Back to Journals » Drug Design, Development and Therapy » Volume 10
Protein kinase C–mediated sodium glucose transporter 1 activation in precondition-induced cardioprotection
Authors kanwal A ![]() , Kasetti S, Putcha UK, Asthana S, Banerjee SK
, Kasetti S, Putcha UK, Asthana S, Banerjee SK
Received 31 January 2016
Accepted for publication 11 March 2016
Published 14 September 2016 Volume 2016:10 Pages 2929—2938
DOI https://doi.org/10.2147/DDDT.S105482
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Abhinav Kanwal,1,2 Sujatha Kasetti,3 Uday Kumar Putcha,4 Shailendra Asthana,2 Sanjay K Banerjee1,2
1Division of Medicinal Chemistry and Pharmacology, Indian Institute of Chemical Technology, Hyderabad, India; 2Drug Discovery Research Center (DDRC), Translational Health Science and Technology Institute (THSTI), Faridabad, Haryana, India; 3Department of Pharmacology, National Institute of Pharmaceutical Education and Research, Hyderabad, India; 4Department of Pathology, National Institute of Nutrition, Hyderabad, India
Abstract: The concept of cardioprotection through preconditioning against ischemia–reperfusion (I/R) injury is well known and established. However, among different proposed mechanisms regarding the concept of ischemic preconditioning, protein kinase C (PKC)-mediated cardioprotection through ischemic preconditioning plays a key role in myocardial I/R injury. Thus, this study was designed to find the relationship between PKC and sodium glucose transporter 1 (SGLT1) in preconditioning-induced cardioprotection, which is ill reported till now. By applying a multifaceted approach, we demonstrated that PKC activates SGLT1, which curbed oxidative stress and apoptosis against I/R injury. PKC activation enhances cardiac glucose uptake through SGLT1 and seems essential in preventing I/R-induced cardiac injury, indicating a possible cross-talk between PKC and SGLT1.
Keywords: ischemic preconditioning, ischemia-reperfusion injury, phorbol-12-myristate, oxidative stress, SGLT1, phlorizin
Introduction
Adequate glucose consumption and its metabolism are very important for the ischemic heart to function properly. Glucose is transported into cardiac myocytes by members of facilitative glucose transporters and sodium glucose cotransporter (SGLT) family. SGLT1 is highly expressed in the heart, but still its role in cardiac function is unknown. The heart utilizes many substrates to generate adenosine triphosphate (ATP) so as to function without fatigue. However, up to 30% of myocardial ATP is generated by glucose.1 Therefore, understanding the underlying mechanism of glucose consumption in ischemic condition is of great importance.
In general, severe ATP deficit occurs during ischemia and leads to irreversible injury or cell death.1 However, during reperfusion, sudden restoration of oxygen and blood flow lead to high ATP generation in the presence of higher calcium concentrations, but also results in the production of excess reactive oxygen species.2 Ischemic preconditioning (IPC), which refers to introduction of repeated brief cycles of ischemia followed by perfusion, enables the heart to resist severe damage to the myocardium resulting from sustained ischemic condition.3 Hence, IPC induces a protective mechanism in the heart.
Different mechanisms were elucidated for precondition-induced cardioprotection. Among all these mechanisms, translocation and activation of protein kinase C (PKC) has gained the maximum scientific interest. An in vitro and in vivo study demonstrates that the blockade of PKC leads to abortion of PC-induced cardioprotection.4 Posttranslational modification, ie, phosphorylation by PKC is a well-known mechanism of cellular regulation. Activation and translocation of PKC might be correlated to increased glucose uptake and cardioprotection.5
SGLTs are well-known transmembrane glucose transporters responsible for the active transport of glucose.6 Interestingly, SGLT1 contains four putative phosphorylation sites for PKC, ie, Thr50, Ser303, Ser418, and Ser562. Among all four sites, Ser418 is the common site for both PKC and PKA.7 However, the question remains whether PKC mediates its effect via direct phosphorylation of one of the putative sites in SGLT1, and so altering its intrinsic activity is still not clear. The presence of PKC phosphorylation sites in SGLT1 proves the importance of the phosphorylation modification on SGLT1 activity.8 It has been shown that the activation of PKC altered the maximum rate of Na+/glucose cotransport in oocytes expressing rabbit SGLT1.8 Transport of glucose is proportional to the number of phosphorylated SGLT1 present on the plasma membrane. Although the activation of PKC is correlated with cardioprotection against ischemic–reperfusion (I/R) injury, its role on SGLT1-mediated cardioprotection has not been investigated so far.
Collectively, this study demonstrated the cardioprotective role of PKC by glucose uptake via SGLT1 transporter. Conversely, SGLT1 inhibition by phlorizin (PZ) abrogated the cardioprotective effect of PKC activator against I/R injury. Therefore, our study underscores the existence of a cross-talk between SGLT1 and PKC activation for glucose uptake in cardioprotection.
Materials and methods
Animal care
All animal experiments were undertaken with the approval of Institutional Animal Ethics Committee (IAEC) of Indian Institute of Chemical Technology, Hyderabad, India. Male Sprague Dawley albino rats (200–250 g) were provided by National Institute of Nutrition, Hyderabad, and had free access to food (pellet diet supplied from National Institute of Nutrition, Hyderabad) and water ad libitum. All the methods were carried out in accordance with the approved guidelines of IAEC.
