Back to Journals » Breast Cancer: Targets and Therapy » Volume 15
Promoter Methylation-Regulated Differentially Expressed Genes in Breast Cancer
Authors Sindi S, Hamdi N, Hassan S, Ganash M ![]() , Alharbi M, Alburae N, Azhari S, Alkhayyat S, Linjawi A, Alkhatabi H, Elaimi A
, Alharbi M, Alburae N, Azhari S, Alkhayyat S, Linjawi A, Alkhatabi H, Elaimi A ![]() , Alrefaei G, Alsubhi N, Alrafiah A
, Alrefaei G, Alsubhi N, Alrafiah A ![]() , Alhazmi S
, Alhazmi S ![]()
Received 9 March 2023
Accepted for publication 21 June 2023
Published 6 July 2023 Volume 2023:15 Pages 435—450
DOI https://doi.org/10.2147/BCTT.S408711
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Pranela Rameshwar
Samar Sindi,1,* Norah Hamdi,1,2,* Sabah Hassan,1,3 Magdah Ganash,1 Mona Alharbi,1 Najla Alburae,1 Sheren Azhari,1 Shadi Alkhayyat,4 Ayman Linjawi,5 Heba Alkhatabi,6,7 Aisha Elaimi,7,8 Ghadeer Alrefaei,9 Nouf Alsubhi,10 Aziza Alrafiah,7 Safiah Alhazmi1,3
1Department of Biological Sciences, King Abdulaziz University, Jeddah, Saudi Arabia; 2Department of Biology, King Khalid University, Abha, Saudi Arabia; 3Princess Dr. Najla Bint Saud Al-Saud Center for Excellence Research in Biotechnology, King Abdulaziz University, Jeddah, Saudi Arabia; 4Department of Internal Medicine, King Abdulaziz University, Jeddah, Saudi Arabia; 5Medical Reference Clinics, Jeddah, Saudi Arabia; 6Hematology Research Unit (HRU), King Fahad Research Center, King Abdulaziz University, Jeddah, Saudi Arabia; 7Department of Medical Laboratory Science, King Abdulaziz University, Jeddah, Saudi Arabia; 8Centre of Excellence in Genomic Medicine Research, King Abdulaziz University, Jeddah, Saudi Arabia; 9Department of Biology, University of Jeddah, Jeddah, Saudi Arabia; 10Biological Sciences Department, College of Science & Arts, King Abdulaziz University, Rabigh, Saudi Arabia
*These authors contributed equally to this work
Correspondence: Aziza Alrafiah, Tel +966‐126401000 (ext. 23495), Fax +966‐126401000 (Ext. 21686), Email [email protected]
Background: Breast cancer is one of the most common malignancies among women. Recent studies revealed that differentially methylated regions (DMRs) are implicated in regulating gene expression. The goal of this research was to determine which genes and pathways are dysregulated in breast cancer when their promoters are methylated in an abnormal way, leading to differential expression. Whole-genome bisulfite sequencing was applied to analyze DMRs for eight peripheral blood samples collected from five Saudi females diagnosed with stages I and II of breast cancer aligned with three normal females. Three of those patients and three normal samples were used to determine differentially expressed genes (DEG) using Illumina platform NovaSeq PE150.
Results: Based on ontology (GO) and KEGG pathways, the analysis indicated that DMGs and DEG are closely related to associated processes, such as ubiquitin-protein transferase activity, ubiquitin-mediated proteolysis, and oxidative phosphorylation. The findings indicated a potentially significant association between global hypomethylation and breast cancer in Saudi patients. Our results revealed 81 differentially promoter-methylated and expressed genes. The most significant differentially methylated and expressed genes found in gene ontology (GO) are pumilio RNA binding family member 1 (PUM1) and zinc finger AN1-type containing 2B (ZFAND2B) also known as (AIRAPL).
Conclusion: The essential outcomes of this study suggested that aberrant hypermethylation at crucial genes that have significant parts in the molecular pathways of breast cancer could be used as a potential prognostic biomarker for breast cancer.
Keywords: breast cancer, DNA methylation, biomarker, differentially methylated regions, differentially expressed gene, whole-genome bisulfite sequencing
Introduction
Breast cancer represents the most frequently diagnosed and fatal malignant neoplasm among women worldwide. In 2020, breast cancer accounted for the fifth-highest rate of cancer death.1 Current data indicates that cancer is both a hereditary and epigenetic disease. Many major aspects of tumor biology are controlled by epigenetic modifications, ranging from the growth, and spread of primary tumors to how the immune system responds throughout the neoplasm microenvironment.2 Interestingly, germline mutations are related to 10% of the hereditary type of breast cancer, while about 90% of breast cancer cases without any somatic mutations revealed the epigenetic mechanisms responsible for this alteration.3
DNA methylation is one of the most researched epigenetic changes in humans, occurring by adding a methyl group to the carbon 5-position (C5) of cytosine to form 5-methylcytosine. Essentially, DNA methyltransferases (DNMTs) are a category of enzymes that catalyze this process. These enzymes convert cytosine into 5-methylcytosine by adding a methyl group at the fifth carbon atom from the S-adenosylmethionine (SAM) molecule.4 The differentially methylated regions (DMRs) in the genomic DNA have been reported to be associated with a variety of disorders and are thought to be involved in gene expression processes. As a result, identifying DMRs is an important and vital step in unraveling the causes of illnesses.5 Moreover, the level of global DNA methylation has decreased as a tumor progresses from normal tissue to invasive cancer, but the abundance of local hypermethylated CpG islands at promoters has increased, leading to transcriptional repression of tumor-suppressive genes. Methylation events of promoters and CpG islands in DNA prevent the expression of target genes by regulating the binding of transcriptional modulators to the promoter.6 In blood, both hypomethylation and hypermethylation mechanisms have been proposed to increase breast cancer risk. Hypomethylation may lead to the activation of proto-oncogenes to become oncogenes, whereas hypermethylation in the promoter region of tumor suppressor genes leads to their inactivation.7 Furthermore, peripheral blood or serum can be used to detect DNA methylation for the early diagnosis and treatment of breast cancer. The higher methylation rate in the promoter region was found in CpG islands, which decreases the expression of the corresponding gene.8
Therefore, the current study aimed to determine novel predictive biomarkers for breast cancer through analysis of DMR in promoter regions and gene expression of the whole genome.
Methodology
Study Population
The research was carried out using eight blood samples from five breast cancer patients and three normal women from Jeddah city, Saudi Arabia, while six of them were selected for gene expression. With ages ranging from 29 to 60 years old. Participants’ blood samples were collected in the EDTA Lavender tube (Franklin Lakes, NJ, USA) from the medical clinic’s reference and the King Abdulaziz University Hospital in Jeddah. The biomedical ethics unit at King Abdulaziz University approved the study (No.349-21).
Whole Genome Bisulfite Sequencing (WBI) Using Novaseq 6000
Genomic DNA was isolated from the whole blood using the DNeasy Blood QIAamp Kit (Qiagen, Hilden, Germany) in accordance with the manufacturer’s guidelines and then stored at −80 C. The whole-genome bisulfite sequencing (WGBS; Novaseq 6000 platform, PE150 read length) was performed by WBI with 10× genome coverage and used according to the manufacturer’s instructions.
