")
Back to Journals » Degenerative Neurological and Neuromuscular Disease » Volume 10
Prostaglandins as the Agents That Modulate the Course of Brain Disorders
Authors Famitafreshi H, Karimian M
Received 2 December 2019
Accepted for publication 30 December 2019
Published 16 January 2020 Volume 2020:10 Pages 1—13
DOI https://doi.org/10.2147/DNND.S240800
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Thomas Müller
Hamidreza Famitafreshi, Morteza Karimian
Physiology Department, Tehran University of Medical Sciences, Tehran, Iran
Correspondence: Morteza Karimian
Department of Physiology, Tehran University of Medical Science, Enghelab St., Shanzdahazar St., Poorsina St, Tehran, Iran
Tel/Fax +98 02166419484
Email [email protected]
Abstract: Neurologic and neuropsychiatric diseases are associated with great morbidity and mortality. Prostaglandins (PGs) are formed by sequential oxygenation of arachidonic acid in physiologic and pathologic conditions. For the production of PGs cyclooxygenase is a necessary enzyme that has two isoforms, that are named COX-1 and COX-2. COX-1 produces type 1 prostaglandins and on the other hand, COX-2 produces type 2 prostaglandins. Recent studies suggest PGs abnormalities are present in a variety of neurologic and psychiatric disorders. In a disease state, type 2 prostaglandins are mostly responsible and type 1 PGs are not so important in the disease state. In this review, the importance of prostaglandins especially type 2 in brain diseases has been discussed and their possible role in the initiation and outcome of brain diseases has been assessed. Overall the studies suggest prostaglandins are the agents that modulate the course of brain diseases in a positive or negative manner. Here in this review article, the various aspects of PGs in the disease state have discussed. It appears more studies must be done to understand the exact role of these agents in the pathophysiology of brain diseases. However, the suppression of prostaglandin production may confer the alleviation of some brain diseases.
Keywords: prostaglandins, depression, Alzheimer, addiction, Parkinson and multiple sclerosis
Introduction
Neuropsychiatric diseases have a wide array of symptoms and related behaviors that have a high prevalence in human societies.1 A great fraction of them occurs in a subset of people that are suffering from other medical diseases.2–5 Many studies have been performed to confirm the basic mechanisms that produce such diseases. Prostaglandins are lipid-derived small molecules that are produced from arachidonic acid by sequential enzymatic reactions.6 Recently it has been given great importance to these small molecules in the occurrence of neuropsychiatric disorders.7 Recently it has been proposed that other than physiologic functions, certain prostaglandins are associated with neuropsychiatric and neurologic disorders.8 New studies aim to establish a relationship between certain types of prostaglandins with a specific disease. Here in this review article, we are going to discuss the important aspects of prostaglandins in the occurrence of brain diseases.
Prostaglandins
Prostaglandins are lipid derives molecules that have important functions in our body. All prostaglandins have 20 carbons but they have different structures that account for diverse physiologic functions.9 The difference in the receptor is responsible for the diverse functions of one prostaglandin that can be inhibitory or stimulatory.10 The synthesis of prostaglandins is a multistep process that begins with the formation of arachidonic acid from diacylglycerol and phospholipase A2. Arachidonic acid can choose the cyclooxygenase pathway or lipoxygenase pathways.11 Prostaglandins are formed by sequential oxygenation of arachidonic acid. Cyclooxygenase has two isoforms COX-1 and COX-2. Recently it has been suggested that a variant of COX-1 can be considered as COX-3.12 Constant production of COX-1 that results in the production of type 1 prostaglandins. On the other hand, COX-2 is activated in certain conditions such as inflammation and growth.13
Prostaglandins Receptors
Eight subtypes of membrane prostanoid receptors have already been discovered: the PGD receptor (DP), four subtypes of the PGE receptor (EP1, EP2, EP3, and EP4), the PGF receptor (FP), the PGI receptor (IP), and the TxA receptor (TP)14 (Table 1). All are G protein-coupled rhodopsin-type receptors with seven transmembrane domains, and each domain is encoded by a different gene. In Table 1 we have summarized the various prostaglandin receptors that are present in the body.15 Cyclooxygenase enzyme releases prostaglandins from arachidonic acid in membrane lipids (Figure 1). There are multiple steps that eventually all types of prostaglandins are produced. The structural difference is to account for their different biological properties. Also different kind of receptors also may account for such differences. One prostaglandin may have a stimulatory effect in a given context and in another context have an inhibitory function. They act as paracrine substances and usually targeted tissue in the vicinity of the site of their production.
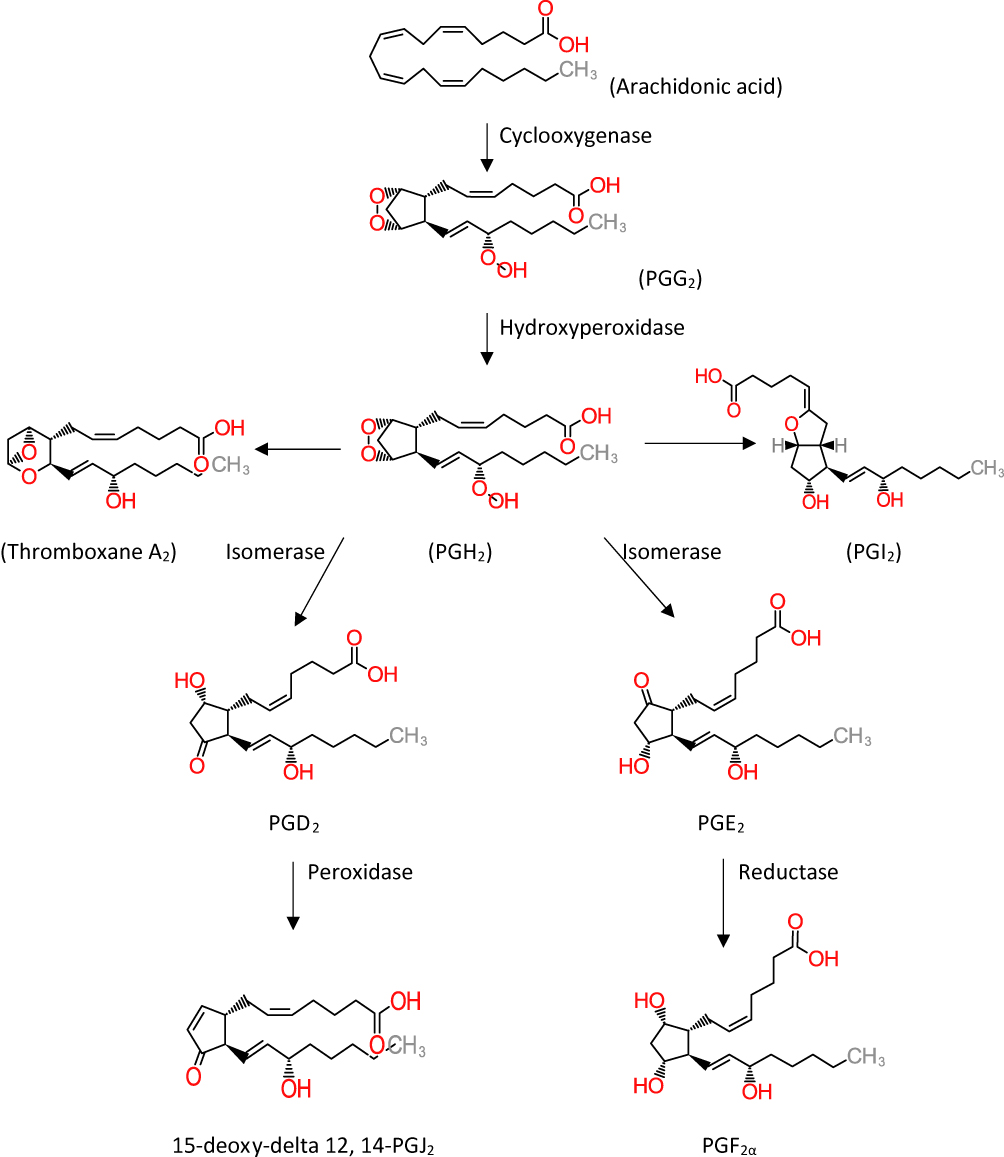 |
Figure 1 Metabolic cascade of the arachidonic acid pathway that leads to prostaglandin production. The cyclooxygenase pathway has been illustrated in the figure. Thromboxane A2 and prostaglandin I2 are synthesized by synthase enzymes. |
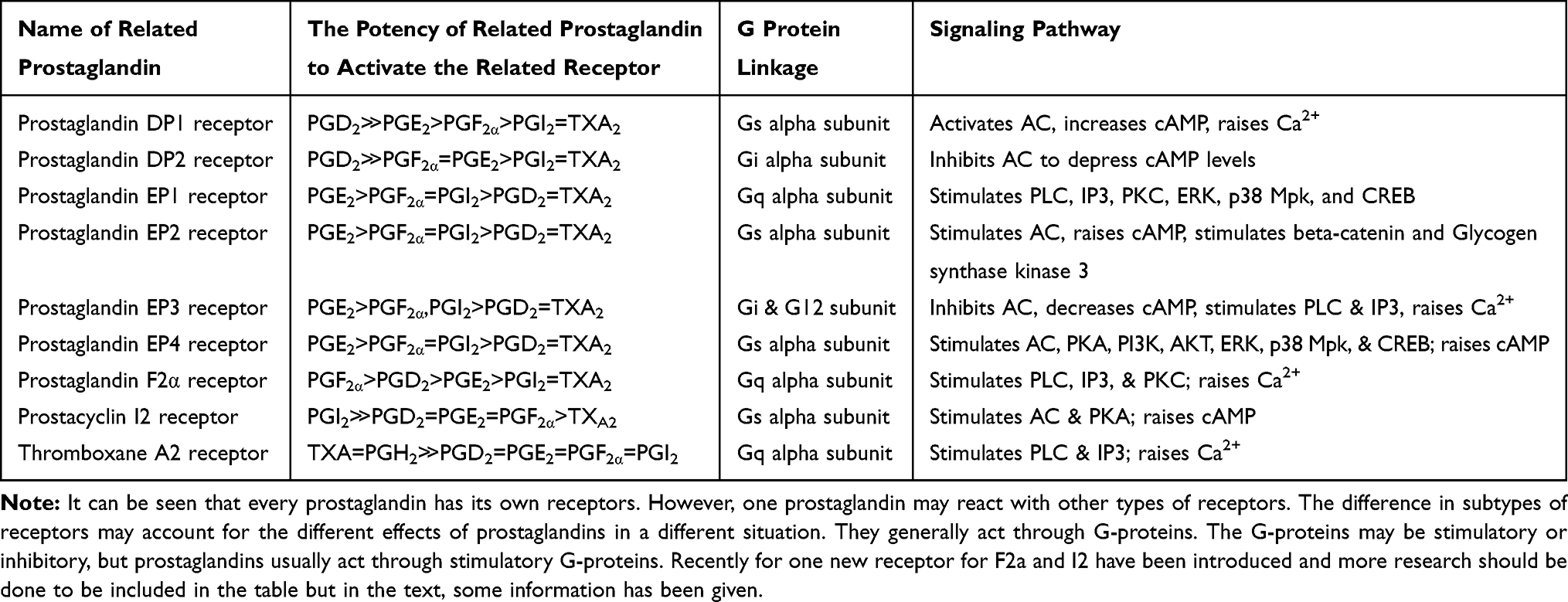 |
Table 1 Different Prostaglandin Receptors and Their Potencies for a Specific Type |
Prostaglandins and Cytokines
Cytokines are small protein molecules released by different inflammatory cell types. Based on the cell type that secretes cytokines, cytokines can be assigned different names such as lymphokines that secrets from lymphocytes and monokines that release from monocytes. Also, they have other names based on their specific activities such as chemokines (chemotactic activities) and interleukins (cytokines that interact with other leukocytes). Cytokines can act as autocrine, paracrine and endocrine. They can mediate inflammatory or anti-inflammatory processes and therefore that can be harmful or not harmful.16 Different cell types produce cytokines but mainly T cells and macrophages secrete cytokines. Pieces of evidence support the role of prostaglandins such as PGEs in the production of certain cytokines.17 The produced cytokines can be inflammatory or anti-inflammatory. However, most studies support the theory that prostaglandins promote cytokine production and the produced cytokines are harmful to neurons.18 In other diseases such as diabetes19 and atopic dermatitis20 also it has been documented the inflammatory role of prostaglandins and increased production of cytokines. In a recent article, the role of cytokines in the production of chronic inflammation has been well discussed and these shreds of evidence support the role of prostaglandins in the pathophysiology of neurodegenerative diseases.21 However other studies support the fact that prostaglandins via production of cytokines may be inflammatory or anti-inflammatory agents.22
