Back to Journals » International Journal of Nanomedicine » Volume 16
Protective Effects of Chitosan-Bilirubin Nanoparticles Against Ethanol-Induced Gastric Ulcers
Authors Huang Z, Shi Y, Wang H, Chun CJ, Chen L, Wang K, Lu Z ![]() , Zhao Y, Li X
, Zhao Y, Li X ![]()
Received 17 October 2021
Accepted for publication 3 December 2021
Published 20 December 2021 Volume 2021:16 Pages 8235—8250
DOI https://doi.org/10.2147/IJN.S344805
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yan Shen
Zhiwei Huang,1– 3,* Yannan Shi,2,* Hengcai Wang,2,* Changju Chun,3 Longwang Chen,1,4 Kang Wang,1,4 Zhongqiu Lu,1,4 Yingzheng Zhao,2 Xinze Li1,4
1Department of Emergency, the First Affiliated Hospital of Wenzhou Medical University, Wenzhou, 325035, People’s Republic of China; 2School of Pharmaceutical Sciences, Wenzhou Medical University, Wenzhou, 325035, People’s Republic of China; 3Research Institute of Pharmaceutical Sciences, College of Pharmacy, Chonnam National University, Gwangju, 61186, Republic of Korea; 4Wenzhou Key Laboratory of Emergency and Disaster Medicine, Wenzhou, 325035, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xinze Li; Yingzheng Zhao
Wenzhou Medical University, University Town, Chashan, Wenzhou, Zhejiang, 325035, People’s Republic of China
Email [email protected]; [email protected]
Purpose: Gastric ulcers (GU) are a disease of the gastrointestinal tract that can be caused by excessive alcohol consumption and heavy use of nonsteroidal anti-inflammatory drugs. GU manifests predominantly as pathological damage, such as extensive inflammatory erosion and superficial bleeding of the gastric mucosa. Oxidative stress damage and the inflammatory response are now considered important predisposing factors for GU, suggesting that antioxidant and anti-inflammatory drugs could be treatments for GU. Nanoparticle drug carriers offer many advantages over conventional drugs, such as improved drug efficiency, increased drug stability, and increased half-life.
Methods: We designed chitosan-bilirubin conjugate (CS-BR) nanoparticles and assessed the anti-inflammatory and antioxidant abilities of CS-BR in gastric epithelial cells. Then, we evaluated the intragastric retention time and the anti-ulcer effects of CS-BR in vivo.
Results: The in vitro data showed that CS-BR nanoparticles protect gastric epithelial cells against oxidative/inflammatory injury. The in vivo study demonstrated that CS-BR nanoparticles accumulate permanently in the stomach and exert powerful antioxidant and anti-inflammatory effects against GU.
Conclusion: This study applied bilirubin to the treatment of GU and confirmed that CS-BR nanoparticles are effective at alleviating acute GU in an experimental model. The findings provide innovative ideas for prophylaxis against or treatment of GU.
Keywords: oxidative stress, inflammation, reactive oxygen species, gastroprotection
Graphical Abstract:

Introduction
Gastric ulcers (GU) are a disease of the digestive system characterized by chronic and recurrent epigastric pain that is caused by an imbalance between mucosa-offensive factors (gastric acid, reactive oxygen species [ROS]) and mucosa-defensive factors (gastric mucus and bicarbonate secretion, prostaglandins, nitric oxide).1 Ethanol is one of the most important causes of gastric mucosal damage. Ethanol stimulation can directly damage gastric mucosal capillary endothelial cells, resulting in intragastric hemorrhage; aggravating tissue ischemia, thereby impacting the normal metabolism and function of the gastric mucosa; and weakening the ability of the mucosa to resist damage and repair.2 Previous studies have shown that ethanol-induced GU mainly result from oxidative stress and an inflammatory reaction, manifested as extensive erosion and bleeding in the gastric mucosa and accompanied by massive infiltration of pro-inflammatory cells and the production of oxygen free radicals as well as immunocytes that aggregate in the ulcerative area and induce lymphocyte differentiation.3 In addition, the accumulation of inflammatory cells and the release of ROS are involved in the inflammatory response to ulcers; this accumulation leads to widespread hemorrhagic ulcers and tissue necrosis, thereby exacerbating the extent of gastric mucosal damage.4 Traditional medications for GU include the histamine receptor antagonist ranitidine, the proton pump inhibitor omeprazole, antacids, and gastric mucosal protection drugs. However, these traditional drugs have unavoidable adverse effects.5 Therefore, development of an effective and safe strategy for protecting the gastric mucosa through anti-inflammatory and antioxidant actions is clinically important and urgent.
Bilirubin (BR) is a metabolite of hemoglobin decomposition in the body. BR is produced in vivo by the degradation of heme, of which 80% to 90% is derived from hemoglobin; the rest is derived from heme-containing proteins, such as myoglobin and cyclooxygenase. Heme in the cytoplasm is degraded by heme oxygenase to biliverdin, which is quickly converted to BR by biliverdin reductase.6,7 BR levels in patients with various diseases have generally been high, so BR is considered a toxic substance in the body; high concentrations thus have represented an important clinical indicator of certain conditions, such as hepatic dysfunction.8 It was not until the 1980s that Stoker et al9 first reported the protective effect of BR against oxidative damage. BR contains an extended system of conjugated double bonds and a pair of reactive hydrogen atoms that is involved in antioxidant activity via hydrogen donation to an incipient radical.10 Recent studies have shown that BR exert powerful anti-inflammatory actions against various oxidative stress-associated diseases, including experimental autoimmune encephalomyelitis, inflammatory bowel disease, hepatic ischemia-reperfusion injury, pulmonary fibrosis, and alum-induced peritonitis.11–15 Other reports have confirmed that physiological concentrations of BR regulate inflammation by inhibiting nuclear factor kappa-B (NF-κB) and inflammasome activation.16 In addition, previous studies in our laboratory showed that BR could modulate macrophages toward the M2-type and enhance the therapeutic effect of islet transplantation in diabetes mellitus.17 However, BR is physiologically insoluble in water, which greatly limits its application in research. Thus, researchers have developed various pharmaceutical preparations for BR that have enhanced water solubility and optimized pharmacokinetics. For example, Lee et al18 used polyethylene glycol (PEG) covalently attached to BR to yield PEGylated BR that self-assembled into nanoscale particles to treat various inflammatory diseases.18 We recently prepared a hyaluronic acid-coated polylysine-BR nanoparticle for acute kidney injury therapy. Reflecting the characteristics of different diseases, many delivery strategies have been designed to exploit the therapeutic potential of BR.19
Chitosan (CS) has been widely used in the field of medical materials and developing drug delivery system for treatment of GU because of its good mucoadhesive properties, biocompatibility, non-antigenicity, biodegradability, and broad-spectrum antimicrobial properties.20,21 When used as a bioadhesive material or drug carrier, CS can adhere to the mucosal lesion and prolong the retention time of the drug at the lesion site.22,23 Combining the natural advantages of CS to construct a drug carrier may be an effective strategy for treating GU.24 In this study, we synthesized a conjugate, CS-BR, and self-assembled CS-BR in solution to form nanoparticles. BR has beneficial effects for treatment of GU via its strong anti-inflammatory and antioxidant capacities. To enhance the therapeutic potential of BR, we developed CS-BR nanoparticles that could scavenge ROS and inhibit the secretion of inflammatory factors in cells and in the body to alleviate GU. We hypothesized that CS-BR ameliorated the cellular uptake and gastric retention of BR then exerted anti-ulcer effects by reducing inflammatory responses and oxidative stress in the region of the GU, thus protecting gastric cells and tissue from additional damage. We detected the relevant indicators and confirmed that CS-BR could reduce the cellular damage caused by the oxidative stress and inflammatory responses. Subsequently, the ethanol-induced GU model was treated with CS-BR, we also found that CS-BR could reduce the damage of GU by exerting good antioxidant and anti-inflammatory effects in gastric tissue. These data demonstrated the excellent cytoprotective and anti-ulcer ability of CS-BR nanoparticles.