Transcriptome Sequencing
Whole genomic RNA was isolated from blood using RNA bler-blood kit (Haven, Jeddah, Saudi Arabia) according to the instructions from the manufacturer and then stored at −80 C. To purify messenger RNA using poly-T oligo-attached magnetic beads. The first strand of cDNA was then generated using random hexamer primers. The libraries were sequenced via Illumina platforms NovaSeq PE150 (12G/sample), NOVOGENE (HK) COMPANY LIMITED.
GO and KEGG Enrichment Analysis of DMR-Related Genes and DEG
Gene Ontology (http://www.geneontology.org/) is an important bioinformatics categorization system that aims to standardize the presentation of gene features across all organisms. It includes three primary sections: cellular component, molecular function, and biological process. Bioconductor (2.13) was used to perform gene ontology (GO) enrichment analysis of DMR-related genes with gene length bias-corrected.9
The Kyoto Encyclopedia of Genes and Genomes (KEGG) (http://www.genome.jp/kegg) is a database for figuring out the high-level functions and benefits of biological systems like cells, organisms, and ecosystems from molecular-level data.10 To examine the statistical enrichment of DMR-related genes in KEGG pathways, the KEGG Orthology-Based Annotation System (KOBAS), whereas clusterProfiler (3.8.1) for DEG with a corrected P-value was less than 0.05.
Whole-Genome Bisulfite Sequencing (WGBS) and mRNA Expression Analysis
The DSS software was used to detect differentially methylated regions (DMRs), while the differential expression gene (DEG) analysis was performed using the DESeq2 R package (1.20.0). Genes with P-value ≤0.05 found by DESeq2 were assigned as differentially expressed.
Results
Genome-Wide DNA Methylation Profiling
Table 1 represents the whole-genome bisulfite sequencing data for breast cancer (BC) and normal samples (N), including clean bases, clean reads, CG content, clean_Q30, bisulfite conversion rate, and mC percentages. For mC results, the most important component, the percentages for breast cancer patients were 0.74%, 0.57%, 0.93%, 1.07%, and 1.01% for BC1–BC5, respectively, while the percentages for normal samples were 1.34%, 0.62%, and 0.65% for N1-N3, respectively.
 |
Table 1 Whole-Genome Bisulfite Sequencing Data for Breast Cancer and Normal Individuals |
Differentially Methylated Regions (DMRs) in Breast Cancer and Normal Individuals
Figure 1A shows the identification of differentially methylated regions (DMRs) between breast cancer patients and normal samples. The DMRs were mostly located at introns for 258 genes as hypermethylated and 837 genes as hypomethylated (258 Hyper and 837 Hypo) followed by other regions (124 Hyper and 447 Hypo), exons (94 Hyper and 272 Hypo), and regulatory regions such as promoters (92 Hyper and 329 Hypo), TSS (11 Hyper and 27 Hypo), 5′UTRs (18 Hyper and 83 Hypo), 3′UTR (16 Hyper and 40 Hypo) and TES (5 Hyper and 5Hypo).
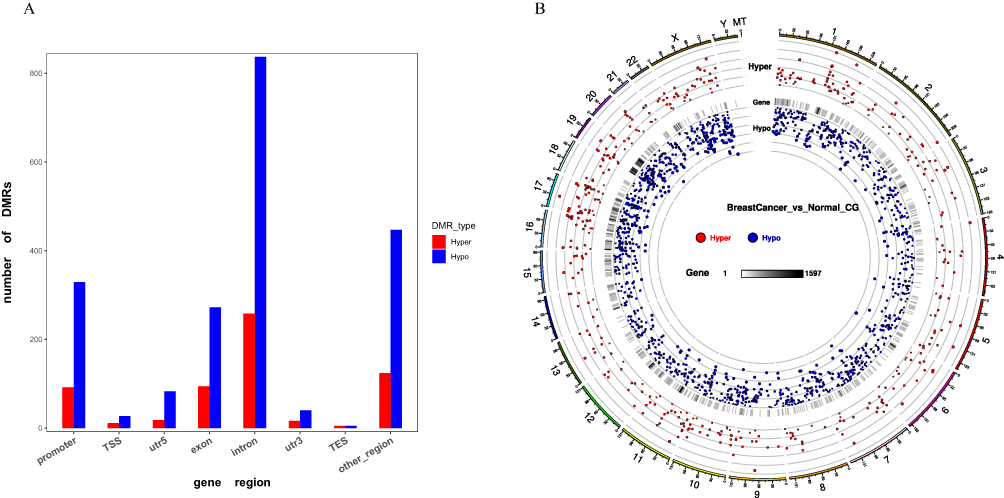 |
Figure 1 DMR distribution in breast cancer samples. (A) Bar graph represents CG context in the different functional regions. (B) Circos plot represents the distribution of Hyper/Hypo DMR methylation level. |
The Distribution of Hyper/Hypo DMR Methylation Level
There were 7406 differentially methylated regions (DMRs) discovered, with 2299 being hyper-methylated and 5107 being hypo-methylated regions. The DMR distributions and their significance to the chromosomes are shown in (Figure 1B), and the circos plot represents from outside (the significance and distribution of DMRs on the chromosome) to inside (the distribution of DMRs based on gene regions).
GO and KEGG Enrichment Analysis of DMR-Related Genes
In Table 2, GO enrichment analysis was carried out utilizing the GOseq R program. According to the significance of the p-value, the hypomethylated genes were primarily participating in the cellular component (CC) including the nucleus, nucleoplasm, and nuclear part. Regarding molecular functions (MF), these genes participate in nucleic acid binding, DNA binding, and heterocyclic compound binding. Moreover, in the biological process (BP), the enrichment was primarily participating in ribosomal small subunit export from the nucleus, and regulation of natural killer cells mediated immune response to tumor cells, indicating that hypomethylated genes may play an important function in the cell cycle. The functional enrichment of hypermethylated genes in BP includes negative regulation of neutrophil degradation and blood vessel remodeling, and CC to interferon-beta. In addition, cellular components such as trans-Golgi network membrane, intracellular component, and heterotrimeric G-protein complex were displayed. Moreover, MF enrichment revealed the presence of (R, R)-butanediol dehydrogenase, L-iditol 2-dehydrogenase, and hexitol dehydrogenase. Furthermore, Figure 2A illustrates the most enriched GO terms for breast cancer patients compared to normal individuals in CG with hypomethylation, and Figure 2B illustrates the GO terms in hyper DMR in the gene promoter.
