Back to Journals » Drug Design, Development and Therapy » Volume 11
Proteinopathy, oxidative stress and mitochondrial dysfunction: cross talk in Alzheimer's disease and Parkinson's disease
Authors Ganguly G, Chakrabarti S, Chatterjee U, Saso L ![]()
Received 17 December 2016
Accepted for publication 11 February 2017
Published 16 March 2017 Volume 2017:11 Pages 797—810
DOI https://doi.org/10.2147/DDDT.S130514
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Qiongyu Guo
Gargi Ganguly,1 Sasanka Chakrabarti,2 Uttara Chatterjee,1 Luciano Saso3
1Department of Pathology, Institute of Post Graduate Medical Education and Research, Kolkata, 2Department of Biochemistry, ICARE Institute of Medical Sciences and Research, Haldia, West Bengal, India; 3Department of Physiology and Pharmacology “Vittorio Erspamer”, Sapienza University of Rome, Rome, Italy
Abstract: Alzheimer’s disease and Parkinson’s disease are two common neurodegenerative diseases of the elderly people that have devastating effects in terms of morbidity and mortality. The predominant form of the disease in either case is sporadic with uncertain etiology. The clinical features of Parkinson’s disease are primarily motor deficits, while the patients of Alzheimer’s disease present with dementia and cognitive impairment. Though neuronal death is a common element in both the disorders, the postmortem histopathology of the brain is very characteristic in each case and different from each other. In terms of molecular pathogenesis, however, both the diseases have a significant commonality, and proteinopathy (abnormal accumulation of misfolded proteins), mitochondrial dysfunction and oxidative stress are the cardinal features in either case. These three damage mechanisms work in concert, reinforcing each other to drive the pathology in the aging brain for both the diseases; very interestingly, the nature of interactions among these three damage mechanisms is very similar in both the diseases, and this review attempts to highlight these aspects. In the case of Alzheimer’s disease, the peptide amyloid beta (Aβ) is responsible for the proteinopathy, while α-synuclein plays a similar role in Parkinson’s disease. The expression levels of these two proteins and their aggregation processes are modulated by reactive oxygen radicals and transition metal ions in a similar manner. In turn, these proteins – as oligomers or in aggregated forms – cause mitochondrial impairment by apparently following similar mechanisms. Understanding the common nature of these interactions may, therefore, help us to identify putative neuroprotective strategies that would be beneficial in both the clinical conditions.
Keywords: proteinopathy, amyloid beta, oxidative stress, α-synuclein, mitochondrial dysfunction
Introduction
Alzheimer’s disease (AD) and Parkinson’s disease (PD) are the two commonest neurodegenerative diseases of the elderly that follow a relentlessly downhill course until the patients succumb to the illness.1–3 Both the diseases appear predominantly in a sporadic fashion, but a small subset of patients in either AD or PD suffer from the familial variety of the disease with well-defined mutations.1–3 The clinical presentations of AD and PD are different, but both are progressive over the years, with PD primarily presenting with motor deficits and AD presenting with dementia and cognitive decline.1–3 Both the diseases show characteristic but complex histopathological findings in the brain at autopsy. However, there are overlapping similarities in AD and PD in terms of molecular pathogenesis, which we attempt to highlight in this review.4–6
The pathogenesis of sporadic AD or PD is highly complex, but several cardinal cellular and molecular mechanisms, such as proteotoxicity, mitochondrial dysfunction and oxidative stress, have been suggested based on postmortem studies in patients and evidence acquired from various experimental models.7–10 Although abnormal accumulation of proteins of altered conformation is considered as the most characteristic molecular signature of AD or PD, it is highly unlikely that only proteinopathy acts as the driving force of the pathology, while others such as oxidative stress and mitochondrial dysfunction are secondary consequences. Instead, it is plausible that they represent interacting damage pathways that culminate in neuronal death and degeneration. Thus, the cross talk among proteinopathy, oxidative stress and mitochondrial dysfunctions on the one hand and the genetic or nongenetic factors that trigger these damage mechanisms on the other have to be analyzed critically to obtain a deeper understanding of the pathogenesis of AD and PD. The literature is replete with studies that demonstrate the varied aspects of proteotoxicity culminating in neurodegeneration in AD or PD, as well as the detailed biophysical aspects of altered protein conformation and aggregation in these disorders; these aspects are not covered in this review.11–14 This review instead analyzes in detail the possible interactions among proteotoxic mechanisms, oxidative stress and mitochondrial functional impairment in relation to the pathogenesis of AD or PD.
Proteinopathy: links with oxidative stress and mitochondrial dysfunction
The accumulation of a specific protein, either the wild-type or a mutant variety, in excess with altered conformations that facilitate aggregation is a hallmark of many neurodegenerative diseases, including AD and PD, which has been often termed as proteinopathy.15 The accumulation of proteins can occur intraneuronally (tau or α-synuclein) or outside the cells (amyloid beta [Aβ] peptide) and can take on varied histopathological forms.15 Some elaboration of this concept in the context of the pathogenesis of AD and PD is necessary before the cross talk of proteinopathy with oxidative stress or mitochondrial function can be analyzed in the individual disease. The accumulation of a specific toxic protein may result from the transcriptional activation or enhanced translation of a specific mRNA or a diminished degradation rate of the protein through impairment of the proteasomal or lysosomal pathway.15–20 In some conditions, the mutant gene produces an abnormal product that is not readily cleared by the protein degradation machinery, leading to its accumulation, and a similar thing can happen if the protein is posttranslationally modified because of changes in the internal milieu of the cell, such as the redox status and the activity of kinases.19,20 The accumulation of the wild-type or the mutant protein in excess amount can trigger conformational alterations, eg, helix to β-strand, which facilitate oligomerization and self-aggregation. The chaperones and co-chaperones, such as heat shock protein (Hsp)70, Hsp90, Hsp40 and others, normally prevent misfolding of intracellular proteins, but excess accumulation, mutations or redox modifications of such proteins may overwhelm this system and, additionally, there may be altered expression levels of these chaperones.21,22 The toxic effect of these protein oligomers or aggregates on diverse functions of the cell organelles defines the term proteotoxicity, but it is also likely that even the monomeric forms of some of these proteins could be toxic when present in excess amounts.
The molecular links of proteinopathy and proteotoxicity with oxidative stress could be varied and complicated. For example, reactive oxygen species (ROS)-responsive transcription factors can alter the expression of genes encoding such toxic proteins or the enzymes involved in the synthesis, processsing or degradation of such proteins.23 Furthermore, the oxidative stress can directly inactivate the proteasomal system to varying degrees, especially the 26S proteasome that is responsible for the degradation of the toxic protein aggregates; many details of such ROS-mediated regulation of proteasomal degradation are currently being elucidated.24,25 The lysosomal removal of the toxic proteins may also be affected by ROS, which are known to have complex interactions with the phenomenon of autophagy.25 Furthermore, ROS may also potentiate the oligomerization of proteins such as α-synuclein and amyloid beta, or these proteins may interact with transition metals or other components to generate further ROS.26,27 Similarly, proteotoxicity and mitochondrial functional impairment are also interdependent. There is evidence to show that aggregated or oligomerized proteins – or even monomers of amyloid beta or α-synuclein – cause varied mitochondrial damages, such as impairment of bioenergetics, altered fusion/fission and impaired mitophagy, as evident in studies with isolated mitochondria, cultured cells and postmortem brain samples.28–31 The dysfunctional mitochondria resulting from proteotoxicity in turn may produce excess ROS, triggering cellular death pathways. These myriad interactions of oxygen radicals, mitochondria and toxic protein oligomers are analyzed in the context of AD and PD separately to understand their importance in the disease pathogenesis.
Alzheimer’s disease
Currently, ~24 million people across the globe suffer from AD, which is characterized by progressive dementia and deficits in multiple cognitive domains.2 The diagnosis of probable AD is made clinically supported by magnetic resonance imaging (MRI) findings, but a more accurate antemortem diagnosis is possible with positron emission tomography (PET) scan of amyloid deposition or decreased 18F-fluoro-2-deoxyglucose uptake or cerebrospinal fluid (CSF) analysis of amyloid beta peptide 1–42 (Aβ42), tau and phosphorylated tau. The diagnosis can be confirmed by postmortem analysis of brain histopathology showing diffuse neuronal and synaptic loss with extracellular amyloid plaques of different varieties and intraneuronal formation of neurofibrillary tangles.2
Proteinopathy: amyloid beta and tau
The toxic proteins that are deposited extracellularly in the brain as amyloid and neuritic plaques are called amyloid beta peptides (Aβ42 and Aβ40 peptides, in particular) derived from a precursor known as amyloid precursor protein (APP) through the sequential actions of two proteases.3,32 These two proteases, β and γ secretases, cleave out the peptides from APP, which is a type I membrane protein expressed as several isoforms in the brain.32,33 The expression, processing and intracellular trafficking of APP and amyloid beta peptides in the trans-Golgi network, endosomes and plasma membrane are well-established events, and the multiple toxicities of the amyloid peptides – especially in the soluble oligomeric form – are thought to spearhead the pathogenesis of AD (amyloid cascade hypothesis).3,34 Thus, based on postmortem data and experimental studies in cell lines, primary culture of hippocampal neurons and transgenic animal models, it has been suggested that oligomers of amyloid beta peptides can lead to mitochondrial dysfunction, calcium dysregulation, inflammatory reaction, endoplasmic reticulum (ER) stress and oxidative damage to trigger the process of neurodegeneration.33,34 Apart from the amyloid beta proteinopathy, another characteristic of AD pathology is the accumulation of phosphorylated tau proteins within the neurons, which also contributes to synaptic dysfunction and axonal degeneration.35,36 The reasons for the abnormal accumulation of amyloid beta protein or phosphorylated tau in the sporadic AD brain are not entirely clear, but it would be interesting to accrue evidence on how oxidative stress may interact with the proteinopathy of the AD brain.