Isolated heart perfusion
Rats were anesthetized with 100 mg/mL of ketamine and 20 mg/mL of xylazine, given intraperitoneally (IP). The rats were given 300 U/kg of heparin (IP) 30 minutes before they were killed. The hearts were then quickly excised and submerged in ice-cold phosphate-buffered saline. The aorta was quickly secured to a cannula, and the heart was perfused with modified Krebs–Henseleit (KH) buffer solution using a nonrecirculating Langendorff preparation (AD Instruments, Sydney, Australia) with a constant flow system (10 mL/min/g). The KH buffer consisted of 4.7 mmol/L KCL, 2.5 mmol/L CaCl2, 1.25 mmol/L MgCl2, 1.25 mmol/L KH2PO4, 0.5 mmol/L ethylenediaminetetraacetic acid, 25 mmol/L NaHCO3, 118 mmol/L NaCl, and 5 mmol/L glucose. During perfusion, the temperature of the buffer was maintained at 37°C and equilibrated with a 95% oxygen–5% carbon dioxide mixture.
Ischemia/reperfusion protocols
The rat hearts were divided into four groups. All hearts were perfused for a minimum 15 minutes for equilibration before the following perfusion protocols were performed (Figure 1). Group 1 (Control): 50 minutes perfusion with modified KH buffer. Group 2 (I/R, injury): 15 minutes perfusion, 15 minutes global no-flow ischemia, followed by 20 minutes reperfusion (I/R) with modified KH buffer. Group 3 (phorbol 12-myristate 13-acetate, PMA): hearts were preconditioned with PKC activator, PMA 500 nmol/L for 15 minutes, followed by I/R. Group 4 (PMA + PZ): preconditioning as in Group 3 pretreated with acute dose of PZ (SGLT1 inhibitor) for a short period of 5 minutes (PMA + PZ). PZ (4.4 mg/mL) was added only to the KH buffer during first 5 minutes of initial perfusion and then washed out with KH buffer during 15 minutes of perfusion (Figure 1). The dose of PZ was based on a previous study.9 Three rat hearts (IPC group) were subjected to ischemic preconditioning (IPC) and IR. IPC was achieved by three cycles of global ischemia for 2 minutes followed by reperfusion for 3 minutes.
 | Figure 1 Schematic representation of I/R injury and PKC-mediated preconditioning protocol. |
Biochemical parameters
At the end of the experiment, the hearts were quickly washed in cold saline and stored at −80°C. Heart tissue samples were thawed and homogenized with ten times (weight/volume) ice-cold 0.1 M phosphate buffer (pH 7.4). Aliquots of heart homogenates were separated and used to measure thiobarbituric acid reactive substances (TBARS), a marker of lipid peroxidation. The remaining homogenates were centrifuged at 10,000 rpm for 15 minutes, and the supernatant was used for other biochemical parameters, ie, catalase, glutathione (GSH), superoxide dismutase (SOD), conjugated dienes, total antioxidant [di(phenyl)-(2,4,6-trinitrophenyl)iminoazanium, DPPH], and vitamin C as per the protocol described.10–13 Protein concentrations were measured according to Bradford’s method.
Histopathological studies
After the ex vivo experiment, rat hearts were fixed in 10% neutral buffered formalin. Paraffin-embedded 5 μm thick sections were obtained and stained with hematoxylin and eosin. Stained sections were examined under a light microscope to assess the extent of cardiac injury.
Apoptosis parameters
Caspase-3 activity was measured using a standard assay kit (Calbichem, San Diego, CA, USA) following the manufacturer’s instruction. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay was performed to measure apoptotic nuclei in hearts for all the groups using TdT-Fragel™ DNA Fragmentation Detection Kit (Calbiochem).
In vitro and in vivo 2-NBDG glucose uptake assay
H9C2 or SGLT1-transfected H9C2 cells were plated at a density of 1×104 cells/well in a 96-well plate and used at subconfluence after 24 hours preincubation. For these experiments, all culture medium was removed from each well and replaced with 100 μL of culture medium in the absence or presence of fluorescent 2-NBDG (50 nm) or 2-NBDG with PKC activators with or without PZ at indicated concentration. Cells were treated with PZ for 30 minutes before treatment with PKC activators. Cells were incubated at 37°C with 5% CO2 for a period of 30 minutes, and 2-NBDG uptake was measured after three washes with phosphate-buffered saline. After lysis, the fluorescence of the cells was measured at excitation/emission maxima of ~465/540 nm.14
For the in vivo study, 2-NBDG (25 μM/kg, IP) and PKC activator (Molsidomine, 10 mg/kg, IP) were administered to mice (20 g) 10 minutes after PZ injection (400 mg/kg, IP). Forty minutes later, mice hearts were excised and washed in cold saline. Hearts were homogenized with 10 volumes (weight/volume) of ice-cold 0.1 M phosphate buffer (pH 7.4). Homogenates were centrifuged at 10,000 rpm for 15 minutes, and 50 μL supernatant was used to measure 2-NBDG glucose uptake. Fluorescence was measured at excitation/emission maxima of ~465/540 nm.14
PKC activity assay
The heart homogenate was used to measure PKC activity. The assay was carried out according to the manufacturer’s protocol (Enzo life Sciences, New York, NY, USA).
Statistical analysis
Data are expressed as mean ± standard error. Differences between two groups were compared by Student’s t-test and among multiple groups by one-way analysis of variance, followed by post hoc Bonferroni test. A P-value ≤0.05 was considered significant. All statistical analyses were performed with GraphPad Prism version 6.0 for Windows (GraphPad Software, La Jolla, CA, USA).
Molecular modeling
The three-dimensional structure of PKC and SGLT1 was produced through molecular modeling. Furthermore, the protein relaxation and protein–protein docking were conducted through NAMD and SwormDock methods, respectively. More details are provided in the Supplementary materials.
Results
Increased cellular glucose uptake is associated with increased PKC activity and increased SGLT1 expression
This study was undertaken to determine whether increased glucose uptake is mediated through SGLT1. We performed in vitro glucose uptake studies using fluorescent glucose analog, 2-NBDG in H9C2 cells overexpressed with SGLT1. Our data indicated that H9C2 cells overexpressing SGLT1 showed a significant (P<0.001) increase in PKC activity compared to control H9C2 cells. This activity was further increased when treated with the PKC activator PMA (Figure 2A). Similarly, PMA treatment significantly increased PKC activity in SGLT1-overexpressed cells (P<0.001) as compared to control cells (Figure 2B). This increased glucose uptake in SGLT1 overexpressed cells was significantly (P<0.001) reduced after treatment with SGLT1 inhibitor, PZ (Figure 2C).