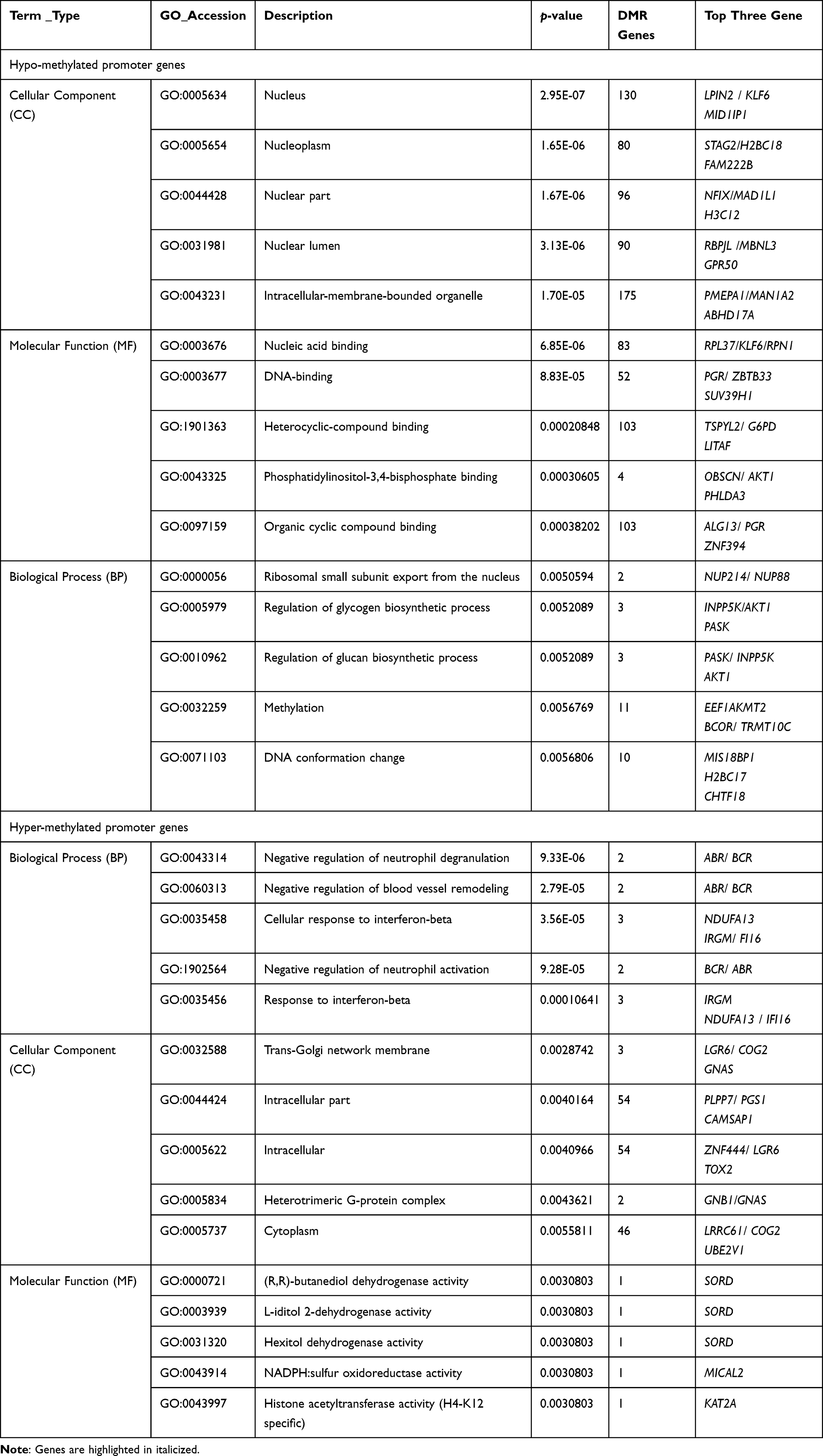 |
Table 2 Functional Enrichment Analysis of Aberrant Hypo- and Hyper-Methylated Promoter Genes in Breast Cancer |
 |
Figure 2 Continued. |
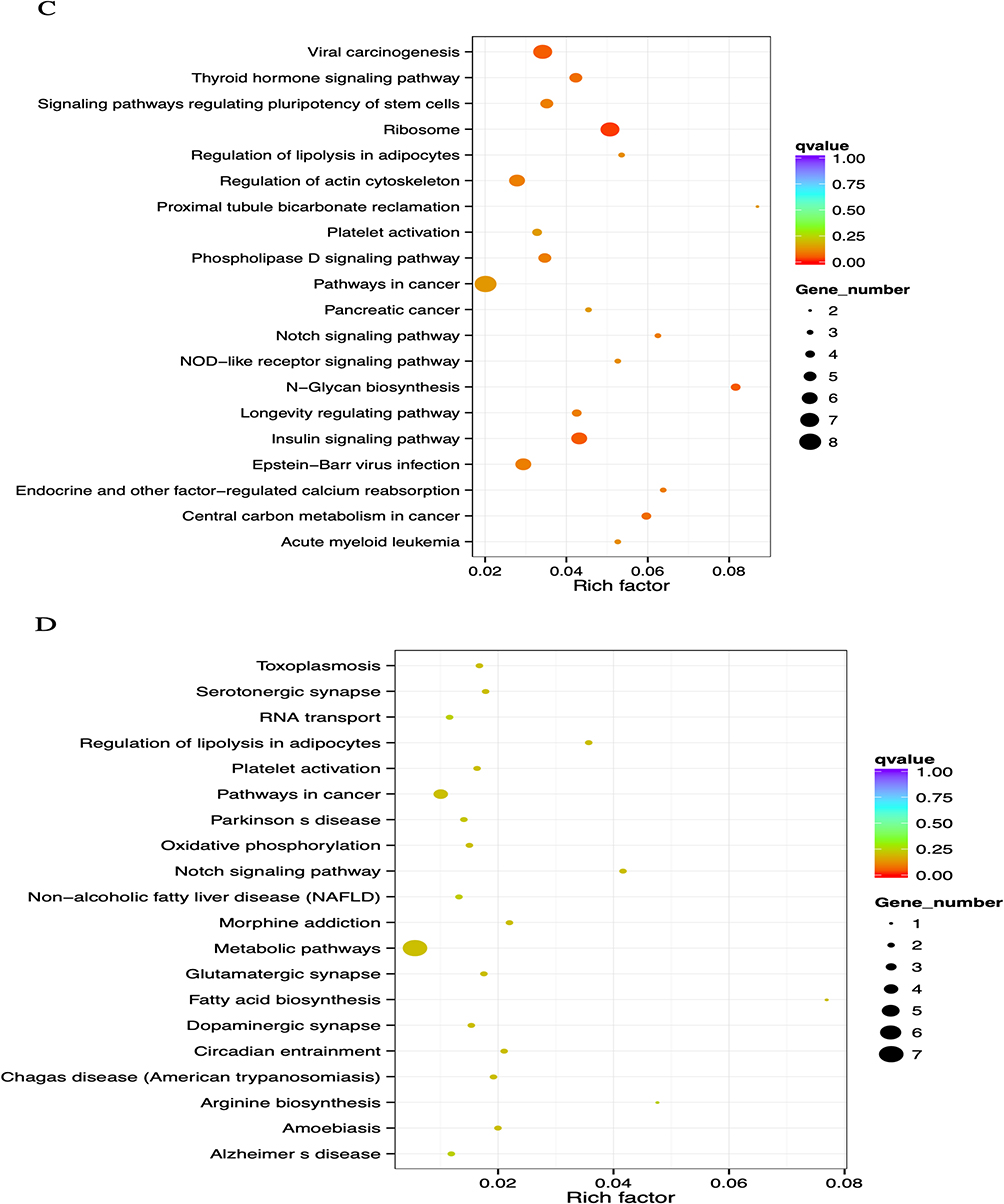 |
Figure 2 GO and KEGG enrichment analysis of DMR-related genes. (A) The Most Enriched of GO terms in CG Hyper DMR promoter genes. (B) The Most Enriched of GO terms in CG Hypo DMR promoter genes. (C) Statistics of Pathway Enrichment CG Hypo DMR promoter genes. (D) Statistics of Pathway Enrichment CG Hyper DMR promoter genes. Note: *Is a significantly enriched term. |
Regarding Table 3, analysis of enrichment of KEGG pathways revealed that the major hypomethylated genes are the genes that play roles in different pathways such as ribosome biosynthesis, N-Glycan biosynthesis, insulin signaling pathway, viral carcinogenesis, and cancer’s central carbon metabolism (Figure 2C). In addition, eight genes were engaged in cancer pathways. The notch signaling system, regulation of lipolysis in adipocytes, pathways in cancer, metabolic pathways, and morphine addiction were affected by hypermethylated genes (Figure 2D).