Prostaglandins in Inflammation
Prostaglandins are considered as the agents that most commonly secreted in inflammation states. Here, we are going to explain the possible role of PGs in neuroinflammation that eventually leads to neurodegeneration.23 However, it is noteworthy to remember that prostaglandins are not always considered as agents that may cause neurodegeneration and will be discussed later. Gilas and neurons are in close talk with each other. Astrocyte development is associated with the development of synaptic structures. Enough level of neuroplasticity can be achieved by the proper function of astrocytes.24 Also in later stages of life astrocytes play an important role in neurotransmitter metabolism.25 Another important type of glial cell is microglia cells that can act in two different ways. They can be destructive to neurons and also they can help to repair neurons. In this sense activation of microglial cells after neuronal destruction can alleviate the severity of disease.26 Prostaglandins as the agents that are mostly produced in inflammation state, can disturb the neuro-glial interaction and eventually disturb the neuronal network. Also, they can impair the microglial function in later stages of the disease.27 The lack of studies about this subject prevents us from further discussion about this issue. However, in different studies, it has been shown that prostaglandins are the cause of inflammation. This is important because prostaglandins are the cause of many abnormalities independent of any mediators. Here we are going to discuss the important aspect of prostaglandins in this context.
Prostaglandin E2 and Inflammation
It is one of the most abundant prostaglandins in the body. It has diverse functions but in inflammation, it mediates the emergence of all classical signs of inflammation such as redness, swelling, and pain.28 The importance of PGE2 in the inflammation process mainly comes from mPGES-1. mPGES-1 is a member of the MAPEG (membrane-associated proteins involved in eicosanoid and glutathione metabolism) superfamily that needs glutathione as a cofactor for PGE2 production.29 In mPGES-1 deficient mice, there was a reduction in severity and incident of rheumatoid arthritis (RA).30,31 Complementary studies revealed that mPGES-1 can cause angiogenesis and formation of granulation tissue.31 Other studies about pain showed similar effects.32 PGE2 acts locally through four different receptors (EP1-EP4). EP3 and EP4 have the highest affinity for PGE2 and almost are found in all tissues and in contrast to EP1 are found in some restricted organs and EP2 is the least abundant.33 EP3 and EP4 are associated with swelling that are one of the symptoms of inflammation34 and EP2 and EP3 causes exudates formation.35 Also, EP4 deficient mice showed an attenuated response in antibody-induced RA.36 In contrast to these effects, anti-inflammatory action can be considered for this type of prostaglandins especially in allergic asthma.37 More studies revealed that this prostaglandin has both inflammatory and anti-inflammatory actions and this is because of the presence of different types of receptors and different kinds of actions of PGE2 on different cell types.38–41 During neuroinflammation also these contrasting roles can be seen.42,43
Prostaglandin I2 and Inflammation
PGI2 is one of the most important regulators of circulation.44 However, besides this important role, PGI2 also is a mediator of edema and pain that is seen in acute inflammation and also mediates extravasations of cells through vessels.45,46 Complementary studies about the PGI2 receptor confirm that this prostaglandin has a pivotal role in acute inflammation and most acute inflammation signs are associated with the emergence of this prostaglandin.47 Furthermore, PGI2 has an important role in pain perception.48 In other animal models of pain assessment, PGI2 was the origin of pain perception that is a symptom of most neurologic diseases.49,50 This prostaglandin also has a selective role through its action on CD4-Th2.51
Prostaglandin D2 and inflammation
PGD2 is found in the brain and also in peripheral tissues.52 In the central nervous system, this prostaglandin regulates pain and also causes pain.53,54 PGD2 is synthesized in the leptomeninges, choroid plexus and oligodendrocytes in the brain and secreted to cerebrospinal fluid (CSF).54 In the peripheral tissues it is mostly secreted by mast cells and other cells also produce PGD2.55,56 This prostaglandin has two types of receptor: DP1 and DP2 (CRTH2). Besides its role in inflammation especially in allergic asthma, this prostaglandin can act to inhibit inflammation.57 However, in contrast to the proinflammatory role, PGD2 may take a role to inhibit inflammation in other areas. This anti-inflammatory role has been reported through the DP1 receptor that is present in dendritic cells.58–60 PGD2 as the precursor of 15d-PGJ2, through this prostaglandin, can suppress inflammation.61–63
Prostaglandin F2α and Inflammation
This is an inflammatory prostaglandin64 that has two receptors: FPA and FPB. This prostaglandin mainly plays an important role in the female reproductive system65,66 but also has a variety of roles in other parts of the body such as brain injury and pain.67,68
Thromboxane and Inflammation
TXA2 normally derived from platelet but also another cell with a half-life of the 30s.69 It reacts mainly through TP (thromboxane/endoperoxide receptor) but also two other isoforms have been reported.70 Besides platelet aggregation and smooth muscle contraction, they have an important role in inflammation.71 TP receptor also is activated with PGH2 and HETEs.72,73 This prostaglandin has a role in inflammation because TP deficient mice will not develop full manifestation of septic shock syndrome.74,75
Prostaglandins in Neuropsychiatric Disease
Neuropsychiatric disease encompasses a wide variety of diseases. Schizophrenia, depression, bipolar disorder, anxiety and addiction to drug abuse and many others are classified as neuropsychiatric disorders. Recently it has been given much interest to find a biomarker for neuropsychiatric diseases.76 Recent studies suggest that prostaglandins can have a role in the occurrence of neuropsychiatric diseases. However because of the lack of enough studies that cover all types of neuropsychiatric disorders, here we cannot discuss all types of neuropsychiatric diseases (Table 2).