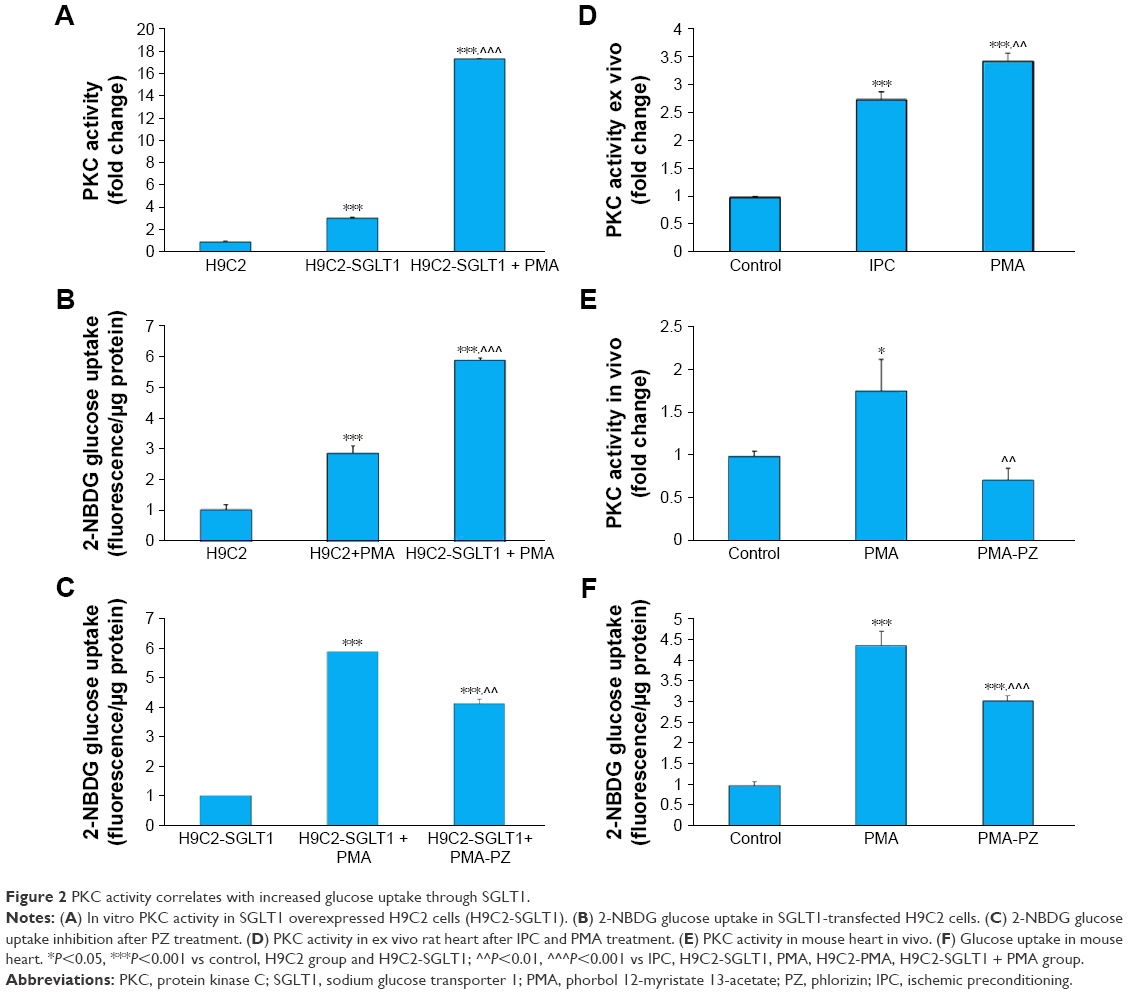 | Figure 2 PKC activity correlates with increased glucose uptake through SGLT1. |
IPC increases PKC activity and enhances glucose uptake
To confirm the activation of PKC in the heart during IPC, we performed IPC in rat heart ex vivo. We observed that PKC activity significantly (P<0.001) increased during IPC (Figure 2D). Mice were treated with PKC activator, PMA, and myocardial PKC activation was measured with or without SGLT1 inhibitor, PZ. We observed a significant increase in myocardial PKC activity in the PMA group, which decreased with PZ treatment (Figure 2E). We also observed that increased PKC activity led to significantly (P<0.001) higher glucose uptake during PKC preconditioning, which was further significantly (P<0.001) reduced with PZ treatment (Figure 2F).
SGLT1 inhibition in the presence of PKC activator diminishes the cardioprotective effect of PKC
There was a significant decrease in myocardial SOD, GSH, and vitamin-C in the I/R group compared to control group. We observed a significant (P<0.001) increase in endogenous antioxidants like catalase, GSH (P<0.05), and SOD (P<0.05) and vitamin C (P<0.01) levels in the PMA group (Figure 3). This beneficial effect of PMA was abrogated after treatment with SGLT1 inhibitor, PZ. A significant decrease in catalase (P<0.01), GSH (P<0.05), SOD (P<0.05), and vitamin C (P<0.001) was observed in PMA + PZ group. An increase in TBARS and conjugated dienes and a decrease in DPPH activity were observed in the I/R heart. However, a significant (P<0.01) reduction in TBARS levels and conjugated dienes levels (P<0.05), and an increase in DPPH activity were observed after treatment with PKC activator, PMA. On the other hand, a significant (P<0.01) increase in the levels of TBARS and conjugated dienes (P<0.05) and a decrease in DPPH activity (P<0.001) were observed after treatment with PZ (Figure 4).
