Back to Journals » OncoTargets and Therapy » Volume 14
Promising Molecular Targets for the Targeted Therapy of Biliary Tract Cancers: An Overview
Received 18 December 2020
Accepted for publication 26 January 2021
Published 25 February 2021 Volume 2021:14 Pages 1341—1366
DOI https://doi.org/10.2147/OTT.S297643
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sanjeev K. Srivastava
Wenwei Yang, Yongkun Sun
National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, 100021, People’s Republic of China
Correspondence: Yongkun Sun Tel +00861087788800
Email [email protected]
Abstract: Biliary tract cancer (BTC) is a leading cause of cancer-related death, due to the limited benefits of current systematic therapies and the heterogeneity of the tumor itself. High heterogeneity means that the clinical and molecular features vary between different subtypes of BTC, while the underlying molecular mechanisms remain unclear. Targeted therapy, where inhibitors are developed to selectively combine with targeted molecules in order to block abnormal signaling pathways in BTC, has shown promise as an emerging form of treatment for various types of cancer. In this article, a comprehensive review is conducted to examine potential molecular targets for BTC targeted therapy and their mechanisms. Furthermore, preliminary data published from clinical trials is utilized to analyze the main drugs used to combat BTC. The collective information presented in this article has provided useful insights into the current understanding of BTC.
Keywords: biliary tract cancer, intrahepatic cholangiocarcinoma, extrahepatic cholangiocarcinoma, gallbladder carcinoma, targeted therapy
Introduction
Biliary tract cancer (BTC), originating from the epithelium of biliary duct systems, is the second most common hepatobiliary cancer and the fifth most common malignancy of digestive system cancers. Based on anatomy, BTC is mainly classified into gallbladder cancers (GBCs), cholangiocarcinomas (CCAs) and the ampullary carcinoma (also called carcinoma of the ampulla of Vater). Cholangiocarcinomas include intrahepatic cholangiocarcinomas (iCCAs) and extrahepatic cholangiocarcinomas (eCCAs), and eCCAs can be further divided into perihilar and distal CCAs.1 Histologically, BTCs include several types: adenocarcinomas, papillary carcinoma, mucinous carcinoma and squamous cancers. Particularly, adenocarcinomas are the most common histologic types (more than 95%) with poor differentiation.1,2
These tumors are rare but malignant with a poor prognosis. The incidence of BTCs varies among different areas of the world. BTCs are relatively common in Southeast Asia and South America, with up to 96 cases per 100,000 people, while their incidence is lower in western countries such as Canada (0.5 to 1.5 cases per 100,000 people).3,4 This phenomenon can be explained by the uneven distribution of risk factors. Moreover, pathogenesis, clinical manifestation and management of different subtypes of BTCs are distinct. Based on some studies, the incidence of iCCAs is increasing, while eCCAs have been in decline.4–6
According to previous studies, a number of risk factors might be significant in biliary tract carcinogenesis. Primary sclerosing cholangitis, choledochal cysts, Caroli’s disease, cirrhosis, congenital fibropolycystic liver disease, Hepatitis B (HBV) and hepatitis C (HCV) – which could cause chronic injury of the hepatobiliary system – are prominent risk factors for BTCs.7,8 Bile duct adenomas, biliary papillomatosis and intrahepatic biliary stones are also demonstrated factors which contribute to BTCs.9 Patients with Lynch syndrome and breast cancer gene 1 (BRCA1) and breast cancer gene 2 (BRCA2) genetic aberrations might also be at higher risk for BTCs.10,11 In northern Thailand, liver fluke infestation, particularly the Opisthorcis viverrini (OV), is considered an enhanced risk of CCA.12 Some other potential contributing factors may include chemicals (eg, Thorotrast), excess alcohol, obesity and smoking.8,9
Patients with BTCs are characterized by weight loss, fever, jaundice and pain, and these tumors aggressively lead to a quick deterioration of patient performance status.13 However, in early stages, most patients with BTCs are asymptomatic with no sensitive biomarker for biliary tract tumors, so it is difficult for the disease to be assessed and treated in time. Accordingly, the global five-year survival rate is only about 10%.14
Current treatments for BTCs mainly include surgery, radiotherapy, chemotherapy, targeted therapy and immunotherapy. Surgery is the first choice for early-stage BTCs. Radical surgery with lymphadenectomy is the only potential treatment to cure localized BTCs. However, less than 35% of BTC patients are diagnosed at an early enough stage to be amenable to surgery.15 Furthermore, even when the early-stage tumors are resected, their relapse rate is very high and the rate of prolonged survival is low.16 Tumor location, pathological type, lymph node invasion and vascular invasion all affect survival after surgical resection. The 5-year overall survival rate for patients after iCCA resection ranges from 39.8% to 48.6%.17,18 Patients with localized biliary tract tumors can also be treated by radio-embolization, chemoembolization and radiotherapy, even though they are not adopted in standard treatment procedures.
Most new cases of BTC are diagnosed at an advanced stage, where the tumors are unresectable and the main treatment option is chemotherapy. Biliary tract cancer is chemotherapy responsive. For first-line treatment, the combination of gemcitabine and cisplatin (GEMCIS) is the standard of care. The superiority of GEMCIS was proved by a Phase III randomized clinical trial, ABC-02. BTC patients in the GEMCIS group had prolonged mOS (11.7 vs 8.1 months, P<0.001) and median progression-free survival (mPFS) (8.0 vs 5.0 months, P<0.001) compared to gemcitabine monotherapy with tolerant toxicity. The rate of tumor control of the GEMCIS group was 81.4%, which was higher than that of the gemcitabine monotherapy control group (71.8%) (NCT00262769).19 In another Phase II study, encouraging antitumor activity suggests gemcitabine plus capecitabine might be an alternative treatment for BTC patients - the mOS was 14 months, the mPFS was 7 months, and patients achieved a disease control rate (DCR) of 73%.20 Gemcitabine plus oxaliplatin (GEMOX) regimen was also assessed in a phase II study as first-line chemotherapy showing marginal improvement.21 Recently, active antitumor activity of oral fluoropyrimidine, S-1, plus gemcitabine (GS) was confirmed for advanced BTC in a phase II clinical trial. The one-year survival, OS, PFS and response rate (RR) were all superior in the experimental arm (S-1 plus gemcitabine) compared to the S-1 monotherapy group.22 Consequently, a phase III randomized clinical trial was conducted to assess and compare the efficacy and safety of the GS and GEMCIS regimens for BTC patients.23 Through March 2016, 354 patients were recruited. The reported mOS was 13.4 months for GEMCIS and 15.1 months for GS therapy, and median PFS also showed the superiority of the GS regimen compared with GEMCIS (6.8 vs 5.8 months). Both regimens had good safety profiles.24 Therefore, S-1 plus gemcitabine might become an emerging standard of care for advanced BTC patients who cannot be treated with platinum agents. A new combination chemotherapy regimen, GEMCIS plus nab-paclitaxel, was tested in a phase II study as first-line treatment in patients with advanced BTC. Based on the published data, nab-paclitaxel plus GEMCIS therapy achieved prolonged mPFS (11.8 months) and mOS (19.2 months) compared to data from previous studies where BTC patients were treated with GEMCIS only. To confirm these findings, a phase III trial will be carried out.25
Currently, there is no standard second-line chemotherapy for BTCs. Due to the quickly worsening performance status after first-line setting, the effectiveness of second-line treatments are limited.26 A randomised phase II study showed prolonged median overall survival (mOS) and median progression-free survival (mPFS) with well-tolerated toxicity indicated an obvious advantage for the second-line XELIRI regimen (irinotecan and capecitabine) compared with irinotecan monotherapy (NCT02558959).27 ABC-06 is a completed phase III clinical trial (NCT01926236) which aimed to determine whether patients with advanced BTC could benefit from chemotherapy (Oxaliplatin, L-folinic acid plus 5 FU) in the second-line treatment. The experimental arm (active symptom control plus chemotherapy) showed an improved mOS (6.2 months vs 5.3 months) and 12-month OS-rate (25.9% vs 11.4%) compared to the control arm (active symptom control only).28
Although chemotherapy is a mainstay of treatment for advanced BTCs, its marginal benefits and relatively severe toxicity may cause adverse effects and diminish the life quality of cancer patients. In the last ten years, targeted therapy has grown increasingly popular due to its better safety profiles and efficiency. The existence of next-generation sequencing and genetic studies shed insight on the molecular mechanism of pathogenesis and its relative molecular signaling pathways in BTCs.
Based on several studies, various genetic aberrations are considered exclusive to the anatomical location of the BTC. In iCCA, the most frequent genomic alterations are TP53 (27%), CDKN2A/B (27%), K-Ras (22%), ARID1A (18%), and IDH1/2 (19%).29,30 K-Ras (42%), TP53 (40%), CDKN2A/B (17%), and SMAD4 (21%) gene aberrations are most common in eCCA, and TP53 (59%), CDKN2A/B (19%), ARID1A (13%), and ERBB2 (16%) are the top four genomic alterations in GBC.29 Particularly, IDH1/2 and FGFR 2 fusion are almost limited to iCCA, with BAP1 gene alteration also being relatively common. On the contrary, ERBB2 and TP53 mutations are more common in eCCA and GBC than in iCCA. PRKACA or PRKACB fusion was exclusively identified in eCCA, and EGFR, ERBB3 and PTEN mutations specifically occurred in GBC.30–32 TP53 and K-Ras mutations indicated poor prognosis of the BTC.29,33
The discovery of the genetic aberrations which might drive the pathogenesis of tumors has promoted the development and application of personalized medicine. Molecular target drugs’ active efficacy has been tested for several kinds of cancers, either as monotherapy or in combination with other antitumor drugs, but a compelling targeted agent for treating BTCs with satisfactory clinical activity has not currently been found, though a large number of basic studies and clinical trials are ongoing.
In this article, we summarized the current targeted therapy of BTCs and reviewed the mechanisms and clinical trials of several promising therapeutic biomarkers which might be targetable in BTCs.
Therapeutic Targets in Biliary Tract Cancers
With targeted therapy becoming the mainstay treatment, identification of molecular alterations and the specific molecules expressed by cancer cells can guide research and treatment. The following represent the most promising targets for BTC targeted therapy.
Fibroblast Growth Factor Receptors (FGFRs)
Fibroblast growth factor receptors (FGFRs) are a family of receptor tyrosine kinases (RTKs), which carry out essential physiologic functions involving cell proliferation, differentiation, migration, and apoptosis. There are four members in the FGFR family: FGFR1, FGFR2, FGFR3 and FGFR4.34,35 The four FGFRs share a high homologous structure, containing an extracellular ligand-binding domain (D1, D2 and D3 immunoglobulin (Ig)-like subunits), a single transmembrane helix and an intracellular tyrosine kinase domain.36,37 There are over 48 receptor isoforms caused by alternative splicing of the four FGFR genes, which differ in ligand-binding and kinase domains.38 FGFRs are located in the cell membrane and can be activated by extracellular signals. The ligand-binding domains can interact with fibroblast growth factors (FGFs), which drive the homodimerization or heterodimerization to subsequently activate the kinase domain and then induce the intracellular cascades. Consequently, FGFRs achieve their physiologic functions in human body.39–41
FGFR2 fusions, the major FGFR gene abbreviations, are frequently found in iCCAs with an incidence of 10–45%, but they are rare in eCCAs (less than 5%).42,43 This phenomenon implies the different pathophysiological features between various anatomical parts in the biliary tract system. FGFR2 fusion proteins could be activated by the dimerization of their respective partners, thus inducing the activation of downstream oncogenic signaling pathways including RAS-RAF-MEK-ERK/MAPK, PI3K/AKT/mTOR and JAK/STAT pathways. Particularly, the MAPK signaling pathway is involved in increased cancer cell motility.44–46
In recent years, numerous studies have illustrated that the FGFR inhibitors play a role in suppressing the growth of biliary tract tumors in cancer patients with FGFR2 gene fusions, particularly inhibiting iCCA development. Several preclinical studies also demonstrated anti-tumor efficacy in murine models, so many FGFR inhibitors have been tested in clinical trials (Table 1).