Materials and Methods
Materials, Cells, and Animals
BR, CS, N-hydroxysuccinimide (NHS), and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) were provided by Macklin Biochemical Co., Ltd. (Shanghai, China). Cell Counting Kit 8 (CCK-8) was obtained from Yeasen Biotechnology Co., Ltd. (Shanghai, China). The superoxide dismutase (SOD) activity kit, the catalase (CAT) assay kit, the malondialdehyde (MDA) assay kit, the glutathione (GSH) assay kit, the hematoxylin and eosin (H&E) staining kit, the glycogen periodic acid–Schiff (PAS) stain kit, fluorescein isothiocyanate (FITC), and lipopolysaccharides (LPS) were purchased from Solarbio Science & Technology Co., Ltd (Beijing, China). Medium 199 (M199), fetal bovine serum (FBS), bovine serum albumin (BSA), and trypsin were purchased from Gibco (Gibco-BRL, Carlsbad, CA, USA). The bicinchoninic acid (BCA) protein assay kit and ROS assay kit were obtained from Beyotime Biotechnology (Shanghai, China). Interleukin-6 (IL-6, 70-EK206/3-96), tumor necrosis factor-α (TNF-α, 70-EK282/4-96), and prostaglandin E2 (PGE2, 70-EK8103/2-48) enzyme-linked immunosorbent assay (ELISA) kits were obtained from Multisciences Biotech Co., Ltd. (Hangzhou, China). The anti-myeloperoxidase (MPO) antibody (ab208670) and the anti-CD68 antibody (ab125212) were obtained from Abcam (Waltham, MA, USA). All other chemicals and buffer solutions were of analytical grade.
The mouse gastric epithelial cells (primary cells were isolated from gastric mucosal tissue of mice) were commercially purchased from Procell Life Science & Technology Co., Ltd (Wuhan, China). The cells were cultured in M199 supplemented with 10% (v/v) FBS and 1% (v/v) penicillin/streptomycin and then incubated in a humidified 5% CO2 incubator at 37 °C.
Male BALB/c mice (6–8 weeks old, 22–26 g) were purchased from Shanghai Laboratory Animal Center (Shanghai, China). Mice were acclimated in a pathogen-free animal facility for 7 days and then randomly grouped for experiments. All animal experiments and study protocols were approved by the Animal Care and Use Committee of Wenzhou Medical University. All procedures involving animals were carried out in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
Synthesis and Characterization of CS-BR
To synthesize CS-BR, 50 μmol of BR, 20 μmol of NHS, and 70 μmol of EDC were dissolved in 3 mL of dimethyl sulfoxide (DMSO), and the mixture was stirred for 30 min at room temperature. Subsequently, CS (0.8% w/v) was dissolved in 1 mL (1% v/v) of aqueous acetic acid solution and magnetically stirred until complete dissolution occurred. The CS solution was elevated to a pH of 6 using 0.01 mol/L of NaOH; CS was maintained in a fully dissolved state. Then, the CS solution was added to the BR solution, and this mixture was stirred under nitrogen gas for 12 h in a dark room. After the reaction, the mixture solution was dialyzed using a 7-kDa dialysis membrane for 6 h against distilled water containing 0.01 mol/L of NaOH. After 6 h, dialysis was again performed against distilled water for another 12 h. The obtained solution was lyophilized to obtain the final conjugated product, CS-BR. The CS-BR nanoparticles were obtained by adding 0.25 mg CS-BR freeze-dried powder into 1 mL distilled water under stirring at 200 rpm for 10 min, then placed it in ultrasonic bath (ultrasonic power = 300 W) for 5 min.
FITC-labeled CS-BR was prepared as previously reported.25,26 In brief, a CS-BR solution was added to an ethanol solution of FITC, and the mixture was stirred at room temperature for 12 h in a dark room. The mixture solution was then dialyzed using a 7-kDa dialysis membrane for 8 h against distilled water under light-proof conditions and was lyophilized to obtain FITC-labeled CS-BR.
CS-BR was characterized by Fourier transform infrared (FTIR, Bruker Ltd. Germany) spectroscopy, X-ray diffraction (XRD, Bruker Ltd. Germany) analysis, proton nuclear magnetic resonance (1H-NMR, 600 Hz, Bruker Ltd., Germany). For 1H-NMR analysis, CS and CS-BR were dissolved in deuterium oxide (D2O) containing 2% (v/v) deuterium chloride (DCl), BR was dissolved in deuterated DMSO (DMSO-d6).
To compare the appearance and ultraviolet-visible (UV/Vis) spectra of BR after complete dissolution, BR was firstly dissolved in DMSO (5 mg/mL) and then diluted with deionized water to 1 mg/mL. The morphology of CS-BR freeze-dried powder and free bilirubin were also visualized with scanning electron microscopy (SEM, Hitachi Ltd., Japan). The particle size, polydispersity index (PDI), and zeta potentials of CS-BR nanoparticles were measured by Particle Size Analyzers (Anton-Paar, Ltd., Austria). The morphology of CS-BR nanoparticles was also observed using a transmission electron microscope (TEM, JEOL Ltd., Japan).
In Vitro Toxicity Study of CS-BR
Cell viability was examined using a CCK-8 assay. In brief, murine gastric epithelial cells were seeded into 96-well plates (3000 cells/well) and incubated in a humidified 5% CO2 incubator at 37 °C for 24 h. Then, the cells were treated with different concentrations of CS (2.5–100 μg), BR (2.5–100 μM) and CS-BR (2.5–100 μM) for another 24 h. Next, 10 μL of 2-[2-methoxy-4-nitrophenyl]-3-[4-nitrophenyl]-5-[2,4-disulfophenyl]-2H-tetrazolium (WST-8) reagent was added to each well and co-cultured for 2 h. Last, the absorbance of the 96-well plates were read at 450 nm using a microplate reader (Tecan Safire, Germany).
In Vitro Assessment of Antioxidant and Anti-Inflammatory Capacities of CS-BR
To evaluate the antioxidant property of CS-BR, gastric epithelial cells were exposed to oxidative stress by incubation with 300 μM of H2O2 for 3 h and then were incubated with BR or CS-BR for 24 h. After incubation, the viability of the cells was determined by CCK-8 assay. In addition, CAT and SOD activities as well as the GSH and MDA content in each group of cells were measured by the reagent kits according to the manufacturers’ instructions. Intracellular ROS production in each group was detected by an ROS fluorescent probe (2,7-dichlorodihydrofluorescein diacetate [DCFH-DA]) and observed with the inverted fluorescence microscope (Olympus Corp., Tokyo, Japan).
To assess the anti-inflammatory effect of CS-BR, gastric epithelial cells were pre-treated with LPS (5 μg/mL) for 6 h and incubated with BR or CS-BR for 24 h. Cell supernatants were then collected to measure the content of pro-inflammatory cytokines (TNF-α and IL-6) using ELISA kits.
In Vitro Cellular Uptake of CS-BR
The gastric epithelial cells were seeded in 12-well plates and then treated with FITC labeled CS-BR for 1 h or 2 h, and cells treated with free FITC as control. Then cells were washed with PBS solution three times. The DAPI solution (10 ug/mL) was added and incubated with cells at room temperature for 10 min, the intracellular fluorescent intensities were visualized by fluorescent microscope (Olympus Corp., Tokyo, Japan).
Ethanol-Induced GU Model and In Vivo Treatment
The ethanol-induced acute GU model was established by oral administration of ethanol, as in previous reports.27 The mice were randomly assigned to the following groups (three rats in each group): GU + saline; GU + BR (10 mg/kg given orally); and GU + CS-BR (10 mg/kg given orally). A group of mice that received only saline was used as the control.
In brief, each group of mice was treated with different orally administered interventions, as noted in the group descriptions, once daily for 7 consecutive days. After 7 days, all mice were strictly prohibited from food intake for 24 h but had free access to water. On day 8, all mice underwent intragastric administration of saline, BR, or CS-BR. After 30 min, the mice in the treatment groups were orally administered anhydrous ethanol (2 mL/kg, 50% v/v), and the mice in the control group were given saline. One hour after intragastric administration of ethanol/saline, all mice were sacrificed. The gastric tissues of mice were rapidly removed and cut along the greater gastric curvature from the cardia to the pylorus. Subsequently, the gastric tissues were rinsed with saline, and the severity of gastric mucosal damage was observed macroscopically.