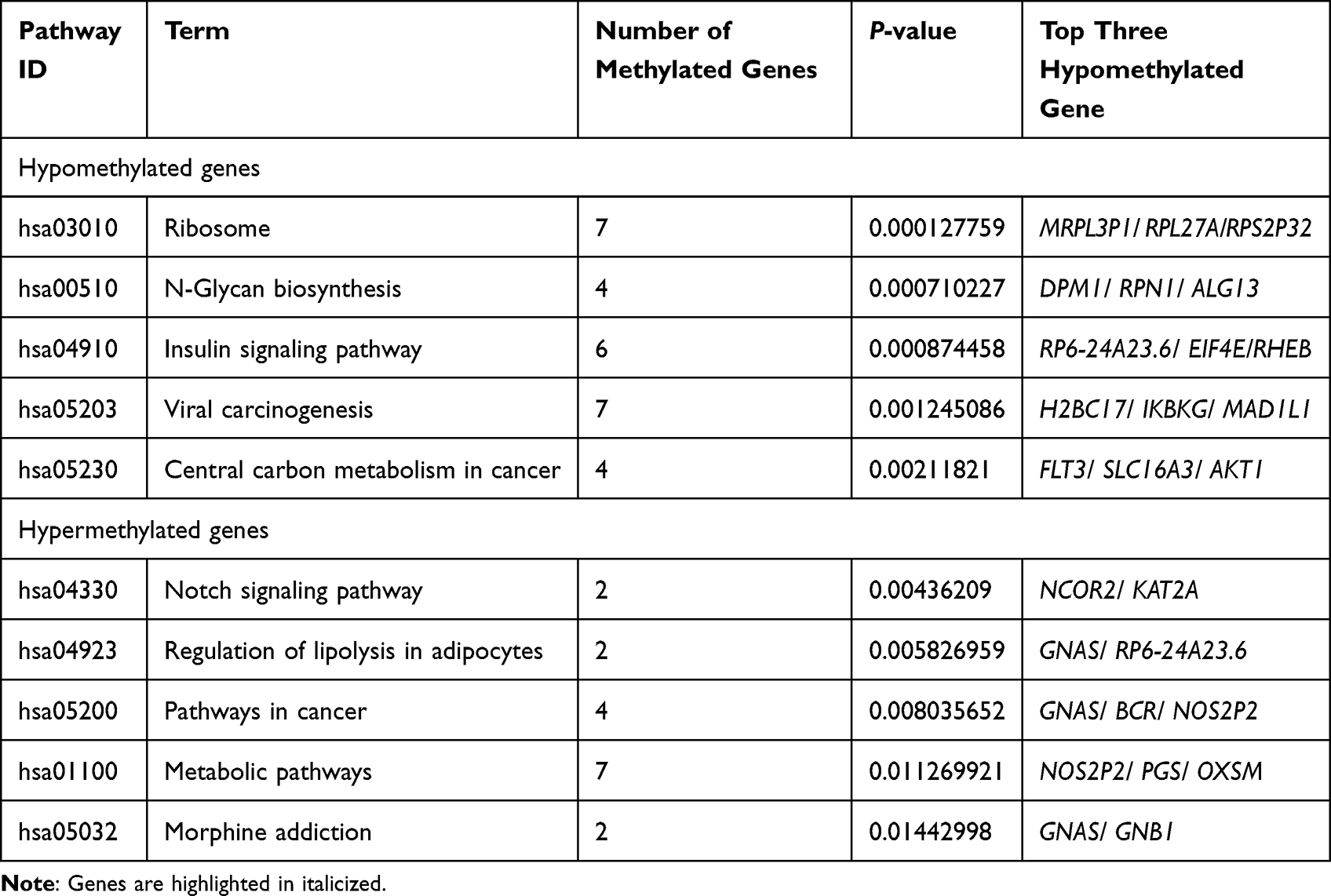 |
Table 3 KEGG Pathway Enrichment of Aberrantly Methylated Genes in Breast Cancer |
The GO and KEGG Analysis of Differentially Expressed Genes
Gene Ontology (GO) enrichment analysis of differentially expressed genes and Kyoto Encyclopedia of Genes and Genomes (KEGG) was implemented by the clusterProfiler R package, in which gene length bias was corrected. GO terms and KEGG pathways with a corrected P value of less than 0.05 were considered significantly enriched by differentially expressed genes. As shown in Table 4, GO of cellular component (CC) results revealed a high enrichment of upregulated differentially expressed genes in chromatin, clear speck, and transcription elongation factor complex, whereas GO of molecular function (MF) results showed that the upregulated DEG were enriched in the activity of DNA-dependent ATPase, ubiquitin-protein transferase, and ubiquitin-like protein transferase. Additionally, the differentially expressed genes were most enriched in the biological processes (BP) including megakaryocyte differentiation, megakaryocyte differentiation, and myeloid cell differentiation (Figure 3A). On the other hand, the downregulated DEG were considerably enriched in CC including the ribosomal subunit, large ribosomal subunit, and the cytosolic ribosome. Meanwhile, these genes were involved in MF such as the structural constituent of ribosome, electron transfer activity, and sulfurtransferase activity. Moreover, BP genes were included in SRP-dependent cotranslational protein targeting to the membrane, cotranslational protein targeting to the membrane, and viral transcription (Figure 3B). As shown in Table 5, the Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis revealed that the upregulated differentially expressed genes are highly enriched in the ubiquitin-mediated proteolysis, leukocyte transendothelial migration, and Ras signaling pathways (Figure 3C). In contrast, the down-regulated differently expressed genes are found in ribosome, oxidative phosphorylation, and Huntington’s disease as presented in (Figure 3D).