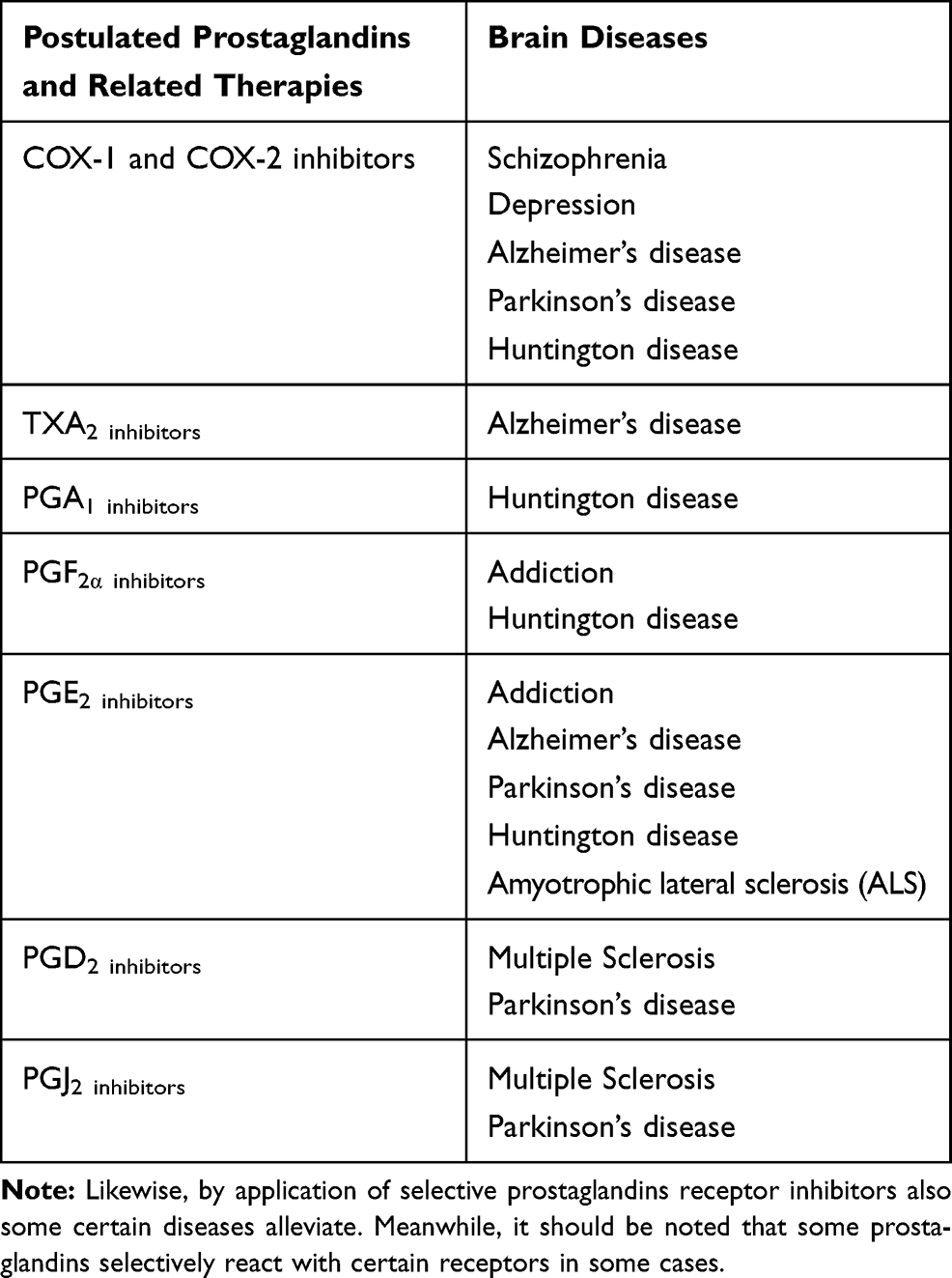 |
Table 2 Protective Roles of NSAIDs and COX Inhibitors in Brain Diseases |
Schizophrenia
Schizophrenia is a mental health illness that globally affects about 15.23/100,000 persons of all adults.77 Schizophrenia is a chronic, severe mental disorder that affects thinking, feelings and behavior. Schizophrenic patients often lose touch with reality.78 Schizophrenic patients come to the clinician with different symptoms. They may have negative or positive symptoms, or have acute symptoms and maybe with exacerbation of preexistent disease.79,80 The evidence that supports prostaglandins plays an important role in schizophrenia comes back to the 70s and 80s. Some studies proposed that prostaglandin deficiency is present in this disease.81 In one study replacement therapy with prostaglandins diminished negative and positive symptoms in schizophrenic patients.82 However, some studies support the increased activity of prostaglandins in schizophrenic patients.83 Thus it can be considered that the reduction of prostaglandins can have a useful effect. Recent studies suggest that treatment with drugs that inhibit prostaglandins may have a palliative role for this disease. In recent years, antipsychotic drugs are administered along with anti-inflammatory such as aspirin to increase the efficacy of treatment in schizophrenic patients.84 The increased inflammatory activity in the prefrontal cortex has been associated with the severity of the disease.85
Depression
Major depressive disorder (MDD), also known simply as depression, is a mental disorder characterized by at least two weeks of low mood state that is present across most situations. It is often accompanied by low self-esteem, loss of interest in normally enjoyable activities, low energy, and pain without a clear cause.86 The lifetime prevalence of depression in the United States is about 15% to 20%.87 This disease affects women more than men.88 It is the leading risk factor for suicide.89 Recent studies suggest that prostaglandins can play a role in depression diagnosis and treatment. Overall all studies support the theory that an increase in prostaglandins and their activity are associated with depression. The existing literature is not so rich about this subject. Based on a few studies, increases in prostaglandins in salivary90 and in serum91 have been associated with this disease. Also, treatment with cyclooxygenase-2 inhibitor celecoxib had a therapeutic effect in major depression.92 However, in one study treatment with ethyl-eicosapentaenoic at a dosage of 1 g/d was effective in treating depression in patients who remained depressed despite adequate standard therapy.93
Addiction
Addiction to drug abuse is a devastating state that has severe harmful effects on all aspects of life from school, workplace city and families to many other areas.94 It is mostly considered as a reward system dysfunction.95,96 Unrestricted abuse of a drug that is rewarding in nature is considered as addiction despite the adverse effects.95 Preoccupation with good memories of the desired effects of the drugs is mostly responsible for relapse to drug abuse.97 The 12-month prevalence of drug addiction in the United States is about 15% to 61%.98 Also, alcohol addiction is another type of addiction that has a high prevalence in western countries.99 The high rates of morbidity and mortality mandate performing different experiments to understand the basic mechanisms that help to control the uncontrolled behaviors. Alcohol consumption increases the production of prostaglandins. Animal studies propose that alcohol consumption increase brain levels of PGE, and PGF.100 Blocking PG production attenuates the behavioral effects of alcohol, and it can be inferred that PGs have a role in the development of alcohol addiction.101 Chronic alcohol consumption also is associated with paradoxical regulation of PGE1 and PGE2.102 In another study overproduction of PGE1 has been associated with cocaine abuse.103 It has been proposed that PGE is mainly involved in the reinstatement of self-administration. It should be noted that prostaglandin receptors are found in the cerebral cortex, hippocampus, and midbrain.104 The reinstatement to drug abuse is mainly mediated by the synergistic effect of the cannabinoid system and arachidonic acid by-products.105 From this view, prostaglandins have an important role in drug addiction.