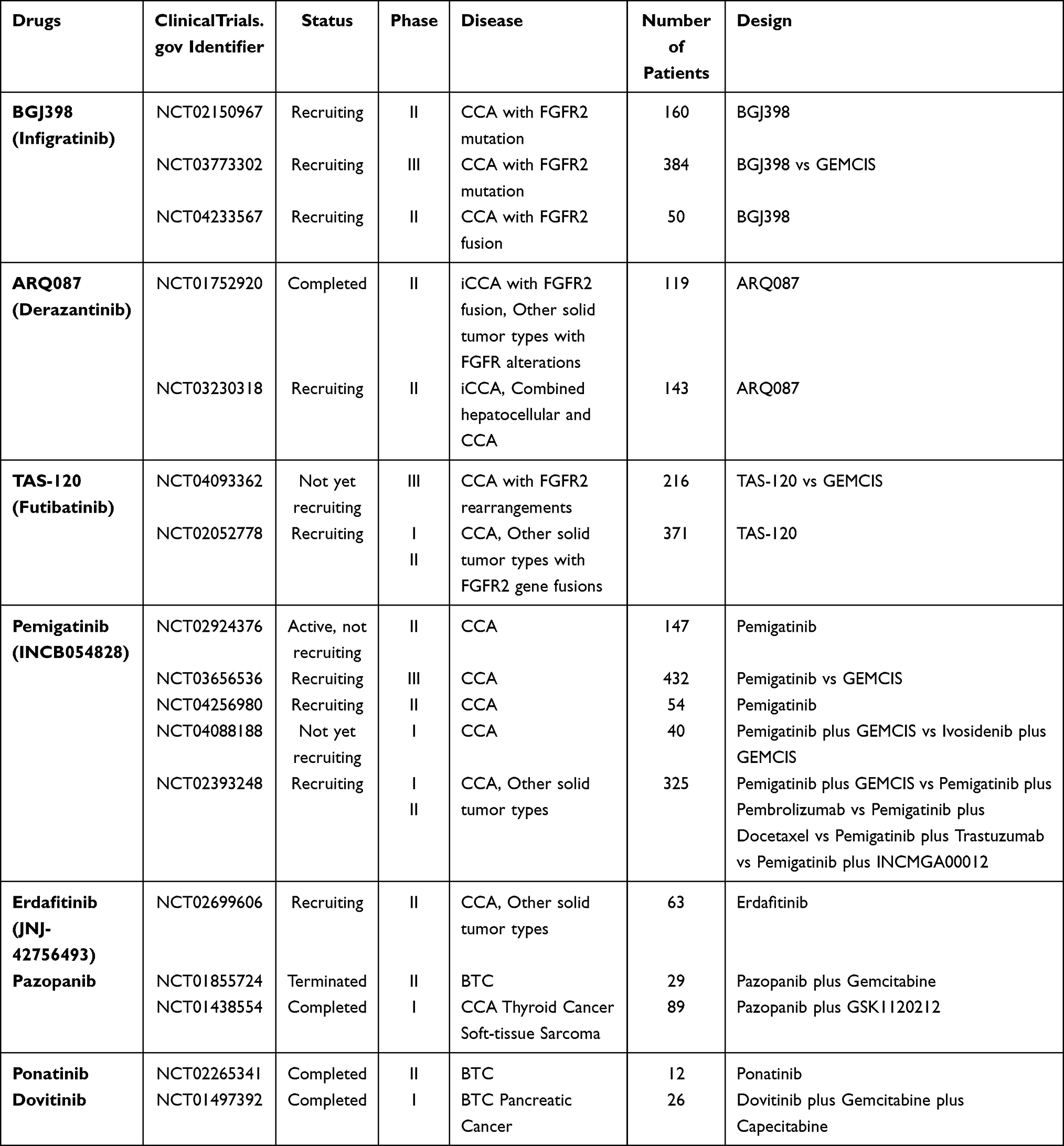 |
Table 1 Clinical Trials Involving FGFR Targeted Therapy in BTC |
So far, the FGFR inhibitors that have entered clinical trials could be divided into three groups: selective tyrosine kinase inhibitors (TKIs), non-selective tyrosine kinase inhibitors (TKIs), and monoclonal antibodies (mAbs).47
Selective TKIs
BGJ398 (Infigratinib), a selective FGFR kinase inhibitor against FGFR 1–4, exhibited effective therapeutic activity against intrahepatic cholangiocarcinoma harboring FGFR2 fusions.48,49 According to several preclinical studies, BGJ398 performed well in suppressing tumor growth in preclinical CCA models with a well-tolerated safety profile.48 A single-arm phase II clinical trial assessed the therapeutic activity of BGJ398 in 61 patients with advanced cholangiocarcinoma containing FGFR alterations, including FGFR2 fusions, mutations and amplifications (NCT02150967). The results reported an overall response rate (ORR) of 14.8% and a disease control rate (DCR) of 75.4%. The mPFS was 5.8 months, which is comparable to first-line chemotherapy. BGJ398 had promising anti-tumor activity especially in patients with FGFR2 fusions.50,51 Recently, based on this encouraging data, a phase III random controlled trial has started to recruit subjects with cholangiocarcinoma containing FGFR2 gene alterations to evaluate the efficacy and safety of BGJ398 versus chemotherapy (NCT03773302). A phase II clinical trial was also initiated to further explore the anti-tumor activity of BGJ398 in CCA patients with FGFR2 fusions (NCT04233567).
ARQ 087, also known as Derazantinib (DZB), is a multi-tyrosine kinase inhibitor targeting FGFR1 to 4.52,53 According to preclinical research, ARQ 087 has displayed prominent inhibitory effects in vivo xenograft models52,54 and in vitro CCA cell lines,54,55 which indicates its potential therapeutic efficacy. The first phase I/II clinical trial for ARQ 087 has completed (NCT01752920). In the beginning, this study recruited 80 patients with advanced solid tumors, including 12 iCCA patients. Among the 12 iCCA patients with FGFR2 fusions, 2 patients had PRs and one confirmed a stable disease (SD). This study showed the tolerant toxicity of ARQ 087 and confirmed its therapeutic effect in advanced cancer patients with FGFR gene alterations, particularly iCCA patients.53 Based on the promising preliminary data, more patients with FGFR2 gene fusion positive advanced iCCA were enrolled; they took derazantinib according to the recommended Phase 2 dose (RP2D). Mazzaferro et al reported the results: among 29 FGFR2 gene fusion-positive iCCA patients, the median PFS was 5.7 months, the ORR was 20.7%, and the DCR was 82.8%.56 This data suggests that Derazantinib might be a good drug for treating iCCA patients. Another phase II study for Derazantinib targeting FGFR2 fusion positive iCCA is ongoing (NCT03230318).
Futibatinib (TAS-120) is an irreversible and highly selective inhibitor which targets all four FGFR subtypes.57 A clinical study reported that TAS-120 showed therapeutic effects in four iCCA patients with FGFR2 fusions who were resistant to the other two FGFR inhibitors (BGJ398 and Debio1347).58 TAS-120 is under phase I/II clinical trials (NCT02052778) investigating its safety and efficacy. According to the recently published data, the disease control rate (DCR) was 75%, indicating promising clinical benefits. The toxicity of TAS-120 is also manageable.59 Another Phase 3 study will begin to assess the efficacy and safety of TAS-120 versus gemcitabine-cisplatin chemotherapy in advanced FGFR2-alteration-positive iCCA patients as first-line treatment (NCT04093362). Futibatinib is a highly selective irreversible FGFR antagonist, which means it has durable activity. Several trials have showed its meaningful benefit in patients with pretreated iCCA with FGFR2 gene aberrations, thus it might be a promising agent for BTCs treatment.60
Pemigatinib (INCB054828) is a reversible and selective inhibitor of FGFR 1, FGFR2 and FGFR3.35,61 Pemigatinib has potential in the treatment of cholangiocarcinoma. A preclinical cell-based study revealed that the cells harboring FGFR2-CLIP1 fusion responded noticeably to Pemigatinib, whereas cells with FGFR2-CLIP1 fusion and N549H mutation both were resistant to this drug.62 There is a large-scale single-arm phase 2 trial (FIGHT-202) assessing the safety and therapeutic activity of Pemigatinib in cholangiocarcinoma patients with and without FGFR2 fusions or rearrangements (NCT02924376). One hundred and seven of the enrolled 146 patients harbored FGFR2 gene fusions or rearrangements, and this group of patients showed a remarkable objective response: 35.5% (95% CI: 26.5–45.4) of patients achieved objective response (3 had complete responses and 35 had partial responses), and the disease control rate (DCR) was 82% (95% CI: 74–89). Median PFS was 6.9 months (95% CI 6.2–9.6) and median OS was 21.1 months. On the contrary, the groups of patients with other FGFR alterations or without FGFR alterations did not achieve any response.63 Based on these encouraging results, a phase 3 clinical trial (FIGHT-302; NCT03656536) is ongoing to compare pemigatinib with chemotherapy (gemcitabine plus cisplatin) for advanced CCA patients with FGFR2 rearrangements.
A host of selective TKIs for FGFR, including AZD4547, CH5183284 (Debio 1347), JNJ-42756493, BAY1163877, and dovitinib, are currently under examination in early-phase trials.
Nonselective TKIs
Apart from selective FGFR inhibitors, there are also several non-selective FGFR inhibitors entering clinical trials.
Pazopanib is a multi-kinase inhibitor mainly targeting VEGFR, PDGFR, c-Kit, FGFR, and c-Fms.64 The anti-tumor effect of pazopanib has been demonstrated in preclinical research. An in vitro study illustrated that the number of cells in gastric cancer cell lines containing FGFR2 gene amplifications would decrease significantly after being treated with pazopanib.65
A phase II multicenter trial was conducted to evaluate the therapeutic efficacy of a gemcitabine and pazopanib combination therapy in 29 advanced biliary tract carcinoma (BTC) patients. 13.8% of enrolled patients in the ITT (intent-to-treat) group and 19.1% in the per protocol (PP) population achieved complete response or partial response. The disappointing objective response rate terminated this trial and prevented more clinical trials from assessing this therapeutic regimen (NCT01855724).66 A Phase I clinical trial evaluated the clinical benefits of the combination of pazopanib with trametinib (an MEK inhibitor) for several kinds of solid tumors including Cholangiocarcinoma (NCT01438554).
Ponatinib is also defined as a multi-TKI because it can target many kinds of tyrosine kinase, such as FGFR 1 to 4, VEGFR, PDGFR, FLT3 and c-SRC.67 In a study, a patient with CCA and FGFR2-MGEA5 fusion took ponatinib, finally achieving preliminary anti-tumor activity.68 Furthermore, meaningful clinical benefits were also verified in another CCA patient with FGFR2-TACC3 fusion who took pazopanib and ponatinib.68 According to the data published on clinical trials.gov, a completed phase II trial of 12 BTC patients with FGFR fusions reported a disease control rate of 45.5% (95% CI: 16.8 to 76.6), progression-free survival (PFS) of 2.4 months and overall survival (OS) of 15.7 months (NCT02265341).
Besides the drugs mentioned above, dovitinib and lenvatinib are also non-selective TKIs entering clinical trials. However, due to the non-selective activity, these drugs may lead to severe toxicities on the cardiovascular system related to VEGFR inhibition, which limits the long-term use of non-selective FGFR inhibitors.69
mAb
In addition to TKIs, monoclonal antibodies (mAbs) are another group of FGFR inhibitors. They can target FGFR with a higher specificity than TKIs, which may result in a better safety profile for patients. However, only a few mAbs have entered clinical trials.47
Bemarituzumab (FPA144) is a humanized IgG1 monoclonal antibody specific to the FGFR2b isoform.70 The specific targeting activity of Bemarituzumab could avoid adverse events like hyperphosphatemia, which occurred in patients treated with pan-FGFR TKIs.71,72
Up to date, there are no clinical trials specifically evaluating BTC patients treated with this drug. A Phase 1 trial demonstrated that bemarituzumab targeted FGFR2b and could be safely used to treat patients with advanced solid tumors (NCT02318329).70
Many FGFR inhibitors are under evaluation, the research about the mechanisms of resistance is ongoing at the same time. On a basis of several studies, secondary mutations in FGFR2 kinase domain, mutations in the TKI domain and emergence of new FGFR2 fusions might all be the reasons for resistance.73–75 Further studies are needed to have a better understand of the mechanisms of resistance and find potential ways to overcome it.
Metabolic Pathway Linked to IDH1/2 Mutations
Isocitrate dehydrogenase (IDH), an essential enzyme for the citric acid cycle, can convert isocitrate to α-ketoglutarate (α-KG) by oxidative decarboxylation, and finally provides ATP and precursors for cellular metabolism.76 In humans, there are 3 isoforms of IDH (IDH1, IDH2, and IDH3) which contribute to regulating cellular metabolism. Several studies have indicated that mutant IDH1 (mIDH1) and mutant IDH2 (mIDH2) are “gain of function” mutations, which means that they gain the ability to catalyze the conversion of α-KG to 2-hydroxyglutarate (2-HG).77,78 The accumulation of 2-HG inhibits the αKG-dependent dioxygenases which play a part in epigenetic regulation, leading to cell proliferation, suppression of cellular differentiation, angiogenesis and invasion.79–84 Therefore, mutations in IDH 1/2 genes are highly related to tumorigenesis. IDH gene mutations are heterozygous point mutations generally occurring in Arginine 132 of IDH1 and Arginine 140 or Arginine 172 of IDH2.78,84 According to genomic profiling, IDH 1/2 mutations were more common in iCCA than in eCCA or GBC, with an incidence ranging from 10% to 36%.30,33,84–86 The occurrence of IDH1 mutation was higher than IDH2.29,87
To examine the mechanism of IDH1/2 gene mutation driving tumorigenesis, two preclinical studies were conducted. AGI-5198, as a tool compound, was proven to target IDH1-mutant glioma cells and suppress the growth of cells, but did not work in IDH1 wild-type glioma cells.88 In another study, a compound named AGI-6780 and hematopoietic cell lines were used to assess the potential utility of mIDH2 inhibitors in treating leukemias with IDH2/R140Q mutations. This study also discovered that AGI-6780 could promote the differentiation of the human IDH2/R140Q mutant hematopoietic cells.82
Although these two compounds showed encouraging effects in preclinical tests, poor pharmacokinetics of AGI-5198 and the lack of in vivo tests to assess AGI-6780 prevent their use in clinical studies.89,90 Several kinds of mIDH inhibitors with good safety and efficacy were developed and have entered clinical trials (Table 2).