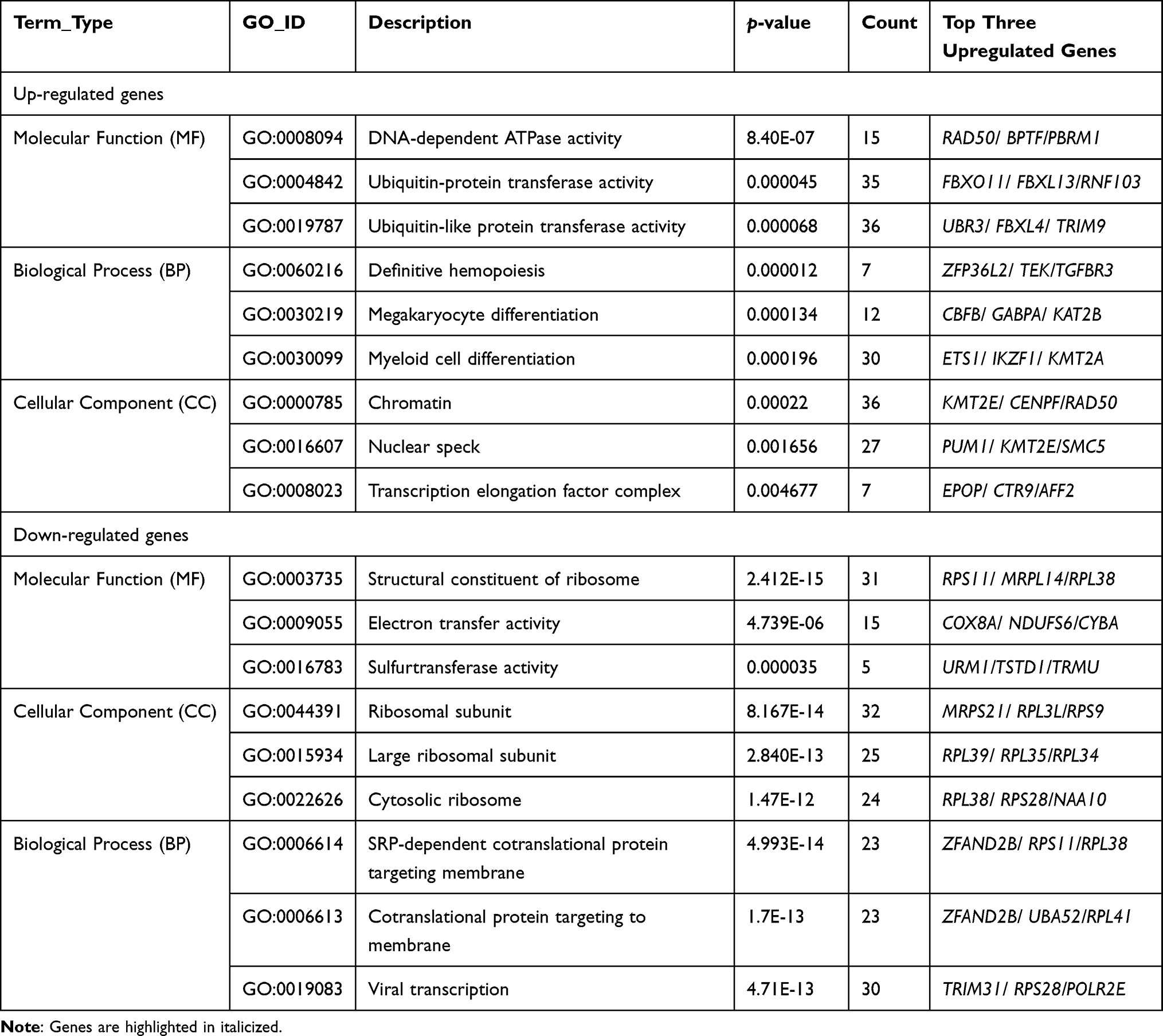 |
Table 4 Functional Enrichment Analysis of Differentially Expressed Genes |
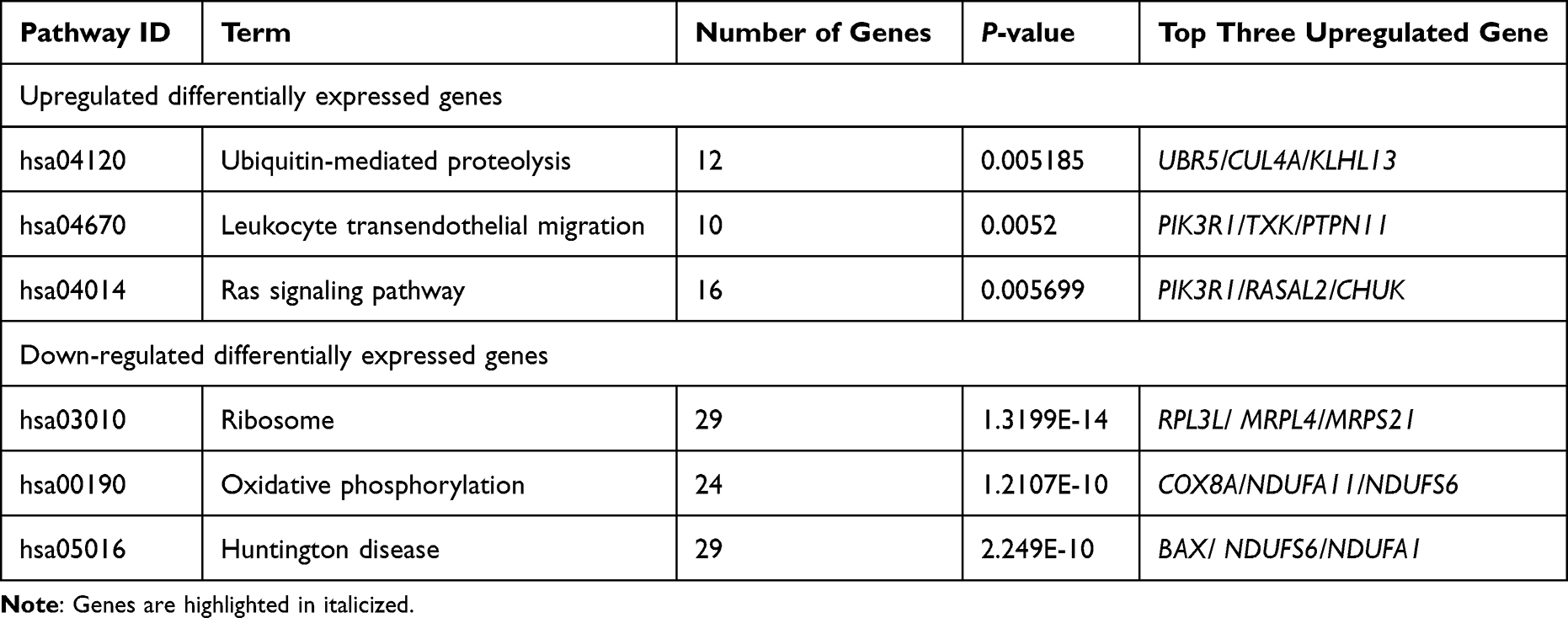 |
Table 5 Analysis of KEGG Pathways of Up/Down Regulated Differentially Expressed Genes |
 |
Figure 3 Continued. |
 |
Figure 3 GO and KEGG enrichment analysis of down and up-regulated differentially expressed genes. (A) GO terms in up-regulated differentially expressed genes. (B) GO terms in down-regulated differentially expressed genes. (C) Statistics of Pathway Enrichment in up-regulated differentially expressed genes. (D) Statistics of Pathway Enrichment in down-regulated differentially expressed genes. |
Differentially Methylated and Expression Genes
In total, 328 differentially methylated and expressed genes were found between normal and breast cancer samples. Of these 328 genes, 247 genes were found in DMR other than the promoter region, while 81 genes were related to promoter regions (Figure 4). The most significant differentially methylated and expressed genes, with a significant P-value of <0.05, which found in gene ontology (GO) are pumilio RNA binding family member 1 (PUM1) and zinc finger AN1-type containing 2B (ZFAND2B) also known as (AIRAPL). The expression level of PUM1 was significantly high in breast cancer compared to normal samples (with a P value of 9.41E-06) and the DMR level at the promoter region was low in breast cancer compared to normal samples. On the contrary, the degree of expression of AIRAPL was significantly low (hypomethylation) in breast cancer compared to normal samples (with a P value of 0.00067) and the DMR level at promoter region was high (hypermethylation) in breast cancer compared to normal samples.