Prostaglandins and Neurologic Diseases
Neurological disorders are increasingly recognized as one of the most prevalent disorders with a high burden to the patients, their families, and society. Statistics showed that globally, in 2016, neurological disorders were the leading cause of DALYs (Years Lived with Disability) (276 [95% UI 247–308] million) and the second leading cause of deaths (9.0 [8.8–9.4] million). So this is very important to introduce new mechanisms that are responsible for the occurrence of these diseases and also introduce new hopes for the treatment of such disorders (Table 2).106
Multiple Sclerosis
Multiple sclerosis (MS) is considered a disabling condition that mainly involves the brain and spinal cord.107 The symptoms of the disease are different and miscellaneous such as numbness or weakness, loss of vision, pain, tremor, slurred speech, fatigue, dizziness and bowel and bladder dysfunction.108 The reaction of the immune system to myelin causes the production of autoantibody and loss of myelin which in turn causes loss of the brain connection to the rest of the body. MS has four types 1) relapsing-remitting (the most common type) 2) primary progressive MS 3) secondary progressive MS 4) progressive relapsing.109 Since the neuroinflammation plays an important role in this disease, new studies have been focused on the assessment of various neuroregulators that causes inflammation. Animal studies bring evidence that prostaglandins can interfere with the disease process. The evidence to support this theory comes from the alleviation of MS by systemic injection of 15d-PGJ2 in animal studies.110 Other evidence support that anti-inflammatory agents such as peroxisome proliferator-activated receptor (PPAR)-γ agonists, including thiazolidinediones (TZDs) and 15-deoxy-Δ 12, 14 prostaglandin J 2 (15d-PGJ 2), have been shown to be effective in the treatment of experimental autoimmune encephalomyelitis (EAE).111 So, it has been suggested that glial cells especially astrocytes have a vital role in controlling autoimmune encephalitis.112,113 15d-PGJ2 a metabolite of PGD2 suppresses the immune response and it is useful for the treatment of MS. Also, the overproduction of this prostaglandin in disease states suggests that this prostaglandin is a necessary factor that controls the progression of the disease. On the other hand, prostaglandin E has a protective role by activating oligodendrocytes that produce myelin.114 Experiments have suggested that prostaglandins are not the same in all types of MS. A more recent study showed that PGD2, PGI2 and 5-lipoxygenase pathways are suppressed in the acute phase of EAE and return to constitutive levels in the chronic phase,114 but in a relapsing-remitting MS, PGD2 does not change.115
Alzheimer’s Disease
Alzheimer’s disease is a neurodegenerative disorder that is defined as the most prevalent cause of dementia (60% to 80%).116 Sever memory loss occurs in this disease that interferes with normal daily life.117 This disease is associated with the accumulation of Amyloidal plaques and Tau tangles in the brain.118 In most cases, it begins in individuals over 65 years old. Different mechanisms have been proposed for Alzheimer’s disease. The first evidence that supports the role of inflammation derives from the reduction of the occurrence of Alzheimer’s disease in the subset of patients that take NSAIDs for rheumatoid arthritis.119 Further experiments showed the overproduction of PGE2 in a subset of patients and NSAID therapy was useful in these patients.120 Astrocyte and microglia interplay plays an important role in the pathophysiology of Alzheimer’s disease and PGE2 is present in some subset of patients. Indeed, PGE2 is a necessary factor that mediates the negative consequence of amyloid-beta and glial cell interaction. Emerging data support the benefit of the application of NSAIDs rather that cox-2 selective inhibitors in controlling Alzheimer’s disease.121 In the later course of Alzheimer’s disease, the negative effect of amyloid-beta aggregation is mediated by PGE2 that its receptor is present in microglial cells that play an important role in Alzheimer’s pathophysiology.122 Recent studies suggest that suppressing prostaglandin’s action on neuronal metabolism is associated with less neuronal injury and lower secretion of proinflammatory cytokines, TNFα, and IL-6.123,124 Also, it seems that prostaglandins may be an agent that alleviates the severity of ischemia-induced dementia.124 The adverse effect of prostaglandins in Alzheimer’s disease is mainly mediated through disturbance that occurs as the consequence of oxidative stress125 and microglia function mediates immune clearance of unwanted by-product materials.126 All these studies suggest that prostaglandins are the agents that may interfere with all the stages that may necessary for both progression and initiation of dementia. Based on these reasons, prostaglandins antagonist have used successfully for the alleviation of Alzheimer’s disease.127 Prostaglandins have been shown to cause the formation and disappearance of APP holoprotein that has a toxic effect on the proteins.128 The positive effect of this treatment is mainly mediated through the selective antagonist of the PGE2 receptor.129 Also, the PGE3 receptor antagonist has a protective role in dementia that is caused by disturbed oxidative stress that eventually leads to amyloid-beta plaque formation (Ikeda-Matsuo). PGE4 antagonist also is an effective treatment for alleviating the cognitive deficit of patients.130 Other prostaglandins such as PGD2 have related to Alzheimer’s disease131 but about PGF2α and TXA2 studies are not confirmatory.
Parkinson’s Disease
Parkinson’s disease is a long-term progressive brain disease that affects movements. The disease usually progresses over time.132 The most prevalent symptoms at the onset of the disease are shaking, rigidity, slowness of movement and difficulty in walking.133 Destruction of dopaminergic neurons in substantial nigra is necessary for the occurrence of the symptoms of the disease. Different treatments have been proposed for this disease.134 An experiment that supports inflammation has a role in this disease is derived from microglial activation. However, prostaglandins in this disease have a negative role. Dopamine is the main target for the treatment of Parkinson’s disease can be considered for prostaglandin- related treatment. A recent study suggests that prostaglandins can cause unfolding and aggregation of Parkinson’s disease associated with UCHL1.135 Also, COX-2 overproduction can cause PGE2 overexpression that results in the death of dopaminergic neurons.136 COX-2 overproduction has been confirmed in another study in this disease.137 NSAID (nonsteroidal anti-inflammatory drugs) intake has been well substantially reduced the severity of Parkinson’s disease by increasing the survival of dopaminergic neurons.138 However, in some studies, this positive effect did not observe. In different experiments, PGE has been shown that is the most putative prostaglandin that interferes with Parkinson’s disease.139 Other prostaglandins such as PGD2 and PGJ2 have been shown to interfere with normal protein metabolism but strong evidence that supports the involvement of such prostaglandins in Parkinson disease is not confirmatory.140
Huntington's Disease
Huntington’s disease is a progressive brain disease that mainly affects individuals in their thirties and forties. It manifests itself through uncontrolled movements, emotional problems, and loss of cognition.141 Many people with Huntington’s disease develop involuntary jerking or twitching movements known as chorea. Also, juvenile Huntington’s disease that is less common than adult-onset tends to progress more quickly than adult form.142 Huntington’s disease affects an estimated 3 to 7 per 100,000 people. Mutations with an autosomal dominant pattern in the HTT gene cause Huntington’s disease.143 COX-2 inhibitors overall improved the outcome of motor function in Huntington’s diseases.144–146 This positive effect was not observed in transgenic animals.147 Enhancement of prostaglandin levels such as PGE2, PGf2α by injection has a negative effect of motor function and neuronal markers and restoration of the normal level of prostaglandins by COX-2 inhibitors had a positive outcome in the treatment of Huntington’s disease.148,149 PGA1 has also suggested having a role in Huntington Disease but further experiments are needed to establish its role.150
Amyotrophic Lateral Sclerosis (ALS)
ALS is a rare neurologic disorder that is characterized by the loss of neurons that are necessary for voluntary movements.151 ALS manifests by stiff muscles, muscle twitching, and muscle weakening. In 90% to 95% of the case, no definite cause is identified.152 The patient has difficulty in all actions that movement of voluntary muscles is necessary such as walking, swallowing and finally breathing. In this disease, prostaglandin alternations also have been observed. In ALS prostaglandins are elevated and have a negative effect on neuronal function. Elevated prostaglandins especially PGE2 has been associated with an increased rate of neuronal death and disability.153 Also, another study confirms that prostaglandin production can cause the disease to progress.154 For this reason, NSAID therapy has been proposed for the alleviation of this disease.155,156
Conclusion
In this review article, the importance of prostaglandins in brain diseases was discussed. Prostaglandins in some diseases act as a protective role and in some diseases act in a negative manner. So NSAID therapy is not recommended in all patients. So, more studies should be done to understand the precise pathophysiology that is responsible for the initiation and progression of brain diseases. Overall these studies suggest prostaglandins are the agents that have an important role in brain disorders’ pathophysiology and these agents modulate the course of the diseases.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Geda YE, Roberts RO, Mielke MM, et al. Baseline neuropsychiatric symptoms and the risk of incident mild cognitive impairment: a population-based study. Am J Psychiatry. 2014;171(5):572–581. doi:10.1176/appi.ajp.2014.13060821
2. Lyketsos CG, Lopez O, Jones B, Fitzpatrick AL, Breitner J, DeKosky S. Prevalence of neuropsychiatric symptoms in dementia and mild cognitive impairment: results from the cardiovascular health study. JAMA. 2002;288(12):1475–1483. doi:10.1001/jama.288.12.1475
3. Modrego PJ, Ferrández J. Depression in patients with mild cognitive impairment increases the risk of developing dementia of Alzheimer type: a prospective cohort study. Arch Neurol. 2004;61(8):1290–1293. doi:10.1001/archneur.61.8.1290
4. Pedersen KF, Larsen JP, Tysnes O-B, Alves G. Prognosis of mild cognitive impairment in early Parkinson disease: the Norwegian ParkWest study. JAMA Neurol. 2013;70(5):580–586. doi:10.1001/jamaneurol.2013.2110
5. Dieperink E, Willenbring M, Ho SB. Neuropsychiatric symptoms associated with hepatitis C and interferon alpha: a review. Am J Psychiatry. 2000;157(6):867–876. doi:10.1176/appi.ajp.157.6.867
6. Kuehl FA, Egan RW. Prostaglandins, arachidonic acid, and inflammation. Science. 1980;210(4473):978–984. doi:10.1126/science.6254151
7. Gądek-Michalska A, Tadeusz J, Rachwalska P, Bugajski J. Cytokines, prostaglandins and nitric oxide in the regulation of stress-response systems. Pharm Rep. 2013;65(6):1655–1662. doi:10.1016/S1734-1140(13)71527-5
8. Bazinet RP, Layé S. Polyunsaturated fatty acids and their metabolites in brain function and disease. Nature Rev Neurosci. 2014;15(12):771. doi:10.1038/nrn3820
9. Rouzer CA, Marnett LJ. Cyclooxygenases: structural and functional insights. J Lipid Res. 2009;50(Supplement):S29–S34. doi:10.1194/jlr.R800042-JLR200
10. Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev. 1999;79(4):1193–1226. doi:10.1152/physrev.1999.79.4.1193
11. Meirer K, Steinhilber D, Proschak E. Inhibitors of the arachidonic acid cascade: interfering with multiple pathways. Basic Clin Pharmacol Toxicol. 2014;114(1):83–91. doi:10.1111/bcpt.2013.114.issue-1
12. Chandrasekharan N, Dai H, Roos KLT, et al. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure, and expression. Proc National Acad Sci. 2002;99(21):13926–13931. doi:10.1073/pnas.162468699
13. Griswold DE, Adams JL. Constitutive cyclooxygenase (COX‐1) and inducible cyclooxygenase (COX‐2): rationale for selective inhibition and progress to date. Med Res Rev. 1996;16(2):181–206. doi:10.1002/(ISSN)1098-1128
14. Narumiya S. Prostanoids in immunity: roles revealed by mice deficient in their receptors. Life Sci. 2003;74(2–3):391–395. doi:10.1016/j.lfs.2003.09.025
15. Tsuboi K, Sugimoto Y, Ichikawa A. Prostanoid receptor subtypes. Prostaglandins Other Lipid Mediat. 2002;68:535–556. doi:10.1016/S0090-6980(02)00054-0
16. Zhang J-M, An J. Cytokines, inflammation and pain. Int Anesthesiol Clin. 2007;45(2):27. doi:10.1097/AIA.0b013e318034194e
17. Haynes D, Whitehouse M, Vernon-Roberts B. The prostaglandin E1 analogue, misoprostol, regulates inflammatory cytokines and immune functions in vitro like the natural prostaglandins E1, E2 and E3. Immunology. 1992;76(2):251.
