MT, et al. Bis-(3′-5′)-cyclic dimeric GMP regulates antimicrobial peptide resistance in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2013;57(5):2066–2075. doi:10.1128/AAC.02499-12
169. da Cunha NB, Cobacho NB, Viana JF, et al. The next generation of antimicrobial peptides (AMPs) as molecular therapeutic tools for the treatment of diseases with social and economic impacts. Drug Discov Today. 2017;22(2):234–248. doi:10.1016/j.drudis.2016.10.017
170. Chiu Y, Kuo TY, Lin CC, Chen WJ. Proteomic analysis reveals responsive proteins of Vibrio parahaemolyticus on exposure to cationic antimicrobial peptides. J Appl Microbiol. 2011;110(1):80–89. doi:10.1111/j.1365-2672.2010.04856.x
171. Chowdhury MH, Diamond G, Ryan LK. 11 Synergy of Antimicrobial Peptides. In: Wang G, editor. Antimicrobial Peptides: Discovery, Design and Novel Therapeutic Strategies. 2017. Vol. 188.
172. Chernov VM, Chernova OA, Mouzykantov AA, et al. Antimicrobial resistance in mollicutes: known and newly emerging mechanisms. FEMS Microbiol Lett. 2018;365(18):fny185. doi:10.1093/femsle/fny185
173. Shen CJ, Kuo TY, Lin CC, Chow LP, Chen WJ. Proteomic identification of membrane proteins regulating antimicrobial peptide resistance in Vibrio parahaemolyticus. J Appl Microbiol. 2010;108(4):1398–1407. doi:10.1111/j.1365-2672.2009.04544.x
174. Maria-Neto S, de Souza Cândido E, Rodrigues DR, et al. Deciphering the magainin resistance process of Escherichia coli strains in light of the cytosolic proteome. Antimicrob Agents Chemother. 2012;56(4):1714–1724. doi:10.1128/AAC.05558-11
175. Poirel L, Jayol A, Nordmann P. Polymyxins: antibacterial activity, susceptibility testing, and resistance mechanisms encoded by plasmids or chromosomes. Clin Microbiol Rev. 2017;30(2):557–596. doi:10.1128/CMR.00064-16
176. Jeannot K, Bolard A, Plesiat P. Resistance to polymyxins in Gram-negative organisms. Int J Antimicrob Agents. 2017;49(5):526–535. doi:10.1016/j.ijantimicag.2016.11.029
177. De Majumdar S, Yu J, Fookes M, et al. Elucidation of the RamA regulon in Klebsiella pneumoniae reveals a role in LPS regulation. PLoS Pathog. 2015;11(1):e1004627. doi:10.1371/journal.ppat.1004627
178. Zhang YF, Han K, Chandler CE, Tjaden B, Ernst RK, Lory S. Probing the sRNA regulatory landscape of P. aeruginosa: post transcriptional control of determinants of pathogenicity and antibiotic susceptibility. Mol Microbiol. 2017;106(6):919–937. doi:10.1111/mmi.13857
179. Dinos G, Athanassopoulos C, Missiri D, et al. Chloramphenicol derivatives as antibacterial and anticancer agents: historic problems and current solutions. Antibiotics. 2016;5(2):20. doi:10.3390/antibiotics5020020
180. Roberts MC, Schwarz S. Tetracycline and chloramphenicol resistance mechanisms. In: Mayers DL, Sobel JD, Ouellette M, Kaye KS, Marchaim, D, editors. Antimicrobial Drug Resistance. Springer; 2017:231–243.