The GU index (UI) was calculated by ImageJ software, as described in a previous report;28 the formula for UI is as follows: UI = (ulcer partial pixel area) ÷ (total gastric pixel area) × 100%. The CAT and SOD activities as well as the GSH and MDA contents in each group of gastric tissues were detected using the corresponding kits. In addition, the levels of PGE2, IL-6, and TNF-α were measured using ELISA kits.
The established model was also used to assess the biosafety of CS-BR after 30 days of treatment. After the mice were sacrificed and the gastric tissues were observed for damage, the major organs (heart, liver, spleen, lung, kidney) were embedded in paraffin and sectioned for subsequent histological analysis experiments. Serum samples were obtained to determine the levels of aspartate aminotransferase (AST), alanine aminotransferase (ALT), serum creatinine (SCr), and blood urea nitrogen (BUN) for assessments of renal and liver function at Department of Laboratory Medicine, The First Affiliated Hospital of Wenzhou Medical University.
In Vivo Distribution of CS-BR
The murine models of GU were also used to evaluate tissue distribution and gastric retention of CS-BR after intragastric administration. FITC-labeled CS-BR nanoparticles were prepared to visualize CS-BR distribution in vivo. After the GU models were established, the mice were orally administered FITC-labeled CS-BR (10 μg/kg of FITC), and free FITC solution was used as a control. The mice were sacrificed at predetermined times (30 min, 60 min, 90 min, and 120 min) after treatments, and the major organs of mice were removed. The fluorescent intensity of collected organs was observed with an imaging system (PerkinElmer, MA, USA). The mean fluorescent intensity in gastric tissue (fundus, corpus, antrum) were quantified with Bruker molecular imaging software.
In an independent study, murine models of GU administered BR or CS-BR orally to mice; the mice were then sacrificed at predetermined times (30 min, 60 min, 90 min, and 120 min), and the gastric tissues were collected. The concentrations of BR in gastric tissues (fundus, corpus, antrum) at different points in time were determined, as previously reported.29
Histological Analysis
The gastric tissues and other organs were cut into appropriate tissue blocks and fixed in a 4% paraformaldehyde solution for 24 h. These samples were removed and rinsed with saline for 2 h and then dehydrated, embedded, and sectioned according to the established procedure, as described in our previous report.30 The sliced samples (5 μm) were deparaffinized and stained with H&E and PAS to detect microscopic gastric injury and the production of mucopolysaccharide in the stomach. The histopathologic scoring criteria were determined to evaluate the severity of the gastric mucosal damage. A previously described scoring method,31,32 with slight modifications, was used: A score of 0 represented no significant inflammatory changes in gastric mucosa; a score of 2 represented a small number of inflammatory cells in the gastric mucosa with local mucosal thickening. A score of 4 represented mucosal edema and thickening with focal hemorrhage; a score of 6 represented mucosal focal necrosis, shedding, or obvious thickening; and a score of 8 represented massive inflammatory cell infiltration with severe mucosal necrosis.
For immunohistochemistry (IHC) analysis, the tissue sections were stained with rabbit polyclonal antibody MPO (ab208670,1:1000, Abcam) and rabbit polyclonal antibody CD68 (ab125212,1:100, Abcam). After overnight exposure to the primary antibodies at 4 °C, the sections were washed with phosphate-buffered saline three times and incubated with horseradish peroxidase polymer secondary antibody for 1 h at 37 °C. Then, dimethylaminoazobenzene was added to manifest positive areas and the nucleus was stained with hematoxylin for 3 min. All stained tissue sections were observed using an optical microscope (Olympus Corp., Tokyo, Japan).
Statistical Analysis
Student’s t-test and the one-way analysis of variance assay were performed for statistical analysis. P values ≤ 0.05 and ≤ 0.01 were considered statistically significant. All data were presented as mean values ± standard deviations in independent experiments.
Results and Discussion
Characterization and Cytotoxicity of CS-BR
CS-BR was synthesized via the EDC/NHS-based carboxyl-ammonia condensation reaction (Figure 1A), as previously reported.17 The obtained CS-BR was confirmed by 1H-NMR analysis. The 1H-NMR spectra of CS exhibited a peak at 5.1 ppm due to anomeric proton (H1), the signals between 3.2 ppm and 3.7 ppm corresponding to protons H3-H6 of the polysaccharide ring, and the signal at 2.9 ppm is due to the H2 proton of the amine group (H-amine). The characteristic peaks of BR were found in the range of 5.1 to 6.9 ppm represented the protons from the C=C bond of BR, and the signals at 1.9 ppm and 4.0 ppm corresponding to protons on methyl and methylene in BR respectively. These characteristic peaks were also observed in the 1H-NMR spectra of CS-BR, which indicated the successful conjugation of CS and BR (Figure 1B). In the FTIR spectrum of CS-BR, the absorption peaks at 1647 cm−1 and 1574 cm−1 represented stretching vibrations of the newly generated amide bond that was derived from the linkage of the amino group in CS and carboxylic acid in BR (Figure 1C). The XRD spectrum of CS and CS-BR showed the distinct diffraction peak at around 20.2°, whereas multiple peaks in the spectrum of BR were observed in the range of 10° to 30°, which demonstrated the unique crystal structure of BR (Figure 1D). When the amino group in CS was attached to BR to form CS-BR, the multiple characteristic peaks of BR were nonexistent, indicating that the conjugation process destroyed the crystal structure of BR. This finding was corroborated by the SEM results. The SEM images confirmed that BR lost its crystalline form after bonding with CS and that CS-BR exhibited an irregular, blocky structure (Figure 1F). The BR solution appeared bright yellow, whereas the CS-BR solution presented as a brownish yellow color; this change might be due to the reaction of CS and BR (Figure 1E). The UV/Vis spectra of BR and CS-BR exhibited distinctive absorption peaks at 452 nm as a result of the characteristic absorption peak for BR (Figure 1I). The grafting ratio of CS-BR was 33.23% ± 2.79% using the measurement of BR.