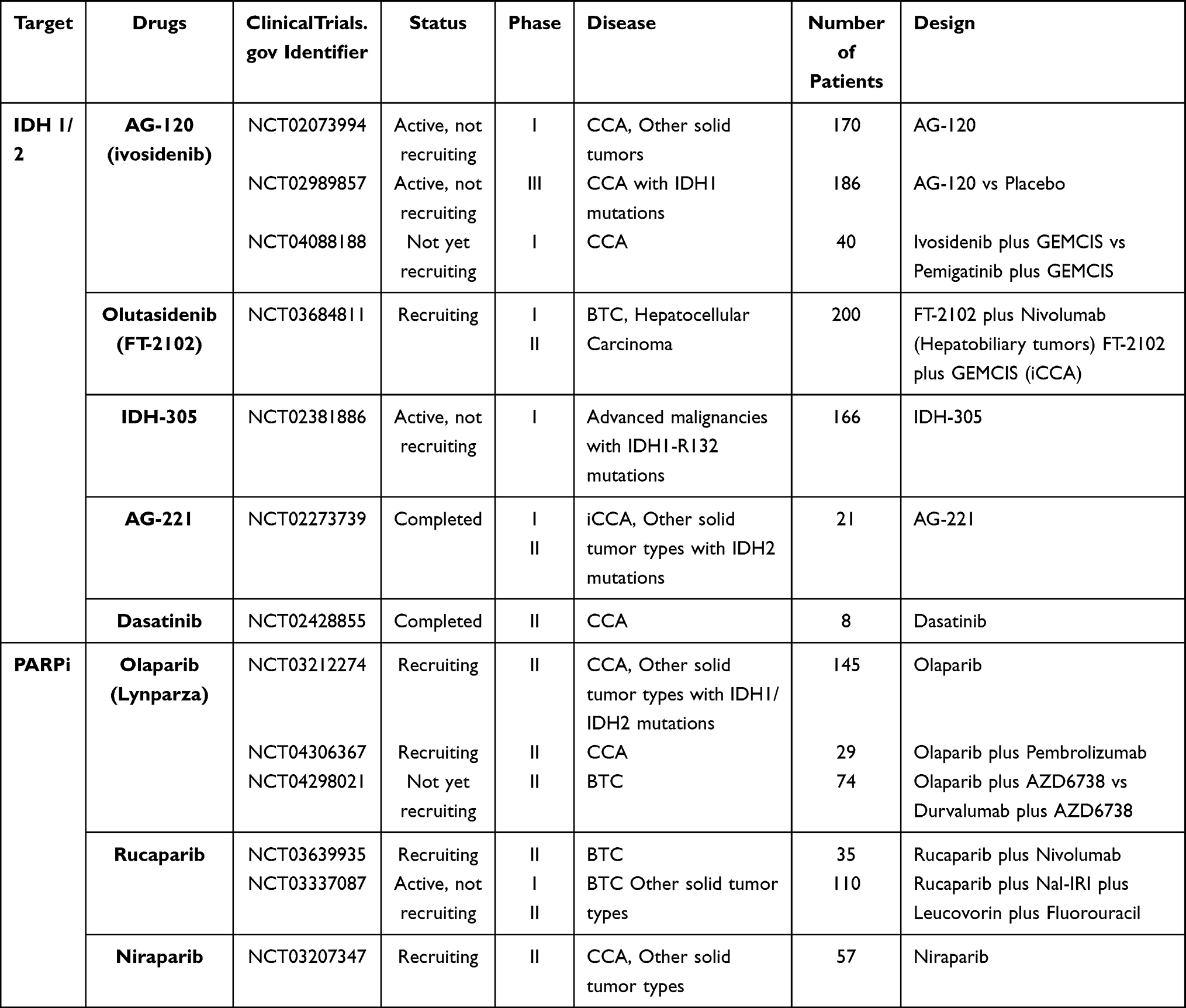 |
Table 2 Clinical Trials for BTC Targeted Therapy Targeting IDH Mutations |
mIDH1 Inhibitors
AG-120 (Ivosidenib), a highly specific inhibitor of mutant IDH1 (mIDH1) enzymes, was developed through optimizing AGI-5198 to enable it to be applied to human therapy.89,90 AG-120 was the first mIDH inhibitor studied in CCA.90 An in vitro study confirmed the ability of AG-120 to selectively decrease the 2-HG levels and restore cell differentiation in mIDH1-positive AML cells by inhibiting the mutant IDH1 enzyme.91 AG-120 also lowered 2-HG levels and showed significant mutant IDH1 enzyme inhibition ability in mice with IDH1-R132 mutations.89 These preclinical studies supported further clinical research of this drug. The published data from a phase I dose escalation study which preliminarily explored the safety and activity of AG-120 in a group of CCA patients with IDH1 mutations was encouraging (NCT02073994). Among 73 pretreated CCA patients, 5% had a partial response and 56% experienced stable disease. Moreover, a 6-month PFS rate of 40.1% and a 12-month PFS rate of 21.8% were achieved, as well as a median OS of 13.8 months (95% CI: 11.1–29.3).92 In addition, when studying the tumor biopsies collected from these patients, scientists found that mutant IDH1 cholangiocarcinoma with a post-dose cytoplasmic decrease upregulated several immune response-related genes such as CTLA4, CXCL10, and CD3G, implying that using AG-120 plus immunotherapies might be a potential regimen.93 A phase III clinical trial named ClarIDHy is under development which compares the efficacy of AG-120 with a placebo in IDH1-mutation-positive CCA patients (NCT02989857). Compared with placebo, ivosidenib showed improved mPFS (2.7 vs 1.4 months) and mOS (10.8 vs 9.7 months). In addition, the group of patients treated with ivosidenib experienced a better quality of life.94 However, 1.3 months of PFS benefit and 1.1 months of OS benefit are limited with a large cost of this drug.
Several other mIDH1 inhibitors that might be effective in CCA are still undergoing testing in clinical trials. Olutasidenib (FT-2102) is a potent inhibitor of mIDH1 whose clinical trials are ongoing in advanced malignancies including CCA (NCT03684811).95 IDH305 is another selective mIDH1 inhibitor developed by Novartis.96 Its activity has been tested by preclinical studies and has moved into clinical trials assessed in patients with advanced malignancies harboring IDHR132 mutations (NCT02381886).97
mIDH2
Enasidenib is a first-in-class selective inhibitor which is specific to the mutant IDH2 enzyme. Its 2-HG suppression ability was demonstrated in multiple in vitro and in vivo preclinical studies. These studies explored the function of Enasidenib in several IDH2-mutant systems, such as cells taken from AML patients and mouse xenograft models. These studies verified that the inhibition of 2-HG led to cellular differentiation, and the research conducted in the AML xenograft mouse model achieved a dose-dependent survival benefit, which promoted the clinical development of Enasidenib.98 Although it has been approved by the FDA, more studies need to be carried out to confirm its efficacy in BTC patients.90 A phase I/II trial of AG-221 in subjects with IDH2-mutant advanced solid tumors, including CCA, was completed in 2018, but results are still unreported (NCT02273739).
Pan-Inhibitor
AG-881 (Vorasidenib) is the first pan-inhibitor of both mIDH1 and mIDH2.90,99,100 However, AG-881 has not been approved by the FDA and there have been no clinical trials evaluating it in biliary tract cancer patients.
Multi‐TKIs
Besides these mIDH inhibitors, a preclinical study discovered that two iCCA cell lines with IDH1 mutations were highly sensitive to multi-tyrosine kinase inhibitors (multi‐TKIs), dasatinib and saracatinib. Both of these inhibitors belong to the SRC family of tyrosine kinases and the subsequent experiments suggested SRC inhibition was of great significance for dasatinib-mediated cytotoxicity. This sensitivity to dasatinib has not occurred in all tumor types with IDH mutations, only in mIDH-positive iCCA tissues.101 A phase II clinical trial testing dasatinib in iCCA patients with IDH mutations was completed, but the results have not yet been published (NCT02428855).
PARPi
Moreover, there were some preclinical studies indicating that poly ADP ribose polymerase (PARP) inhibitors could kill the tumor cells with IDH mutations.102,103 As mentioned before, mutant IDH can cause the accumulation of 2-HG, which can significantly decrease homologous recombination repair (HRR) activity by inhibiting the αKG-dependent dioxygenases and subsequently improving the sensitivity to PARP inhibitors.102 Based on these promising results, several PARP inhibitors are being investigated in mIDH CCA patients, including olaparib (NCT03212274, NCT04306367, NCT04298021), rucaparib (NCT03639935, NCT03337087) and niraparib (NCT03207347).
Furthermore, a recent report studied tumor samples from 1292 BTC patients showed Breast Cancer Susceptibility Gene (BRCA) mutations with higher rate in subjects with microsatellite instability high (MSI-H) and tumors with higher tumor mutational burden (TMB). PARP inhibitor is a possible treatment for BRCA-mutated cancers. Cancers with high TMB and MSI-H showed a better response to immunotherapy. Therefore, the combination of PARPi plus immune checkpoint inhibitors is of high interest in treatment of BTCs.104
In the future, the combination of IDH inhibitors and other agents (eg, chemotherapy, targeted therapy, immunotherapy) may become the first-line treatment. However, the mechanism of resistance is still unclear and how to overcome the resistance needs to be explored.
Epidermal Growth Factor Receptor (EGFR)/HER2
Epidermal growth factor receptor (EGFR) and HER2, members of the ErbB family, are two common receptors involved in the tumorigenesis of BTCs. The ErbB family consists of four members: ERBB1 (EGFR), ERBB2 (HER2), ERBB3 and ERBB4, which are all receptor tyrosine kinases. They have a similar molecular structure composed of an intracellular tyrosine kinase domain, a single transmembrane lipophilic region and an extracellular ligand-binding domain.105,106
Besides epidermal growth factors (EGFs), transforming growth factor-α (TGF-α) and amphiregulin specifically bind to EGFR. Binding of the ligands to EGFR is followed by dimerization, which successively stimulates its tyrosine kinase domain autophosphorylation and activates downstream signal transduction cascades. Specifically, none of the EGFs can interact with HER2. Although there is no soluble ligand for HER2, it is the preferential partner of another member of the same family during heterodimerization, which subsequently induces the activation of its tyrosine kinase domain and downstream signaling pathways.105 The main signaling pathways activated by ErbBs are the MAPK, PI3K/AKT/mTOR, and JAK/STAT pathways that control and regulate cell proliferation, differentiation, metabolism, stress reaction and migration.106–109
In various human cancers, EGFR gene amplification commonly takes place, resulting in EGFR overexpression and making tumor cells sensitive to epidermal growth factors. This phenomenon enables the downstream signaling pathway to be continuously activated, causing cancer cells to gain proliferative and metastatic advantage.105,110 In many tumors, EGFs and cytokines can be produced by tumor cells, stromal cells or macrophages that interact with tumor cells inducing constitutive EGFR activation and tumor cell metastasis.111 Amplification of HER2 also leads to HER2 overexpression in several kinds of tumors, which is highly related to tumorigenesis, tumor cell invasion and metastasis. Regarding genetic mutations, EGFR mutations are rare, and HER2 mutations have only been identified in a small number of cancers.105
Based on a previous study, EGFR expression occurs in iCCAs with an incidence of 100%, followed by eCCAs with an expression level of 52.6% and GBCs at 38.5%. HER2 is mainly overexpressed in eCCAs (ranging from 5.1% to 26.3%) and GBCs (ranging from 5.1% to 10%).112–114 The EGFR mutations were tested in up to 15% of BTCs and the incidence of HER2 mutations in iCCAs was only 0.9%.106,114,115
To date, several preclinical studies tested and confirmed the potential therapeutic effect of EGFR or HER2 inhibitors for BTCs. A study carried out by Weidmann et al demonstrated that NVP-AEE788, a dual EGFR/HER2 inhibitor, could more effectively suppress the proliferation of human CCA cell lines in vitro compared to gefitinib and erlotinib (EGFR inhibitors). Furthermore, this team also tested the antitumor activity of NVP-AEE788 in vivo. In the experiment group, NVP-AEE788 was administered in nude mice which were injected with EGl-1 eCCA cell lines, significantly reducing the volume of tumors compared with the control group.116 Another preclinical study examined the effect of gefitinib (a selective EGFR inhibitor) and GW2974 (a dual EGFR/HER2 inhibitor) in mice with gallbladder carcinoma. The results showed that both two inhibitors acted as promising chemopreventive and therapeutic agents for GBCs in mice models.117 These results from preclinical studies suggest that EGFR and HER2 might be targetable and promising receptors in BTC targeted therapy.