Oxidative stress affects amyloid beta accumulation
The evidence of oxidative damage in postmortem AD brain is quite compelling, with significant accumulation of oxidative damage markers of lipid, protein and DNA, increased accumulation of transition metals such as Fe, Cu and Zn as well as impaired antioxidant defense.37 The recent redox proteomics analysis of the postmortem AD brain has demonstrated oxidative damage to key enzymes involved in energy metabolism, neurotransmitter-related proteins, mitochondrial proteins and proteasomal components.38
The cross talk between oxidative stress and amyloid beta proteinopathy may occur via multiple ways affecting transcription of the APP gene or translation of APP mRNA, processing and degradation of APP and amyloid beta peptides as well as interactions of APP with transition metals. The promoter sites of the APP gene have been mapped in different species, and several transcription factors such as HSF-1 and NF-kB, which are responsive to ROS, can bind here and induce APP expression.39,40 The involvement of NF-kB and ROS in upregulating APP expression has been demonstrated in various experimental models.41,42 The posttranscriptional control of APP expression has been elucidated in great detail, and APP mRNA contains a 5′-untranslated region (UTR) stem–loop arrangement of the iron-responsive element (IRE), where the IRE-binding protein (IREBP) binds and downregulates translation. In the presence of increased level of intracellular iron, IREBP dissociates from the binding site at the 5′-UTR of APP mRNA, leading to translational upregulation in a manner similar to the translational regulation of ferritin by iron.43 Transition metals such as Fe are important catalysts for ROS production, and elevated levels of iron have been reported from postmortem analyses of AD brains; thus, an obvious connection is implied between oxidative stress and APP production in the AD brain.44 The major processing enzymes of APP in the amyloidogenic pathway are β-secretase (BACE1) and γ-secretase.3,32 There are several binding sites for redox-responsive transcription factors, such as Sp1, NF-kB, and HIF-1α, near the promoter region of BACE1 and the expression of BACE1 may be modulated by ROS.45,46 However, the effect on gene expression of BACE1 by such transcription factors is often very complex, and both up- and downregulation of BACE1 gene have been reported after binding of such transcription factors depending on the cell types and the physiological or experimental conditions of the cells.45,47 There are multiple reasons for such varied response following binding of these transcription factors. For example, the nature of the subunits binding to the NF-kB binding sites may determine whether up- or downregulation of BACE1 gene takes place.45 Similarly, though NF-kB is a redox-sensitive transcription factor, its interaction with ROS is complex and may lead to either activation and enhanced nuclear translocation in certain experimental conditions or decreased nuclear binding in others.48 However, oxidative stress-induced increased BACE1 activity has been reported in many experimental systems.46,49 In several studies, products of oxidative damage (such as 4-hydroxynonenal [4-HNE]) or oxidants (such as H2O2 and iron–ascorbate mixture) have been shown to increase BACE1 activity through activation of stress-activated protein kinase.50,51 In a more extensive study, it has been shown that oxidative stress-induced increase in BACE1 activity both in cultured cells and in experimental animals requires the involvement of γ-secretase and activation of the JNK/c-jun pathway.52 Additionally, oxidative stress increases BACE1 activity at the translational level involving double-stranded RNA-dependent protein kinase (PKR) and eukaryotic initiation factor-2 (eIF2) phosphorylation.53 Such data do suggest that oxidative stress contributes to the increase in BACE1 activity in AD. The γ-secretase enzyme complex, composed of presenilin1 (PS1), nicastrin, PEN2 and APH1, is also essential for the release of Aβ42 from APP, and its activity is enhanced by oxidative stress through upregulation of PS1.52
In different experimental models, oxidative stress has been shown to increase the production and accumulation of Aβ42, which could be accounted for by its effect on APP expression and processing.54 Additionally, in AD, the clearance of amyloid beta from the brain is affected as a result of oxidative damage. The clearance of amyloid beta from the brain is regulated by the low-density lipoprotein receptor-related protein 1 (LRP1) and the receptor for advanced glycation end products (RAGE).55–57 The LRP1 exists in two forms; the membrane-bound form is responsible for the removal of cerebral amyloid beta to the circulation across the blood–brain barrier and is expressed in neurons, astrocytes, vascular endothelial cells and smooth muscle cells, while the soluble form binds the amyloid beta in the peripheral circulation.55,56 RAGE, present in the blood–brain barrier, on the other hand, may allow the reentry of amyloid beta from the peripheral circulation. Under oxidative stress, the membrane-bound LRP1 is altered, preventing the clearance of amyloid beta from the brain; furthermore, the oxidized soluble LRP1 fails to bind the circulating amyloid beta properly, allowing its reentry into the brain.57
Amyloid beta induces oxidative stress
Multiple mechanisms are operative in initiating the oxidative damage in the AD brain, but in particular, amyloid beta-induced ROS generation has been well documented in a large number of experimental studies with cultured cells, subcellular fractions, experimental animals and cell-free chemical systems.58–60 Both Aβ42 and Aβ40 can bind transition metals in a redox-active form through several amino acid residues that include His6, His13 and His14, and the related coordination chemistry has been elaborately described.61 The bound metal ions can take part in redox-cycling reactions that generate ROS, and the mechanism – though not fully understood – may require Met35 of the peptide or another reducing agent in the surroundings.61 These experimental studies are significant because of the postmortem evidence of increased levels of transition metals such as Fe, Cu and Zn in the AD brain, especially near the plaque, which implies that amyloid beta-mediated ROS production could be important in AD pathogenesis. However, based on scattered but substantial experimental evidence, an opposite view posits an antioxidant and protective role of amyloid beta, whereby the peptide scavenges the reactive radicals of lipid oxidation, prevents the formation of ROS through sequestration of the transition metals or even blocks the mitochondrial production of oxygen free radicals.62 Apart from this direct production of ROS by bound redox-active metals, amyloid beta may lead to increased production of ROS intracellularly and to subsequent neuronal death through the involvement of ASK 1.58 In neuronal culture, amyloid beta increases ROS production presumably through the activation of reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, but enhanced mitochondrial production of oxygen radicals, which is preventable by mitochondria-targeted antioxidants, has been demonstrated.63,64 The other mechanisms of amyloid beta-induced ROS production involve microglial activation and priming by the soluble or fibrillar form of the peptide, as observed in primary cultures of microglia or coculture of microglia and neurons.65,66 The activated microglial cells produce both ROS and proinflammatory cytokines such as interleukin (IL)-6, IL-1β, tumor necrosis factor (TNF)-α and initiate the inflammatory response in AD brain following activation by amyloid beta acting through a variety of receptors.67 Thus, the microglia contain a B-type of scavenger receptor, called CD36, which has been shown to act as a receptor for fibrillar amyloid beta, and the activation of this receptor by the peptide leads to increased microglial ROS production, cytokine expression and phagocytosis.67,68 Others have shown that amyloid beta-induced microglial activation and ROS production is mediated by MAC-1 receptor and PI3K, causing activation of NADPH oxidase.69 The central role of NADPH oxidase in microglial production of ROS induced by amyloid beta has been highlighted in many other studies.65,69
Oxidative stress and tau phosphorylation