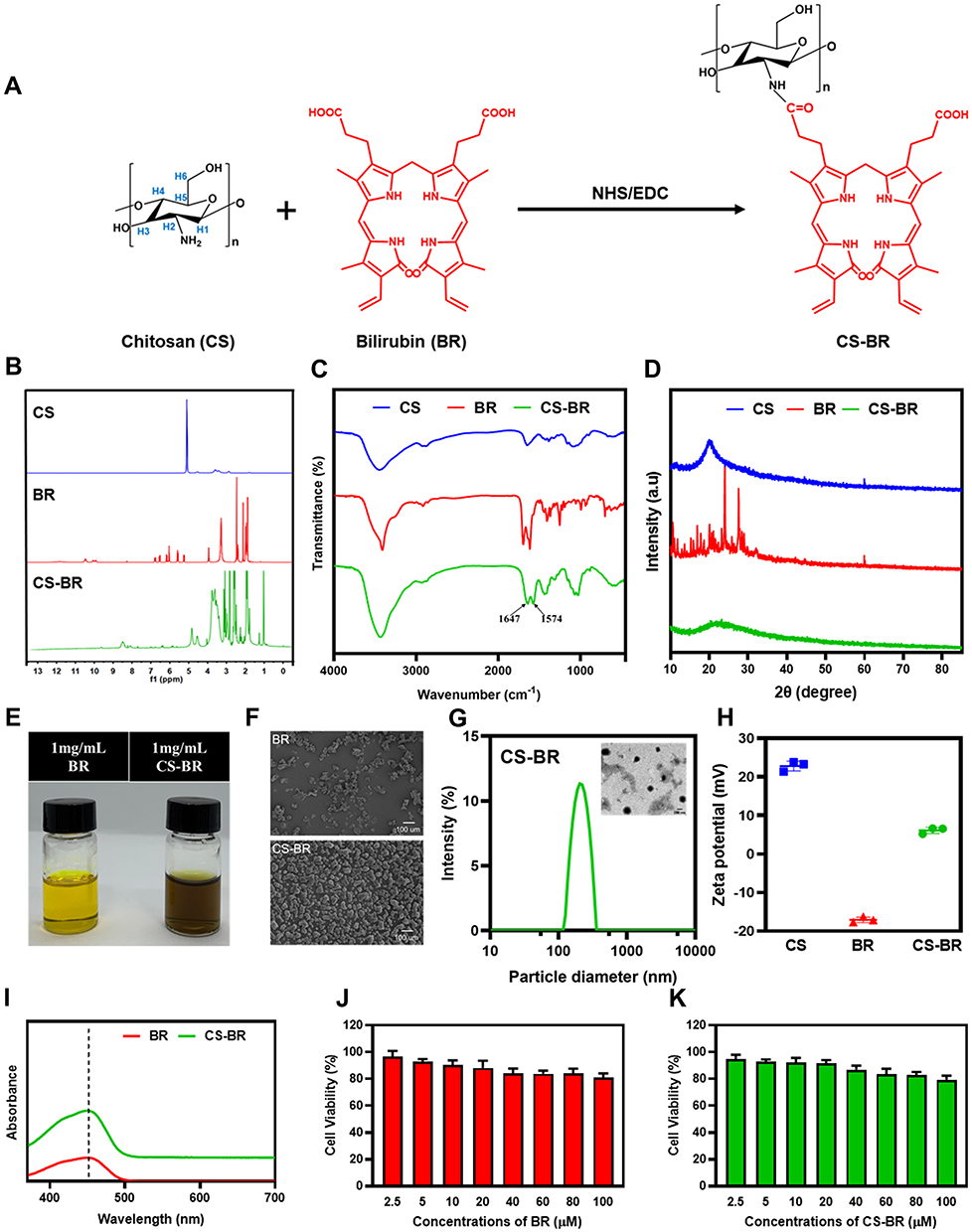 |
Figure 1 Synthesis and characterization of the chitosan-bilirubin conjugate (CS-BR). (A) Synthetic route of CS-BR, (B) proton nuclear magnetic resonance (1H-NMR) spectra, (C) Fourier transform infrared (FTIR) spectra, and (D) X-ray diffraction (XRD) spectra of chitosan (CS), bilirubin (BR), and CS-BR. (E) Photograph images of BR and CS-BR solutions, (F) scanning electron microscope (SEM) images of BR and CS-BR powders. (G) Hydrodynamic size distributions and transmission electron microscope (TEM) images of CS-BR nanoparticles. (H) Zeta potential of CS, BR, and CS-BR. (I) The ultraviolet-visible (UV/Vis) spectra of BR and CS-BR. The viability of gastric epithelial cells after incubation with the different concentrations of (J) BR and (K) CS-BR for 24 h. Data are expressed as the mean ± standard deviation (SD) (n=3). |
We next evaluated the particle size distribution, polydispersity index (PDI), and zeta potential of CS-BR. As shown in Figure 1G and H, the hydrodynamic diameter of CS-BR was 233.9 ± 6.6 nm, and its PDI value was 0.188 ± 0.023. In addition, the TEM image of CS-BR showed that the nanoparticles exhibited spherical shapes, had smooth surfaces, and had good dispersibility. These results showed that we had succeeded in preparing CS-BR nanoparticles with suitable particle sizes and homogeneous dispersibility. The zeta potential of CS is positive at 22.8 ± 1.3 mV, whereas BR has a negative charge at −17.1 ± 0.7 mV; therefore, CS-BR exhibited a positive zeta potential at 6.2 ± 0.8 mV, which meant that positively charged CS-BR could bind to a negatively charged membrane via electrostatic interaction.
To assess the cytotoxicity of CS-BR on gastric epithelial cells, the cells were treated with BR or CS-BR (2.5–100 μM) for 24 h, and the cell viability was then determined under different concentrations of CS-BR treatment (Figure 1J and K). The viability of cells was maintained at more than 90% after treatment with CS-BR (2.5–20 μM, BR equivalent). Unexpectedly, the cell viability still exceeded 80% when exposed to high levels of CS-BR (20–100 μM). In addition, chitosan (2.5 μg–100 μg) also exhibited good cytocompatibility (Figure S2). These evidences suggests that CS-BR has low toxicity to gastric epithelial cells; therefore, we selected a slightly higher physiological concentration of CS-BR (20 μM) for subsequent experiments.
Assessment of the Antioxidant Effect of CS-BR
In general, the mucus secreted by gastric epithelial cells prevented damage to the gastric mucosa by gastric acid and pepsin.33 However, ethanol stimulation and subsequent oxidative stress caused massive apoptosis in the gastric mucosa, which was an important mechanism for gastric mucosal damage and the process of GU formation. Oxidative stress was also a major detrimental factor in the pathogenesis and progression of acute GU.34 Epithelial cells in the gastric mucosa produced extensive ROS after stimulation with ethanol. These ROS molecules not only were detrimental to the healing of gastric mucosal wounds but could also exacerbate the enlargement of ulcer wounds.4 Therefore, our therapeutic goal for GU was a decrease in the cellular damage from oxidative stress and inflammatory responses in the gastric mucosa. As a potent endogenous antioxidant, BR could scavenge excessive ROS and exert a strong anti-inflammatory effect. BR upregulates the expression levels of antioxidant enzymes, including heme oxygenase-1 and SOD-1, via activation of the nuclear factor E2-related factor 2 pathway and suppresses the inflammatory response by inhibiting activation of the NF-κB signaling pathway.35,36
Therefore, we evaluated the antioxidative capacity of CS-BR on elevated oxidative stress in H2O2-stimulated gastric epithelial cells. The production of ROS in cells was detected by a fluorescent probe, DCFH-DA. As shown in Figure 2A, the H2O2 group exhibited significantly higher fluorescent intensity than the control group did. The intervention of BR or CS-BR decreased ROS generation after H2O2 injury. Semi-quantitative analysis of fluorescent intensity was also performed to visually compare the amount of ROS generated in each group; compared with BR, CS-BR showed greater resistance to ROS production and better protected cells from oxidative stress–related damage (Figure 2B). Similarly, the cell viability declined when exposed to H2O2, but CS-BR effectively enhanced cell viability (Figure 2C). The intracellular antioxidant defense system was severely damaged after oxidative stress injury, and levels of SOD and CAT were detected. After H2O2 treatment, the activities of SOD and CAT decreased sharply compared with control group activities, which indicated that the antioxidant capacity of cells was severely impaired in the presence of H2O2. However, BR and CS-BR significantly improved intracellular antioxidant enzyme activities to counteract oxidative stress injury (Figure 2D and E). In addition, after H2O2 administration, cells accumulated the lipid peroxide MDA and exhibited reduced levels of the antioxidant GSH. The CS-BR group showed reduced MDA production and elevated GSH levels in cells (Figure 2F and G). These results demonstrated that CS-BR reduced the production of toxic substances associated with oxidative stress and increased the antioxidant capacity of cells to resist the damage caused by oxygen free radicals.