EGFR Inhibitors
The selective EGFR inhibitors primarily include Erlotinib, Cetuximab, Panitumumab and Gefitinib.
Erlotinib is a selective and reversible EGFR inhibitor, which has been under clinical evaluation for a long time. A Phase II clinical trial preliminarily evaluating the efficacy of erlotinib in patients with unresectable BTCs revealed that the disease control rate was 50% with 52% of patients achieving 6-month overall survival.118 Based on the modest benefits showed in phase II studies, a large phase III trial comprised of 268 patients compared the efficacy and safety of erlotinib plus gemcitabine plus oxaliplatin (GEMOX) regimen with GEMOX regimen alone in patients with metastatic BTC (NCT01149122). The group treated with chemotherapy plus erlotinib achieved a higher objective response rate (30% vs 16%, p=0.005). However, there was no survival benefit in either group with a median OS of 9.5 months in both groups (p=0.611) and an mPFS slightly longer in the GEMOX plus erlotinib group (5.8 vs 4.2 months). Particularly, in subgroup analysis, patients with CCAs achieved significantly longer mPFS after taking the erlotinib plus GEMOX regimen (5.9 months vs 3.0 months, p=0.049).119
Cetuximab is another monoclonal antibody selectively targeting EGFR, which has been assessed in combination with chemotherapy in several phase II studies with BTC patients. A phase II trial compared the efficacy and safety of GEMOX with and without Cetuximab in patients with advanced BTCs (NCT01216345). For patients taking GEMOX plus cetuximab, the overall response rate was 63% and the disease control rate was 80%, which indicated the encouraging antitumor activity of the GEMOX plus cetuximab regimen. Compared with results from other studies, cetuximab plus GEMOX demonstrated a better overall response rate.120 However, data from another phase II trial, BINGO study, suggested that the potential antitumor activity of cetuximab did not provide any clinical benefit when used in combination with GEMOX in patients with biliary cancer compared with the GEMOX regimen alone (NCT00552149). mOS was 11.0 months in the chemotherapy plus cetuximab arm, which is lower than that of the chemotherapy alone arm (12.4 months).121
K-Ras mutations are regarded as a negative predictive factor for cancer prognosis and the therapeutic efficacy of EGFR inhibitors in colorectal cancer patients.122 In a phase II trial, BTC patients stratified by K-Ras status were administered with GEMOX with or without cetuximab (NCT01267344). GEMOX plus cetuximab only achieved marginal therapeutic benefits, and the overall survival of the GEMOX plus cetuximab group did not improve significantly (10.6 vs 9.8 months, P=0.91). The data also suggested that K-Ras mutations did not affect the survival of BTC patients.123
All in all, most of the clinical trials failed to verify any compelling therapeutic effect of the addition of cetuximab to GEMOX.
Panitumumab, a selective EGFR inhibitor, has been tested in combination with chemotherapy in several phase II trials. Vecti-BIL study was designed to compare the therapeutic efficacy of GEMOX with and without panitumumab in chemotherapy-naïve BTC patients possessing a wild-type K-Ras status (NCT01389414). The addition of panitumumab did not improve mPFS significantly (5.3 vs 4.4 months), and no survival benefit was observed (9.9 vs 10.2 months).124 Similarly, data from the PICCA study also confirmed that there was no survival benefit gained from the addition of panitumumab to gemcitabine and cisplatin chemotherapy (GEMCIS) in K-Ras wild-type BTC patients (NCT01320254).125
So far, though results from a meta-analysis indicated that anti-EGFR inhibitors could prolong PFS and response rate, several completed randomized clinical trials all failed to confirm the therapeutic effects of EGFR inhibitors in BTC patients with little clinical benefit. Therefore, further exploration in this field is needed.
HER2 Inhibitors
HER2 overexpression and gene amplification are the common occurrences in BTCs, leading to the development of specific HER2 inhibitors.
Trastuzumab is an antibody specifically targeting HER2. Although trastuzumab has not been approved for the treatment of BTC, its anti-proliferative activity in HER2-overexpressing BTC cell lines was verified in a preclinical study.126 Another preclinical study demonstrated the antitumor effect in a mouse xenograft model through increasing apoptosis.127 A retrospective analysis conducted by Javle et al found that trastuzumab had a disease control rate (including partial response, stable disease, or complete response) of 100% in gallbladder cancer group patients. On the contrary, there were no responses in CCA patients after taking trastuzumab.128 These promising results promoted further investigation of HER2 inhibitors in BTCs. This drug is under clinical research and exploration, with several phase II studies in progress.
On the basis of preclinical studies, the inhibitory effect of pertuzumab was confirmed both in BTC cell lines which overexpressed HER2 and HER3 and in vivo.129 Two case reports showed that dual-anti-HER2 therapy pertuzumab and trastuzumab significantly improved the survival benefits of BTC patients. Therefore, dual anti-HER2 therapy might become a potent treatment option against BTC.130,131 Currently, a phase II trial evaluating a trastuzumab plus pertuzumab regimen in patients with advanced solid tumors, including BTCs, is ongoing (NCT02091141).
There is still some disagreement on the therapeutic effects of HER2 inhibitors, so further data from clinical studies is expected.
EFGR and HER2 Double Inhibitors
Dual EFGR and HER2 tyrosine kinase inhibitors are inhibitors of both EGFR and HER2, including lapatinib, afatinib, neratinib, AEE788, varlitinib, and dacomitinib.
So far, two phase II clinical trials assessing the therapeutic efficacy of lapatinib in BTC patients have been completed. A phase II study beginning in 2004 evaluated lapatinib in 17 advanced BTC patients and 40 hepatocellular cancer (HCC) patients (NCT00101036). Results were poor: no objective response, as well as PFS and mOS for BTC patients of only 1.8 months and 5.2 months, respectively.132 Later, similar poor data were obtained from another phase II study, and this study was terminated early (NCT00107536).133
A phase I study investigated the efficacy of afatinib in combination with gemcitabine and cisplatin (GEMCIS) in patients with advanced BTC (NCT01679405). Only 9 participants enrolled and this study was discontinued due to futility.134 Afatinib combined with capecitabine is currently under evaluation in a phase I trial in patients with bile duct carcinoma and pancreatic cancer (NCT02451553).
So far, there are few clinical studies evaluating neratinib. SUMMIT, a basket trial, explored the efficacy of neratinib in EGFR/HER2 mutation-positive cancer patients, including BTC patients (NCT01953926). The preliminary data presented at American Association for Cancer Research’s Annual Meeting 2017 indicated promising antitumor activity of neratinib in BTC patients with an ORR of 22%.135
For AEE788, varlitinib and dacomitinib, though they lack clinical trials to verify their therapeutic efficacy, preclinical studies have implied bright prospects for them as treatments for biliary tract cancers. The preclinical study for AEE788 has been mentioned before. Varlitinib (ASLAN001) is a new promising therapeutic inhibitor for CCA treatment. Its anti-tumor effect was confirmed both in vitro and in vivo, and the effect was improved when used in combination with the PI3K inhibitor BKM-120.136 Currently there are several ongoing phase I and phase II clinical trials with varlitinib in BTC patients. The efficacy of dacomitinib (PF00299804) was assessed in eight BTC cell lines. As monotherapy, dacomitinib showed good inhibitory effects in two of the eight cell lines. Furthermore, dacomitinib in combination with gemcitabine showed improved anti-tumor effects in seven of the eight cell lines.137
These promising results support further studies to be carried out for the treatment of BTCs (Table 3).
 |
Table 3 Clinical Trials for BTC Targeted Therapy Targeting ErbB Family |
Neurotrophic Tropomyosin Receptor Kinase (NTRK)
Recently, neurotrophic tropomyosin receptor kinase (NTRK) gene fusion has become a promising avenue for cancer targeted therapy. NTRK genes encode for tropomyosin receptor kinase (TRK) receptors, which are transmembrane receptors structured with an extracellular ligand-binding domain, a transmembrane region and an intracellular kinase domain. There are three TRK receptors in the TRK receptor family: TRK A, TRK B and TRK C receptors, encoded by NTRK1, NTRK2 and NTRK3 genes, respectively.138,139 TRK receptors play an essential role in nervous system development and function. The ligands for TRK receptors, neurotrophins (NTs), activate downstream signaling pathways regulating cellular proliferation, differentiation and survival when they bind to TRK receptors. However, when NTRK gene fusion occurs, chimeric TRK proteins are produced which are constitutively activated conferring an oncogenic potential.140,141
With the development of next-generation sequencing (NGS) and fluorescence in situ hybridization (FISH) techniques, NTRK fusions have been detected in various types of tumors, such as salivary gland carcinoma, sarcoma, and thyroid carcinoma.142 Ross et al identified NTRK fusion (RABGAP1L-NTRK1) in one of the 28 iCCA patient samples (3.5%).86 Another study reported an incidence rate of 0.25% in 787 CCA patients.142 A recent report presented at ESMO World Congress on Gastrointestinal Cancer 2020 studied the incidence of NTRK gene fusions in biliopancreatic malignancies, which showed the percentage of NTRK gene fusions was only 0.67% among patients with BTC.143 Moreover, TRK inhibitors can also suppress the abnormal activity induced by ROS1 and ALK fusions, which also occur in CCA patients.144 Although their incidence in BTCs is still low, selective TRK inhibitors have been developed and the concept of precision medicine has gradually become popular, implying that NTRK fusions might become a promising target for biliary tumor treatment.
The efficacy of a few selective TRK inhibitors for BTCs are under evaluation in preclinical and clinical studies (Table 4).
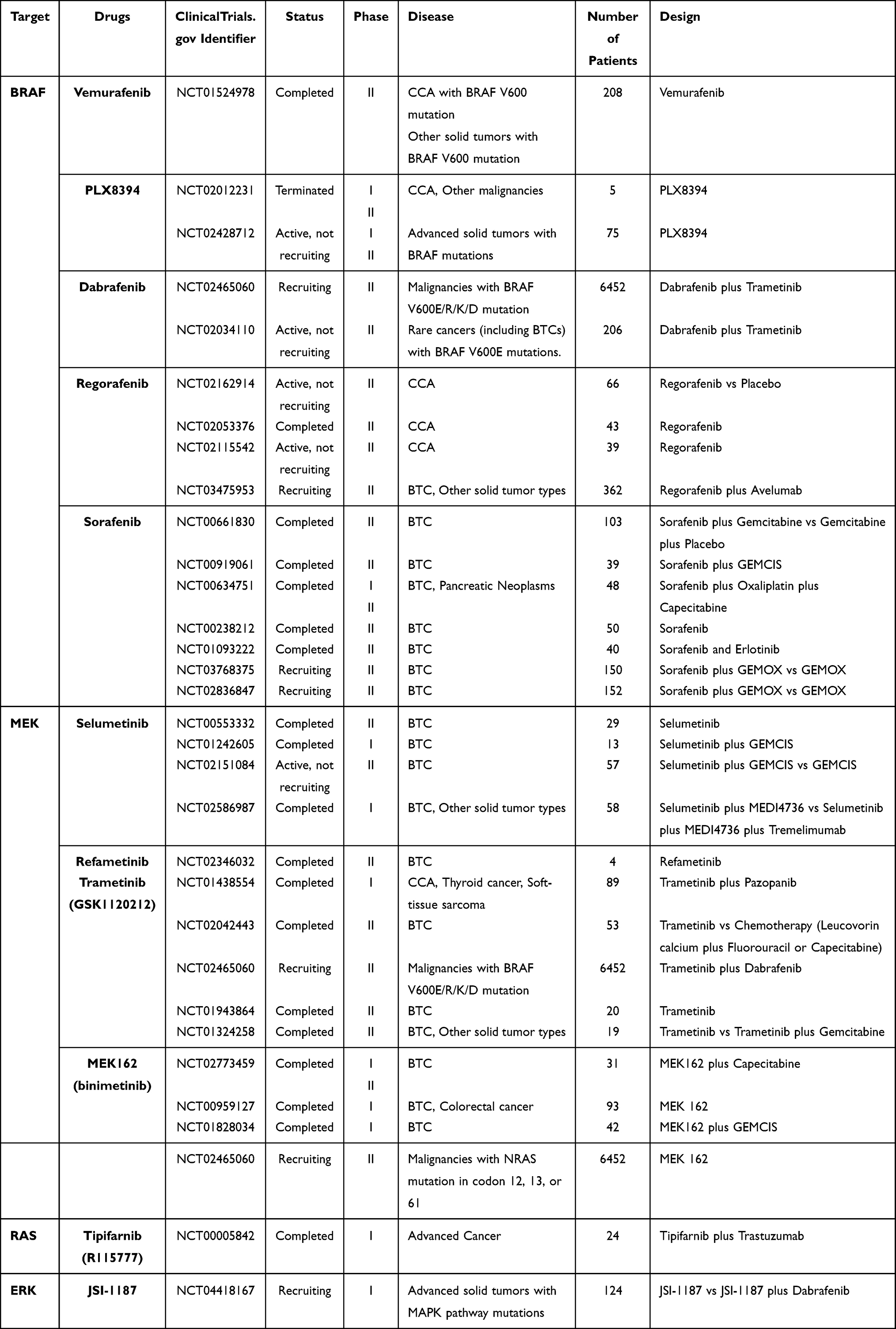 |
Table 4 Clinical Trials for BTC Targeted Therapy Targeting the Ras/Raf/MEK/Erk Signaling Pathway |
Two preclinical studies used mouse models to verify that TRK inhibitors effectively control tumor growth and confirm that NTRK gene fusions (Etv6-NTRK3 fusion and Bcan-Ntrk1 fusion) can initiate tumorigenesis.145,146
Entrectinib (RDX-101, NMS-P626) is an oral inhibitor against the activity induced by the TRK family, C-ros oncogene 1 (ROS1) and anaplastic lymphoma kinase (ALK). It has proven effective in several clinical trials involving patients with NTRK gene fusions.147–149 Larotrectinib (VITRAKVI) is the first pan-TRK inhibitor approved by the US Food and Drug Administration for treating patients with solid tumors harboring NTRK gene fusions. Promising data published from three multicenter clinical trials (NCT02122913, NCT02637687, NCT02576431) contributed to the accelerated approval of larotrectinib. The efficacy was assessed in 55 participants covering 12 cancer types, including 2 with CCA. The reported ORR was 75%, including 22% CR and 53% PR. A phase II MATCH trial also evaluated the efficacy of Larotrectinib in cancer patients with NTRK1, NTRK2, or NTRK3 gene fusions (NCT02465060).