The formation of neurofibrillary tangles from intracellular accumulation of hyperphosphorylated tau protein is another characteristic feature of AD pathogenesis, which contributes significantly to the axonal degeneration and synaptic dysfunction associated with these disorders.70,71 The microtubule-associated tau protein has multiple phosphorylation sites in the C-terminal and in the proline-rich regions of the protein, and a number of kinases – especially GSK3β and CDK5 – can phosphorylate the latter.71 Several phosphatases, predominantly PP2A, PP1 and PP2B, can dephosphorylate tau.71 In AD, the increased phosphorylation of tau protein is the result of increased activities of GSK3β and CDK5, with an associated decrease in PP2A activity, but the reasons for such alterations in the activities of kinases and phosphatases in this disease condition are not clear. In different experimental models, the role of oxidative stress on tau phosphorylation has been investigated. Chronic oxidative stress in the form of glutathione depletion has been shown to increase tau phosphorylation in cultured M17 neuroblastoma cells through activation of JNK and p38 as well as diminished activity of PP2A.72 In cultured cortical neurons, amyloid beta-induced tau phosphorylation is mediated by oxidative stress through the involvement of p38 kinase and the phenomenon is blocked by the antioxidant trolox.73 Similarly, in rat primary cortical neuronal culture, a combination of Fe2+ and H2O2 causes increased phosphorylation of tau, presumably through the activation of GSK3.74 On the contrary, there are reports of decreased phosphorylation of tau under oxidative stress in different experimental models through modulations of kinases (such as GSK3 or CDK5) or phosphatases (such as PP1), and thus, the relationship between oxidative stress and tau phosphorylation is somewhat uncertain at present.75,76
Amyloid beta and mitochondrial functions
Mitochondrial dysfunction is a key mechanism in the pathogenesis of AD, and studies with both postmortem AD brains and various experimental models have corroborated this.77 For example, a variety of structural changes such as fragmented mitochondria with abnormal cristae, impairment of bioenergetics along with diminished cytochrome oxidase, α-ketodehydrogenase, pyruvate dehydrogenase activities, decreased adenosine triphosphate (ATP) synthesis, mitochondrial membrane depolarization, increased ROS production and altered mitochondrial biogenesis and dynamics have been reported.77,78 Since mitochondrial oxidative phosphorylation is the major source of ROS, it is reasonable to assume that mitochondrial dysfunction contributes significantly to genesis of oxidative stress in AD brain. However, the interrelationship between mitochondrial dysfunction and proteotoxicity is the subject of intense research. In particular, extensive studies have been conducted using transgenic AD models, postmortem AD brain, cultured cells and isolated mitochondria to elucidate how amyloid beta or its soluble oligomers interact with mitochondria through multiple mechanisms. In transgenic AD mice, progressive accumulation of amyloid beta occurs in the brain mitochondria, with diminished activities of respiratory chain enzymes and decreased oxygen consumption rate; similar accumulations of amyloid beta also take place in different AD-affected brain regions in postmortem AD brain.79 Immunofluorescence staining and confocal microscopy, as well as other techniques, have identified that amyloid beta (Aβ) binds to mitochondrial short-chain alcohol dehydrogenase, which is known as Aβ-binding alcohol dehydrogenase (ABAD).79 The other protein with which amyloid beta may remain in association is Hsp60, which is a marker of the mitochondrial matrix.79 The accumulation of amyloid beta may lead to inhibition of mitochondrial peptidasome (PreP), which removes the presequences from the N-terminals of mitochondrially targeted proteins.80 The impaired processing of these preproteins may change the protein profile of mitochondria, leading to multiple functional anomalies of this organelle in AD.80 Another possible mechanism of multiple functional impairments of mitochondria in AD is the blockage of protein import channels of this organelle by APP, preventing the entry of nuclear DNA-coded proteins of mitochondria, including the subunits of the respiratory chain complexes.30,81 The soluble oligomers of amyloid beta, however, can impair functions of isolated mitochondria incubated in vitro, which may possibly be related to its interactions with many other mitochondrial proteins, such as adenine nucleotide translocase (ANT), components of translocase of outer membrane (TOM) or translocase of inner membrane (TIM), cyclophilin D, uncoupler protein (UCP) and others.82 The interactions of soluble oligomers of amyloid beta with lipid biomembranes have been studied in depth by several groups, which indicates that membrane-spanning channels (Aβ channels) could be created by such oligomers, allowing different ions to pass through these channels.83,84 If such a mechanism is operative in the interactions of amyloid beta oligomers with mitochondria, it may explain some of the toxic effects of amyloid beta on this organelle. The other interesting finding in the context of amyloid beta-induced mitochondrial dysfunction in astrocytes is the involvement of cytosolic and calcium-independent phospholipase A2 in this process.85 Furthermore, in astrocytes, amyloid beta has been reported to activate NADPH oxidase to enhance ROS production, which in turn may cause mitochondrial dysfunction and glutathione depletion in both neurons and astrocytes.86
Figure 1 summarizes the various possible mechanisms in AD brain through which oxidative stress leads to an accumulation of amyloid β42, which in turn impairs mitochondrial function and interacts with metal ions in microglia.
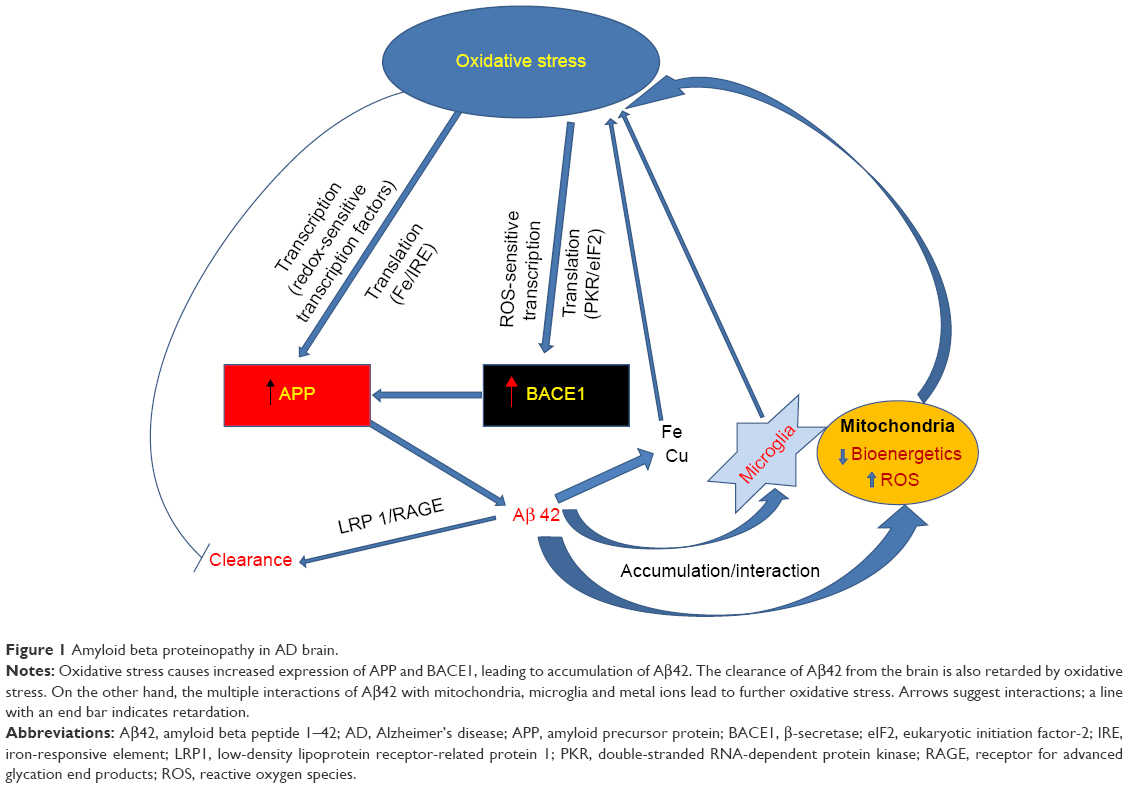 | Figure 1 Amyloid beta proteinopathy in AD brain. |
Parkinson’s disease
The disease presents clinically as a triad of bradykinesia, muscular rigidity and resting tremor in elderly people, with a prevalence of 425 per 100,000 in the age group of 64–75 years, which increases further with advancing age.1,87 As the disease gradually worsens, many secondary symptoms develop, such as postural instability; difficulty in swallowing, breathing and speaking; sleep disturbance; depression and dementia. Majority of PD patients (>95%) suffer from the sporadic variety of the disease, while mutations in 18 chromosomal loci (PARK 1, PARK 2, PARK 3, PARK 4, PARK 5 and so on) lead to familial PD (<5%).1,88 Some mutations as in α-synuclein (PARK 1), Parkin (PARK 2), PINK1 (PARK 6) or DJ-1 (PARK 7) give rise to monogenic forms of familial PD, with either autosomal dominant or recessive inheritance.1,88 The pathological hallmark of the disease is the degeneration of the dopaminergic (DAergic) neurons of the substantia nigra projecting into the striatum, but extensive postmortem studies have now established that the neurodegeneration affects many other areas of the brain, beginning in the olfactory bulb and dorsal nucleus of the vagus and progressing sequentially to the pons, medulla, midbrain, mesocortical areas and, finally, the neocortex.89 In sporadic and some types of familial PD, the neurodegeneration is associated with the development of round eosinophilic inclusion bodies called Lewy bodies within the soma of the neurons and the appearance of spindle-shaped or thread-like structures within the cell processes called Lewy neurites.89,90 Both Lewy bodies and Lewy neurites are immunoreactive for α-synuclein.89,91
Proteinopathy in PD
The major protein component of the intraneuronal Lewy bodies that are present in the degenerating neurons of sporadic PD is α-synuclein, which is a small protein of 140 amino acids (14.1 kDa) abundantly present in the brain.4,90 The physiological function of this protein is not fully known, but it may play a pivotal role in synaptic plasticity and the vesicular transport and release of neurotransmitters.90,92 This protein, in the native state, remains in an unfolded form and has a propensity to bind to liposomes, synthetic membranes and membranes of the organelles.90,92,93 The binding to synthetic membranes through the interaction of the acidic phospholipids and the N-terminal lysine-rich segment of the peptide causes a conformational change from the unfolded form to partially helical structures, as observed through a variety of biophysical techniques in vitro.90,92,93 The oligomerization and subsequent fibrillization of α-synuclein is initiated and propagated through the conformational change of the natively unfolded or the membrane-bound helical form of the protein to the β-strand conformation, and dimerization may be a key step in this process.90,92–94 The process occurs in a concentration-dependent manner and is also affected by membrane binding or posttranslational modifications such as phosphorylation, proteolysis, oxidative modification and metal binding.90,92–94