18. Tzeng S-F, Hsiao H-Y, Mak O-T. Prostaglandins and cyclooxygenases in glial cells during brain inflammation. Curr Drug Targets-Inflammation Allergy. 2005;4(3):335–340. doi:10.2174/1568010054022051
19. Schoenberger SD, Kim SJ, Sheng J, Rezaei KA, Lalezary M, Cherney E. Increased prostaglandin E2 (PGE2) levels in proliferative diabetic retinopathy, and correlation with VEGF and inflammatory cytokines. Invest Ophthalmol Vis Sci. 2012;53(9):5906–5911. doi:10.1167/iovs.12-10410
20. Chan SC, Kim JW, Henderson WR, Hanifin JM. Altered prostaglandin E2 regulation of cytokine production in atopic dermatitis. J Immunol. 1993;151(6):3345–3352.
21. Aoki T, Narumiya S. Prostaglandins and chronic inflammation. Trends Pharmacol Sci. 2012;33(6):304–311. doi:10.1016/j.tips.2012.02.004
22. Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31(5):986–1000. doi:10.1161/ATVBAHA.110.207449
23. Teeling J, Perry V. Systemic infection and inflammation in acute CNS injury and chronic neurodegeneration: underlying mechanisms. Neuroscience. 2009;158(3):1062–1073. doi:10.1016/j.neuroscience.2008.07.031
24. Dzyubenko E, Gottschling C, Faissner A. Neuron-glia interactions in neural plasticity: contributions of neural extracellular matrix and perineuronal nets. Neural Plast. 2016;2016.
25. Terni B, López-Murcia FJ, Llobet A. Role of neuron-glia interactions in developmental synapse elimination. Brain Res Bull. 2017;129:74–81. doi:10.1016/j.brainresbull.2016.08.017
26. Amor S, Peferoen LA, Vogel DY, et al. Inflammation in neurodegenerative diseases–an update. Immunology. 2014;142(2):151–166. doi:10.1111/imm.12233
27. Block ML, Hong J-S. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol. 2005;76(2):77–98. doi:10.1016/j.pneurobio.2005.06.004
28. Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294(5548):1871–1875. doi:10.1126/science.294.5548.1871
29. Jakobsson P-J, Thorén S, Morgenstern R, Samuelsson B. Identification of human prostaglandin E synthase: a microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc National Acad Sci. 1999;96(13):7220–7225. doi:10.1073/pnas.96.13.7220
30. Trebino CE, Stock JL, Gibbons CP, et al. Impaired inflammatory and pain responses in mice lacking an inducible prostaglandin E synthase. Proc National Acad Sci. 2003;100(15):9044–9049. doi:10.1073/pnas.1332766100
31. Kamei D, Yamakawa K, Takegoshi Y, et al. Reduced pain hypersensitivity and inflammation in mice lacking microsomal prostaglandin e synthase-1. J Biol Chem. 2004;279(32):33684–33695. doi:10.1074/jbc.M400199200
32. Brenneis C, Coste O, Schmidt R, et al. Consequences of altered eicosanoid patterns for nociceptive processing in mPGES‐1‐deficient mice. J Cell Mol Med. 2008;12(2):639–648. doi:10.1111/j.1582-4934.2007.00110.x
33. Sugimoto Y, Narumiya S. Prostaglandin E receptors. J Biol Chem. 2007;282(16):11613–11617. doi:10.1074/jbc.R600038200
34. Honda T, Segi-Nishida E, Miyachi Y, Narumiya S. Prostacyclin-IP signaling and prostaglandin E2-EP2/EP4 signaling both mediate joint inflammation in mouse collagen-induced arthritis. J Exp Med. 2006;203(2):325–335. doi:10.1084/jem.20051310
35. Yuhki K-I, Ueno A, Naraba H, et al. Prostaglandin receptors EP2, EP3, and IP mediate exudate formation in carrageenin-induced mouse pleurisy. J Pharmacol Exp Ther. 2004;311(3):1218–1224. doi:10.1124/jpet.104.071548
36. McCoy JM, Wicks JR, Audoly LP. The role of prostaglandin E2 receptors in the pathogenesis of rheumatoid arthritis. J Clin Invest. 2002;110(5):651–658. doi:10.1172/JCI0215528
37. Kunikata T, Yamane H, Segi E, et al. Suppression of allergic inflammation by the prostaglandin E receptor subtype EP3. Nat Immunol. 2005;6(5):524. doi:10.1038/ni1188
38. Egan KM, Lawson JA, Fries S, et al. COX-2-derived prostacyclin confers atheroprotection on female mice. Science. 2004;306(5703):1954–1957. doi:10.1126/science.1103333
39. Harris SG, Padilla J, Koumas L, Ray D, Phipps RP. Prostaglandins as modulators of immunity. Trends Immunol. 2002;23(3):144–150. doi:10.1016/S1471-4906(01)02154-8
40. Krause P, Bruckner M, Uermösi C, Singer E, Groettrup M, Legler DF. Prostaglandin E2 enhances T-cell proliferation by inducing the costimulatory molecules OX40L, CD70, and 4-1BBL on dendritic cells. Blood. 2009;113(11):2451–2460. doi:10.1182/blood-2008-05-157123
41. Legler DF, Krause P, Scandella E, Singer E, Groettrup M. Prostaglandin E2 is generally required for human dendritic cell migration and exerts its effect via EP2 and EP4 receptors. J Immunol. 2006;176(2):966–973. doi:10.4049/jimmunol.176.2.966
42. Noda M, Kariura Y, Pannasch U, et al. Neuroprotective role of bradykinin because of the attenuation of pro‐inflammatory cytokine release from activated microglia. J Neurochem. 2007;101(2):397–410. doi:10.1111/jnc.2007.101.issue-2
43. Brenneis C, Coste O, Altenrath K, et al. Anti-inflammatory role of microsomal prostaglandin E synthase-1 in a model of neuroinflammation. J Biol Chem. 2011;286(3):2331–2342. doi:10.1074/jbc.M110.157362
44. Liou J-Y, Shyue S-K, Tsai M-J, Chung C-L, Chu K-Y, Wu KK. Colocalization of prostacyclin synthase with prostaglandin H synthase-1 (PGHS-1) but not phorbol ester-induced PGHS-2 in cultured endothelial cells. J Biol Chem. 2000;275(20):15314–15320. doi:10.1074/jbc.275.20.15314
45. Murata T, Ushikubi F, Matsuoka T, et al. Altered pain perception and inflammatory response in mice lacking prostacyclin receptor. Nature. 1997;388(6643):678. doi:10.1038/41780
46. Berkenkopf JW, Weichman BM. Production of prostacyclin in mice following intraperitoneal injection of acetic acid, phenylbenzoquinone and zymosan: its role in the writhing response. Prostaglandins. 1988;36(5):693–709. doi:10.1016/0090-6980(88)90014-7
47. Ueno A, Naraba H, Ikeda Y, et al. Intrinsic prostacyclin contributes to exudation induced by bradykinin or carrageenin: a study on the paw edema induced in IP-receptor-deficient mice. Life Sci. 2000;66(12):PL155–PL160. doi:10.1016/S0024-3205(00)00420-3
48. Doi Y, Minami T, Nishizawa M, Mabuchi T, Mori H, Ito S. Central nociceptive role of prostacyclin (IP) receptor induced by peripheral inflammation. Neuroreport. 2002;13(1):93–96. doi:10.1097/00001756-200201210-00022
49. Bley KR, Bhattacharya A, Daniels DV, et al. RO1138452 and RO3244794: characterization of structurally distinct, potent and selective IP (prostacyclin) receptor antagonists. Br J Pharmacol. 2006;147(3):335–345. doi:10.1038/sj.bjp.0706554
50. Pulichino A-M, Rowland S, Wu T, et al. Prostacyclin antagonism reduces pain and inflammation in rodent models of hyperalgesia and chronic arthritis. J Pharmacol Exp Ther. 2006;319(3):1043–1050. doi:10.1124/jpet.106.110387
51. Jaffar Z, Wan K-S, Roberts K. A key role for prostaglandin I2 in limiting lung mucosal Th2, but not Th1, responses to inhaled allergen. J Immunol. 2002;169(10):5997–6004. doi:10.4049/jimmunol.169.10.5997
52. JOWSEY IR, THOMSON AM, FLANAGAN JU, et al. Mammalian class Sigma glutathione S-transferases: catalytic properties and tissue-specific expression of human and rat GSH-dependent prostaglandin D2 synthases. Biochem J. 2001;359(3):507–516. doi:10.1042/bj3590507
53. Eguchi N, Minami T, Shirafuji N, et al. Lack of tactile pain (allodynia) in lipocalin-type prostaglandin D synthase-deficient mice. Proc National Acad Sci. 1999;96(2):726–730. doi:10.1073/pnas.96.2.726
54. Urade Y, Hayaishi O. Prostaglandin D2 and sleep/wake regulation. Sleep Med Rev. 2011;15(6):411–418. doi:10.1016/j.smrv.2011.08.003
55. Michimata T, Tsuda H, Sakai M, et al. Accumulation of CRTH2-positive T-helper 2 and T-cytotoxic 2 cells at implantation sites of human decidua in a prostaglandin D2-mediated manner. Mol Hum Reprod. 2002;8(2):181–187. doi:10.1093/molehr/8.2.181
56. Lewis R, Soter N, Diamond P, Austen K, Oates J, Roberts L. Prostaglandin D2 generation after activation of rat and human mast cells with anti-IgE. J Immunol. 1982;129(4):1627–1631.