Additional TRK inhibitors are under preclinical and clinical development, but few of them have been tested in BTC patients. ONO-7579 is a pan-TRK inhibitor whose anti-tumor effect was demonstrated in a preclinical study using two GBC cell lines: NOZ (harboring K-Ras mutant) and TYGBK-1 (wild-type K-Ras). The results indicated that ONO-7579 could effectively suppress proliferation in the TYGBK-1 cell line, but not in the NOZ cell line, suggesting that ONO-7579 may have a potent anti-tumor effect on GBC cells without K-Ras mutation.150 However, the only phase I clinical study of ONO-7579 for patients with NTRK gene fusion-positive solid tumors was terminated for commercial reasons (NCT03182257).
Even though the incidence of NTRK gene fusion is low, the testing of NTRKgene fusion is of high interest due to the development of several specific TRK inhibitors. Further studies evaluating TRK inhibitors specific to BTC treatment are expected.
Ras/Raf/MEK/Erk Signaling Pathway Inhibitors
The Ras/Raf/MEK/Erk signaling pathway is one of the main signaling pathways for BTC carcinogenesis. Ras is a kinase which is encoded by the Ras gene, a proto-oncogene. When the Ras gene mutates, it expresses abnormal Ras oncoproteins, which leads to consecutive activations of itself and its downstream signaling pathways. Consequently, the unlimited proliferation and suppressed apoptosis of cells occurs.151,152
There are at least three downstream signaling pathways of the Ras oncoprotein. Raf kinase was the first discovered Ras effector. This signaling pathway has a generic name: mitogen-activated protein kinase (MAPK) pathway. In this signal transduction cascade, Ras combines with a GTP molecule to activate the Raf kinase (a MAPKKK); Raf then activates MEK (a MAPKK), which subsequently activates ERK1/2 (MAPKs). Erk1 and Erk2 are able to phosphorylate some transcription factors (eg, Ets, Elk-1, SAP-1) and kinase which is responsible for protein synthesis (eg, Mnk1 kinase). Besides activating several growth-promoting genes, this pathway also causes cells to lose anchorage and contact inhibition properties. Furthermore, it plays an important role in Ras oncogene-associated cell shape changing.153,154 B-Raf is a homology of the Raf protein and its mutant form is observed in about 66% of human melanomas.155,156
K-Ras, N-Ras and B-Raf mutations have been commonly detected in various cancer types such as gastrointestinal cancers, lung cancers and melanomas. For biliary tract carcinomas, studies in different countries show a variance in the frequency of K-Rasmutations, ranging from 15.3% to 67% in eCCAs and from 9% to 45% in iCCAs. Moreover, in a Japanese test group, the incidence of K-Ras mutations in GBC was relatively higher than in other regions. The presence of K-Ras mutations is correlated to worse prognosis.157–159 By contrast, N-Ras mutations exist in 3.6% of iCCAs and 2.6% of eCCAs, while B-Raf mutations are only found in 3% to 5% of iCCAs.46,157,160
These mutated proteins and their downstream signal proteins have gradually become the new targets for BTC targeted therapy. Many novel inhibitors targeting the Ras–Raf–MEK–ERK pathway have been evaluated by a large number of studies (Table 4).
K-Ras
Currently, there are no specific inhibitors targeting the K-Ras mutated form, so this pathway can only be suppressed by inhibiting the downstream functional proteins.
B-Raf
As mentioned before, the Raf protein has a homology called B-Raf which exists in various types of cancer.
Vemurafenib is a specific inhibitor of the B-Raf V600 mutated protein. Up to date, only one published clinical trial has evaluated the therapeutic effects of vemurafenib in BTC patients (NCT01524978). This phase II study assessed the clinical benefits of vemurafenib in multiple nonmelanoma cancers with B-Raf V600 mutations, including 8 CCA patients. The released results showed that, among the 8 CCA patients, only one patient had partial response (12%) and 4 patients experienced stable disease (50%).161 A reported case showed that a CCA patient with B-Raf V600 mutations achieved complete response after taking vemurafenib, panitumumab, and irinotecan therapy.162
Another B-Raf inhibitor, PLX8394, was investigated by two phase I/II clinical trials in patients with advanced solid tumors including CCA (NCT02428712, NCT02012231). However, one study was terminated while the other has not yet released any data.
Dabrafenib is also a specific B-Raf V600 inhibitor. According to a published case report, a patient with B-Raf V600-mutated iCCA performed exceptional response, including symptomatic and radiological improvement, to dabrafenib plus trametinib (an MEK1/2 inhibitor) dual therapy, causing more clinical trials to be conducted.163 An ongoing phase II clinical trial, Rare Oncology Agnostic Research (ROAR) basket trial, has provided extremely important results for dabrafenib and trametinib. Among 43 patients with B-Raf V600E-mutated BTCs, this regimen achieved an investigator-assessed ORR of 51% and an independent reviewer-assessed ORR of 47%, with a tolerant safety profile. Therefore, the authors suggested that B-Raf V600E mutation testing should be considered in all patients with BTCs.164
Additionally, regorafenib (BAY 73-4506) and sorafenib (Bay 43-9006) are both multi-kinase inhibitors against Raf-1 protein kinase and B-Raf kinase. However, a phase II trial described an intolerant toxicity and no achievable survival benefit by adding sorafenib to GEMCIS in BTC patients.165
MEK
MEK is another contributor to the MAPK pathway, and several kinase inhibitors for MEK are under investigation.
A phase II multicenter trial led by The Ohio State University was designed to determine the safety and efficacy of selumetinib monotherapy, a MEK1/2 inhibitor, in metastatic biliary tract cancer. The results revealed that 12% of the participants had an objective response and another 68% experienced stable disease. Furthermore, the mPFS of 3.7 months and mOS of 9.8 months both compare favorably with previously published data. The encouraging response and well-tolerated safety profile indicate that selumetinib might be a promising inhibitor for BTC treatment.166 Another phase Ib trial, the ABC-04 trial, evaluated the combination of selumetinib with GEMCIS in patients with advanced BTC (NCT01242605). A median PFS of 6.4 months and acceptable toxicity profile indicated this regimen could achieve a modest efficacy in BTC patients.167 A phase II trial evaluating the combination of GEMCIS and selumetinib versus GEMCIS alone is ongoing (NCT02151084).
Trametinib, another MEK inhibitor, has been tested in a group of Japanese patients, showing a 12-week stable disease rate of 10%, mPFS of 10.6 weeks and a rate of 1-year OS of 20% (NCT01943864).168 Trametinib has also been tested with pazopanib, a VEGFR TKI, though this combination did not offer any survival benefit (NCT01438554).169 Other clinical trials of trametinib monotherapy or combination therapy have not shown any priorities, so this drug warrants further research.
Binimetinib (MEK162) is a potent MEK 1/2 inhibitor whose preliminary antitumor activity has been demonstrated in several Phase I clinical trials in patients with BTC.170,171 A phase I/II trial assessing the combination of binimetinib and capecitabine showed promising antitumor efficacy in BTC patients with MAPK pathway mutations (NCT02773459).172 However, another phase I/II trial assessing binimetinib in combination with chemotherapy (GEMCIS) in BTC patients did not show any priority compared to chemotherapy alone (NCT01828034).173
A phase II trial of refametinib assessed in BTC patients has completed, but the results are still unknown (NCT02346032).
ERK
Up to date, there have not been any Erk inhibitors approved in the world. JSI-1187 is an oral, highly selective Erk 1/2 inhibitor which is mainly used to treat tumors with MAPK pathway mutations. Other Erk inhibitors including LY3214996, LTT462 and Ulixertinib are under clinical evaluation.
PI3K/Akt/mTOR Signaling Pathway Inhibitors
A second important downstream signaling pathway driven by the Ras protein is the PI3K/Akt/mTOR pathway, which is involved in BTC tumorigenesis and progression.
Phosphatidylinositol 3-kinase (PI3K) is a direct downstream effector of Ras. It is an essential kinase adding phosphates to phospholipids, which contributes to the formation of phosphatidylinositol (3,4,5)-triphosphate (PIP3). PIP3 subsequently combines with Akt, a serine/threonine kinase also known as protein kinase B (PKB), and activates it.174 Once activated, Akt can phosphorylate several proteins which affect the cells. Firstly, Akt can prolong the cell life cycle by inactivating the pro-apoptotic proteins, such as Bad and Caspase-9. Secondly, activated Akt promotes cell proliferation by inactivating the anti-proliferative proteins GSK-3β, FOXO4 and p21Cip1. Finally, Akt is able to activate mammalian target of rapamycin (mTOR), a protein promoting protein synthesis and stimulating cell growth in size.175–177
Normally, activation of the PI3K/Akt/mTOR pathway is under tight control. The PTEN gene is a tumor suppressor gene playing an essential role in regulating the activity of the PI3K/Akt/mTOR pathway. The PTEN gene encodes the PTEN protein, a phosphatase, which then reverses the actions of PI3K to control the level of PIP3, thus controlling the activation of the PI3K/Akt/mTOR pathway.178 However, in various types of tumors, the hyperactivation of PI3K or inactivation of PTEN occurs, which deregulates this signaling pathway and confers the cells oncogenic potential.179
According to several studies, PI3K mutations were detected in 4.4% of iCCA patients and 6.5% of eCCA patients. PTEN mutations were observed in 4.4% of iCCAs and 3.9% of eCCAs.157 Blocking this signaling pathway by the use of specific inhibitors might inhibit tumor growth, including biliary tract cancers. As a result, many inhibitors targeting the effector proteins in the PI3K/Akt/mTOR pathway are under development and investigation (Table 5).