The accumulation of α-synuclein in vulnerable regions in the PD brain could be because of overexpression through transcriptional (eg, DNA methylation status) and posttranscriptional mechanisms (iron acting through IRE and IREBP at the 5′-UTR of the mRNA) as well as by the diminished degradation of the protein through proteasomal and lysosomal pathways.18,95–98 The ubiquitin–proteasomal system (UPS) is a major pathway of intracellular protein degradation and is particularly responsible for the removal of damaged or misfolded proteins.99 The UPS uses a group of enzymes (E1, E2 and E3) to conjugate a chain of multiple ubiquitin (a small protein of 76 amino acids with a highly conserved sequence) units to target proteins destined for degradation, and subsequently these tagged proteins are delivered to the 26S/20S proteasomal complex for ATP-dependent degradation.100 The proteasomal impairment has been suggested to be an important mechanism of neurodegeneration in PD and has been studied extensively in experimental models as well as in postmortem brains, especially because mutations in several genes, such as Parkin and ubiquitin C-terminal hydrolase L1 (UCH-L1) coding for the UPS enzymes, give rise to some varieties of familial PD.88,101–104 In the substantia nigra of the postmortem PD brain, significant loss of the α-subunit of the 26S/20S proteasomal complex, coupled with impaired activity of the 20S subunit, and diminished levels of endogenous proteasomal activators have been observed.99 Moreover, the systemic administration of natural and synthetic proteasomal inhibitors to rats has been shown to give rise to striatal dopamine (DA) depletion and nigral DAergic neuronal death.102 Similarly, the proteasomal inhibitor lactacystin induces apoptosis in cultured SH-SY5Y cells through translocation of the cytosolic Bax to mitochondria.105 Furthermore, the overexpression of mutant α-synuclein in differentiated PC12 cells causes decreased proteasomal activity and increased sensitivity of the cells to apoptotic death following exposure to subtoxic concentrations of the proteasomal inhibitor.104 Thus, proteasomal impairment has been suggested to play a pivotal role in the pathogenesis of PD, but several inconsistencies have also been pointed out with regard to neurodegeneration caused by proteasomal inhibitors.98 However, in general, it is plausible that proteasomal inactivation could be the mechanism for the accumulation of α-synuclein in the sporadic PD brain, and in turn, the latter in its aggregated form may further downregulate the 26S proteasomal activity presumably by interacting with the 19S cap. The other relevant mechanisms for the impairment of UPS in PD may include DA-derived ROS and quinones.106,107
Several missense mutations (A53T, A30P and E46K) or duplication or triplication of the α-synuclein gene results in the familial form of PD with autosomal dominance.4,88 A genome-wide association study has also indicated that common variations of the α-synuclein gene constitute a risk factor for PD.108 Lentivirus-mediated overexpression of wild-type or mutant human α-synuclein after injection into substantia nigra in rats leads to DAergic neurodegeneration, with the formation of inclusion bodies immunoreactive for α-synuclein.109 Recombinant adeno-associated virus-mediated expression of α-synuclein (wild or mutant) in mouse substantia nigra triggers progressive neurodegeneration, and interestingly, both wild and mutant varieties of α-synuclein cause similar degrees of pathology.110 Similarly, fibrillar rat α-synuclein injected into mice striatum causes neurodegeneration, with Lewy body-like structures in substantia nigra and other areas.111 When Lewy body-enriched fractions from postmortem PD brains are injected into substantia nigra or striatum of mice or monkeys, human α-synuclein is seen to be internalized within neurons, triggering a progressive neurodegeneration that is dependent upon the expression of endogenous α-synuclein of the animal.112 Furthermore, in cell-based models, overexpression or accumulation of α-synuclein induces cell death or potentiates DA-induced loss of cell viability.113 Thus, there is a growing body of evidence of α-synucleinopathy as the pivotal mechanism of PD neurodegeneration, even though the mechanism of toxicity is not clearly understood.
Alpha-synuclein and mitochondrial dysfunction in PD
A most characteristic pathology of PD, as mentioned already, is the impairment of mitochondrial functions at different levels.4,5,114 The original postmortem finding was a decrease in complex I activity in the substantia nigra of PD patients, and subsequently, the activities of other respiratory chain complexes have been shown to be decreased to a variable extent in the platelets and skeletal muscles of PD subjects.115–117 Furthermore, mitochondrial fusion and fission, responsible for the dynamic morphology and functional quality of the organelle, are possibly altered in sporadic PD, as suggested from studies in cell-based models of this disease, including cybrids.5,118,119 Cybrids are created by the fusion of human neuroblastoma cells or teratocarcinoma cells depleted of endogenous mitochondrial DNA (mtDNA) with platelets from PD subjects.119,120 Such cybrids also show a variety of defects in respiratory chain activity, ROS production and susceptibility to mitochondrial toxins.119 Several toxins and endogenously available molecules, such as rotenone, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), paraquat, 6-hydroxy-DA (6-OHDA) and DA, which are known to impair mitochondrial functions, can produce PD-like neurodegeneration in animal models coupled with behavioral deficits.7,121–124 In cultured DAergic cell lines such as PC12 and SH-SY5Y, MPTP, DA or 6-OHDA trigger apoptotic cell death with profound mitochondrial dysfunctions.122,124 Accumulation of mitochondrial DNA mutations has been observed in laser-microdissected nigral neurons from postmortem PD brains.125 Mutations in several genes, such as Parkin and PINK1, cause familial forms of PD, and the proteins coded by these genes are known to maintain the functional quality of mitochondria through mitophagy.126 Thus, altered mitophagy could be a possible mechanism of the mitochondrial dysfunction observed in PD, and this topic needs some elaborate analysis. The detailed mechanism of mitophagy, a type of macroautophagy to remove dysfunctional and damaged mitochondria, has been extensively studied in both yeast and mammalian systems. In yeast, mitophagy requires several proteins, including Uth1, ancient ubiquitous protein 1 (Aup1), autophagy 32 (Atg32) and so on; Atg32, which is considered the mitophagy-specific receptor of mitochondria, interacts with Atg11 during the formation of the autophagosome.126,127 In mammals, including humans, mitophagy is a more complex phenomenon and, along with mitochondrial fusion and fission processes, it regulates the quality control of the mitochondria. Several proteins, such as BCL2, adenovirus E1B 19 kDa-interacting protein 3-like (BniP3), and BNIP3-like protein X (Nix), which are proapoptotic proteins belonging to the Bcl-2 family, are involved in the process of mitophagy, and these proteins through their C-terminal transmembrane domains get embedded in the mitochondrial outer membrane.126,127 Many other proteins, such as the Fun14 domain-containing protein 1 (Fundc1), Atg7, Parkin and PINK1, have been shown to be involved in mitophagy under different experimental conditions.127,128 In particular, the PINK1 and Parkin signaling for mitophagy is important in the context of PD pathogenesis, and the inducer for mitophagy could be mitochondrial membrane depolarization.127,128 It has been shown conclusively that PINK1 accumulates on damaged and depolarized mitochondria and further recruits Parkin by translocating it from the cytosol to the mitochondrial surface, triggering mitophagy.128–131 There is some evidence, though not conclusively established, that Parkin is phosphorylated by PINK1, which also activates the E3 ubiquitin ligase activity of Parkin, leading to ubiquitination of the mitochondrial membrane proteins.128,129,131 Some recent studies have indicated that PINK1 accumulation, along with Parkin recruitment on damaged mitochondria, depends on the bioenergetic state of the cell, and ATP may have a key involvement in this process.132,133 Furthermore, other studies have indicated complex interactions of Parkin, PINK1 and α-synuclein in regulating mitochondrial dynamics and mitophagy.134,135 Overall, it appears that altered mitophagy and mitochondrial dynamics play a key role in PD pathogenesis, with the involvement of Parkin, PINK1 and α-synuclein, but the molecular scenario is far from clear as of now.