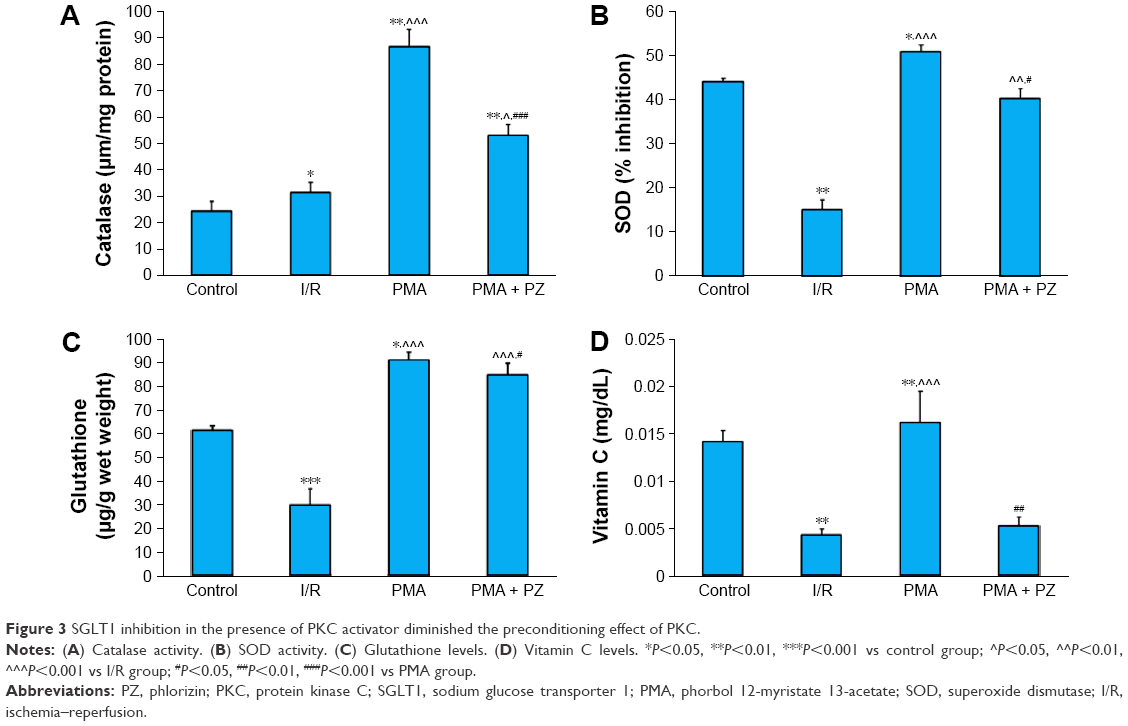 | Figure 3 SGLT1 inhibition in the presence of PKC activator diminished the preconditioning effect of PKC. |
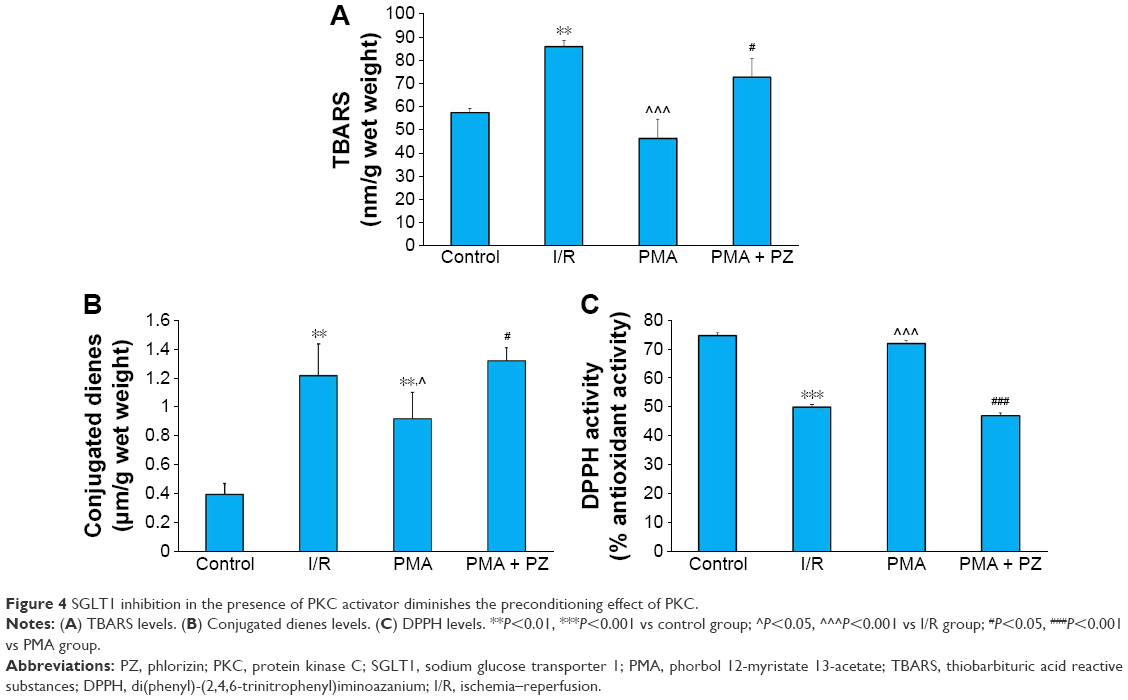 | Figure 4 SGLT1 inhibition in the presence of PKC activator diminishes the preconditioning effect of PKC. |
Inhibition of SGLT1 in the presence of PKC activator induces apoptosis
It is well known that caspase-3 activity is increased in ischemic conditions and leads to apoptosis. We measured caspase-3 activity in all the groups. We observed a significant (P<0.01) increase in caspase-3 activity in the I/R heart. Caspase-3 activity was attenuated significantly (P<0.01) in PMA group. However, myocardial caspase-3 activity increased (P<0.01) in PMA + PZ group compared to that in I/R group (Figure 5A). To examine the extent of apoptotic cell death, TUNEL staining was also performed. A count of TUNEL-positive nuclei showed that there was an increase in apoptotic nuclei in I/R group. An increase in apoptosis was attenuated in PMA group. However, SGLT1 inhibition in PMA-treated hearts (PMA + PZ group) significantly increased the TUNEL-positive nuclei when compared to PMA group (Figure 5B).