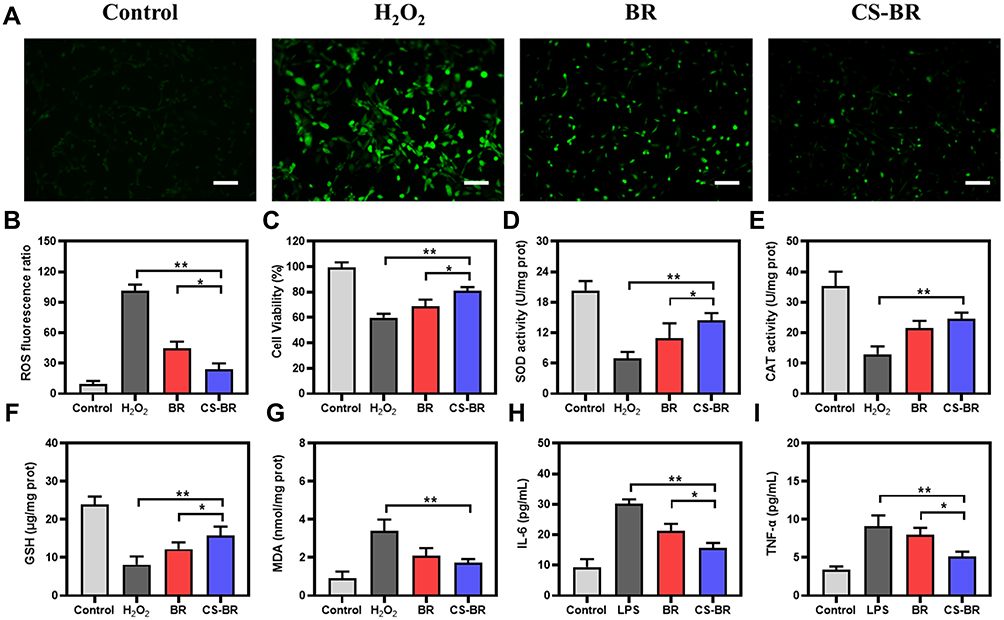 |
Figure 2 The antioxidant and anti-inflammatory effects of the chitosan-bilirubin conjugate (CS-BR). (A) The intracellular reactive oxygen species (ROS) of H2O2-stimulated gastric epithelial cells treated with bilirubin (BR) or CS-BR for 24 h were observed by fluorescent microscopy. Scale bar = 100 μm. (B) The semi-quantitative analysis of fluorescence intensity of ROS. (C) The cell viability of H2O2-stimulated gastric epithelial cells treated with BR or CS-BR for 24 h. The levels of (D) superoxide dismutase (SOD), (E) catalase (CAT), (F) glutathione (GSH), (G) malondialdehyde (MDA), (H) interleukin 6 (IL-6), and (I) tumor necrosis factor α (TNF-α) in each group. Data are expressed as the mean ± standard deviation (SD) (n = 3). *P < 0.05, **P < 0.01. |
The inflammatory response that is concomitant with oxidative stress could be equally devastating to cells. To evaluate the anti-inflammatory action of CS-BR, the cells were stimulated with LPS for 6 h and incubated with BR or CS-BR for 24 h. Then, the culture supernatant was collected to measure the levels of inflammatory cytokines (IL-6 and TNF-α). As illustrated in Figure 2H and I, the LPS group showed high levels of inflammatory factors, whereas CS-BR effectively inhibited the secretion of IL-6 and TNF-α. The results of these experiments show that CS-BR is a cytoprotective agent that could substantially reduce the cellular damage caused by the oxidative stress and the inflammatory response.
As shown in Figure S1, CS-BR internalized into gastric epithelial cells after 1 h and 2 h. Compared with the control group, the CS-BR group exhibited much stronger fluorescent intensities, which indicated that CS-BR was faster internalized into cells due to the interaction between positive-charged CS-BR and negative-charged membrane.
Drug transport to cells is a key step in drug efficacy. CS-BR nanoparticles bind to proteoglycans on the cell surface via a nonspecific electrostatic interaction, and an interaction occurs between CS ligands and receptors on the surface of cell membranes, including massive amino groups in CS as the active sites that enhance the adhesion of cells and nanoparticles.37,38 All of these binding advantages significantly enhance the cellular uptake and transmembrane transport of CS-BR. Therefore, CS-BR exerts better anti-inflammatory and antioxidant activities than free BR does.
Intragastric Retention of CS-BR in the GU Model
Oral administration is one of the most convenient and compliant methods of administering medication for patients with GU. Nanotechnology-based drug delivery systems have been widely available for oral medicine.39,40 However, when administered orally, these drugs had a short residence time in the stomach because of fast gastric emptying. Thus, we urgently need strategies to prolong the retention time of drugs in the stomach to maximize the anti-ulcer effect of the drug.
FITC-labeled CS-BR was used to observed the intragastric retention of BR in the stomach, and free FITC was used as a control. After intragastric administration of FITC-labeled CS-BR or free FITC, the mice with GU were sacrificed at various predetermined time points, and the main organs, including the stomach, were collected. As shown in Figure 3A, most of the CS-BR accumulated in the stomach after oral administration and rarely distributed into other organs during the observation period. The fluorescent intensities in the stomach of the control group and the CS-BR group were similar 30 min after administration. The fluorescent intensity in the control group decreased rapidly over time, and fluorescent distribution in the stomach was hardly observed at 120 min. Importantly, the apparent fluorescent distribution in the CS-BR group suggested that a significant portion of CS-BR still remained in the stomach at 60 min. Additional quantification of the fluorescent intensity showed that fluorescent signals in the CS-BR group were obviously stronger than those in the control group at 60, 90, and 120 min; this finding demonstrated that CS-BR extended the retention time of BR in the stomach (Figure 3B). Significantly, the fluorescent intensities of the control group were quite weak in corpus and antrum at 90 min and 120 min, whereas the fluorescent signal of the CS-BR group was relatively strong in ulcer-prone areas (corpus and antrum) that meant a considerable amount of CS-BR was still accumulated in these sites after 90 minutes (Figure S3). These results showed that CS-BR could longer accumulate in gastric tissue, especially in ulcer-prone areas such as the corpus and antrum after 90 minutes of administration, the CS-BR significantly prolonged the retention time in the gastric tissue when compared to the control group.
 |
Figure 3 The retention time of the chitosan-bilirubin conjugate (CS-BR) in gastric tissue after oral delivery. (A) The fluorescent distribution to gastric tissue and other organs in mice with gastric ulcers after oral administration with fluorescein isothiocyanate (FITC)–labeled CS-BR or free FITC (as the control) at predetermined times. (B) The semi-quantitation of fluorescent intensity in the stomach of each group. (C) The concentration of bilirubin (BR) in the stomach after oral administration with BR or CS-BR at different time points. Data are expressed as the mean ± standard deviation (SD) (n = 3). |
We also measured the content of BR in the gastric tissue after the mice with GU were gavaged with BR or CS-BR (Figure 3C). The concentration of BR in the CS-BR group was higher than that in the free BR group at different time points. These results all showed that CS-BR effectively retained the BR in the stomach and exerted sustained anti-ulcer activity.
In addition, the BR concentrations in the CS-BR group were higher than that in the BR group in different areas of the stomach. Compared with the rapid decrease of BR concentration of the BR group in the stomach within 120 minutes of administration, the CS-BR effectively improved the accumulation of BR in gastric tissues. In the inflamed area (corpus and antrum), the concentration of BR was quite low after 90 minutes of free BR administration whereas CS-BR group exhibited relatively high BR concentration ((Figure S4). These data indicated that CS-BR could enhance the accumulation of BR and prolong the retention time of BR in gastric tissue (especially in an inflamed area) that was beneficial for BR to exert the anti-ulcer effects.
As a natural cationic polysaccharide and linear long-chain polymeric molecule, CS shows good bioadhesive properties. CS is exposed to the mucin in the mucus layer, which results in mutual adsorption—an important condition for strong and lasting adhesion. In an acidic environment, the amino group on the CS molecule is protonated to form the positively charged –NH3+, and then the protonated amino group adheres to the negatively charged mucin by electrostatic interaction.41 Bioadhesive CS offers a tight contact with the mucosal layer at the target site, prolongs the retention time of the drug at the lesion site, and improves the local efficacy of the drug.42 Therefore, compared with free BR, CS-BR nanoparticles exhibited a more powerful retention capacity in gastric tissue.
In Vivo Therapeutic Efficacy of CS-BR in Mice with GU
The pathogenesis of GU is related to oxygen free radicals; ethanol severely affects the expression of several oxidoreductases, such as ethanol dehydrogenase, which accelerates the production of acetaldehyde. Under the action of xanthine oxidase, acetaldehyde converts into ROS, such as the hydroxyl radical and the superoxide anion, causing gastric mucosal injury.43 Ethanol-induced GU triggers oxidative stress that damages the gastric mucus layer and gastric defense system and activates related redox-sensitive transcription factors, thereby increasing the degree of inflammatory response in gastric tissue. In addition, the ethanol could directly damage gastric mucosa through destruction, dehydration, and mucosal cytotoxicity, then ethanol-induced inflammation, oxidative stress, and apoptosis through leukocyte recruitment, which further indirectly damaged gastric mucosa. Consequently, attenuating oxidative damage and inflammatory injury in lesion sites is critical to the treatment and prevention of GU.