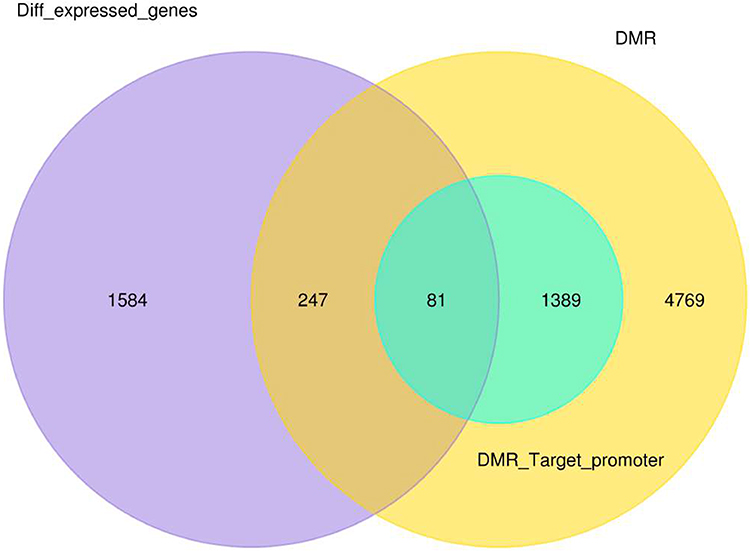 |
Figure 4 Venn diagram represents the number of differential methylated and expressed gene between normal and breast cancer samples. |
PUM1 and AIRAPL Promoter Region and Transcription Factors (TFs)
MethPrimer online software (http://www.urogene.org/methprimer/) was used to explore the possible impact of DNA methylation on CpG islands at promoter regions in regulating the expression of PUM1 and AIRAPL. Two expected CpG islands were discovered in promoter regions of both genes (Figure 5A). Moreover, gene Cards (https://www.genecards.org) were used to identify the significant TFs for both genes. Specificity protein 1 (Sp1) was shown to be one of the most significant transcription factors (TFs) of both the PUM1 and AIRAPL genes. It has been found that one TF binding site of SP1 is in the PUM1 promoter region and two of them are in the promoter and first exon of the AIRAPL gene (Figure 5B).
 |
Figure 5 PUM1 and AIRAPL promoter region and transcription factors. (A) CpG islands in PUM1 and AIRAPL. (B) SP1 binding sites in PUM1 and AIRAPL. |
Discussion
Globally, breast cancer has a significant death rate, mostly due to late diagnosis. DNA methylation patterns are connected with gene suppression or activation in numerous kinds of cancer. Emerging research reveals that aberrant DNA methylation is strongly related to the diagnosis and prognosis of breast cancer.11 Moreover, cancers in humans are characterized by widespread changes in DNA methylation patterns. The current study aimed to determine novel prognostic biomarkers for breast cancer in promoter regions. Whole-genome bisulfite sequencing was done to analyze DMRs and DEG for breast cancer-related genes.
According to the present results, 116 million clean readings per sample were acquired using filtering the WBI Novaseq 6000 raw data. About 0.86% of cytosine sites were methylated. This result is lower than a previous study which found that the rate of 5mC ranged from 1.9 to 4.3%).12 The explanation for the greater drop in methylcytosine compared to the previous study is that most of the breast cancer samples in our study were hypomethylated. These changes could be related to aberrant hypo- and hypermethylation distribution in the tumor cells of numerous (CpG) islands, which are mainly associated with gene promoters.13 Consequently, DNA hypomethylation is frequently detected at the early onset of carcinogenesis, although it may also be related to tumor development.14 In addition, DNA hypomethylation is often detected in the early stages of carcinogenesis or aberrant non-cancerous tissue.15 In a previous study, hypomethylation was suggested as a potential risk factor associated with breast cancer among women.16 Similarly, our bioinformatic findings revealed that DMRs are associated with differences in hypomethylation in different regions such as introns, promoters, and other regions, which revealed a correlation between hypomethylation and breast cancer patients in Saudi women since our patients were at an early stage (I and II). Correspondingly, Shen et al evaluated the methylation levels in the placenta in 22 pairs of chromosomes and discovered that chromosome 6 included a high number of DMRs accounting for 15% of all DMRs, while no DMR was detected on chromosome 9. Furthermore, the greatest incidence of hypermethylation and hypomethylation was seen on chromosome 12, while the lowest rate of hypomethylation was observed on chromosome 17.17 On the contrary, Zhou et al determined that chromosome 17 had the highest mean frequency of hypomethylated sites, whereas chromosome 6 had the highest frequency of hypermethylated sites.18 The present study revealed that chromosome 17 had both hypo and hypermethylation events, while chromosome 19 and chromosome X had hypomethylation events only. This suggested that environmental factors and lifestyle have been implicated in the variation in an individual’s DNA methylation patterns.