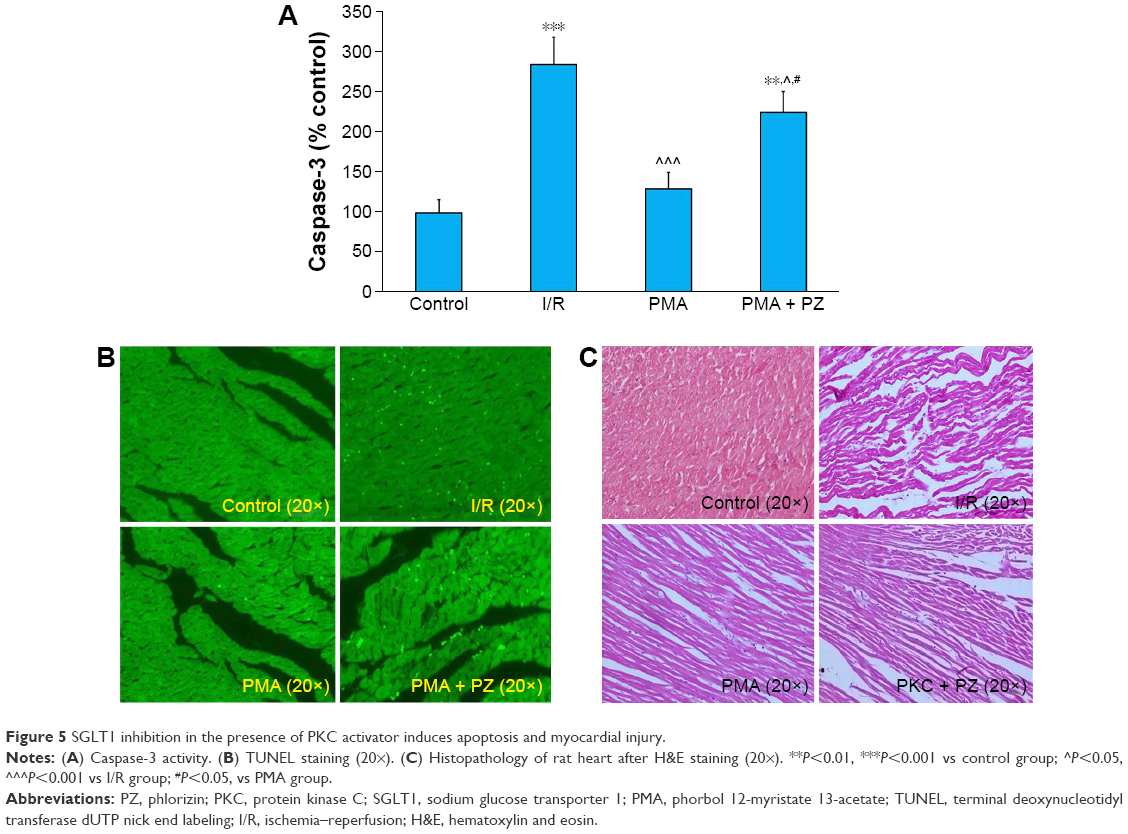 | Figure 5 SGLT1 inhibition in the presence of PKC activator induces apoptosis and myocardial injury. |
Protective effect of PKC activation on myocardial injury as nullified by SGLT1 inhibition: histopathological analysis
In this study, hearts from the control group showed no edema or loss of myocytes (Figure 5C). Increased separation of myofibers along with edema and loss of myocytes were present in I/R hearts. However, cardiac muscle fibers in the PMA group alone showed more protection. In PMA + PZ group, the protection of PMA was lost as interstitial edema along with focal lysis of cardiomyocytes was observed (Figure 5C).
Computational modeling and docking
The crystal structure of PKC which constitute it all four domains, C1, C2 and catalytic C3 and C4 domains is not yet reported. A complete active form of rat SGLT1 is also not available; therefore, homology models were generated and analyzed through the modeller15 (the Supplementary materials provides more detailed information).
Molecular dynamics was employed to allow conformational relaxation of the protein structures prior to subjecting them to protein–protein docking calculations. From Figures 6 and 7, it can be seen that both protein structures displayed an initial structural rearrangement that was followed by convergence to an RMSD plateau, indicating minimal structural fluctuation during the latter part of the simulation. The overall structural fluctuation of SGLT1 was observed to be less than that of PKC, with an RMSD of 2.2 and 3.5 Å, respectively. This is expected as the protein structure (template) of the former has high resolution and covers more space, while the latter has multiple domains that are poorly connected.