We evaluated the anti-ulcer capacity of CS-BR in vivo. As reported in many studies on GU, the mice were pretreated with a therapeutic substance for several days before the ethanol-induced GU model was established. Considering the clearance result of CS-BR in vivo, the mice were orally administered daily for 7 days to progressively increase the anti-ulcer capacity in the body by CS-BR, thereby reduced the damage of oxidative stress and inflammatory response caused by GU when ethanol was suddenly poured into the stomach to cause damage. The last administration was 30 min before treatment with ethanol (Figure 4A). We evaluated the severity of GU in these groups. The gastric tissues were removed from the mice, and the entire interior of the stomach was exposed to observe the pathological changes in gastric tissue. As illustrated in Figure 4B, the gastric mucosa of mice in the control group was intact and smooth, without ulcer or hemorrhagic spots. In contrast, the mice in the GU group had extensive ulceration and multiple bleeding spots concentrated in the glandular stomach (white arrows), and the medial gastric mucosa of these mice showed obvious submucosal edema, mucosal erosion, ulceration, necrosis, and linear and striated hemorrhagic ulceration with large bleeding areas. Hemorrhage and ulceration were obvious in the gastric tissue of the BR group, with slight improvements in ulceration compared with the GU group, but gastric mucosal damage and edema were still evident. In contrast, the degree of mucosal edema and congestion in gastric tissues of mice were significantly reduced with the intervention of CS-BR. In addition, the ulcer area was decreased and showed sporadic, punctate shapes in the stomachs of mice in the CS-BR group. These results demonstrated that CS-BR could effectively alleviate the symptoms associated with GU. Additionally, the UI was calculated, and the CS-BR group exhibited a lower ulcer index than the BR group did (Figure 4C).
 |
Figure 4 The anti-ulcer effect of the chitosan-bilirubin conjugate (CS-BR) via inhibition of oxidative stress and the inflammatory response. (A) The timeline of the experimental design. (B) Photograph of gastric tissue in each group. (C) The gastric ulcer (GU) index in each group. The levels of (D) prostaglandin E2 (PGE2), (E) superoxide dismutase (SOD), (F) catalase (CAT), (G) glutathione (GSH), (H) malondialdehyde (MDA), (I) interleukin 6 (IL-6), and (J) tumor necrosis factor α (TNF-α) in the gastric tissue of mice. Data are expressed as the mean ± standard deviation (SD) (n = 3). *P < 0.05, **P < 0.01. |
content of PGE2 in the gastric tissue of each group were determined by ELISA. PEG2, synthesized from arachidonic acid or linoleic acid in the stomach, as catalyzed by cyclooxygenase, is a recognized protective factor in the gastric mucosa. The protective mechanism of PGE2 involves promotion of mucus secretion, inhibition of gastric acid secretion, and increases to the blood flow in the local gastric mucosa.44 In normal physiological conditions, adequate PGE2 facilitates the secretion of mucus and bicarbonate from gastric mucosal epithelial cells to maintain mucosal integrity. In the GU group, the level of PGE2 in gastric tissue decreased sharply, whereas PGE2 content increased greatly in the CS-BR treatment group (Figure 4D). This finding showed that CS-BR could accelerate the synthesis and secretion of PGE2 by epithelial cells, enhance the defense function of the gastric mucosa, and improve the function of the regenerated mucosa—changes that together might be a mechanism that promotes GU healing.
Massive ROS production occurs in GU, and these ROS cause lipid peroxidation in the gastric mucosa that leads to gastric mucosal blood flow disorder and injury. MDA is a lipid peroxide formed by polyunsaturated fatty acids in cells or tissues after oxidative stress; its formation causes protein denaturation, DNA damage, and enzyme inactivation.45 The level of MDA indirectly reflects the severity of oxygen free radical attacks on organisms and cells. Antioxidant enzymes, such as SOD and CAT, are an important line of defense for oxygen radical scavenging systems in the body. SOD is an antioxidant enzyme that exists in organisms to specifically remove the superoxide anion and catalyze the decomposition of superoxide radicals into H2O2, which then decompose into H2O and oxygen molecules under the action of CAT.46 The ethanol could directly damage gastric mucosa through destruction, dehydration, and mucosal cytotoxicity, then ethanol-induced inflammation, oxidative stress, and apoptosis through leukocyte recruitment, which further indirectly damaged gastric mucosa. Thus, we believed that CS-BR exerted anti-ulcer effects via reducing inflammatory responses and oxidative stress in the region of the GU, then protected gastric cells and tissue from further damage.
We assessed oxidative stress-related indicators (MDA, SOD, CAT) in gastric tissue of each group. In the CS-BR group, the amount of MDA in the gastric mucosa significantly increased, whereas the activity of SOD and CAT as well as the level of GSH decreased, indicating that lipid peroxidation was involved in the pathophysiological process of ethanol-induced GU in mice. CS-BR exhibited gastroprotective effects against oxidative stress by increasing GSH production, increasing SOD and CAT activities, and reducing MDA content (Figure 4E–H). Excessive ROS induced severe inflammation, causing more damage to the gastric mucosa. CS-BR effectively inhibited the secretion of inflammatory cytokines (IL-6 and TNF-α) and then reduced the degree of the inflammatory response in GU tissue (Figure 4I and J). These results show that CS-BR exerted a good curative effect for GU via enhancement of antioxidant capacity and suppression of the inflammatory reaction in the lesion. Crucially, CS-BR showed better therapeutic effectiveness than free BR did, and this improvement might be attributed to the longer retention time of CS-BR in the stomach.
Histological Analysis
We used H&E staining to observe the pathological changes in the gastric tissue and then calculated the pathological score in each group (Figure 5B). As shown in Figure 5A, the control group showed dense and intact gastric mucosa as well as regular rows of epithelial cells without mucosal defects or inflammatory cell infiltration. In contrast, the gastric mucosa of mice in the GU group showed severe disruption and histological incompleteness, with visible inflammatory cell infiltration, disorganized glandular structure, and obvious epithelial cell detachment. Free BR alleviated the pathological changes of GU to some extent, but some tissue structure destruction and gland loss were still observed. Compared with BR, CS-BR was associated with a gastric mucosa that was only slightly damaged and destroyed, which indicated that CS-BR exhibited a better protective effect than BR did on the gastric mucosa. In addition, PAS staining was performed to assess the level of gastric mucus secretion; purple portions represented PAS-positive areas. Quantitative analysis of PAS staining showed that mice in the control group secreted sufficient mucus to protect the gastric mucosa, whereas gastric mucus production was greatly reduced after GU. CS-BR vastly increased the secretion of mucus to alleviate damage to the stomach (Figure 5C).
 |
Figure 5 The histological analysis of gastric tissue. (A) The images of hematoxylin and eosin (H&E) staining and periodic acid–Schiff (PAS) staining as well as the representative immunohistochemical images of myeloperoxidase (MPO) and CD68 in the gastric tissue of each group. Scale bar = 100 μm. (B) The pathological score in each group. (C) Quantitative analysis of the PAS-positive area. The quantitation of positive cells of (D) MPO and (E) CD68 in each group. Data are expressed as the mean ± standard deviation (SD) (n = 3). *P < 0.05, **P < 0.01. |
MPO, a heme protein contained in neutrophils, is synthesized in bone marrow and stored in cyanophage granules before the granulocyte enters circulation. External stimulation causes neutrophil aggregation and release of MPO; its level is often used to assess the extent of neutrophil infiltration and inflammatory damage in tissues. MPO has potent pro-inflammatory effects and might be directly involved in tissue damage.47,48 Macrophages are involved in a variety of inflammatory reactions and the occurrence of ulcers;49 we used CD68 antibody to observe macrophage aggregation in gastric tissue. All tissue sections were stained by IHC, and quantitative analysis of the positive expression was conducted in each group. The substantial expression of MPO and CD68 in the gastric tissue of the GU group illustrated that severe inflammation occurred in the process of ulceration. BR and CS-BR both effectively reduced the inflammatory response in gastric tissue, but CS-BR exhibited a superior anti-inflammatory ability (Figure 5D and E). These data all show that CS-BR exerted an excellent anti-ulcer effect via attenuation of inflammation in the ulcer lesion site.