Previous research highlighted the significance of the GO enrichment analysis of differentially methylated genes and identified relevant GO keywords for cell adhesion-related biological processes, such as hemophilic cell adhesion through plasma membrane adhesion molecules. In addition, genes involved in the sister chromatid cohesion, mitotic nuclear division, chromosomal segregation, apoptotic processes, and cell cycle were hypomethylated.19,20 In this present study, hypermethylation-stimulated biological processes including negative regulation of neutrophil degradation and blood vessel remodeling were identified. Meanwhile, hypomethylation in CC, including the nucleus, nucleoplasm, nuclear part, nuclear lumen, and intracellular membrane-bounded organelle, was detected. Moreover, the most significant functional enrichment analysis (GO) was MF in up/down-regulated genes. Upregulated genes were involved in the activity of DNA-dependent ATPase, ubiquitin-protein transferase, and ubiquitin-like protein transferase, whereas downregulated genes were involved in the structural constituent of ribosome, electron transfer activity, and sulfurtransferase activity. These findings were also reported by Chen et al, who found that the upregulated differentially expressed genes were enriched in transcription factor activity, sequence-specific DNA binding, and nucleic acid binding transcription factor activity, and the downregulated differentially expressed genes were enriched in receptor binding and identical protein binding.21
DNA methylation may be variably regulated in genes that impact signaling pathways such as the cell cycle and the p53 signaling pathway. According to our results, hypomethylated genes are related to pathways that control ribosome biosynthesis, N-Glycan biosynthesis, and the insulin signaling pathway; therefore, it was assumed that glucose metabolism contributed to resistance to anti-cancer drugs and cell proliferation.20 Furthermore, hypermethylated genes contributed to the Notch signaling pathway, regulation of lipolysis in adipocytes, and pathways in cancer which are significantly affected in breast cancer.
Otherwise, the KEGG pathway analyses for upregulated genes were detected in ubiquitin-mediated proteolysis, leukocyte transendothelial migration, and Ras signaling pathway. Downregulated genes were found to be involved in the ribosome, oxidative phosphorylation, and Huntington’s disease. Despite prior results by Zhang et al, the study showed that downregulated genes enriched in cytokine–cytokine receptor interaction, calcium signaling pathway, and T cell receptor signaling pathway.22
The most striking observation to emerge from our data was identifying two coding-protein genes (PUM1 and AIRAPL) which are significantly differentially methylated and expressed genes, with a P value of <0.05, which are also found in gene ontology (GO) analysis. PUM1 is engaged in a variety of physiological processes, including the cell cycle, DNA repair, and cell renewal. It has been demonstrated that PUM1 functions as an oncogene in ovarian cancer and other types of malignancies.23 As per our findings, PUM1 was upregulated in breast cancer lineage cells and tissues.24 The encoded protein of this gene consists of eight repeats and N- and C-terminal flanking regions to form a sequence-specific RNA binding domain that acts as a post-transcriptional repressor by binding to the 3’-UTR of mRNA targets where the Pumilio Response Element (PRE), 5’-UGUANAUA-3’ leading to translational inhibition and mRNA degradation. PUM1 targets 48 genes such as DNA polymerase processivity factor proliferating cell nuclear antigen (PCNA) and Ubiquitin-conjugating enzyme E2 A (UBE2A) which are related to DNA repair and apoptosis, therefore decreasing the expression of PMUI by mediating its methylated prompter could be induced increasing its target gene expression and enhancing apoptosis and responding to anti-cancer drugs, such as cisplatin.25 Research in this area has shown that PUM1 is overexpressed in breast cancer lineage cells and tissues.24
Likewise, AIRAPL is a ubiquitin-binding protein found on the endoplasmic reticulum (ER) membranes necessary for maintaining cellular folding capacity, and its dysfunction leads to accelerated aging and protein aggregation.26 The upregulation of AIRAPL was most significant between normal and breast cancer due to lower methylation levels as hypomethylation. On the other hand, the crystal three-dimensional structure of AIRAPL protein was solved by Simin Rahighi who found that AIRAPL has three molecules; each domain comprises two ubiquitin-interacting motifs in tandem (tUIMs) and zinc motif in the medal.27 As a result, the protein encoded in this gene plays a part in maintaining the homeostasis of target proteins by controlling the translocation and proteasomal degradation of developing proteins at the endoplasmic reticulum. Additionally, it participates in the destruction of target proteins by ubiquitin-mediated proteasomes when signal-mediated translocation to the endoplasmic reticulum is unsuccessful. The zinc-finger domains are crucial in regulating the immunological response, and loss of AIRAPL function accelerates senescence and protein aggregation.26 Our findings showed that the methylation level within the promoter region of this gene was increased in breast cancer patients compared to normal individuals. Consequently, the overexpression of genes was decreased in breast cancer patients compared to the normal. In addition, Specificity protein 1 (Sp1) was shown to be one of the most significant transcription factors (TFs) on the PUM1 and AIRAPL genes. SP1 is a zinc-finger protein belonging to the SP family of transcription factors. The standard sequence of the SP1-binding site in GpC-rich promoter regions is 5’-(GGGGCGGC)-3’ where a high level of DNA methylation at that site could prevent the binding of SP1 to the target sequence resulting in the repression of gene expression. Similarly, a previous study revealed that the CpG island hypermethylation of the proximal promoter region of the FOXF2 inhibited the affinity of SP1 to its binding site resulting in the downregulation of FOXF2 expression.28 In our view, the most compelling explanation for the present set of findings is that the promoted hypomethylation on CpG islands may increase the expression of PUM1 which is contrary to the previous study, while the promoted hypermethylation on CpG islands could decrease the expression of the AIRAPL gene by controlling the affinity of SP1 to its binding site at the promoter region.
Conclusions
In the current study, a potentially significant connection between breast cancer patients and global hypomethylation has been detected. In addition, DMGs and DEG are closely related to ubiquitin-protein transferase activity, ubiquitin-mediated proteolysis, and oxidative phosphorylation. The essential outcomes of this study suggested that aberrant hypermethylation at critical genes that have significant roles in the molecular pathways of breast cancer could be used as a potential prognostic biomarker for breast cancer. The most significantly differentially methylated and expressed genes are PUM1 and AIRAPL. These findings added critical information to our understanding of the epigenetic mechanisms that have the potential to be used as breast cancer biomarkers.
Participation Approval and Ethical Approval
The investigation was conducted in accordance with the declaration of Helsinki and was approved by the King Abdulaziz University biomedical ethics unit’s ethics committee (No.349-21). The participants signed informed consent forms.
Acknowledgments
The authors wish to express gratitude to the Central Laboratory for Biological Sciences at the Science College at King Abdulaziz University for providing advice and support.