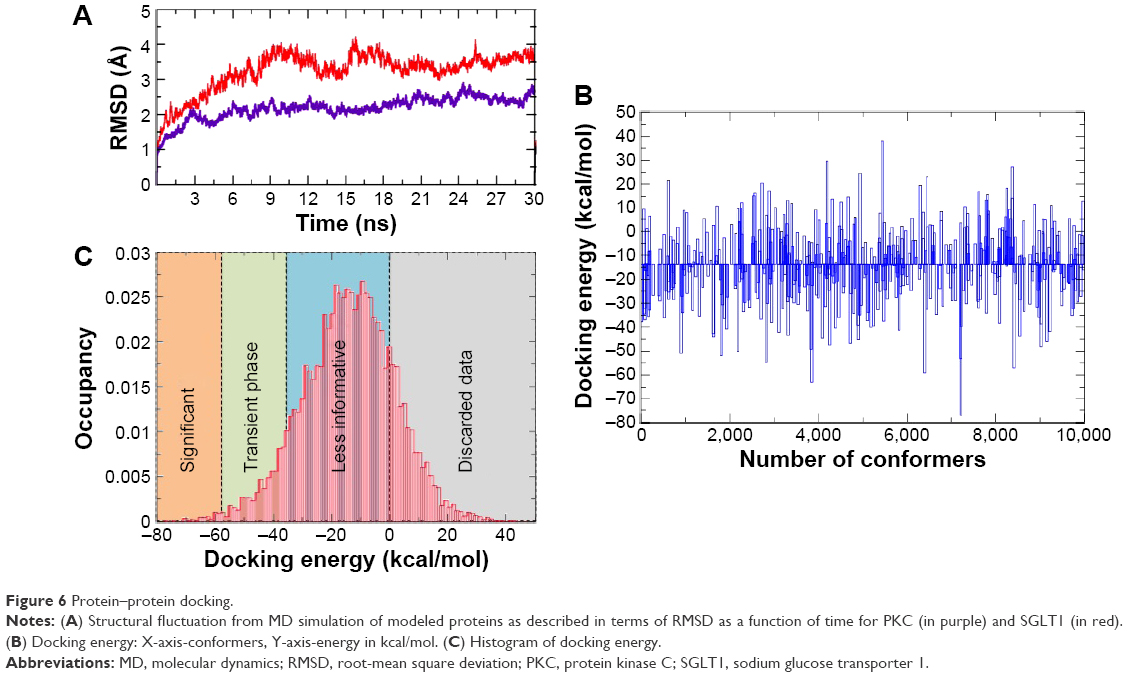 | Figure 6 Protein–protein docking. |
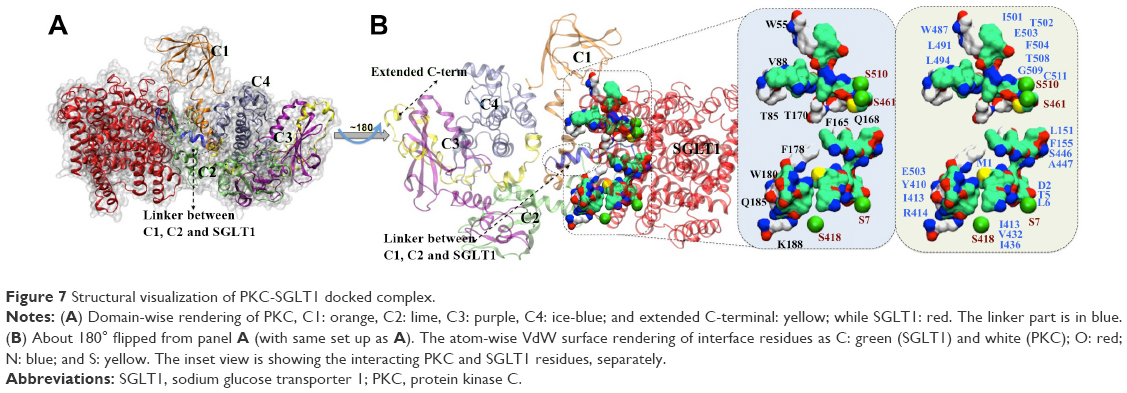 | Figure 7 Structural visualization of PKC-SGLT1 docked complex. |
The docking procedure aimed to generate a set of solutions for candidates with at least one near native structure. The rigid docking allows some steric clashes because proteins in their unbound conformation can collide when placed in their native interaction positions. Therefore, apart from rigid docking through pyDock, the flexible docking by SwormDock16 was done on relaxed structures of proteins. Furthermore, the candidate solutions were scored and ranked according to different parameters such as clusters, lowest binding energy, number of conformers, and agreement with known binding sites. Only the best docked pose, which ranged above −75 to −80 kcal/mol, was used for further analysis (Figure 6B and C). We quantified the docking results by dividing them into four sections: lowest energy zone (above −60 kcal/mol), transient energy zone (−60 to −40), low energy zone (−20 to 0), and discarded data zone (from 0 to 40 kcal/mol; Figure 6C).
Clustering of docked poses was conducted to filter out the most likely complex of PKC–SGLT1, as it was reported earlier.17 We found that the C1 and C2 domains of PKC were mainly involved in interaction with SGLT1 (Figures 7 and S1). It is already known from previous studies that the C2 domain of PKC is involved in transmembrane interacton,18 which corroborates well with our findings. However, some residues of C1 domain are also involved in the interaction. From the lowest docking energy pose of complex and through guided data from the literature, the most likely binding mode of the complex was selected for quantitative analysis. Three types of interactions were noticed, ie, HB, HpH contacts, and Pi–Pi interactions at the interface site of PKC–SGLT1. The most frequent HBs are PKC@Asp124:OD1-SGLT1@Thr508:N =2.3 Å, PKC@Asp124:OD2-SGLT1@Gly509:N =2.8 Å, PKC@Asp126:OD1-SGLT1@Ala447:N =3.1 Å, PKC@Asp126:OD2-SGLT1@Ala447:N =3.3 Å, PKC@Lys128:NZ-SGLT1@Ser446:N =3.2Å, PKC@Ser130:N-SGLT1@Ser446:OG =3.5 Å, PKC@Gln185:O-SGLT1@Arg414:NH1 =3.1 Å, PKC@Gln185:OD1-SGLT1@Arg414:NH2 =3.4 Å, PKC@Gly186:O-SGLT1@Arg414:NH1 =3.3 Å, and PKC@Lys188:NZ-SGLT1@Thr410:OH =2.8 Å. Additionally, the Pi–Pi interaction between PKC@Phe121:SGLT1@Phe155 (centroid distance 3.8 Å) and PKC@Trp55:SGLT1@Phe504 (4.2 Å) also contributed to the higher stability of the complex. Additional stability was gained by hydrophobic interactions between the residues of PKC: Trp55, Val88, Trp85, Phe165, Tyr170rak, Phe178, Trp180, and Lys188, and SGLT1: Met1, Tyr5, Lys6, Lys151, Phe155, Tyr410, Ile413, Val432, Ile436, Trp487, Lys491, Lys494, Ile501, Trp502, and Phe504.