57. Kabashima K, Narumiya S. The DP receptor, allergic inflammation and asthma. Prostaglandins Leukotrienes Essential Fatty Acids. 2003;69(2–3):187–194. doi:10.1016/S0952-3278(03)00080-2
58. Hammad H, De Heer HJ, Soullie T, Hoogsteden HC, Trottein F, Lambrecht BN. Prostaglandin D2 inhibits airway dendritic cell migration and function in steady state conditions by selective activation of the D prostanoid receptor 1. J Immunol. 2003;171(8):3936–3940. doi:10.4049/jimmunol.171.8.3936
59. Angeli V, Faveeuw C, Roye O, et al. Role of the parasite-derived prostaglandin D2 in the inhibition of epidermal Langerhans cell migration during schistosomiasis infection. J Exp Med. 2001;193(10):1135–1148. doi:10.1084/jem.193.10.1135
60. Spik I, Brénuchon C, Angéli V, et al. Activation of the prostaglandin D2 receptor DP2/CRTH2 increases allergic inflammation in mouse. J Immunol. 2005;174(6):3703–3708. doi:10.4049/jimmunol.174.6.3703
61. Bell-Parikh LC, Ide T, Lawson JA, McNamara P, Reilly M, FitzGerald GA. Biosynthesis of 15-deoxy-Δ 12, 14-PGJ 2 and the ligation of PPARγ. J Clin Invest. 2003;112(6):945–955. doi:10.1172/JCI200318012
62. Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-γ is a negative regulator of macrophage activation. Nature. 1998;391(6662):79. doi:10.1038/34178
63. Straus DS, Pascual G, Li M, et al. 15-Deoxy-Δ12, 14-prostaglandin J2 inhibits multiple steps in the NF-κB signaling pathway. Proc National Acad Sci. 2000;97(9):4844–4849. doi:10.1073/pnas.97.9.4844
64. Basu S. Oxidative injury induced cyclooxygenase activation in experimental hepatotoxicity. Biochem Biophys Res Commun. 1999;254(3):764–767. doi:10.1006/bbrc.1998.9956
65. Sugimoto Y, Yamasaki A, Segi E, et al. Failure of parturition in mice lacking the prostaglandin F receptor. Science. 1997;277(5326):681–683. doi:10.1126/science.277.5326.681
66. Saito O, Guan Y, Qi Z, et al. Expression of the prostaglandin F receptor (FP) gene along the mouse genitourinary tract. Am J Physiol Renal Physiol. 2003;284:F1164–F1170. doi:10.1152/ajprenal.00441.2002
67. Kunori S, Matsumura S, Mabuchi T, et al. Involvement of prostaglandin F2α receptor in ATP-induced mechanical allodynia. Neuroscience. 2009;163(1):362–371. doi:10.1016/j.neuroscience.2009.05.069
68. Saleem S, Ahmad AS, Maruyama T, Narumiya S, Doré S. PGF 2α FP receptor contributes to brain damage following transient focal brain ischemia. Neurotox Res. 2009;15(1):62–70. doi:10.1007/s12640-009-9007-3
69. Félétou M, Verbeuren TJ, Vanhoutte PM. Endothelium‐dependent contractions in SHR: a tale of prostanoid TP and IP receptors. Br J Pharmacol. 2009;156(4):563–574. doi:10.1111/bph.2009.156.issue-4
70. Félétou M, Vanhoutte PM, Verbeuren TJ. The thromboxane/endoperoxide receptor (TP): the common villain. J Cardiovasc Pharmacol. 2010;55(4):317–332. doi:10.1097/FJC.0b013e3181d8bc8a
71. Nakahata N. Thromboxane A2: physiology/pathophysiology, cellular signal transduction and pharmacology. Pharmacol Ther. 2008;118(1):18–35.
72. Gluais P, Lonchampt M, Morrow JD, Vanhoutte PM, Feletou M. Acetylcholine‐induced endothelium‐dependent contractions in the SHR aorta: the Janus face of prostacyclin. Br J Pharmacol. 2005;146(6):834–845. doi:10.1038/sj.bjp.0706390
73. Montuschi P, Barnes PJ, Roberts LJ. Isoprostanes: markers and mediators of oxidative stress. FASEB J. 2004;18(15):1791–1800. doi:10.1096/fj.04-2330rev
74. Yamada T, Fujino T, Yuhki K-I, et al. Thromboxane A2 regulates vascular tone via its inhibitory effect on the expression of inducible nitric oxide synthase. Circulation. 2003;108(19):2381–2386. doi:10.1161/01.CIR.0000093194.21109.EC
75. Boffa -J-J, Just A, Coffman TM, Arendshorst WJ. Thromboxane receptor mediates renal vasoconstriction and contributes to acute renal failure in endotoxemic mice. J Am Society Nephrol. 2004;15(9):2358–2365. doi:10.1097/01.ASN.0000136300.72480.86
76. Quinones MP, Kaddurah-Daouk R. Metabolomics tools for identifying biomarkers for neuropsychiatric diseases. Neurobiol Dis. 2009;35(2):165–176. doi:10.1016/j.nbd.2009.02.019
77. McGrath J, Saha S, Chant D, Welham J. Schizophrenia: a concise overview of incidence, prevalence, and mortality. Epidemiol Rev. 2008;30(1):67–76. doi:10.1093/epirev/mxn001
78. Howard R, Rabins PV, Seeman MV, Jeste DV. Late-Onset tI. Late-onset schizophrenia and very-late-onset schizophrenia-like psychosis: an international consensus. Am J Psychiatry. 2000;157(2):172–178. doi:10.1176/appi.ajp.157.2.172
79. Ventura J, Nuechterlein KH, Subotnik KL, Gutkind D, Gilbert EA. Symptom dimensions in recent-onset schizophrenia and mania: a principal components analysis of the 24-item brief psychiatric rating scale. Psychiatry Res. 2000;97(2–3):129–135. doi:10.1016/S0165-1781(00)00228-6
80. Sikich L, Frazier JA, McClellan J, et al. Double-blind comparison of first-and second-generation antipsychotics in early-onset schizophrenia and schizo-affective disorder: findings from the treatment of early-onset schizophrenia spectrum disorders (TEOSS) study. Am J Psychiatry. 2008;165(11):1420–1431. doi:10.1176/appi.ajp.2008.08050756
81. Horrobin D. Schizophrenia as a prostaglandin deficiency disease. Lancet. 1977;309(8018):936–937. doi:10.1016/S0140-6736(77)92228-0
82. Puri BK, Richardson AJ. Sustained remission of positive and negative symptoms of schizophrenia following treatment with eicosapentaenoic acid. Arch Gen Psychiatry. 1998;55(2):188–189. doi:10.1001/archpsyc.55.2.188
83. Arvindakshan M, Sitasawad S, Debsikdar V, et al. Essential polyunsaturated fatty acid and lipid peroxide levels in never-medicated and medicated schizophrenia patients. Biol Psychiatry. 2003;53(1):56–64. doi:10.1016/S0006-3223(02)01443-9
84. Dean B, Gibbons A, Gogos A, Udawela M, Thomas E, Scarr E. Studies on prostaglandin-endoperoxide synthase 1: lower levels in schizophrenia and after treatment with antipsychotic drugs in conjunction with aspirin. Int J Neuropsychopharmacol. 2017;21(3):216–225. doi:10.1093/ijnp/pyx092
85. Fillman S, Cloonan N, Catts V, et al. Increased inflammatory markers identified in the dorsolateral prefrontal cortex of individuals with schizophrenia. Mol Psychiatry. 2013;18(2):206. doi:10.1038/mp.2012.110
86. Fava M, Kendler KS. Major depressive disorder. Neuron. 2000;28(2):335–341. doi:10.1016/S0896-6273(00)00112-4
87. Moussavi S, Chatterji S, Verdes E, Tandon A, Patel V, Ustun B. Depression, chronic diseases, and decrements in health: results from the World Health Surveys. Lancet. 2007;370(9590):851–858. doi:10.1016/S0140-6736(07)61415-9
88. Belle D, Doucet J. Poverty, inequality, and discrimination as sources of depression among US women. Psychol Women Q. 2003;27(2):101–113. doi:10.1111/1471-6402.00090
89. Dumais A, Lesage A, Alda M, et al. Risk factors for suicide completion in major depression: a case-control study of impulsive and aggressive behaviors in men. Am J Psychiatry. 2005;162(11):2116–2124. doi:10.1176/appi.ajp.162.11.2116