Funding
This research work was funded by Institutional Fund Projects under grant no. (IFPHI-096-247-2020). Therefore, the authors gratefully acknowledge technical and financial support from the Ministry of Education and King Abdulaziz University, DSR, Jeddah, Saudi Arabia.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Soerjomataram I, Bray F. Planning for tomorrow: global cancer incidence and the role of prevention 2020–2070. Nat Rev Clin Oncol. 2021;18(10):663–672. doi:10.1038/s41571-021-00514-z
2. Garcia-Martinez L, Zhang Y, Nakata Y, Chan HL, Morey L. Epigenetic mechanisms in breast cancer therapy and resistance. Nat Commun. 2021;12(1). doi:10.1038/s41467-021-22024-3
3. Al-Moghrabi N, Al-Qasem AJS, Aboussekhra A. Methylation-related mutations in the BRCA1 promoter in peripheral blood cells from cancer-free women. Int J Oncol. 2011;39(1):129–135. doi:10.3892/ijo.2011.1021
4. Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38(1):23–38. doi:10.1038/npp.2012.112
5. Chen DP, Lin YC, Fann CSJ. Methods for identifying differentially methylated regions for sequence- and array-based data. Brief Funct Genomics. 2016;15(6):485–490. doi:10.1093/bfgp/elw018
6. Park SM, Choi EY, Bae M, Choi JK, Kim YJ. A long-range interactive DNA methylation marker panel for the promoters of HOXA9 and HOXA10 predicts survival in breast cancer patients. Clin Epigenetics. 2017;9(1). doi:10.1186/s13148-017-0373-z
7. Wielsøe M, Tarantini L, Bollati V, Long M, Bonefeld-Jørgensen EC. DNA methylation level in blood and relations to breast cancer, risk factors and environmental exposure in Greenlandic Inuit women. Basic Clin Pharmacol Toxicol. 2020;127(4):338–350. doi:10.1111/bcpt.13424
8. Xu Z, Sandler DP, Taylor JA. Blood DNA methylation and breast cancer: a prospective case-cohort analysis in the sister study. J Natl Cancer Inst. 2020;112(1):87–94. doi:10.1093/jnci/djz065
9. Young MD, Wakefield MJ, Smyth GK, Oshlack A. Open access METHOD gene ontology analysis for RNA-Seq: accounting for selection bias GOseq GOseq Is a Method for GO analysis of RNA-Seq data that takes into account the length bias inherent in RNA-Seq. Genome Biol. 2010;11:1–2.
10. Kanehisa M, Araki M, Goto S, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36(SUPPL. 1). doi:10.1093/nar/gkm882
11. Hong MX, Ye Q, Bing ZG, et al. Identification of differentially methylated genes as diagnostic and prognostic biomarkers of breast cancer. World J Surg Oncol. 2021;19(1). doi:10.1186/s12957-021-02124-6
12. Olivia OY, Jianga P. Genome-wide detection of cytosine methylation by single molecule real-time sequencing. Proc Natl Acad Sci. 2021;118(5):2019768118. doi:10.1073/pnas.2019768118
13. Pfeifer GP. Defining driver DNA methylation changes in human cancer. Int J Mol Sci. 2018;19(4):1166. doi:10.3390/ijms19041166
14. Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics. 2009;1(2):239–259. doi:10.2217/EPI.09.33
15. Park SY, Yoo EJ, Cho NY, Kim N, Kang GH. Comparison of CpG island hypermethylation and repetitive DNA hypomethylation in premalignant stages of gastric cancer, stratified for Helicobacter pylori infection. J Pathol. 2009;219(4):410–416. doi:10.1002/path.2596
16. Choi JY, James SR, Link PA, et al. Association between global DNA hypomethylation in leukocytes and risk of breast cancer. Carcinogenesis. 2009;30(11):1889–1897. doi:10.1093/carcin/bgp143
17. Shen Z, Tang Y, Song Y, Shen W, Zou C. Differences of DNA methylation patterns in the placenta of large for gestational age infant. Medicine. 2020;99(39):e22389. doi:10.1097/MD.0000000000022389
18. Zhou Q, Chen Q, Chen X, Hao L. Bioinformatics analysis to screen DNA methylation-driven genes for prognosis of patients with bladder cancer. Transl Androl Urol. 2021;10(9):3604–3619. doi:10.21037/tau-21-326
19. Ikuno A, Akeda K, Takebayashi SI, Shimaoka M, Okumura K, Sudo A. Genome-wide analysis of DNA methylation profile identifies differentially methylated loci associated with human intervertebral disc degeneration. PLoS One. 2019;14(9):e0222188. doi:10.1371/journal.pone.0222188
20. Cai C, Wang W, Tu Z. Aberrantly DNA methylated-differentially expressed genes and pathways in hepatocellular carcinoma. J Cancer. 2019;10(2):355–366. doi:10.7150/jca.27832
21. Chen B, Tang H, Chen X, et al. Transcriptomic analyses identify key differentially expressed genes and clinical outcomes between triple-negative and non-triple-negative breast cancer. Cancer Manag Res. 2019;11:179–190. doi:10.2147/CMAR.S187151
22. Zhang R, Chang C, Jin Y, et al. Identification of DNA methylation-regulated differentially expressed genes in RA by integrated analysis of DNA methylation and RNA-Seq data. J Transl Med. 2022;20(1):481. doi:10.1186/s12967-022-03664-5
23. Yang Y, Su X, Shen K, et al. PUM1 is upregulated by DNA methylation to suppress antitumor immunity and results in poor prognosis in pancreatic cancer. Transl Cancer Res. 2021;10(5):2153–2168. doi:10.21037/tcr-20-3295
24. Silva ILZ, Kohata AA, Shigunov P. Modulation and function of Pumilio proteins in cancer. Semin Cancer Biol. 2022;86:298–309. doi:10.1016/j.semcancer.2022.03.010
25. Yamada T, Imamachi N, Imamura K, et al. Systematic analysis of targets of pumilio-mediated mRNA decay reveals that PUM1 repression by DNA damage activates translesion synthesis. Cell Rep. 2020;31(5):107542. doi:10.1016/j.celrep.2020.107542
26. Glinka T, Alter J, Braunstein I, et al. Signal-peptide-mediated translocation is regulated by a p97-AIRAPL complex. Biochem J. 2014;457(2):253–261. doi:10.1042/BJ20130710
27. Rahighi S, Braunstein I, Ternette N, et al. Selective binding of AIRAPL tandem UIMs to Lys48-linked tri-ubiquitin chains. Structure. 2016;24(3):412–422. doi:10.1016/j.str.2015.12.017
28. Tian HP, Lun SM, Huang HJ, et al. DNA methylation affects the SP1-regulated transcription of FOXF2 in breast cancer cells. J Biol Chem. 2015;290(31):19173–19183. doi:10.1074/jbc.M114.636126
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
SEMA7A as a Novel Prognostic Biomarker and Its Correlation with Immune Infiltrates in Breast Cancer
Zhang S, Kong F, Zheng L, Li X, Jia L, Yang L
International Journal of General Medicine 2024, 17:4081-4099
Published Date: 14 September 2024
Serum Direct Bilirubin as a Biomarker for Breast Cancer
Hu J, Cai Y, Chen Y, Zhu X
Breast Cancer: Targets and Therapy 2024, 16:735-743
Published Date: 7 November 2024
The Role of Actin-Binding Proteins in Breast Cancer Progression
Alfarafisa NM, Herriyanto RI, Alhaq KF, Fauzi MB, Khairani AF
Breast Cancer: Targets and Therapy 2026, 18:566309
Published Date: 19 February 2026
Clinical Relevance of USP44 Expression and DNA Methylation Status in Breast Cancer Cell Lines, Tumor Tissues, and Circulating Tumor DNA
Park KT, Lee NR, Jeong YJ
Breast Cancer: Targets and Therapy 2026, 18:599533
Published Date: 28 April 2026
The Regulatory Mechanism and Value in Early Diagnosis and Prognosis Assessment of miR-4448 in Breast Cancer
Zhang Z, Liu Z, Li C, Wang F, Tian Z, Liu X, Yu Z
Breast Cancer: Targets and Therapy 2026, 18:592003
Published Date: 21 May 2026