Furthermore, in an effort to dissect these interactions from the docking simulations, total interaction energy between PKC and SGLT1 was calculated, as described in Figure S2, within the frame work of Amber-force field19 description. The residue-wise contribution revealed that F504, T508, G509, K151, F155, W487, K491, and W502 of SGLT1 and W55, W85, F121, D124, S130, F165, and K188 of PKC contribute maximally either in form of Van der Waals forces and/or electrostatically to establish the interactions.
Discussion
Our observations in this study showed a novel mechanism by which the activity of sodium-coupled glucose transporter, SGLT1, is regulated in the heart. Posttranslational phosphorylation of multiple different proteins are responsible for the regulation of diverse cellular activities like transport, signal transduction, cytoskeletal regulation, and metabolism.20 Here, we present evidence that PKC plays an important role in upregulation of SGLT1 activity. Previous literature showed SGLT1 contains four putative sites for PKC (Thr50, Ser303, Ser418, and Ser562) and one putative site for PKA (Ser418).8 The presence of these phosphorylation sites indicates that SGLT1 can be posttranslationally modified to modulate its activity. In this study, we demonstrated that PKC showed cardioprotection through SGLT1 activation.
To determine the effect of PKC on glucose uptake, we demonstrated that PKC activation enhanced glucose uptake in H9C2 cells and mouse heart. PKC-induced glucose uptake was also demonstrated previously.21 To confirm whether increased SGLT1 activity through PKC is responsible for increased glucose uptake, we performed glucose uptake studies in the presence of SGLT1 inhibitor, PZ. Our data clearly showed that PZ blocked the PKC-induced increased glucose uptake both in vitro and in vivo.
After confirming the relationship between PKC activation and SGLT1-induced glucose uptake, we wanted to explore whether increased glucose uptake through PKC activation is beneficial against I/R injury in rat heart. The concept of PKC-induced preconditioning is well known and is shown to have beneficial effect against I/R injury.22 We hypothesize that increased glucose uptake via SGLT1 might be responsible for PKC-induced cardioprotection. To prove our hypothesis, we used an ex vivo I/R injury model where hearts were pretreated with PKC activator in the presence and absence of SGLT1 inhibitor, PZ. Here, we treated ex vivo hearts with a dose of PZ, which is nontoxic and previously reported.9 We chose to treat the ex vivo heart for only 5 minutes before induction of preconditioning by PKC activator. The concept was to pass the PZ solution in the heart for a short time so that direct interaction between SGLT1 and PZ could be attained in the absence of PZ in KH buffer during I/R injury. We assumed that this interaction may inhibit cardiac glucose uptake during I/R injury.
Increased oxidative stress in I/R heart was associated with an increase in lipid peroxidation and a decrease in endogenous antioxidants. However, induction of preconditioning by PKC activation significantly attenuated the oxidative stress. All these beneficial effects of PKC were abrogated after inhibition of SGLT1 by PZ. We also looked at the induction of apoptosis as a measure of cardiac injury. Increased apoptosis as measured by increased caspase-3 activity and TUNEL-positive nuclei was observed in I/R heart. PKC activation in heart reduced apoptosis that was induced by I/R. However, this protection was lost when PKC-activated hearts were inhibited by PZ. Similarly, histopathological findings also showed that protection in PMA-treated I/R heart was abrogated in the presence of SGLT1 inhibitor. Our data demonstrated that PKC activation increased cardioprotective effect, while SGLT1 inhibition diminished the cardioprotective effect induced by PKC.
After confirming the relationship between PKC and SGLT1, molecular modeling studies were conducted to elucidate the interaction of PKC–SGLT1 complex. We have identified key serine residues involved in phosphorylation through protein–protein docking. The vicinity of interface site between PKC and SGLT1, and the PKC phosphorylation site in SGLT1 indicated that PKC activates SGLT1. The different multifaceted computational approaches from molecular modeling to docking also confirmed the formation of a stable adduct between PKC and SGLT1. We identified a region, which we mentioned as Linker, originates from C2 domain and showing maximum interaction at interface site (Figures 7 and S1). Our finding corroborates well with earlier literature18 as the best model of the complex has shown that the C2 and C1 domain of PKC interacts with SGLT1. The significant interaction of PKC and SGLT1 gives an impression that PKC activates SGLT1; indeed, the structural quantification of such a binding complex has elucidated the stable adduct of PKC–SGLT1. Finally, an ensemble of structures obtained from the final 8 ns of MD simulation was cross-docked to yield a subsequent set of possible PKC–SGLT1 complexes. Postanalysis of these top ten complexes provided pertinent information on the binding interface of PKC–SGLT1, particularly that their interaction surface is not flat and protrudes well into each binding partner. Interestingly, docking results have shown that some serine residues, S7, S418, S461, and Ser510, are present at the vicinity of PKC–SGLT1’s interface site (Figure S1). It is noteworthy that among these serine residues, S418 is already known as phosphorylation site in PKA. This model supports our hypothesis that increased glucose uptake in cardiac injury is mediated by PKC via SGLT1.