The trigger for mitochondrial alterations in sporadic PD in the absence of environmental toxins such as paraquat or MPTP is not so far convincingly established, but ROS and toxic quinones derived from oxidation of DA could be important in this respect.124 On the other hand, there is enough evidence to suggest that the interactions of mitochondria with α-synuclein through multiple mechanisms could play a determining role in the PD-associated dysfunction of this organelle.114,136 The N-terminal region of α-synuclein contains a cryptic sequence containing several positively charged residues for targeting the mitochondria, and the protein enters the organelle through interaction with TOM40 and remains associated with the inner membrane.114,136,137 The accumulation of α-synuclein within mitochondria has been shown to inhibit mitochondrial complex I activity.114,137 In postmortem PD brain, increased accumulation of α-synuclein within the mitochondria has been shown in the regions of the substantia nigra and striatum, but not in the cerebellum, and this study has further demonstrated the interaction of α-synuclein with complex I both in postmortem PD brain tissue and in cultured cell lines.138 Similarly, aggregated α-synuclein has been shown to inhibit complex I activity in cybrids, leading to impaired ATP synthesis and mitochondrial respiration.139 On the other hand, the interaction of α-synuclein with mitochondria, which leads to membrane depolarization and impaired ATP synthesis but without changes in the activities of the respiratory complexes, has been clearly demonstrated in vitro, and apparently, α-synuclein interacts with components of the mitochondrial permeability transition pore (mPTP).28,140 It has been further shown that this interaction of α-synuclein with mPTP in cultured SH-SY5Y cells triggers cell death, which could be prevented by α-synuclein gene knockdown.140 Furthermore, α-synuclein has been shown to inhibit mitochondrial fusion, resulting in fragmented mitochondria in cultured cells overexpressing the protein, and the phenomenon can be blocked by coexpression of Parkin, PINK1 and DJ-1.141
Alpha-synuclein and oxidative stress: interactions in PD pathogenesis
Oxidative stress is considered to be an important element of PD pathogenesis as is evident from the accumulation of oxidative damage markers and transition metals such as Fe in the postmortem PD brain, especially in the substantia nigra.142,143 In experimental models of PD, enhanced production of ROS has been observed, with alterations of the antioxidant enzyme levels.144 Mitochondrial dysfunction caused by α-synuclein could be a source of enhanced production of ROS, as has been observed in several experimental models.140,145 Overexpression of wild or mutant α-synuclein in SH-SY5Y cells has also been shown to increase the intracellular level of ROS.146 On the other hand, iron – which facilitates ROS production and catalyzes peroxidative damage to biomolecules – accumulates in the substantia nigra of PD brains in excess amounts and causes translational upregulation of α-synuclein.96 Furthermore, the binding of transition metals such as Fe and Cu to α-synuclein has been studied with a variety of biophysical techniques in different studies, and apparently, such binding may lead to enhanced cytotoxicity of α-synuclein through multiple mechanisms.147 For example, Fe can trigger the formation of large sodium dodecyl sulfate (SDS)-resistant oligomers of α-synuclein capable of forming membrane-spanning channels, which may partly explain the toxicity of α-synuclein on intracellular organelles.147 The binding of iron to α-synuclein may also lead to the formation of H2O2 through redox reactions.147 The binding of Cu to α-synuclein also enhances the toxicity of the latter, as has been shown in cultured SH-SY5Y cells overexpressing α-synuclein.146 Since DAergic neurons are particularly affected in PD, it has been suggested that DA oxidation products such as ROS and toxic quinones could contribute to PD pathology.124,148 In this context, it is interesting to note that the interaction of α-synuclein with DA has been studied by several groups, indicating a modulatory role of DA oxidation products on α-synuclein oligomerization and cytotoxicity.149 In a very elaborate study, it has been shown that in human DAergic neurons and rat DAergic cell lines exposed to paraquat, increased accumulation and aggregation of α-synuclein occurs, which is crucially dependent upon the activity of NADPH oxidase, implicating the role of ROS in the process.150 This study has further shown that the systemic injection of paraquat in rats causes increased protein expression of α-synuclein and NADPH oxidase, along with the accumulation of oxidative damage markers in the substantia nigra, which can be abolished by knocking down NADPH oxidase (Nox1) gene by adeno-associated virus-mediated overexpression of a specific short hairpin RNA (shRNA).150 The knockdown of Nox1 also prevents the nigral DAergic neuronal loss after paraquat treatment of rat.150 Another interesting cross talk between oxidative stress and α-synuclein is presumable when oxidatively modified protein becomes partially resistant to degradation by the UPS or chaperone-mediated autophagy, leading to accumulation of the misfolded protein.151 Several types of oxidative modifications of α-synuclein have been demonstrated, such as nitration of tyrosine residues, oxidation of methionine residues and covalent adduct formation with 4-hydroxynonenal.151 Such oxidatively modified α-synuclein proteins, in general, inhibit oligomerization and fibril formation by the native monomer and instead may give rise to “off-pathway” oligomers, but the toxic consequence of this has not been clearly established.151 The various mechanisms leading to the accumulation of α-synuclein in the PD brain and the toxicity of this protein in monomeric or oligomeric form in the mitochondria are highlighted in Figure 2.
 | Figure 2 Pathways of α-synuclein accumulation and toxicity in PD. |
Conclusion
This review has shown that the three cardinal processes of molecular pathogenesis of AD and PD, such as proteinopathy, mitochondrial dysfunction and oxidative stress, are interdependent phenomena with extensive and reinforcing cross talking, but there is no discernible arrangement of upstream and downstream events. Thus, targeting one of the three processes separately with a putative drug is unlikely to be successful as a neuroprotective measure. On the other hand, multiple drugs or other measures targeted to these pathways taken in combination may be beneficial in preventing the progress of these diseases. Currently, intensive research is going on using animal and cell-based models to elucidate how these molecular damage pathways finally lead to the programmed cell death of neurons in AD and PD. It is perhaps more prudent to identify the triggers that lead an aging brain to follow the pathway of AD or PD pathology, and apparently, the metabolic and environmental risk factors of these diseases identified in clinical cases may provide us the clue.