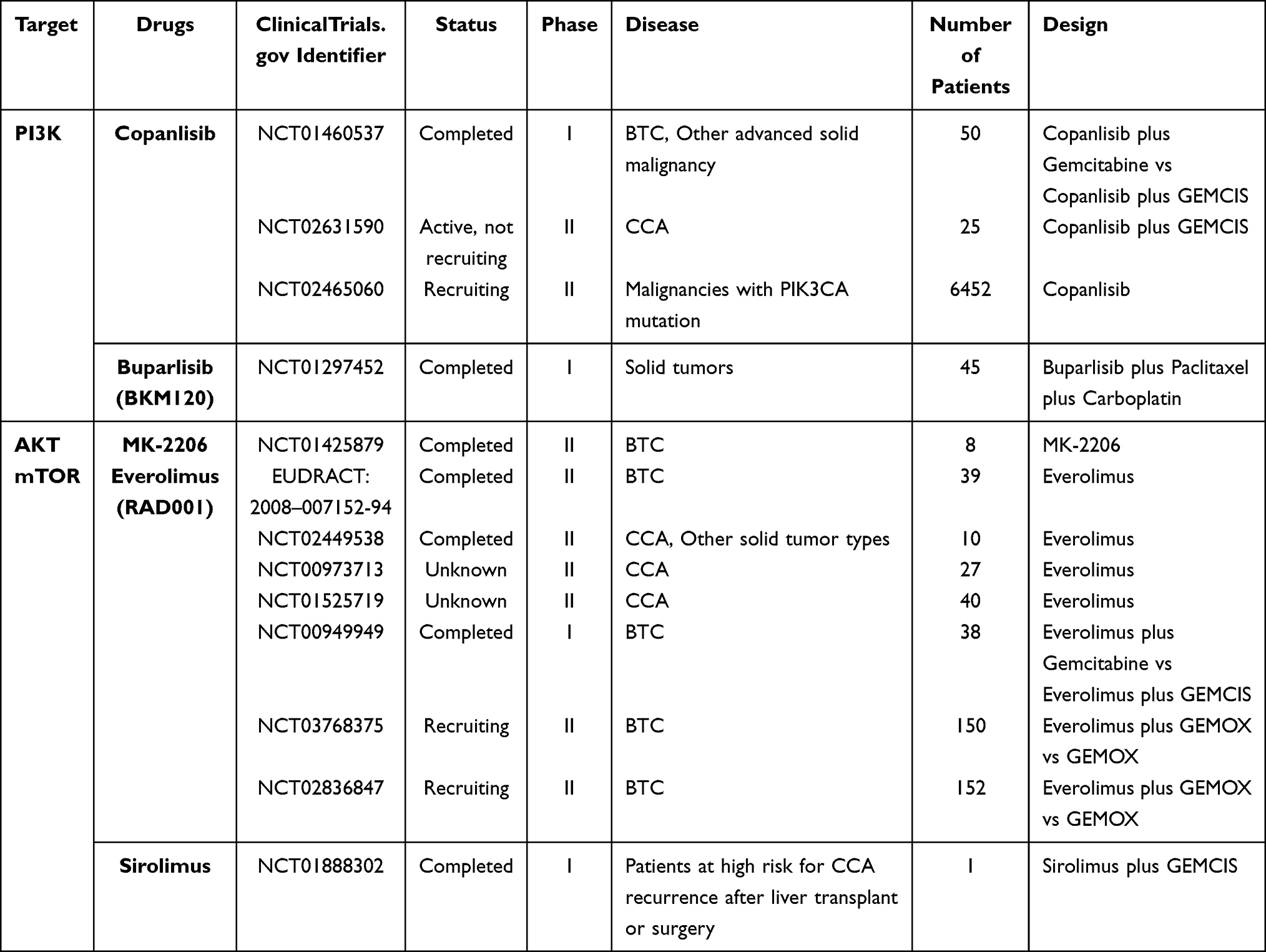 |
Table 5 Clinical Trials for BTC Targeted Therapy Targeting the PI3K/Akt/mTOR Signaling Pathway |
PI3K Inhibitors
Copanlisib (BAY 80–6946) is a selective pan-class I PI3K inhibitor which has been evaluated in a phase I study with 50 patients with advanced solid tumors, including 23 BTC patients (NCT01460537). Response rate was 6.3% in the copanlisib with gemcitabine and 12% in the copanlisib with cisplatin plus gemcitabine (GEMCIS) arm. Among the 23 BTC patients, response rate was 17%. The safety profile of copanlisib is acceptable.180 Currently, the therapeutic effects of the copanlisib plus GEMCIS regimen is being assessed in a phase II trial with CCA patients (NCT02631590), and the results are highly anticipated.
The first study demonstrating the anti-tumor effects of BKM120, also named Buparlisib, in BTC cells was conducted by Jin et al. They found that BKM120 could suppress the proliferation and migration of BTC cells in vitro. Furthermore, inhibiting both PI3K and MEK by BKM120 plus MEK162 showed inhibitory effects in BTC cells with both K-Ras mutations and PI3K mutations, which was not achievable by BKM120 alone.181 However, a phase I trial of BKM120 plus mFOLFOX6 (5-FU/LV plus oxaliplatin) did not show a tolerant safety profile in advanced gastrointestinal tumors, including 4 CCA patients (NCT01571024). Specifically, 13 of the 17 participants experienced grade 3/4 adverse events.182 On the contrary, another phase I study demonstrated that BKM120 is safe with a clear evidence of antitumor activity in patients with advanced cancers, including GBC.183 In addition, a phase I trial verified the good safety profile and therapeutic efficacy of buparlisib in combination with paclitaxel and carboplatin in solid tumors (NCT01297452). For patients without PTEN expression, the preliminary antitumor activity was notable.184
Sakamoto et al used BTC cell lines as a preclinical model to verify that LY3023414, a PI3K/mTOR dual inhibitor, possessed anti-proliferative activity, marking it as a potential new agent for BTC treatment.185
AKT Inhibitors
The main Akt inhibitors, including MK-2206, FPA124 and A-443654, were confirmed to inhibit cell proliferation and migration in various BTC cell lines.186 Moreover, in a preclinical study, genistein was found to suppress CCA cell growth by inhibiting the activation of Akt.187
Among these Akt inhibitors, MK-2206 is considered the most promising, as it is the only one to have entered clinical trials so far. Wilson et al confirmed that MK-2206 could suppress the CCA cell growth by inducting apoptosis in vitro.188 A phase II clinical trial recruiting 8 BTC patients evaluated the safety and efficacy of MK-2206 (NCT01425879). However, only 2 patients achieved stable disease (25%), which was the best observed response. Even though the toxicity was tolerant, there were no notable clinical benefits existing in this small group of participants.189 Further research needs to be conducted for MK-2206 development.
mTOR
Everolimus, sirolimus (also called rapamycin), and temsirolimus are all first-generation inhibitors of mTOR. The superiority of everolimus and sirolimus has been evidenced by preclinical researches in CCA, promoting the commencement of clinical studies.
Several clinical studies of everolimus in BTC have been carried out. A retrospective study showed that, among 22 BTC patients, everolimus achieved a DCR of 50% with a higher incidence of adverse events (64%).190 A phase I study evaluated the safety and antitumor activity of everolimus in combination with gemcitabine and everolimus plus GEMCIS regimen in patients with solid tumors. Among 37 participants, 10 participants enrolled in Cohort III were all BTC patients, with 6 patients achieving stable disease (60%). The toxicities of both regimens were manageable.191 A phase II clinical trial (I.T.M.O. study) was conducted in Italy (EUDRACT: 2008-007152-94) to evaluate the therapeutic efficacy and safety of RAD001 (everolimus) in 39 advanced BTC patients who were previously treated with chemotherapy. It reported a DCR of 44.7%, ORR of 5.1% and mOS of 7.7 months with tolerable drug toxicity.192 Another relevant phase II clinical trial of everolimus in cancer patients with PI3K abbreviation or PTEN loss did not show any general clinical benefit, though the only CCA patient achieved stable disease (NCT02449538).193
In recent years, the data of the RADiChol study, a phase II clinical trial, was published (NCT00973713). Twenty-seven patients with advanced BTC were enrolled in this study. The primary endpoint DCR at 12 weeks was 48%, with an mPFS of 5.5 months and mOS of 9.5 months. Generally, everolimus monotherapy was well tolerated and did show a clinical benefit in advanced BTC patients.194 Presently, the therapeutic efficacy of everolimus in BTC treatment has not been confirmed, so more clinical data are expected to be released.
Sirolimus is another mTOR inhibitor under clinical investigation. Pilot studies have not demonstrated obvious clinical activity of sirolimus, but partial participants also achieved partial response and stable disease.195,196 The only clinical study evaluating sirolimus in combination with gemcitabine plus cisplatin for patients at high risk for CCA after liver transplant or surgery has finished, but results are unknown so far (NCT01888302).
In conclusion, mTOR inhibitors achieved modest clinical benefits in advanced BTC patients, and they should be validated by more randomized controlled trial (RCT) studies.
Wnt Signaling Pathaway
Wnt signaling pathway is an intracellular signaling pathway. A study indicated that, in human CCA, the expression of Wnt signaling was significantly increased. Therefore, suppression of Wnt signaling pathway might be an option for inhibition of CCA growth.197
RNF43 is a RING domain E3 ubiquitin ligase, which could suppress p53-mediated apoptosis and inhibit Wnt signaling. When this gene mutated, Wnt signaling was increased.198,199 Recently, RNF43 mutations have been highlighted in BTC patients, with an incidence of 9.3% in CCA cases.200 Therefore, the blockades of this signaling pathway were developed and their activity and safety are assessed in clinical trial.
Other several Wnt signaling pathway inhibitors, including DKN-01, ICG-001, C-59 and CGX1321, are also under development and research. We are looking forward to unfolding this area in the next years.
Future Prospects
Biliary tract cancer is a highly fatal disease and a challenge for clinical treatment due to its “silent” symptoms, fast progression and high recurrence rate. As for systematic therapy, the options and therapeutic effects are limited.
Despite great advances made to uncover the molecular mechanism of BTC tumorigenesis, many obstacles remain. The major roadblock is that there are different entities included in BTC (ie, iCCA, eCCA, GBC) with different clinical and molecular features, which is also termed heterogeneity. Different signaling pathways and complex molecular interactions underlie the cancer heterogeneity and individual’s susceptibility to different drugs. Heterogeneity is a limitation for studies with targeted agents and BTC targeted therapy, since various activated or inhibited pathways are strongly influenced by the molecular features of the tumor, which vary among the entities. In addition, as most clinical trials are currently in Phase II, more credible phase III randomized clinical trials are warranted to verify therapeutic efficacy and safety. Another tricky problem is that these promising targets only exist in a small proportion of patients; therefore, many approved inhibitors cannot be used in the majority of patients. The identification of novel targets is required to carry out individualized treatment in most patients.
Gene aberrations are regarded as the drivers of tumors. With the concept of “precision medicine” and the continuing development of sequencing technology, molecular targeted therapy can offer new ideas. Since oncogenesis and tumor progression are regulated and controlled by a large number of signal molecules and signaling pathways, there are many promising targets for targeted therapy. Numerous preclinical studies and clinical trials are ongoing to develop and evaluate new inhibitors, and some have achieved an encouraging therapeutic efficacy. IDH1/2 mutations and FGFR2 fusions are some of the most promising current targets for BTC targeted therapy, and more data are expected to verify their efficacy in the future. Furthermore, immunotherapy has also been applied in the treatment of BTCs. Immune checkpoint inhibitors nivolumab and pembrolizumab have been approved by the FDA for microsatellite instability-high (MSI-H) tumors.
With the coming era of big data and the emergence of next-generation sequencing, the implementation of individualized treatment becomes possible. Timely diagnosis (eg, liquid biopsy) and targeted therapy will significantly improve the prognosis of cancer patients. Liquid biopsy is an emerging tool for earlier cancer diagnosis with minimal invasiveness. It tests circulating tumor DNA (ctDNA), a tumor-derived fragmented DNA which exists in blood, to diagnose cancers. The difficulty in obtaining sufficient biopsy samples to confirm the diagnosis is still challenging in BTC, therefore, ctDNA could play an important role in BTC patients.201,202 Targeted therapy has become one of the mainstay treatments for cancer patients. More therapeutic strategies, such as immunotherapy, focusing on common molecular targets and epigenetic alterations have emerged, and even the non-coding RNA and miRNA may eventually become new targets for BTC treatments.
In the future, precision treatment may become a reality for patients with malignant biliary tract cancer through the combination of clinical therapy with the molecular profile of tumors.
Abbreviations
BTC, biliary tract cancer; GBCs, gallbladder cancers; CCAs, cholangiocarcinomas; iCCAs, intrahepatic cholangiocarcinomas; eCCAs, extrahepatic cholangiocarcinomas; HBV, hepatitis B; HCV, hepatitis C; BRCA1, breast cancer gene 1; BRCA2, breast cancer gene 2; OV, Opisthorcis viverrine; mOS, median overall survival; mPFS, median progression-free survival; DCR, disease control rate; GEMCIS, gemcitabine and cisplatin; GEMOX, gemcitabine plus oxaliplatin; GS, S-1 plus gemcitabine; FGFRs, fibroblast growth factor receptors; RTKs, receptor tyrosine kinases; FGFs, fibroblast growth factors; TKIs, tyrosine kinase inhibitors; mAbs, monoclonal antibodies; ORR, overall response rate; DZB, derazantinib; SD, stable disease; IDH, isocitrate dehydrogenase; PARP, poly ADP ribose polymerase; HRR, homologous recombination repair; EGFR, epidermal growth factor receptor; EGFs, epidermal growth factors; TGF-α, transforming growth factor-α; HCC, hepatocellular cancer; NTRK, neurotrophic tropomyosin receptor kinase; TRK, tropomyosin receptor kinase; NTs, neurotrophins; NGS, next generation sequencing; FISH, fluorescence in situ hybridization; ROS1, C-ros oncogene 1; ALK, anaplastic lymphoma kinase; MAPK, mitogen-activated protein kinase; PI3K, phosphatidylinositol 3-kinase; PIP3, phosphatidylinositol (3,4,5)-triphosphate; PKB, protein kinase B; mTOR, mammalian target of rapamycin; RCT, randomized controlled trial; MSI-H, microsatellite instability-high.