181. Li H, Lin X-M, Wang S-Y, Peng X-X. Identification and antibody-therapeutic targeting of chloramphenicol-resistant outer membrane proteins in Escherichia coli. J Proteome Res. 2007;6(9):3628–3636. doi:10.1021/pr070307y
182. Zhang QS, Ye C, Yang X. Anti-outer membrane vesicle antibodies increase antibiotic sensitivity of pan-drug-resistant Acinetobacter baumannii. Front Microbiol. 2019;10:1379. doi:10.3389/fmicb.2019.01379
183. Biot FV, Valade E, Garnotel E, et al. Involvement of the efflux pumps in chloramphenicol selected strains of Burkholderia thailandensis: proteomic and mechanistic evidence. PLoS One. 2011;6(2):e16892. doi:10.1371/journal.pone.0016892
184. Peng B, Wang C, Li H, et al. Outer membrane proteins form specific patterns in antibiotic-resistant Edwardsiella tarda. Front Microbiol. 2017;8:69. doi:10.3389/fmicb.2017.00069
185. Long KS, Vester B. Resistance to linezolid caused by modifications at its binding site on the ribosome. Antimicrob Agents Chemother. 2012;56(2):603–612. doi:10.1128/AAC.05702-11
186. Voigt B, Albrecht D, Dalhoff A. Mode of action of MCB3681 in Staphylococcus aureus–a proteomic study. Arch Clin Microbiol. 2016;7(6). doi:10.4172/1989-8436.100061
187. Feng J, Billal DS, Lupien A, et al. Proteomic and transcriptomic analysis of linezolid resistance in Streptococcus pneumoniae. J Proteome Res. 2011;10(10):4439–4452. doi:10.1021/pr200221s
188. Grossman TH. Tetracycline antibiotics and resistance. Cold Spring Harb Perspect Med. 2016;6(4):a025387. doi:10.1101/cshperspect.a025387
189. Falagas ME, Vardakas KZ, Kapaskelis A, Triarides NA, Roussos NS. Tetracyclines for multidrug-resistant Acinetobacter baumannii infections. Int J Antimicrob Agents. 2015;45(5):455–460. doi:10.1016/j.ijantimicag.2014.12.031
190. Yun SH, Park EC, Lee S-Y, et al. Antibiotic treatment modulates protein components of cytotoxic outer membrane vesicles of multidrug-resistant clinical strain, Acinetobacter baumannii DU202. Clin Proteomics. 2018;15(1):28. doi:10.1186/s12014-018-9204-2
191. Lin X, Kang L, Li H, Peng X. Fluctuation of multiple metabolic pathways is required for Escherichia coli in response to chlortetracycline stress. Mol Biosyst. 2014;10(4):901–908. doi:10.1039/C3MB70522F
192. Kudo F, Eguchi T. Aminoglycoside antibiotics: new insights into the biosynthetic machinery of old drugs. Chem Rec. 2016;16(1):4–18. doi:10.1002/tcr.201500210
193. Jackson J, Chen C, Buising K. Aminoglycosides: how should we use them in the 21st century? Curr Opin Infect Dis. 2013;26(6):516–525. doi:10.1097/QCO.0000000000000012
194. Ma Y, Guo C, Li H, Peng X-X. Low abundance of respiratory nitrate reductase is essential for Escherichia coli in resistance to aminoglycoside and cephalosporin. J Proteomics. 2013;87:78–88. doi:10.1016/j.jprot.2013.05.019
195. Nabu S, Lawung R, Isarankura-Na-Ayudhya P, Isarankura-Na-Ayudhya C, Roytrakul S, Prachayasittikul V. Reference map and comparative proteomic analysis of Neisseria gonorrhoeae displaying high resistance against spectinomycin. J Med Microbiol. 2014;63(3):371–385. doi:10.1099/jmm.0.067595-0
196. Al-Majdoub ZM, Owoseni A, Gaskell SJ, Barber J. Effects of gentamicin on the proteomes of aerobic and oxygen-limited Escherichia coli. J Med Chem. 2013;56(7):2904–2910. doi:10.1021/jm301858u
197. Zhang D-F, Li H, Lin X-M, Peng X-X. Outer membrane proteomics of kanamycin-resistant Escherichia coli identified MipA as a novel antibiotic resistance-related protein. FEMS Microbiol Lett. 2015;362(11). doi:10.1093/femsle/fnv074
198. Hu Y, Zhang M, Lu B, Dai J. Helicobacter pylori and antibiotic resistance, a continuing and intractable problem. Helicobacter. 2016;21(5):349–363. doi:10.1111/hel.12299
199. Wilson DN. Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat Rev Microbiol. 2014;12(1):35. doi:10.1038/nrmicro3155
200. Cornick J, Bentley S. Streptococcus pneumoniae: the evolution of antimicrobial resistance to beta-lactams, fluoroquinolones and macrolides. Microbes Infect. 2012;14(7–8):573–583. doi:10.1016/j.micinf.2012.01.012
201. Cash P, Argo E, Ford L, Lawrie L, McKenzie H. A proteomic analysis of erythromycin resistance in Streptococcus pneumoniae. Electrophoresis. 1999;20(11):2259–2268. doi:10.1002/(SICI)1522-2683(19990801)20:11<2259::AID-ELPS2259>3.0.CO;2-F
202. Smiley R, Bailey J, Sethuraman M, Posecion N, Ali MS. Comparative proteomics analysis of sarcosine insoluble outer membrane proteins from clarithromycin resistant and sensitive strains of Helicobacter pylori. J Microbiol. 2013;51(5):612–618. doi:10.1007/s12275-013-3029-5
203. Redgrave LS, Sutton SB, Webber MA, Piddock LJ. Fluoroquinolone resistance: mechanisms, impact on bacteria, and role in evolutionary success. Trends Microbiol. 2014;22(8):438–445. doi:10.1016/j.tim.2014.04.007
204. Hooper DC, Jacoby GA. Topoisomerase inhibitors: fluoroquinolone mechanisms of action and resistance. Cold Spring Harb Perspect Med. 2016;6(9):a025320. doi:10.1101/cshperspect.a025320
205. Drlica K, Zhao X, Malik M, Hiasa H, Mustaev A, Kerns R. Fluoroquinolone Resistance. In: Bonev BB, Brown NM,editors. Bacterial Resistance to Antibiotics: From Molecules to Man. 2019:8125.