PKC is important in different signal transduction pathways for the regulation of cardiac function in both normal and disease condition. Our data has shown that increased SGLT1-mediated cardiac glucose uptake via PKC activation is beneficial to reduce cardiac injury induced by I/R. Thus, there is a scope to explore drug development by using PKC activator against myocardial ischemia where SGLT1-mediated cardiac glucose uptake can be enhanced.
In summary, our data showed that PKC activation enhances cardiac glucose uptake through SGLT1, indicating a possible cross-talk between PKC and SGLT1, which is essential to prevent I/R-induced cardiac injury. Furthermore, we also confirmed that inhibition of SGLT1 abrogated PKC-induced beneficial effects.
Acknowledgments
This work was supported by Department of Biotechnology (DBT) (BT/PR13768/MED/30/300/2010) and Ramalingaswami fellowship funds to SKB. Authors are also thankful to Council of Scientific and Industrial Research (CSIR) for providing Senior Research Fellowship to AK.
Author contributions
Abhinav Kanwal, Shailendra Asthana, and Sanjay K Banerjee participated in the research design. Abhinav Kanwal, Sujatha Kasetti, Shailendra Asthana, and Uday Kumar Putcha conducted the experiments. Abhinav Kanwal, Sanjay K Banerjee, Shailendra Asthana, and Uday Kumar Putcha performed data analysis. Abhinav Kanwal, Sanjay K Banerjee, and Shailendra Asthana wrote or contributed to the writing of the manuscript. All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflict of interest in this work.
References
Misra MK, Sarwat M, Bhakuni P, Tuteja R, Tuteja N. Oxidative stress and ischemic myocardial syndromes. Med Sci Monit Basic Res. 2009;15(10):RA209–RA219. | ||
Murry CE, Richard VJ, Reimer KA, Jennings RB. Ischemic preconditioning slows energy metabolism and delays ultrastructural damage during a sustained ischemic episode. Circ Res. 1990;66(4):913–931. | ||
Steenbergen C, Perlman ME, London RE, Murphy E. Mechanism of preconditioning. Ionic alterations. Circ Res. 1993;72(1):112–125. | ||
Ytrehus K, Liu Y, Downey JM. Preconditioning protects ischemic rabbit heart by protein kinase C activation. Am J Physiol Heart Circ Physiol. 1994;35(3):H1145. | ||
Nishino Y, Miura T, Miki T, et al. Ischemic preconditioning activates AMPK in a PKC-dependent manner and induces GLUT4 up-regulation in the late phase of cardioprotection. Cardiovasc Res. 2004;61(3):610–619. | ||
Wright EM. The intestinal Na+/glucose cotransporter. Annu Rev Physiol. 1993;55(1):575–589. | ||
Inagaki K, Churchill E, Mochly-Rosen D. Epsilon protein kinase C as a potential therapeutic target for the ischemic heart. Cardiovasc Res. 2006;70(2):222–230. | ||
Wright EM, Hirsch JR, Loo DD, Zampighi GA. Regulation of Na+/glucose cotransporters. J Exp Biol. 1997;200(Pt 2):287–293. | ||
Moran JK, Lee HB, Blaufox MD. Optimization of urinary FDG excretion during PET imaging. J Nucl Med. 1999;40(8):1352–1357. | ||
Gupta P, Kanwal A, Putcha UK, et al. Cardioprotective effect of ritonavir, an antiviral drug, in isoproterenol induced myocardial necrosis: a new therapeutic implication. J Transl Med. 2013;11(1):80. | ||
Pijoan M, Gerjovich HJ. The use of 2,4-dinitrophenylhydrazine for the determination of ascorbic acid. Science. 1946;103(2668):202–203. | ||
Szabo M, Idiţoiu C, Chambre D, Lupea A. Improved DPPH determination for antioxidant activity spectrophotometric assay. Chemical Papers. 2007;61(3):214–216. | ||
Vaya J, Aviram M. Nutritional antioxidants mechanisms of action, analyses of activities and medical applications. Curr Med Chem Immunol Endocr Metab Agents. 2001;1(1):99–117. | ||
Kanwal A, Singh SP, Grover P, Banerjee SK. Development of a cell-based nonradioactive glucose uptake assay system for SGLT1 and SGLT2. Anal Biochem. 2012;429(1):70–75. | ||
Eswar N, Webb B, Marti-Renom MA, et al. Comparative protein structure modeling using Modeller. Curr Protoc Bioinformatics. 2006;Chapter 5:Unit 5.6. | ||
Torchala M, Moal IH, Chaleil RA, Fernandez-Recio J, Bates PA. SwarmDock: a server for flexible protein-protein docking. Bioinformatics (Oxford). 2013;29(6):807–809. | ||
Asthana S, Shukla S, Ruggerone P, Vargiu AV. Molecular mechanism of viral resistance to a potent non-nucleoside inhibitor unveiled by molecular simulations. Biochemistry. 2014;53(44):6941–6953. | ||
Farah CA, Sossin WS. The role of C2 domains in PKC signaling. Adv Exp Med Biol. 2012;740:663–683. | ||
Case DA, Cheatham TE 3rd, Darden T, et al. The Amber biomolecular simulation programs. J Comput Chem. 2005;26(16):1668–1688. | ||
Graves JD, Krebs EG. Protein phosphorylation and signal transduction. Pharmacol Ther. 1999;82(2–3):111–121. | ||
Davidoff AJ, Davidson MB, Carmody MW, Davis ME, Ren J. Diabetic cardiomyocyte dysfunction and myocyte insulin resistance: role of glucose-induced PKC activity. Mol Cell Biochem. 2004;262(1–2):155–163. | ||
Muller BA, Dhalla NS. Mechanisms of the beneficial actions of ischemic preconditioning on subcellular remodeling in ischemic-reperfused heart. Curr Cardiol Rev. 2010;6(4):255–264. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.