CS as oral drug delivery carriers have good biocompatibility and bioadhesiveness, and they improve drug stability and controlled release. BR, as an endogenous antioxidant, may exert unexpected curative effects for GU. Leveraging these advantages, BR was conjugated to CS to form CS-BR nanoparticles that could enhance the potential therapeutic effect of BR on GU. The application of nanotechnology increased the efficacy of CS-BR. GU with severe inflammation increased tissue permeability that contributed to enhanced accumulation and cellular uptake of nanoparticles in inflamed areas. Given the experimental results of this study, we believed that drug carriers matched with BR as a therapeutic substance have promising applications in mucosal ulcerative diseases.
In Vivo Safety Evaluation of CS-BR
During the past decades, studies on the biological effects of BR have mainly focused on neurotoxicity, especially neonatal jaundice. These studies also reported high levels of BR in other diseases, such as hepatitis and brain damage, especially in newborns.50 Our previous studies also confirmed that excessively high concentrations of BR have caused serious damage to islet cells. Thus, the therapeutic concentration of BR is relatively narrow, and researchers must pay attention to the adverse effects of BR during disease treatments. We have screened for the optimal BR concentration (20 μM) in islet cells17 and then used a relatively conservative dose (5 mg/kg) to treat acute kidney injury.19 In this study, we also selected a low concentration of bilirubin for GU to allow BR to exert curative effects without causing significant toxic effects to the body.
To investigate the biological toxicity of BR and CS-BR nanoparticles in a GU model, mice in this study were treated with BR or CS-BR for 30 days; then, major organs and serum samples were collected. The results of H&E staining showed that BR and CS-BR did not cause pathological damage to these organs (Figure 6A). Ethanol-treated mice developed abnormal liver function, with elevated ASL and ALT levels caused by damage to liver cells from ethanol (Figure 6B and C). The SCr and BUN levels were not significantly different in these groups, which indicated that renal function was normal in all mice (Figure 6D and E). These results showed that BR and CS-BR had negligible adverse effects on the mice in our GU model. As a promising therapeutic agent for GU, CS-BR nanoparticles not only effectively ameliorated GU in mice but also demonstrated high biosafety during in vivo applications.
 |
Figure 6 The evaluation of biocompatibility for the chitosan-bilirubin conjugate (CS-BR). (A) The images of hematoxylin and eosin (H&E) staining of the major organ in each group. Scale bar = 50 μm. The levels of (B) aspartate aminotransferase (AST), (C) alanine aminotransferase (ALT), (D) serum creatinine (SCr), and (E) blood urea nitrogen (BUN) in serum samples. Data are expressed as the mean ± standard deviation (SD) (n = 3). *P < 0.05. Abbreviations: n.s, indicates no statistical significance. |
Conclusion
BR is widely applied in the treatment of various diseases, such as cardiovascular diseases, Crohn’s disease, and diabetes mellitus, because of its strong capacity to scavenge free radicals. In this study, we synthesized a conjugate, CS-BR, and applied it in the form of nanoparticles to the treatment of GU. The positively charged CS is instrumental in improving cellular uptake of BR via electrostatic interaction between CS and the cell membrane. Additionally, the bioadhesive properties of CS can achieve a long duration of gastric retention and promote the long-lasting therapeutic effects of BR against GU, thus alleviating the damage from oxidative stress and inflammation. Together, the data from this study show that CS-BR nanoparticles effectively improve the antioxidant capacity of BR and inhibit the secretion of inflammatory cytokines in the cells and tissues. Compared with free BR, CS-BR exhibited better anti-ulcer effects, which indicated that our strategy for BR delivery amplified the curative effects of BR. We believe that BR has good therapeutic potential and that development of drug carriers for BR against gastrointestinal diseases is worthwhile.
Acknowledgment
This research was supported by the National Science Foundation of China (Grant No. 81772316 and 81772112), Xinmiao Talent Program of Zhejiang Province (Grant No. 2020R413067).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Lanas A, Chan FKL. Peptic ulcer disease. Lancet. 2017;390(10094):613–624. doi:10.1016/S0140-6736(16)32404-7
2. Rocco A, Compare D, Angrisani D, Sanduzzi Zamparelli M, Nardone G. Alcoholic disease: liver and beyond. World J Gastroenterol. 2014;20(40):14652–14659. doi:10.3748/wjg.v20.i40.14652
3. Chakraborty S, Stalin S, Das N, Choudhury ST, Ghosh S, Swarnakar S. The use of nano-quercetin to arrest mitochondrial damage and MMP-9 upregulation during prevention of gastric inflammation induced by ethanol in rat. Biomaterials. 2012;33(10):2991–3001. doi:10.1016/j.biomaterials.2011.12.037
4. Yeo D, Hwang SJ, Kim WJ, Youn H-J, Lee H-J. The aqueous extract from Artemisia capillaris inhibits acute gastric mucosal injury by inhibition of ROS and NF-kB. Biomed Pharmacother. 2018;99:681–687. doi:10.1016/j.biopha.2018.01.118
5. Scally B, Emberson JR, Spata E, et al. Effects of gastroprotectant drugs for the prevention and treatment of peptic ulcer disease and its complications: a meta-analysis of randomised trials. Lancet Gastroenterol Hepatol. 2018;3(4):231–241. doi:10.1016/S2468-1253(18)30037-2
6. Jangi S, Otterbein L, Robson S. The molecular basis for the immunomodulatory activities of unconjugated bilirubin. Int J Biochem Cell Biol. 2013;45(12):2843–2851. doi:10.1016/j.biocel.2013.09.014
7. Kamisako T, Kobayashi Y, Takeuchi K, et al. Recent advances in bilirubin metabolism research: the molecular mechanism of hepatocyte bilirubin transport and its clinical relevance. J Gastroenterol. 2000;35(9):659–664. doi:10.1007/s005350070044
8. Sullivan JI, Rockey DC. Diagnosis and evaluation of hyperbilirubinemia. Curr Opin Gastroenterol. 2017;33(3):164–170. doi:10.1097/MOG.0000000000000354
9. Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science. 1987;235(4792):1043–1046. doi:10.1126/science.3029864
10. Stocker R. Antioxidant activities of bile pigments. Antioxid Redox Signal. 2004;6(5):841–849. doi:10.1089/ars.2004.6.841
11. Kim TW, Kim Y, Jung W, et al. Bilirubin nanomedicine ameliorates the progression of experimental autoimmune encephalomyelitis by modulating dendritic cells. J Control Release. 2021;331:74–84. doi:10.1016/j.jconrel.2021.01.019
12. Lee Y, Sugihara K, Gillilland MG, Jon S, Kamada N, Moon JJ. Hyaluronic acid-bilirubin nanomedicine for targeted modulation of dysregulated intestinal barrier, microbiome and immune responses in colitis. Nat Mater. 2020;19(1):118–126. doi:10.1038/s41563-019-0462-9
13. Kim JY, Lee DY, Kang S, et al. Bilirubin nanoparticle preconditioning protects against hepatic ischemia-reperfusion injury. Biomaterials. 2017;133:1–10. doi:10.1016/j.biomaterials.2017.04.011
14. Keum H, Kim D, Kim J, et al. A bilirubin-derived nanomedicine attenuates the pathological cascade of pulmonary fibrosis. Biomaterials. 2021;275:120986. doi:10.1016/j.biomaterials.2021.120986