Acknowledgments
The authors are alone responsible for the content and writing of this paper. No financial support was obtained for this particular manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Davie CA. A review of Parkinson’s disease. Br Med Bull. 2008;86(1):109–127. | ||
Chakrabarti S, Khemka VK, Banerjee A, Chatterjee G, Ganguly A, Biswas A. Metabolic risk factors of sporadic Alzheimer’s disease: implications in the pathology, pathogenesis and treatment. Aging Dis. 2015;6(4):282–299. | ||
Nussbaum RL, Ellis CE. Alzheimer’s disease and Parkinson’s disease. N Engl J Med. 2003;348(14):1356–1364. | ||
Xie A, Gao J, Xu L, Meng D. Shared mechanisms of neurodegeneration in Alzheimer’s disease and Parkinson’s disease. Biomed Res Int. 2014;2014:648740. | ||
Perier C, Vila M. Mitochondrial biology and Parkinson’s disease. Cold Spring Harb Perspect Med. 2011;2(2):a009332. | ||
Bonda DJ, Smith MA, Perry G, Lee HG, Wang X, Zhu X. The mitochondrial dynamics of Alzheimer’s disease and Parkinson’s disease offer important opportunities for therapeutic intervention. Curr Pharm Des. 2011;17(31):3374–3380. | ||
Subramaniam SR, Chesselet M-F. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog Neurobiol. 2013;106–107:17–32. | ||
Xie W, Wan OW, Chung KK. New insights into the role of mitochondrial dysfunction and protein aggregation in Parkinson’s disease. Biochim Biophys Acta. 2010;1802(11):935–941. | ||
Butterfield DA, Lauderback CM. Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic Biol Med. 2002;32(11):1050–1060. | ||
Reddy PH, Beal MF. Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol Med. 2008;14(2):45–53. | ||
Cookson MR. α-Synuclein and neuronal cell death. Mol Neurodegener. 2009;4:9. | ||
Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8(6):595–608. | ||
Friedrich RP, Tepper K, Ronicke R, et al. Mechanism of amyloid plaque formation suggests an intracellular basis of a pathogenicity. Proc Natl Acad Sci U S A. 2010;107(5):1942–1947. | ||
Uversky VN, Eliezer D. Biophysics of Parkinson’s Disease: structure and aggregation of α-synuclein. Curr Protein Peptide Sci. 2009;10:483–499. | ||
Kovacs GG, Budka H. Current concepts of neuropathological diagnostics in practice: neurodegenerative diseases. Clin Neuropathol. 2010;29(5):271–288. | ||
O’brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer’s disease. Ann Rev Neurosci. 2011;34:185–204. | ||
Cahill CM, Lahiri DK, Huang X, Rogers JT. Amyloid precursor protein and alpha synuclein translation, implications for iron and inflammation in neurodegenerative diseases. Biochim Biophys Acta. 2009;1790(7):615–628. | ||
Jowaed A, Schmitt I, Kaut O, Wüllner U. Methylation regulates alpha-synuclein expression and is decreased in Parkinson’s disease patients’ brains. J Neurosci. 2010;30(18):6355–6359. | ||
Takalo M, Salminen A, Soininen H, Hiltunen M, Haapasalo A. Protein aggregation and degradation mechanisms in neurodegenerative diseases. Am J Neurodegener Dis. 2013;2(1):1–14. | ||
Ciechanover A, Kwon YT. Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med. 2015;47:e147. | ||
Morimoto RI. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008;22(11):1427–1438. | ||
Niforou K, Cheimonidou C, Trougakos IP. Molecular chaperones and proteostasis regulation during redox imbalance. Redox Biol. 2014;2:323–332. | ||
Sen CK, Packer L. Antioxidant and redox regulation of gene transcription. FASEB J. 1996;10(7):709–720. | ||
Aiken CT, Kaake RM, Wang X, Huang L. Oxidative stress-mediated regulation of proteasome complexes. Mol Cell Proteomics. 2011;10(5):R110.006924. | ||
Pajares M, Jiménez-moreno N, Dias IH, et al. Redox control of protein degradation. Redox Biol. 2015;6:409–420. | ||
Xiang W, Menges S, Schlachetzki J, et al. Posttranslational modification and mutation of histidine 50 trigger alpha synuclein aggregation and toxicity. Mol Neurodegener. 2015;10:8. | ||
Snead D, Eliezer D. Alpha-synuclein function and dysfunction on cellular membranes. Exp Neurobiol. 2014;23(4):292–313. | ||
Banerjee K, Sinha M, Pham Cle L, et al. Alpha-synuclein induced membrane depolarization and loss of phosphorylation capacity of isolated rat brain mitochondria: implications in Parkinson’s disease. FEBS Lett. 2010;584(8):1571–1576. | ||
Wang X, Su B, Lee H, et al. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci. 2009;29(28):9090–9103. | ||
Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J Neurosci. 2006;26(35):9057–9068. | ||
Protter D, Lang C, Cooper AA. α-Synuclein and mitochondrial dysfunction: a pathogenic partnership in Parkinson’s disease. Parkinson’s Dis. 2012;2012. | ||
Dong S, Duan Y, Hu Y, Zhao Z. Advances in the pathogenesis of Alzheimer’s disease: a re-evaluation of amyloid cascade hypothesis. Transl Neurodegener. 2012;1(1):18. | ||
Armstrong RA. The pathogenesis of Alzheimer’s disease: a reevaluation of the Amyloid cascade Hypothesis. Int J Alzheimers Dis. 2011;2011:1–6. | ||
Jiang S, Li Y, Zhang X, Bu G, Xu H, Zhang Y. Trafficking regulation of proteins in Alzheimer’s disease. Mol Neurodegener. 2014;9:6. | ||
Mondragón-rodríguez S, Perry G, Zhu X, Moreira PI, Acevedo-aquino MC, Williams S. Phosphorylation of tau protein as the link between oxidative stress, mitochondrial dysfunction, and connectivity failure: implications for Alzheimer’s disease. Oxid Med Cell Longev. 2013;2013:940603. | ||
Alonso AC, Grundke-Iqbal I, Iqbal K. Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med. 1996;2(7):783–787. | ||
Butterfield DA, Swomley AM, Sultana S. Amyloid β-peptide (1–42)-induced oxidative stress in Alzheimer disease: importance in disease pathogenesis and progression. Antioxid Redox Signal. 2013;19:823–835. | ||
Butterfield DA, Di Domenico F, Swomley AM, Head E, Perluigi M. Redox proteomics analysis to decipher the neurobiology of Alzheimer-like neurodegeneration: overlaps in down’s syndrome and Alzheimer’s disease brain. Biochem J. 2014;463(2):177–189. | ||
Theuns J, Van Broeckhoven C. Transcriptional regulation of Alzheimer’s disease genes: implications for susceptibility. Hum Mol Genet. 2000;9(16):2383–2394. | ||
Hoffman PW, Chernak JM. DNA binding and regulatory effects of transcription factors SP1 and USF at the rat amyloid precursor protein gene promoter. Nucleic Acids Res. 1995;23(12):2229–2235. | ||
Picone P, Nuzzo D, Caruana L, et al. Metformin increases APP expression and processing via oxidative stress, mitochondrial dysfunction and NF-κB activation: use of insulin to attenuate metformin’s effect. Biochim Biophys Acta. 2015;1853(5):1046–1059. | ||
Ko SY, Lin YP, Lin YS, Chang SS. Advanced glycation end products enhance amyloid precursor protein expression by inducing reactive oxygen species. Free Radic Biol Med. 2010;49(3):474–480. | ||
Rogers JT, Randall JD, Cahill CM, et al. An iron-responsive element type II in the 5′-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J Biol Chem. 2002;277(47):45518–45528. | ||
Huang X, Moir RD, Tanzi RE, Bush AI, Rogers JT. Redox-active metals, oxidative stress, and Alzheimer’s disease pathology. Ann N Y Acad Sci. 2004;1012:153–163. | ||
Chen XF, Zhang YW, Xu H, Bu G. Transcriptional regulation and its misregulation in Alzheimer’s disease. Mol Brain. 2013;6:44. | ||
Chami L, Checler F. BACE1 is at the crossroad of a toxic vicious cycle involving cellular stress and β-amyloid production in Alzheimer’s disease. Mol Neurodegener. 2012;7:52. | ||
Guglielmotto M, Aragno M, Autelli R, et al. The up-regulation of BACE1 mediated by hypoxia and ischemic injury: role of oxidative stress and HIF1alpha. J Neurochem. 2009;108(4):1045–1056. | ||
Kaur U, Banerjee P, Bir A, Sinha M, Biswas A, Chakrabarti S. Reactive oxygen species, redox signaling and neuroinflammation in Alzheimer’s disease: the NF-κB connection. Curr Top Med Chem. 2015;15(5):446–457. | ||
Tong Y, Zhou W, Fung V, et al. Oxidative stress potentiates BACE1 gene expression and Abeta generation. J Neural Transm. 2004;112(3):455–469. | ||
Tamagno E, Bardini P, Obbili A, et al. Oxidative stress increases expression and activity of BACE in NT2 neurons. Neurobiol Dis. 2002;10(3):279–288. | ||
Tamagno E, Parola M, Bardini P, et al. Beta-site APP cleaving enzyme up-regulation induced by 4-hydroxynonenal is mediated by stress-activated protein kinases pathways. J Neurochem. 2005;92(3):628–636. | ||
Tamagno E, Guglielmotto M, Aragno M, et al. Oxidative stress activates a positive feedback between the γ- and β-secretase cleavages of the β-amyloid precursor protein. J Neurochem. 2008;104(3):683–695. | ||