206. Coldham NG, Randall LP, Piddock LJ, Woodward MJ. Effect of fluoroquinolone exposure on the proteome of Salmonella enterica serovar Typhimurium. J Antimicrob Chemother. 2006;58(6):1145–1153. doi:10.1093/jac/dkl413
207. Vranakis I, De Bock P-J, Papadioti A, et al. Identification of potentially involved proteins in levofloxacin resistance mechanisms in Coxiella burnetii. J Proteome Res. 2011;10(2):756–762. doi:10.1021/pr100906v
208. Lin X-M, Li H, Wang C, Peng X-X. Proteomic analysis of nalidixic acid resistance in Escherichia coli: identification and functional characterization of OM proteins. J Proteome Res. 2008;7(6):2399–2405. doi:10.1021/pr800073c
209. Reeves PT. Antibiotics: groups and properties. In: Wang J, MacNeil JD, Kay JF, editors. Chemical Analysis of Antibiotic Residues in Food. New Jersey (USA): Wiley Publishing; 2012:30–31.
210. Huang C-H, Chuang M-H, Lo W-L, et al. Alkylhydroperoxide reductase of Helicobacter pylori as a biomarker for gastric patients with different pathological manifestations. Biochimie. 2011;93(7):1115–1123. doi:10.1016/j.biochi.2011.03.008
211. Isidro J, Mendes AL, Serrano M, Henriques AO, Oleastro M. Overview of clostridium difficile infection: life cycle, epidemiology, antimicrobial resistance and treatment. In: Clostridium Difficile-A Comprehensive Overview. IntechOpen; 2017.
212. Chong PM, Lynch T, McCorrister S, et al. Proteomic analysis of a NAP1 Clostridium difficile clinical isolate resistant to metronidazole. PLoS One. 2014;9(1):e82622. doi:10.1371/journal.pone.0082622
213. Goldstein BP. Resistance to rifampicin: a review. J Antibiot. 2014;67(9):625. doi:10.1038/ja.2014.107
214. Cai X-C, Xi H, Liang L, et al. Rifampicin-resistance mutations in the rpoB gene in Bacillus velezensis CC09 have pleiotropic effects. Front Microbiol. 2017;8:178. doi:10.3389/fmicb.2017.00178
215. Fajardo-Cavazos P, Leehan JD, Nicholson WL. Alterations in the spectrum of spontaneous rifampicin-resistance mutations in the Bacillus subtilis rpoB gene after cultivation in the human spaceflight environment. Front Microbiol. 2018;9:192. doi:10.3389/fmicb.2018.00192
216. Neri A, Mignogna G, Fazio C, Giorgi A, Schininà ME, Stefanelli P. Neisseria meningitidis rifampicin resistant strains: analysis of protein differentially expressed. BMC Microbiol. 2010;10(1):246. doi:10.1186/1471-2180-10-246
217. Sandalakis V, Psaroulaki A, De Bock P-J, et al. Investigation of rifampicin resistance mechanisms in Brucella abortus using MS-driven comparative proteomics. J Proteome Res. 2012;11(4):2374–2385. doi:10.1021/pr201122w
218. Johansen TB, Scheffer L, Jensen VK, Bohlin J, Feruglio SL. Whole-genome sequencing and antimicrobial resistance in Brucella melitensis from a Norwegian perspective. Sci Rep. 2018;8(1):8538. doi:10.1038/s41598-018-26906-3
219. Provenzano JC, JF S, Rôças IN, Domingues RR, Leme AFP, Silva MR. Metaproteome analysis of endodontic infections in association with different clinical conditions. PLoS One. 2013;8(10):e76108. doi:10.1371/journal.pone.0076108
220. Wang Y, Huang K-Y, Huo Y. Proteomic comparison between Salmonella Typhimurium and Salmonella Typhi. J Microbiol. 2014;52(1):71–76. doi:10.1007/s12275-014-3204-3
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.