15. Lin Y, Wang S, Yang Z, et al. Bilirubin alleviates alum-induced peritonitis through inactivation of NLRP3 inflammasome. Biomed Pharmacother. 2019;116:108973. doi:10.1016/j.biopha.2019.108973
16. Li Y, Huang B, Ye T, Wang Y, Xia D, Qian J. Physiological concentrations of bilirubin control inflammatory response by inhibiting NF-κB and inflammasome activation. Int Immunopharmacol. 2020;84:106520. doi:10.1016/j.intimp.2020.106520
17. Zhao Y-Z, Huang Z-W, Zhai -Y-Y, et al. Polylysine-bilirubin conjugates maintain functional islets and promote M2 macrophage polarization. Acta Biomater. 2021;122:172–185. doi:10.1016/j.actbio.2020.12.047
18. Lee Y, Kim H, Kang S, Lee J, Park J, Jon S. Bilirubin nanoparticles as a nanomedicine for anti-inflammation therapy. Angew Chem Int Ed Engl. 2016;55(26):7460–7463. doi:10.1002/anie.201602525
19. Huang Z-W, Shi Y, Zhai -Y-Y, et al. Hyaluronic acid coated bilirubin nanoparticles attenuate ischemia reperfusion-induced acute kidney injury. J Control Release. 2021;334:275–289. doi:10.1016/j.jconrel.2021.04.033
20. Ahsan SM, Thomas M, Reddy KK, Sooraparaju SG, Asthana A, Bhatnagar I. Chitosan as biomaterial in drug delivery and tissue engineering. Int J Biol Macromol. 2018;110:97–109. doi:10.1016/j.ijbiomac.2017.08.140
21. Wei S, Ching YC, Chuah CH. Synthesis of chitosan aerogels as promising carriers for drug delivery: a review. Carbohydr Polym. 2020;231:115744. doi:10.1016/j.carbpol.2019.115744
22. Pawde DM, Viswanadh MK, Mehata AK, et al. Mannose receptor targeted bioadhesive chitosan nanoparticles of clofazimine for effective therapy of tuberculosis. Saudi Pharm J. 2020;28(12):1616–1625. doi:10.1016/j.jsps.2020.10.008
23. Aksungur P, Sungur A, Unal S, Iskit AB, Squier CA, Senel S. Chitosan delivery systems for the treatment of oral mucositis: in vitro and in vivo studies. J Control Release. 2004;98(2):269–279. doi:10.1016/j.jconrel.2004.05.002
24. Anandan R, Nair PGV, Mathew S. Anti-ulcerogenic effect of chitin and chitosan on mucosal antioxidant defence system in HCl-ethanol-induced ulcer in rats. J Pharm Pharmacol. 2004;56(2):256–269. doi:10.1211/0022357023079
25. Caprifico AE, Polycarpou E, Foot PJS, Calabrese G. Biomedical and pharmacological uses of fluorescein isothiocyanate chitosan-based nanocarriers. Macromol Biosci. 2021;21(1):2000312. doi:10.1002/mabi.202000312
26. Cheon JY, Lee HM, Park WH. Formation of silver nanoparticles using fluorescence properties of chitosan oligomers. Mar Drugs. 2018;16(1):11. doi:10.3390/md16010011
27. Swarnakar S, Mishra A, Ganguly K, Sharma AV. Matrix metalloproteinase-9 activity and expression is reduced by melatonin during prevention of ethanol-induced gastric ulcer in mice. J Pineal Res. 2007;43(1):56–64. doi:10.1111/j.1600-079X.2007.00443.x
28. Mousa AM, El-Sammad NM, Hassan SK, et al. Antiulcerogenic effect of Cuphea ignea extract against ethanol-induced gastric ulcer in rats. BMC Complement Altern Med. 2019;19(1):345. doi:10.1186/s12906-019-2760-9
29. Yao Q, Jiang X, Zhai -Y-Y, et al. Protective effects and mechanisms of bilirubin nanomedicine against acute pancreatitis. J Control Release. 2020;322:312–325. doi:10.1016/j.jconrel.2020.03.034
30. Yao Q, Huang Z-W, Zhai -Y-Y, et al. Localized controlled release of bilirubin from β-cyclodextrin-conjugated ε-polylysine to attenuate oxidative stress and inflammation in transplanted islets. ACS Appl Mater Interfaces. 2020;12(5):5462–5475. doi:10.1021/acsami.9b18986
31. Laine L, Weinstein WM. Histology of alcoholic hemorrhagic “gastritis”: a prospective evaluation. Gastroenterology. 1988;94(6):1254–1262. doi:10.1016/0016-5085(88)90661-0
32. Chen P, Shen Y, Shi H, et al. Gastroprotective effects of Kangfuxin-against ethanol-induced gastric ulcer via attenuating oxidative stress and ER stress in mice. Chem Biol Interact. 2016;260:75–83. doi:10.1016/j.cbi.2016.10.021
33. Tulassay Z, Herszényi L. Gastric mucosal defense and cytoprotection. Best Pract Res Clin Gastroenterol. 2010;24(2):99–108. doi:10.1016/j.bpg.2010.02.006
34. Pérez S, Taléns-Visconti R, Rius-Pérez S, Finamor I, Sastre J. Redox signaling in the gastrointestinal tract. Free Radic Biol Med. 2017;104:75–103. doi:10.1016/j.freeradbiomed.2016.12.048
35. Qaisiya M, Coda Zabetta CD, Bellarosa C, Tiribelli C. Bilirubin mediated oxidative stress involves antioxidant response activation via Nrf2 pathway. Cell Signal. 2014;26(3):512–520. doi:10.1016/j.cellsig.2013.11.029
36. Yao Q, Jiang X, Kou L, Samuriwo AT, Xu H-L, Zhao Y-Z. Pharmacological actions and therapeutic potentials of bilirubin in islet transplantation for the treatment of diabetes. Pharmacol Res. 2019;145:104256. doi:10.1016/j.phrs.2019.104256
37. Aguilar A, Zein N, Harmouch E, et al. Application of chitosan in bone and dental engineering. Molecules. 2019;24(16):3009. doi:10.3390/molecules24163009
38. Hozumi K, Nomizu M. Mixed peptide-conjugated chitosan matrices as multi-receptor targeted cell-adhesive scaffolds. Int J Mol Sci. 2018;19(9):2713. doi:10.3390/ijms19092713
39. Wang Y, Pi C, Feng X, Hou Y, Zhao L, Wei Y. The influence of nanoparticle properties on oral bioavailability of drugs. Int J Nanomedicine. 2020;15:6295–6310. doi:10.2147/IJN.S257269
40. Kim KS, Suzuki K, Cho H, Youn YS, Bae YH. Oral nanoparticles exhibit specific high-efficiency intestinal uptake and lymphatic transport. ACS Nano. 2018;12(9):8893–8900. doi:10.1021/acsnano.8b04315
41. Stenger Moura FC, Perioli L, Pagano C, et al. Chitosan composite microparticles: a promising gastroadhesive system for taxifolin. Carbohydr Polym. 2019;218:343–354. doi:10.1016/j.carbpol.2019.04.075
42. Islam MA, Park T-E, Reesor E, et al. Mucoadhesive chitosan derivatives as novel drug carriers. Curr Pharm Des. 2015;21(29):4285–4309. doi:10.2174/1381612821666150901103819
43. Bandyopadhyay D, Chattopadhyay A. Reactive oxygen species-induced gastric ulceration: protection by melatonin. Curr Med Chem. 2006;13(10):1187–1202. doi:10.2174/092986706776360842
44. Zhang C, Gao F, Gan S, et al. Chemical characterization and gastroprotective effect of an isolated polysaccharide fraction from Bletilla striata against ethanol-gastric ulcer. Food Chem Toxicol. 2019;131:110539. doi:10.1016/j.fct.2019.05.047
45. Elshazly SM, Abd El Motteleb DM, Ibrahim IAAE-H. Hesperidin protects against stress induced gastric ulcer through regulation of peroxisome proliferator activator receptor gamma in diabetic rats. Chem Biol Interact. 2018;291:153–161. doi:10.1016/j.cbi.2018.06.027
46. Mao GD, Thomas PD, Lopaschuk GD, Poznansky MJ. Superoxide dismutase (SOD)-catalase conjugates: role of hydrogen peroxide and the Fenton reaction in SOD toxicity. J Biol Chem. 1993;268(1):416–420. doi:10.1016/S0021-9258(18)54167-3
47. Aratani Y. Myeloperoxidase: its role for host defense, inflammation, and neutrophil function. Arch Biochem Biophys. 2018;640:47–52. doi:10.1016/j.abb.2018.01.004
48. Ndrepepa G. Myeloperoxidase: abridge linking inflammation and oxidative stress with cardiovascular disease. Clin Chim Acta. 2019;493:36–61. doi:10.1016/j.cca.2019.02.022
49. Zhang J, Zhao Y, Hou T, et al. Macrophage-based nanotherapeutic strategies in ulcerative colitis. J Control Release. 2020;320:363–380. doi:10.1016/j.jconrel.2020.01.047
50. Alexandra Brito M, Silva RFM, Brites D. Bilirubin toxicity to human erythrocytes: a review. Clin Chim Acta. 2006;374(1–2):46–56. doi:10.1016/j.cca.2006.06.012
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.