Mouton-liger F, Paquet C, Dumurgier J, et al. Oxidative stress increases BACE1 protein levels through activation of the PKR-eIF2α pathway. Biochem Biophys Acta. 2012;1822(6):885–896. | ||
Misonou H, Morishima-kawashima M, Ihara Y. Oxidative stress induces intracellular accumulation of amyloid beta-protein (Abeta) in human neuroblastoma cells. Biochemistry. 2000;39(23):6951–6959. | ||
Deane R, Bell RD, Sagare A, Zlokovic BV. Clearance of amyloid-beta peptide across the blood-brain barrier: implication for therapies in Alzheimer’s disease. CNS Neurol Disord Drug Targets. 2009;8(1):16–30. | ||
Bates KA, Verdile G, Li Q-X, et al. Clearance mechanisms of Alzheimer’s amyloid-β peptide: implications for therapeutic design and diagnostic tests. Mol Psychiatr. 2008;14(5):469–486. | ||
Ramanathan A, Nelson AR, Sagare AP, Zlokovic BV. Impaired vascular-mediated clearance of brain amyloid beta in Alzheimer’s disease: the role, regulation and restoration of LRP1. Front Aging Neurosci. 2015;7:136. | ||
Kadowaki H, Nishitoh H, Urano F, et al. Amyloid β induces neuronal cell death through ROS-mediated ASK1 activation. Cell Death Differ. 2005;12(1):19–24. | ||
Behl C. Hydrogen peroxide mediates amyloid β protein toxicity. Cell. 1994;77(6):817–827. | ||
Chakrabarti S, Sinha M, Thakurta IG, Banerjee P, Chattopadhyay M. Oxidative stress and amyloid beta toxicity in Alzheimer’s disease: intervention in a complex relationship by antioxidants. Curr Med Chem. 2013;20(37):4648–4664. | ||
Smith DG, Cappai R, Barnham KJ. The redox chemistry of the Alzheimer’s disease amyloid β peptide. Biochim Biophys Acta. 2007;1768(8):1976–1990. | ||
Sinha M, Bhowmick P, Banerjee A, Chakrabarti S. Antioxidant role of amyloid β protein in cell-free and biological systems: implication for the pathogenesis of Alzheimer disease. Free Radic Biol Med. 2013;56:184–192. | ||
Shelat PB, Chalimoniuk M, Wang JH, et al. Amyloid beta peptide and NMDA induce ROS from NADPH oxidase and AA release from cytosolic phospholipase A2 in cortical neurons. J Neurochem. 2008;106(1):45–55. | ||
Hu H, Li M. Mitochondria-targeted antioxidant mitotempo protects mitochondrial function against amyloid beta toxicity in primary cultured mouse neurons. Biochem Biophys Res Commun. 2016;478(1):174–180. | ||
Qin L, Liu Y, Cooper C, Liu B, Wilson B, Hong J-S. Microglia enhance β-amyloid peptide-induced toxicity in cortical and mesencephalic neurons by producing reactive oxygen species. J Neurochem. 2002;83(4):973–983. | ||
Qin B, Cartier L, Dubois-dauphin M, Li B, Serrander L, Krause KH. A key role for the microglial NADPH oxidase in APP-dependent killing of neurons. Neurobiol Aging. 2006;27(11):1577–1587. | ||
Doens D, Fernández PL. Microglia receptors and their implications in the response to amyloid β for Alzheimer’s disease pathogenesis. J Neuroinflammation. 2014;11:48. | ||
Coraci IS, Husemann J, Berman JW, et al. CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer’s disease brains and can mediate production of reactive oxygen species in response to beta-amyloid fibrils. Am J Pathol. 2002;160(1):101–112. | ||
Zhang D, Hu X, Qian L, et al. Microglial MAC1 receptor and PI3K are essential in mediating β-amyloid peptide-induced microglial activation and subsequent neurotoxicity. J Neuroinflammation. 2011;8(1):3. | ||
Mondragón-rodríguez S, Perry G, Luna-muñoz J, Acevedo-aquino MC, Williams S. Phosphorylation of tau protein at sites Ser (396-404) is one of the earliest events in Alzheimer’s disease and Down syndrome. Neuropathol Appl Neurobiol. 2014;40(2):121–135. | ||
Alavi Naini SM, Soussi-yanicostas N. Tau hyperphosphorylation and oxidative stress, a critical vicious circle in neurodegenerative tauopathies? Oxid Med Cell Longev. 2015;2015:151979. | ||
Su B, Wang X, Lee HG, et al. Chronic oxidative stress causes increased tau phosphorylation in M17 neuroblastoma cells. Neurosci Lett. 2010;468(3):267–271. | ||
Giraldo E, Lloret A, Fuchsberger T, Viña J. Aβ and tau toxicities in Alzheimer’s are linked via oxidative stress-induced p38 activation: protective role of vitamin E. Redox Biol. 2014;2:873–877. | ||
Lovell MA, Xiong S, Xie C, Davies P, Markesbery WR. Induction of hyperphosphorylated tau in primary rat cortical neuron cultures mediated by oxidative stress and glycogen synthase kinase-3. J Alzheimers Dis. 2004;6(6):659–671. | ||
Egaña JT, Zambrano C, Nuñez MT, Gonzalez-billault C, Maccioni RB. Iron-induced oxidative stress modify tau phosphorylation patterns in hippocampal cell cultures. Biometals. 2003;16(1):215–223. | ||
Zambrano CA, Egaña JT, Núñez MT, Maccioni RB, González-billault C. Oxidative stress promotes tau dephosphorylation in neuronal cells: the roles of cdk5 and PP1. Free Radic Biol Med. 2004;36(11):1393–1402. | ||
Moreira PI, Carvalho C, Zhu X, Smith MA, Perry G. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim Biophys Acta. 2010;1802(1):2–10. | ||
Onyango IG, Dennis J, Khan SM. Mitochondrial dysfunction in Alzheimer’s disease and the rationale for bioenergetics based therapies. Aging Dis. 2016;7(2):201–214. | ||
Chen JX, Yan SD. Amyloid-beta-induced mitochondrial dysfunction. J Alzheimers Dis. 2007;12(2):177–184. | ||
Mossmann D, Vögtle F, Taskin AA, et al. Amyloid-β peptide induces mitochondrial dysfunction by inhibition of preprotein maturation. Cell Metab. 2014;20(4):662–669. | ||
Spuch C, Ortolano S, Navarro C. New insights in the amyloid-beta interaction with mitochondria. J Aging Res. 2012;2012:324968. | ||
Sinha M, Behera P, Bhowmick P, Banerjee K, Basu S, Chakrabarti S. Aging promotes amyloid-β peptide induced mitochondrial dysfunctions in rat brain: a molecular link between aging and Alzheimer’s disease. J Alzheimers Dis. 2011;27(4):753–765. | ||
Arispe N, Diaz JC, Simakova O. Abeta ion channels. Prospects for treating Alzheimer’s disease with Abeta channel blockers. Biochim Biophys Acta. 2007;1768(8):1952–1965. | ||
Lal R, Lin H, Quist AP. Amyloid beta ion channel: 3D structure and relevance to amyloid channel paradigm. Biochim Biophys Acta. 2007;1768(8):1966–1975. | ||
Zhu D, Lai Y, Shelat PB, Hu C, Sun GY, Lee JC. Phospholipases A2 mediate amyloid-beta peptide-induced mitochondrial dysfunction. J Neurosci. 2006;26(43):11111–11119. | ||
Abramov AY, Canevari L, Duchen MR. Beta-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J Neurosci. 2004;24(2):565–575. | ||
Pringsheim T, Jette N, Frolkis A, Steeves TDL. The prevalence of Parkinson’s disease: a systematic review and meta-analysis. Mov Disord. 2014;29(13):1583–1590. | ||
Klein C, Westenberger A. Genetics of Parkinson’s disease. Cold Spring Harb Perspect Med. 2012;2(1):a008888. | ||
Braak H, Ghebremedhin E, Rüb U, Bratzke H, Del Tredici K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004;318(1):121–134. | ||
Stefanis L. Synuclein in Parkinson’s disease. Cold Spring Harb Perspect Med. 2011;2:a009399. | ||
Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett. 1998;251(3):205–208. | ||
Dikiy I, Eliezer D. Folding and misfolding of alpha-synuclein on membranes. Biochim Biophys Acta. 2012;1818(4):1013–1018. | ||
Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci. 2013;14(1):38–48. | ||
Roostaee A, Beaudoin S, Staskevicius A, Roucou X. Aggregation and neurotoxicity of recombinant α-synuclein aggregates initiated by dimerization. Mol Neurodegener. 2013;8:5. | ||
Matsumoto L, Takuma H, Tamaoka A, et al. CpG demethylation enhances alpha-synuclein expression and affects the pathogenesis of Parkinson’s disease. PLoS One. 2010;5(11):e15522. | ||
Febbraro F, Giorgi M, Caldarola S, Loreni F, Romero-ramos M. α-Synuclein expression is modulated at the translational level by iron. Neuroreport. 2012;23(9):576–580. | ||
Dehay B, Martinez-vicente M, Caldwell GA, et al. Lysosomal impairment in Parkinson’s disease. Mov Disord. 2013;28(6):725–732. | ||
Cook C, Petrucelli L. A critical evaluation of the ubiquitin-proteasome system in Parkinson’s disease. Biochim Biophys Acta. 2009;1792(7):664–675. | ||
Mcnaught KS, Belizaire R, Isacson O, Jenner P, Olanow CW. Altered proteasomal function in sporadic Parkinson’s disease. Exp Neurol. 2003;179(1):38–46. | ||
Myung J, Kim KB, Crews CM. The ubiquitin-proteasome pathway and proteasome inhibitors. Med Res Rev. 2001;21(4):245–273. | ||
McNaught KS, Olanow CW, Halliwell B, Isacson O, Jenner P. Failure of the ubiquitin–proteasome system in Parkinson’s disease. Nat Rev Neurosci. 2001;2(8):589–594. | ||