Funding
This work was supported by Wu Jieping Medical Foundation (project number: 320.6790.17198-4)
Disclosure
The authors report no conflicts of interest for this work.
References
1. de Groen PC, Gores GJ, LaRusso NF, Gunderson LL, Nagorney DM. Biliary tract cancers. N Engl J Med. 1999;341(18):1368–1378. doi:10.1056/NEJM199910283411807
2. Lombardi P, Marino D, Fenocchio E, Chila G, Aglietta M, Leone F. Emerging molecular target antagonists for the treatment of biliary tract cancer. Expert Opin Emerg Drugs. 2018;23(1):63–75. doi:10.1080/14728214.2018.1444749
3. Mosconi S, Beretta GD, Labianca R, Zampino MG, Gatta G, Heinemann V. Cholangiocarcinoma. Crit Rev Oncol Hematol. 2009;69(3):259–270. doi:10.1016/j.critrevonc.2008.09.008
4. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–86. doi:10.1002/ijc.29210
5. Torre LA, Siegel RL, Islami F, Bray F, Jemal A. Worldwide burden of and trends in mortality from gallbladder and other biliary tract cancers. Clin Gastroenterol Hepatol. 2018;16(3):427–437. doi:10.1016/j.cgh.2017.08.017
6. Shaib YH, Davila JA, McGlynn K, El-Serag HB. Rising incidence of intrahepatic cholangiocarcinoma in the United States: a true increase? J Hepatol. 2004;40(3):472–477. doi:10.1016/j.jhep.2003.11.030
7. Chapman RW. Risk factors for biliary tract carcinogenesis. Ann Oncol. 1999;10(Suppl 4):308–311. doi:10.1093/annonc/10.suppl_4.S308
8. Charbel H, Al-Kawas FH. Cholangiocarcinoma: epidemiology, risk factors, pathogenesis, and diagnosis. Curr Gastroenterol Rep. 2011;13(2):182–187. doi:10.1007/s11894-011-0178-8
9. Khan SA, Toledano MB, Taylor-Robinson SD. Epidemiology, risk factors, and pathogenesis of cholangiocarcinoma. HPB (Oxford). 2008;10(2):77–82. doi:10.1080/13651820801992641
10. Shigeyasu K, Tanakaya K, Nagasaka T, et al. Early detection of metachronous bile duct cancer in Lynch syndrome: report of a case. Surg Today. 2014;44(10):1975–1981. doi:10.1007/s00595-013-0669-3
11. Golan T, Raitses-Gurevich M, Kelley RK, et al. Overall survival and clinical characteristics of BRCA-associated cholangiocarcinoma: a multicenter retrospective study. Oncologist. 2017;22(7):804–810. doi:10.1634/theoncologist.2016-0415
12. Sriamporn S, Pisani P, Pipitgool V, Suwanrungruang K, Kamsa-ard S, Parkin DM. Prevalence of Opisthorchis viverrini infection and incidence of cholangiocarcinoma in Khon Kaen, Northeast Thailand. Trop Med Int Health. 2004;9(5):588–594. doi:10.1111/j.1365-3156.2004.01234.x
13. Harrington J, Carter L, Basu B, Cook N. Drug development and clinical trial design in pancreatico-biliary malignancies. Curr Probl Cancer. 2018;42(1):73–94. doi:10.1016/j.currproblcancer.2018.01.003
14. Everhart JE, Ruhl CE. Burden of digestive diseases in the United States Part III: liver, biliary tract, and pancreas. Gastroenterology. 2009;136(4):1134–1144. doi:10.1053/j.gastro.2009.02.038
15. Horgan AM, Amir E, Walter T, Knox JJ. Adjuvant therapy in the treatment of biliary tract cancer: a systematic review and meta-analysis. J Clin Oncol. 2012;30(16):1934–1940. doi:10.1200/JCO.2011.40.5381
16. Endo I, Gonen M, Yopp AC, et al. Intrahepatic cholangiocarcinoma: rising frequency, improved survival, and determinants of outcome after resection. Ann Surg. 2008;248(1):84–96. doi:10.1097/SLA.0b013e318176c4d3
17. Jiang W, Zeng ZC, Tang ZY, et al. A prognostic scoring system based on clinical features of intrahepatic cholangiocarcinoma: the Fudan score. Ann Oncol. 2011;22(7):1644–1652. doi:10.1093/annonc/mdq650
18. Ribero D, Pinna AD, Guglielmi A, et al. Surgical approach for long-term survival of patients with intrahepatic cholangiocarcinoma: a multi-institutional analysis of 434 patients. Arch Surg. 2012;147(12):1107–1113. doi:10.1001/archsurg.2012.1962
19. Valle J, Wasan H, Palmer DH, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med. 2010;362(14):1273–1281. doi:10.1056/NEJMoa0908721
20. Knox JJ, Hedley D, Oza A, et al. Combining gemcitabine and capecitabine in patients with advanced biliary cancer: a phase II trial. J Clin Oncol. 2005;23(10):2332–2338. doi:10.1200/JCO.2005.51.008
21. André T, Tournigand C, Rosmorduc O, et al. Gemcitabine combined with oxaliplatin (GEMOX) in advanced biliary tract adenocarcinoma: a GERCOR study. Ann Oncol. 2004;15(9):1339–1343. doi:10.1093/annonc/mdh351
22. Morizane C, Okusaka T, Mizusawa J, et al. Randomized phase II study of gemcitabine plus S-1 versus S-1 in advanced biliary tract cancer: a Japan Clinical Oncology Group trial (JCOG 0805). Cancer Sci. 2013;104(9):1211–1216. doi:10.1111/cas.12218
23. Mizusawa J, Morizane C, Okusaka T, et al. Randomized Phase III study of gemcitabine plus S-1 versus gemcitabine plus cisplatin in advanced biliary tract cancer: japan Clinical Oncology Group Study (JCOG1113, FUGA-BT). Jpn J Clin Oncol. 2016;46(4):385–388. doi:10.1093/jjco/hyv213
24. Morizane C, Okusaka T, Mizusawa J, et al. Combination gemcitabine plus S-1 versus gemcitabine plus cisplatin for advanced/recurrent biliary tract cancer: the FUGA-BT (JCOG1113) randomized phase III clinical trial. Ann Oncol. 2019;30(12):1950–1958. doi:10.1093/annonc/mdz402
25. Shroff RT, Javle MM, Xiao L, et al. Gemcitabine, cisplatin, and nab-paclitaxel for the treatment of advanced biliary tract cancers: a Phase 2 clinical trial. JAMA Oncol. 2019;5(6):824–830. doi:10.1001/jamaoncol.2019.0270
26. Lamarca A, Hubner RA, David Ryder W, Valle JW. Second-line chemotherapy in advanced biliary cancer: a systematic review. Ann Oncol. 2014;25(12):2328–2338. doi:10.1093/annonc/mdu162
27. Zheng Y, Tu X, Zhao P, et al. A randomised phase II study of second-line XELIRI regimen versus irinotecan monotherapy in advanced biliary tract cancer patients progressed on gemcitabine and cisplatin. Br J Cancer. 2018;119(3):291–295. doi:10.1038/s41416-018-0138-2
28. Martinez FJ, Shroff RT. Biliary tract cancers: systemic therapy for advanced disease. Chin Clin Oncol. 2020;9(1):5. doi:10.21037/cco.2019.12.07
29. Javle M, Bekaii-Saab T, Jain A, et al. Biliary cancer: utility of next-generation sequencing for clinical management. Cancer. 2016;122(24):3838–3847. doi:10.1002/cncr.30254
30. Lee H, Ross JS. The potential role of comprehensive genomic profiling to guide targeted therapy for patients with biliary cancer. Therap Adv Gastroenterol. 2017;10(6):507–520. doi:10.1177/1756283X17698090
31. Jain A, Javle M. Molecular profiling of biliary tract cancer: a target rich disease. J Gastrointest Oncol. 2016;7(5):797–803. doi:10.21037/jgo.2016.09.01
32. Nakamura H, Arai Y, Totoki Y, et al. Genomic spectra of biliary tract cancer. Nat Genet. 2015;47(9):1003–1010. doi:10.1038/ng.3375
33. Churi CR, Shroff R, Wang Y, et al. Mutation profiling in cholangiocarcinoma: prognostic and therapeutic implications. PLoS One. 2014;9(12):e115383. doi:10.1371/journal.pone.0115383
34. Krook MA, Lenyo A, Wilberding M, et al. Efficacy of FGFR inhibitors and combination therapies for acquired resistance in FGFR2-fusion cholangiocarcinoma. Mol Cancer Ther. 2020;19(3):847–857. doi:10.1158/1535-7163.MCT-19-0631
35. Dai S, Zhou Z, Chen Z, Xu G, Chen Y. Fibroblast growth factor receptors (FGFRs): structures and small molecule inhibitors. Cells. 2019;8(6):614. doi:10.3390/cells8060614
36. Itoh N, Ornitz DM. Evolution of the Fgf and Fgfr gene families. Trends Genet. 2004;20(11):563–569. doi:10.1016/j.tig.2004.08.007
37. Farrell B, Breeze AL. Structure, activation and dysregulation of fibroblast growth factor receptor kinases: perspectives for clinical targeting. Biochem Soc Trans. 2018;46(6):1753–1770. doi:10.1042/BST20180004
38. Coutts JC, Gallagher JT. Receptors for fibroblast growth factors. Immunol Cell Biol. 1995;73(6):584–589. doi:10.1038/icb.1995.92
39. Belov AA, Mohammadi M. Molecular mechanisms of fibroblast growth factor signaling in physiology and pathology. Cold Spring Harb Perspect Biol. 2013;5(6). doi:10.1101/cshperspect.a015958
40. Ornitz DM, Itoh N. The fibroblast growth factor signaling pathway. Wiley Interdiscip Rev Dev Biol. 2015;4(3):215–266.
41. Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov. 2009;8(3):235–253.
42. Graham RP, Barr Fritcher EG, Pestova E, et al. Fibroblast growth factor receptor 2 translocations in intrahepatic cholangiocarcinoma. Hum Pathol. 2014;45(8):1630–1638. doi:10.1016/j.humpath.2014.03.014
43. Mertens JC, Rizvi S, Gores GJ. Targeting cholangiocarcinoma. Biochim Biophys Acta Mol Basis Dis. 2018;1864(4 Pt B):1454–1460. doi:10.1016/j.bbadis.2017.08.027
44. Wu YM, Su F, Kalyana-Sundaram S, et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. 2013;3(6):636–647. doi:10.1158/2159-8290.CD-13-0050
45. Arai Y, Totoki Y, Hosoda F, et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology. 2014;59(4):1427–1434. doi:10.1002/hep.26890
46. Chong DQ, Zhu AX. The landscape of targeted therapies for cholangiocarcinoma: current status and emerging targets. Oncotarget. 2016;7(29):46750–46767. doi:10.18632/oncotarget.8775
47. Ghedini GC, Ronca R, Presta M, Giacomini A. Future applications of FGF/FGFR inhibitors in cancer. Expert Rev Anticancer Ther. 2018;18(9):861–872. doi:10.1080/14737140.2018.1491795
48. Guagnano V, Kauffmann A, Wohrle S, et al. FGFR genetic alterations predict for sensitivity to NVP-BGJ398, a selective pan-FGFR inhibitor. Cancer Discov. 2012;2(12):1118–1133. doi:10.1158/2159-8290.CD-12-0210
49. Guagnano V, Furet P, Spanka C, et al. Discovery of 3-(2,6-dichloro-3,5-dimethoxy-phenyl)-1-{6-[4-(4-ethyl-piperazin-1-yl)-phenylamino]-pyrimidin-4-yl}-1-methyl-urea (NVP-BGJ398), a potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase. J Med Chem. 2011;54(20):7066–7083. doi:10.1021/jm2006222
50. Javle M, Lowery M, Shroff RT, et al. Phase II Study of BGJ398 in patients with FGFR-altered advanced cholangiocarcinoma. J Clin Oncol. 2018;36(3):276–282. doi:10.1200/JCO.2017.75.5009
51. Gilbert JA. BGJ398 for FGFR-altered advanced cholangiocarcinoma. Lancet Oncol. 2018;19(1):e16. doi:10.1016/S1470-2045(17)30902-6
52. Chila R, Hall GT, Abbadessa G, Broggini M, Damia G. Multi-chemotherapeutic schedules containing the pan-FGFR inhibitor ARQ 087 are safe and show antitumor activity in different xenograft models. Transl Oncol. 2017;10(2):153–157. doi:10.1016/j.tranon.2016.12.003
53. Papadopoulos KP, El-Rayes BF, Tolcher AW, et al. A Phase 1 study of ARQ 087, an oral pan-FGFR inhibitor in patients with advanced solid tumours. Br J Cancer. 2017;117(11):1592–1599. doi:10.1038/bjc.2017.330
54. Hall TG, Yu Y, Eathiraj S, et al. Preclinical activity of ARQ 087, a novel inhibitor targeting FGFR dysregulation. PLoS One. 2016;11(9):e0162594. doi:10.1371/journal.pone.0162594
55. Raggi C, Fiaccadori K, Pastore M, et al. Antitumor activity of a novel fibroblast growth factor receptor inhibitor for intrahepatic cholangiocarcinoma. Am J Pathol. 2019;189(10):2090–2101. doi:10.1016/j.ajpath.2019.06.007
56. Mazzaferro V, El-Rayes BF, Droz Dit Busset M, et al. Derazantinib (ARQ 087) in advanced or inoperable FGFR2 gene fusion-positive intrahepatic cholangiocarcinoma. Br J Cancer. 2019;120(2):165–171. doi:10.1038/s41416-018-0334-0
57. Kalyukina M, Yosaatmadja Y, Middleditch MJ, Patterson AV, Smaill JB, Squire CJ. TAS-120 cancer target binding: defining reactivity and revealing the first fibroblast growth factor receptor 1 (FGFR1) irreversible structure. ChemMedChem. 2019;14(4):494–500. doi:10.1002/cmdc.201800719
58. Goyal L, Shi L, Liu LY, et al. TAS-120 overcomes resistance to ATP-competitive FGFR inhibitors in patients with FGFR2 fusion-positive intrahepatic cholangiocarcinoma. Cancer Discov. 2019;9(8):1064–1079. doi:10.1158/2159-8290.CD-19-0182
59. Bahleda R, Meric-Bernstam F, Goyal L, et al. Phase I, first-in-human study of futibatinib, a highly selective, irreversible FGFR1-4 inhibitor in patients with advanced solid tumors. Ann Oncol. 2020;31(10):1405–1412. doi:10.1016/j.annonc.2020.06.018
60. Rizzo A, Ricci AD, Brandi G. Futibatinib, an investigational agent for the treatment of intrahepatic cholangiocarcinoma: evidence to date and future perspectives. Expert Opin Investig Drugs. 2020:1–8.