90. Ohishi K, Ueno R, Nishino S, Sakai T, Hayaishi O. Increased level of salivary prostaglandins in patients with major depression. Biol Psychiatry. 1988;23(4):326–334. doi:10.1016/0006-3223(88)90283-1
91. Lieb J, Karmali R, Horrobin D. Elevated levels of prostaglandin E2 and thromboxane B2 in depression. Prostaglandins Leukotrienes Med. 1983;10(4):361–367. doi:10.1016/0262-1746(83)90048-3
92. Müller N, Schwarz M, Dehning S, et al. The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol Psychiatry. 2006;11(7):680. doi:10.1038/sj.mp.4001805
93. Peet M, Horrobin DF. A dose-ranging study of the effects of ethyl-eicosapentaenoate in patients with ongoing depression despite apparently adequate treatment with standard drugs. Arch Gen Psychiatry. 2002;59(10):913–919. doi:10.1001/archpsyc.59.10.913
94. Effertz T, Mann K. The burden and cost of disorders of the brain in Europe with the inclusion of harmful alcohol use and nicotine addiction. Eur Neuropsychopharmacol. 2013;23(7):742–748. doi:10.1016/j.euroneuro.2012.07.010
95. Volkow N, Li T-K. The neuroscience of addiction. Nat Neurosci. 2005;8(11):1429. doi:10.1038/nn1105-1429
96. Koob GF, Le Moal M. Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology. 2001;24(2):97. doi:10.1016/S0893-133X(00)00195-0
97. Julien RM. A Primer of Drug Action: A Concise Nontechnical Guide to the Actions, Uses and side effects of psychoactive drugs, revised and updated. Holt Paperbacks; 2013.
98. Sussman S, Lisha N, Griffiths M. Prevalence of the addictions: a problem of the majority or the minority? Eval Health Prof. 2011;34(1):3–56. doi:10.1177/0163278710380124
99. Hasin DS, Stinson FS, Ogburn E, Grant BF. Prevalence, correlates, disability, and comorbidity of DSM-IV alcohol abuse and dependence in the United States: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Arch Gen Psychiatry. 2007;64(7):830–842. doi:10.1001/archpsyc.64.7.830
100. Rotrosen J, Mandio D, Segarnick D, Traficante L, Gershon S. Ethanol and prostaglandin E1: biochemical and behavioral interactions. Life Sci. 1980;26(22):1867–1876. doi:10.1016/0024-3205(80)90615-3
101. George FR, Meisch RA. Cyclooxygenase inhibitors antagonize the rate-depressant effects of ethanol on fixed-ratio responding. Alcohol. 1990;7(4):355–360. doi:10.1016/0741-8329(90)90095-T
102. Backon J. Vasopressin inhibition via combined head-out water immersion and a prostaglandin E-1 precursor in the treatment of male reproductive failure due to chronic alcohol abuse. Med Hypotheses. 1989;29(2):81–83. doi:10.1016/0306-9877(89)90066-2
103. Leishman E, Kokesh KJ, Bradshaw HB. Lipids and addiction: how sex steroids, prostaglandins, and cannabinoids interact with drugs of abuse. Ann N Y Acad Sci. 2013;1282(1):25–38. doi:10.1111/nyas.12081
104. Akaneya Y. The remarkable mechanism of prostaglandin E2 on synaptic plasticity. Gene Regul Syst Bio. 2007;1:117762500700100009. doi:10.1177/117762500700100009
105. Yamamoto T, Anggadiredja K, Hiranita T. New perspectives in the studies on endocannabinoid and cannabis: a role for the endocannabinoid-arachidonic acid pathway in drug reward and long-lasting relapse to drug taking. J Pharmacol Sci. 2004;96(4):382–388. doi:10.1254/jphs.FMJ04003X5
106. Feigin VL, Vos T. Global burden of neurological disorders: from global burden of disease estimates to actions. Neuroepidemiology. 2019;52(1–2):1–2. doi:10.1159/000495197
107. Hemmer B, Archelos JJ, Hartung H-P. New concepts in the immunopathogenesis of multiple sclerosis. Nature Rev Neurosci. 2002;3(4):291. doi:10.1038/nrn784
108. Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69(2):292–302. doi:10.1002/ana.22366
109. Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83:278–286. doi:10.1212/WNL.0000000000000560
110. Natarajan C, Bright J. Peroxisome proliferator-activated receptor-gamma agonists inhibit experimental allergic encephalomyelitis by blocking IL-12 production, IL-12 signaling and Th1 differentiation. Genes Immun. 2002;3(2):59. doi:10.1038/sj.gene.6363832
111. Storer PD, Xu J, Chavis J, Drew PD. Peroxisome proliferator-activated receptor-gamma agonists inhibit the activation of microglia and astrocytes: implications for multiple sclerosis. J Neuroimmunol. 2005;161(1–2):113–122. doi:10.1016/j.jneuroim.2004.12.015
112. Drew PD, Xu J, Storer PD, Chavis JA, Racke MK. Peroxisome proliferator-activated receptor agonist regulation of glial activation: relevance to CNS inflammatory disorders. Neurochem Int. 2006;49(2):183–189. doi:10.1016/j.neuint.2006.04.003
113. Xu J, Chavis JA, Racke MK, Drew PD. Peroxisome proliferator-activated receptor-α and retinoid X receptor agonists inhibit inflammatory responses of astrocytes. J Neuroimmunol. 2006;176(1–2):95–105. doi:10.1016/j.jneuroim.2006.04.019
114. Kihara Y, Matsushita T, Kita Y, et al. Targeted lipidomics reveals mPGES-1-PGE2 as a therapeutic target for multiple sclerosis. Proc National Acad Sci. 2009;106(51):21807–21812. doi:10.1073/pnas.0906891106
115. Ayoub SS, Wood EG, Hassan S-U, Bolton C. Cyclooxygenase expression and prostaglandin levels in central nervous system tissues during the course of chronic relapsing experimental autoimmune encephalomyelitis (EAE). Inflammation Res. 2011;60(10):919. doi:10.1007/s00011-011-0352-3
116. Barnes DE, Yaffe K. The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011;10(9):819–828. doi:10.1016/S1474-4422(11)70072-2
117. Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dementia. 2011;7(3):280–292. doi:10.1016/j.jalz.2011.03.003
118. Scheuner D, Eckman C, Jensen M, et al. Secreted amyloid β–protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med. 1996;2(8):864. doi:10.1038/nm0896-864
119. Mcgeer P, Mcgeer E, Rogers J, Sibley J. Anti-inflammatory drugs and Alzheimer disease. Lancet. 1990;335(8696):1037. doi:10.1016/0140-6736(90)91101-F
120. Chaudhry UA, Zhuang H, Crain BJ, Doré S. Elevated microsomal prostaglandin-E synthase–1 in Alzheimer’s disease. Alzheimer’s Dementia. 2008;4(1):6–13. doi:10.1016/j.jalz.2007.10.015
121. Hoozemans JJ, Veerhuis R, Janssen I, van Elk E-J, Rozemuller AJ, Eikelenboom P. The role of cyclo-oxygenase 1 and 2 activity in prostaglandin E2 secretion by cultured human adult microglia: implications for Alzheimer’s disease. Brain Res. 2002;951(2):218–226. doi:10.1016/S0006-8993(02)03164-5
122. Jin J, Shie F-S, Liu J, et al. Prostaglandin E 2 receptor subtype 2 (EP2) regulates microglial activation and associated neurotoxicity induced by aggregated α-synuclein. J Neuroinflammation. 2007;4(1):2. doi:10.1186/1742-2094-4-2
123. Keene CD, Chang R, Stephen C, et al. Protection of hippocampal neurogenesis from toll-like receptor 4-dependent innate immune activation by ablation of prostaglandin E2 receptor subtype EP1 or EP2. Am J Pathol. 2009;174(6):2300–2309. doi:10.2353/ajpath.2009.081153
124. Shimamura M, Zhou P, Casolla B, et al. Prostaglandin E2 type 1 receptors contribute to neuronal apoptosis after transient forebrain ischemia. J Cereb Blood Flow Metab. 2013;33(8):1207–1214. doi:10.1038/jcbfm.2013.69