McNaught KS, Perl DP, Brownell AL, Olanow CW. Systemic exposure to proteasome inhibitors causes a progressive model of Parkinson’s disease. Ann Neurol. 2004;56(1):149–162. | ||
Eriksen JL, Wszolek Z, Petrucelli L. Molecular pathogenesis of Parkinson disease. Arch Neurol. 2005;62(3):353–357. | ||
Tanaka Y, Engelender S, Igarashi S, et al. Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum Mol Genet. 2001;10(9):919–926. | ||
Perez-Alvarez S, Solesio ME, Manzanares J, Jordán J, Galindo MF. Lactacystin requires reactive oxygen species and BAX redistribution to induce mitochondria mediated cell death. Br J Pharmacol. 2009;158(4):1121–1130. | ||
Zhou ZD, Lim TM. Dopamine (DA) induced irreversible proteasome inhibition via DA derived quinones. Free Radic Res. 2009;43(4):417–430. | ||
Snyder H, Mensah K, Theisler C, Lee J, Matouschek A, Wolozin B. Aggregated and monomeric alpha-synuclein bind to the S6′ proteasomal protein and inhibit proteasomal function. J Biol Chem. 2003;278(14):11753–11759. | ||
Simón-sánchez J, Schulte C, Bras JM, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet. 2009;41(12):1308–1312. | ||
Lo Bianco C, Ridet JL, Schneider BL, Deglon N, Aebischer P. Alpha-synucleinopathy and selective dopaminergic neuron loss in a rat lentiviral-based model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2002;99(16):10813–10818. | ||
Oliveras-salvá M, Van der Perren A, Casadei N, et al. rAAV2/7 vector-mediated overexpression of alpha-synuclein in mouse substantia nigra induces protein aggregation and progressive dose-dependent neurodegeneration. Mol Neurodegener. 2013;8:44. | ||
Paumier KL, Luk KC, Manfredsson FP, et al. Intrastriatal injection of pre-formed mouse α-synuclein fibrils into rats triggers α-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol Dis. 2015;82:185–199. | ||
Recasens A, Dehay B, Bové J, et al. Lewy body extracts from Parkinson disease brains trigger α-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol. 2014;75(3):351–362. | ||
Bisaglia M, Greggio E, Maric D, Miller DW, Cookson MR, Bubacco L. Alpha-synuclein overexpression increases dopamine toxicity in BE2-M17 cells. BMC Neurosci. 2010;11:41. | ||
Mullin S, Schapira A. A-synuclein and mitochondrial dysfunction in Parkinson’s disease. Mol Neurobiol. 2013;47(2):587–597. | ||
Schapira AHV, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD. Mitochondrial complex I deficiency in Parkinson’s disease. J Neurochem. 1990;54(3):823–827. | ||
Blake CI, Spitz E, Leehey M, Hoffer BJ, Boyson SJ. Platelet mitochondrial respiratory chain function in Parkinson’s disease. Mov Disord. 1997;12(1):3–8. | ||
Blin O, Desnuelle C, Rascol O, et al. Mitochondrial respiratory failure in skeletal muscle from patients with Parkinson’s disease and multiple system atrophy. J Neurol Sci. 1994;125(1):95–101. | ||
Santos D, Esteves AR, Silva DF, Januário C, Cardoso SM. The impact of mitochondrial fusion and fission modulation in sporadic Parkinson’s disease. Mol Neurobiol. 2015;52(1):573–586. | ||
Swerdlow RH, Parks JK, Miller SW, et al. Origin and functional consequences of the complex I defect in Parkinson’s disease. Ann Neurol. 1996;40(4):663–671. | ||
Trimmer PA, Bennett JP. The cybrid model of sporadic Parkinson’s disease. Exp Neurol. 2009;218(2):320–325. | ||
Dauer W, Przedborski S. Parkinson’s disease. Neuron. 2003;39(6):889–909. | ||
Iglesias-gonzález J, Sánchez-iglesias S, Méndez-Álvarez E, et al. Differential toxicity of 6-hydroxydopamine in SH-SY5Y human neuroblastoma cells and rat brain mitochondria: protective role of catalase and superoxide dismutase. Neurochem Res. 2012;37(10):2150–2160. | ||
Winklhofer KF, Haass C. Mitochondrial dysfunction in Parkinson’s disease. Biochim Biophys Acta. 2010;1802(1):29–44. | ||
Jana S, Sinha M, Chanda D, et al. Mitochondrial dysfunction mediated by quinone oxidation products of dopamine: implications in dopamine cytotoxicity and pathogenesis of Parkinson’s disease. Biochim Biophys Acta. 2011;1812(6):663–673. | ||
Bender A, Krishnan KJ, Morris CM, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. 2006;38(5):515–517. | ||
Deas E, Wood NW, Plun-favreau H. Mitophagy and Parkinson’s disease: the PINK1-parkin link. Biochim Biophys Acta. 2011;1813(4):623–633. | ||
Ding WX, Yin XM. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem. 2012;393(7):547–564. | ||
Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85(2):257–273. | ||
Narendra DP, Jin SM, Tanaka A, et al. PINK1 is selectively stabilized on impaired mitochondria to activate parkin. PLoS Biol. 2010;8(1):e1000298. | ||
Vives-Bauza C, Zhou C, Huang Y, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A. 2010;107(1):378–383. | ||
Matsuda N, Sato S, Shiba K, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189(2):211–221. | ||
Lee S, Zhang C, Liu X. Role of glucose metabolism and ATP in maintaining PINK1 levels during Parkin-mediated mitochondrial damage responses. J Biol Chem. 2015;290(2):904–917. | ||
Van Laar VS, Arnold B, Cassady SJ, Chu CT, Burton EA, Berman SB. Bioenergetics of neurons inhibit the translocation response of Parkin following rapid mitochondrial depolarization. Hum Mol Genet. 2011;20(5):927–940. | ||
Norris KL, Hao R, Chen LF, et al. Convergence of Parkin, PINK1, and α-synuclein on stress-induced mitochondrial morphological remodeling. J Biol Chem. 2015;290(22):13862–13874. | ||
Liu W, Vives-Bauza C, Acín-Peréz R, et al. PINK1 defect causes mitochondrial dysfunction, proteasomal deficit and alpha-synuclein aggregation in cell culture models of Parkinson’s disease. PLoS One. 2009;4(2):e4597. | ||
Devi L, Anandatheerthavarada HK. Mitochondrial trafficking of APP and alpha synuclein: relevance to mitochondrial dysfunction in Alzheimer’s and Parkinson’s diseases. Biochim Biophys Acta. 2010;1802(1):11–19. | ||
Chinta SJ, Mallajosyula JK, Rane A, Andersen JK. Mitochondrial α-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci Lett. 2010;486(3):235–239. | ||
Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem. 2008;283(14):9089–9100. | ||
Reeve AK, Ludtmann MH, Angelova PR, et al. Aggregated α-synuclein and complex I deficiency: exploration of their relationship in differentiated neurons. Cell Death Dis. 2015;6:e1820. | ||
Bir A, Sen O, Anand S, et al. α-Synuclein-induced mitochondrial dysfunction in isolated preparation and intact cells: implications in the pathogenesis of Parkinson’s disease. J Neurochem. 2014;131(6):868–877. | ||
Kamp F, Exner N, Lutz AK, et al. Inhibition of mitochondrial fusion by α-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. 2010;29(20):3571–3589. | ||
Jenner P, Olanow CW. Oxidative stress and the pathogenesis of Parkinson’s disease. Neurology. 1996;47(6 suppl 3):161S–170S. | ||
Sofic E, Riederer P, Heinsen H, et al. Increased iron (III) and total iron content in post mortem substantia nigra of parkinsonian brain. J Neural Transm. 1988;74(3):199–205. | ||
Cassarino DS, Fall CP, Swerdlow RH, et al. Elevated reactive oxygen species and antioxidant enzyme activities in animal and cellular models of Parkinson’s disease. Biochim Biophys Acta. 1997;1362(1):77–86. | ||
Di Maio R, Barrett PJ, Hoffman EK, et al. α-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci Transl Med. 2016;8(342):342ra78. | ||
Anandhan A, Rodriguez-rocha H, Bohovych I, et al. Overexpression of alpha-synuclein at non-toxic levels increases dopaminergic cell death induced by copper exposure via modulation of protein degradation pathways. Neurobiol Dis. 2015;81:76–92. | ||
Carboni E, Lingor P. Insights on the interaction of alpha-synuclein and metals in the pathophysiology of Parkinson’s disease. Metallomics. 2015;7(3):395–404. | ||
Hastings TG. The role of dopamine oxidation in mitochondrial dysfunction: implications for Parkinson’s disease. J Bioenerg Biomembr. 2009;41(6):469–472. | ||
Leong SL, Cappai R, Barnham KJ, Pham CL. Modulation of alpha-synuclein aggregation by dopamine: a review. Neurochem Res. 2009;34(10):1838–1846. | ||
Cristóvão AC, Guhathakurta S, Bok E, et al. NADPH oxidase 1 mediates α-synucleinopathy in Parkinson’s disease. J Neurosci. 2012;32(42):14465–14477. | ||
Schildknecht S, Gerding HR, Karreman C, et al. Oxidative and nitrative alpha-synuclein modifications and proteostatic stress: implications for disease mechanisms and interventions in synucleinopathies. J Neurochem. 2013;125(4):491–511. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.