125. Keene CD, Chang RC, Lopez-Yglesias AH, et al. Suppressed accumulation of cerebral amyloid β peptides in aged transgenic Alzheimer’s disease mice by transplantation with wild-type or prostaglandin E2 receptor subtype 2-null bone marrow. Am J Pathol. 2010;177(1):346–354. doi:10.2353/ajpath.2010.090840
126. Johansson JU, Woodling NS, Wang Q, et al. Prostaglandin signaling suppresses beneficial microglial function in Alzheimer’s disease models. J Clin Invest. 2015;125(1):350–364. doi:10.1172/JCI77487
127. Abe T, Kunz A, Shimarnura M, Zhou P, Anrather J, Iadecola C. The neuroprotective effect of prostaglandin E2 EP1 receptor inhibition has a wide therapeutic window, is sustained in time and is not sexually dimorphic. J Cereb Blood Flow Metab. 2009;29(1):66–72. doi:10.1038/jcbfm.2008.88
128. Pooler AM, Arjona AA, Lee RK, Wurtman RJ. Prostaglandin E2 regulates amyloid precursor protein expression via the EP2 receptor in cultured rat microglia. Neurosci Lett. 2004;362(2):127–130. doi:10.1016/j.neulet.2004.03.013
129. Birrell MA, Maher SA, Buckley J, et al. Selectivity profiling of the novel EP2 receptor antagonist, PF‐04418948, in functional bioassay systems: atypical affinity at the guinea pig EP2 receptor. Br J Pharmacol. 2013;168(1):129–138. doi:10.1111/j.1476-5381.2012.02088.x
130. Hoshino T, Namba T, Takehara M, et al. Improvement of cognitive function in Alzheimer’s disease model mice by genetic and pharmacological inhibition of the EP4 receptor. J Neurochem. 2012;120(5):795–805. doi:10.1111/jnc.2012.120.issue-5
131. Kanekiyo T, Ban T, Aritake K, et al. Lipocalin-type prostaglandin D synthase/β-trace is a major amyloid β-chaperone in human cerebrospinal fluid. Proc National Acad Sci. 2007;104(15):6412–6417. doi:10.1073/pnas.0701585104
132. Beitz JM. 1. Abstract 2. Introduction and epidemiology 3. Pathophysiology 4. Risk factors/diagnosis 5. Clinical Presentation 5.1. Motor. Front Biosci. 2014;6(6574.31).
133. Jankovic J. Parkinson’s disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry. 2008;79(4):368–376. doi:10.1136/jnnp.2007.131045
134. Lang AE, Obeso JA. Challenges in Parkinson’s disease: restoration of the nigrostriatal dopamine system is not enough. Lancet Neurol. 2004;3(5):309–316. doi:10.1016/S1474-4422(04)00740-9
135. Koharudin LM, Liu H, Di Maio R, Kodali RB, Graham SH, Gronenborn AM. Cyclopentenone prostaglandin-induced unfolding and aggregation of the Parkinson disease-associated UCH-L1. Proc National Acad Sci. 2010;107(15):6835–6840. doi:10.1073/pnas.1002295107
136. Teismann P, Tieu K, Choi D-K, et al. Cyclooxygenase-2 is instrumental in Parkinson’s disease neurodegeneration. Proc National Acad Sci. 2003;100(9):5473–5478. doi:10.1073/pnas.0837397100
137. Wang T, Pei Z, Zhang W, et al. MPP+-induced COX-2 activation and subsequent dopaminergic neurodegeneration. FASEB j. 2005;19(9):1134–1136. doi:10.1096/fj.04-2457fje
138. Chen H, Jacobs E, Schwarzschild MA, et al. Nonsteroidal antiinflammatory drug use and the risk for Parkinson’s disease. Ann Neurol. 2005;58(6):963–967. doi:10.1002/(ISSN)1531-8249
139. Mattammal MB, Strong R, Lakshmi VM, Chung HD, Stephenson AH. Prostaglandin H synthetase‐mediated metabolism of dopamine: implication for Parkinson’s disease. J Neurochem. 1995;64(4):1645–1654. doi:10.1046/j.1471-4159.1995.64041645.x
140. Li Z, Melandri F, Berdo I, et al. Δ12-Prostaglandin J2 inhibits the ubiquitin hydrolase UCH-L1 and elicits ubiquitin–protein aggregation without proteasome inhibition. Biochem Biophys Res Commun. 2004;319(4):1171–1180. doi:10.1016/j.bbrc.2004.05.098
141. Novak MJ, Tabrizi SJ. Huntington’s disease: clinical presentation and treatment. Int Rev Neurobiol. 2011;98:297–323.
142. Nance MA, Myers RH. Juvenile onset Huntington’s disease—clinical and research perspectives. Ment Retard Dev Disabil Res Rev. 2001;7(3):153–157. doi:10.1002/mrdd.1022
143. Pringsheim T, Wiltshire K, Day L, Dykeman J, Steeves T, Jette N. The incidence and prevalence of Huntington’s disease: a systematic review and meta‐analysis. Mov Disorders. 2012;27(9):1083–1091. doi:10.1002/mds.25075
144. Kalonia H, Kumar P, Kumar A, Nehru B. Protective effect of rofecoxib and nimesulide against intra-striatal quinolinic acid-induced behavioral, oxidative stress and mitochondrial dysfunctions in rats. Neurotoxicology. 2010;31(2):195–203. doi:10.1016/j.neuro.2009.12.008
145. Kumar P, Padi S, Naidu P, Kumar A. Cyclooxygenase inhibition attenuates 3‐nitropropionic acid‐induced neurotoxicity in rats: possible antioxidant mechanisms. Fundam Clin Pharmacol. 2007;21(3):297–306. doi:10.1111/fcp.2007.21.issue-3
146. Trottier Y, Lutz Y, Stevanin G, et al. Polyglutamine expansion as a pathological epitope in Huntington’s disease and four dominant cerebellar ataxias. Nature. 1995;378(6555):403. doi:10.1038/378403a0
147. Norflus F, Nanje A, Gutekunst C-A, et al. Anti-inflammatory treatment with acetylsalicylate or rofecoxib is not neuroprotective in Huntington’s disease transgenic mice. Neurobiol Dis. 2004;17(2):319–325. doi:10.1016/j.nbd.2004.07.011
148. Kumar P, Kalonia H, Kumar A. Role of LOX/COX pathways in 3‐nitropropionic acid‐induced Huntington’s Disease‐like symptoms in rats: protective effect of licofelone. Br J Pharmacol. 2011;164(2b):644–654. doi:10.1111/j.1476-5381.2011.01418.x
149. Anglada-Huguet M, Xifro X, Giralt A, Zamora-Moratalla A, Martin ED, Alberch J. Prostaglandin E2 EP1 receptor antagonist improves motor deficits and rescues memory decline in R6/1 mouse model of Huntington’s disease. Mol Neurobiol. 2014;49(2):784–795. doi:10.1007/s12035-013-8556-x
150. Qin Z-H, Wang Y, Chen R-W, et al. Prostaglandin A1 protects striatal neurons against excitotoxic injury in rat striatum. J Pharmacol Exp Ther. 2001;297(1):78–87.
151. Zarei S, Carr K, Reiley L, et al. A comprehensive review of amyotrophic lateral sclerosis. Surg Neurol Int. 2015;6.
152. Andersen PM, Al-Chalabi A. Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nature Rev Neurol. 2011;7(11):603. doi:10.1038/nrneurol.2011.150
153. Almer G, Teismann P, Stevic Z, et al. Increased levels of the pro-inflammatory prostaglandin PGE2 in CSF from ALS patients. Neurology. 2002;58(8):1277–1279. doi:10.1212/WNL.58.8.1277
154. Liang X, Wang Q, Shi J, et al. The prostaglandin E2 EP2 receptor accelerates disease progression and inflammation in a model of amyotrophic lateral sclerosis. Ann Neurol. 2008;64(3):304–314. doi:10.1002/ana.21437
155. Pompl P, Ho L, Bianchi M, McManus T, Qin W, Pasinetti G. A therapeutic role for cyclooxygenase-2 inhibitors in a transgenic mouse model of amyotrophic lateral sclerosis. FASEB J. 2003;17(6):725–727. doi:10.1096/fj.02-0876fje
156. Kiaei M, Kipiani K, Petri S, et al. Integrative role of cPLA2 with COX‐2 and the effect of non‐steriodal anti‐inflammatory drugs in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurochem. 2005;93(2):403–411. doi:10.1111/j.1471-4159.2005.03024.x
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.