61. Roskoski R
62. Krook MA, Bonneville R, Chen HZ, et al. Tumor heterogeneity and acquired drug resistance in FGFR2-fusion-positive cholangiocarcinoma through rapid research autopsy. Cold Spring Harb Mol Case Stud. 2019;5(4):a004002. doi:10.1101/mcs.a004002
63. Abou-Alfa GK, Sahai V, Hollebecque A, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol. 2020;21(5):671–684. doi:10.1016/S1470-2045(20)30109-1
64. Harris PA, Boloor A, Cheung M, et al. Discovery of 5-[[4-[(2,3-dimethyl-2H-indazol-6-yl)methylamino]-2-pyrimidinyl]amino]-2-methyl-b enzenesulfonamide (Pazopanib), a novel and potent vascular endothelial growth factor receptor inhibitor. J Med Chem. 2008;51(15):4632–4640. doi:10.1021/jm800566m
65. Kim ST, Jang HL, Lee SJ, et al. Pazopanib, a novel multitargeted kinase inhibitor, shows potent in vitro antitumor activity in gastric cancer cell lines with FGFR2 amplification. Mol Cancer Ther. 2014;13(11):2527–2536. doi:10.1158/1535-7163.MCT-14-0255
66. Sgouros J, Aravantinos G, Koliou GA, et al. First line gemcitabine/pazopanib in locally advanced and/or metastatic biliary tract carcinoma. A Hellenic Cooperative Oncology Group Phase II Study. Anticancer Res. 2020;40(2):929–938. doi:10.21873/anticanres.14026
67. Tan FH, Putoczki TL, Stylli SS, Luwor RB. Ponatinib: a novel multi-tyrosine kinase inhibitor against human malignancies. Onco Targets Ther. 2019;12:635–645. doi:10.2147/OTT.S189391
68. Borad MJ, Champion MD, Egan JB, et al. Integrated genomic characterization reveals novel, therapeutically relevant drug targets in FGFR and EGFR pathways in sporadic intrahepatic cholangiocarcinoma. PLoS Genet. 2014;10(2):e1004135. doi:10.1371/journal.pgen.1004135
69. Nicolini FE, Gagnieu M-C, Heiblig M, et al. Cardio-vascular events occurring on ponatinib in chronic phase chronic myeloid leukemia patients, preliminary analysis of a multicenter cohort. Blood. 2013;122(21):4020. doi:10.1182/blood.V122.21.4020.4020
70. Catenacci DVT, Rasco D, Lee J, et al. Phase I escalation and expansion study of bemarituzumab (FPA144) in patients with advanced solid tumors and FGFR2b-selected gastroesophageal adenocarcinoma. J Clin Oncol. 2020;38(21):Jco1901834. doi:10.1200/JCO.19.01834
71. Tabernero J, Bahleda R, Dienstmann R, et al. Phase I dose-escalation study of JNJ-42756493, an oral pan-fibroblast growth factor receptor inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2015;33(30):3401–3408. doi:10.1200/JCO.2014.60.7341
72. Bahleda R, Italiano A, Hierro C, et al. Multicenter Phase I study of erdafitinib (JNJ-42756493), oral pan-fibroblast growth factor receptor inhibitor, in patients with advanced or refractory solid tumors. Clin Cancer Res. 2019;25(16):4888–4897. doi:10.1158/1078-0432.CCR-18-3334
73. Goyal L, Saha SK, Liu LY, et al. Polyclonal secondary FGFR2 mutations drive acquired resistance to FGFR inhibition in patients with FGFR2 fusion-positive cholangiocarcinoma. Cancer Discov. 2017;7(3):252–263. doi:10.1158/2159-8290.CD-16-1000
74. Byron SA, Chen H, Wortmann A, et al. The N550K/H mutations in FGFR2 confer differential resistance to PD173074, dovitinib, and ponatinib ATP-competitive inhibitors. Neoplasia. 2013;15(8):975–988. doi:10.1593/neo.121106
75. Kim SY, Ahn T, Bang H, et al. Acquired resistance to LY2874455 in FGFR2-amplified gastric cancer through an emergence of novel FGFR2-ACSL5 fusion. Oncotarget. 2017;8(9):15014–15022. doi:10.18632/oncotarget.14788
76. Dalziel K. Isocitrate dehydrogenase and related oxidative decarboxylases. FEBS Lett. 1980;117(Suppl):K45–K55. doi:10.1016/0014-5793(80)80569-2
77. Kipp BR, Voss JS, Kerr SE, et al. Isocitrate dehydrogenase 1 and 2 mutations in cholangiocarcinoma. Hum Pathol. 2012;43(10):1552–1558. doi:10.1016/j.humpath.2011.12.007
78. Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462(7274):739–744. doi:10.1038/nature08617
79. Lu C, Ward PS, Kapoor GS, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483(7390):474–478. doi:10.1038/nature10860
80. Chowdhury R, Yeoh KK, Tian YM, et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011;12(5):463–469. doi:10.1038/embor.2011.43
81. Evans B, Griner E. Registered report: oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Elife. 2015;4:e07420. doi:10.7554/eLife.07420
82. Wang F, Travins J, DeLaBarre B, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340(6132):622–626. doi:10.1126/science.1234769
83. Turcan S, Rohle D, Goenka A, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483(7390):479–483. doi:10.1038/nature10866
84. Wang P, Dong Q, Zhang C, et al. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene. 2013;32(25):3091–3100. doi:10.1038/onc.2012.315
85. Farshidfar F, Zheng S, Gingras MC, et al. Integrative genomic analysis of cholangiocarcinoma identifies distinct IDH-mutant molecular profiles. Cell Rep. 2017;19(13):2878–2880. doi:10.1016/j.celrep.2017.06.008
86. Ross JS, Wang K, Gay L, et al. New routes to targeted therapy of intrahepatic cholangiocarcinomas revealed by next-generation sequencing. Oncologist. 2014;19(3):235–242. doi:10.1634/theoncologist.2013-0352
87. Borger DR, Tanabe KK, Fan KC, et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist. 2012;17(1):72–79. doi:10.1634/theoncologist.2011-0386
88. Rohle D, Popovici-Muller J, Palaskas N, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340(6132):626–630. doi:10.1126/science.1236062
89. Popovici-Muller J, Lemieux RM, Artin E, et al. Discovery of AG-120 (Ivosidenib): a first-in-class mutant IDH1 inhibitor for the treatment of IDH1 mutant cancers. ACS Med Chem Lett. 2018;9(4):300–305. doi:10.1021/acsmedchemlett.7b00421
90. Golub D, Iyengar N, Dogra S, et al. Mutant isocitrate dehydrogenase inhibitors as targeted cancer therapeutics. Front Oncol. 2019;9:417. doi:10.3389/fonc.2019.00417
91. Hansen E, Quivoron C, Straley K, et al. AG-120, an oral, selective, first-in-class, potent inhibitor of mutant IDH1, reduces intracellular 2HG and induces cellular differentiation in TF-1 R132H cells and primary human IDH1 mutant AML patient samples treated ex vivo. Blood. 2014;124(21):3734. doi:10.1182/blood.V124.21.3734.3734
92. Lowery MA, Burris HA
93. Ishii Y, Sigel C, Lowery MA, et al. editor AG-120 (ivosidenib), a first-in-class mutant IDH1 inhibitor, promotes morphologic changes and upregulates liver-specific genes in IDH1 mutant cholangiocarcinoma.
94. Ghassan K, Abou-Alfa TM, Javle M, editor ClarIDHy: a global, phase 3, randomized, double-blind study of ivosidenib vs placebo in patients with advanced cholangiocarcinoma with an isocitrate dehydrogenase 1 (IDH1) mutation; 2019.
95. Caravella JA, Lin J, Diebold RB, et al. Structure-based design and identification of FT-2102 (Olutasidenib), a potent mutant-selective IDH1 inhibitor. J Med Chem. 2020;63(4):1612–1623. doi:10.1021/acs.jmedchem.9b01423
96. Cho YS, Levell JR, Liu G, et al. Discovery and evaluation of clinical candidate IDH305, a brain penetrant mutant IDH1 inhibitor. ACS Med Chem Lett. 2017;8(10):1116–1121. doi:10.1021/acsmedchemlett.7b00342
97. DiNardo CD, Schimmer AD, Yee KWL, et al. A Phase I Study of IDH305 in patients with advanced malignancies including relapsed/refractory AML and MDS that harbor IDH1R132 mutations. Blood. 2016;128(22):1073. doi:10.1182/blood.V128.22.1073.1073
98. Yen K, Travins J, Wang F, et al. AG-221, a first-in-class therapy targeting acute myeloid leukemia harboring oncogenic IDH2 mutations. Cancer Discov. 2017;7(5):478–493. doi:10.1158/2159-8290.CD-16-1034
99. Chen J, Yang J, Cao P. The evolving landscape in the development of isocitrate dehydrogenase mutant inhibitors. Mini Rev Med Chem. 2016;16(16):1344–1358. doi:10.2174/1389557516666160609085520
100. Ma R, Yun CH. Crystal structures of pan-IDH inhibitor AG-881 in complex with mutant human IDH1 and IDH2. Biochem Biophys Res Commun. 2018;503(4):2912–2917. doi:10.1016/j.bbrc.2018.08.068
101. Saha SK, Gordan JD, Kleinstiver BP, et al. Isocitrate dehydrogenase mutations confer dasatinib hypersensitivity and SRC dependence in intrahepatic cholangiocarcinoma. Cancer Discov. 2016;6(7):727–739. doi:10.1158/2159-8290.CD-15-1442
102. Sulkowski PL, Corso CD, Robinson ND, et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci Transl Med. 2017;9:375. doi:10.1126/scitranslmed.aal2463
103. Molenaar RJ, Radivoyevitch T, Nagata Y, et al. IDH1/2 mutations sensitize acute myeloid leukemia to PARP inhibition and this is reversed by IDH1/2-mutant inhibitors. Clin Cancer Res. 2018;24(7):1705–1715. doi:10.1158/1078-0432.CCR-17-2796
104. Spizzo G, Puccini A, Xiu J, et al. Molecular profile of BRCA-mutated biliary tract cancers. ESMO Open. 2020;5(3):e000682. doi:10.1136/esmoopen-2020-000682
105. Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5(5):341–354. doi:10.1038/nrc1609
106. Sirica AE. Role of ErbB family receptor tyrosine kinases in intrahepatic cholangiocarcinoma. World J Gastroenterol. 2008;14(46):7033–7058. doi:10.3748/wjg.14.7033
107. Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2(2):127–137. doi:10.1038/35052073
108. Olayioye MA, Neve RM, Lane HA, Hynes NE. The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO j. 2000;19(13):3159–3167. doi:10.1093/emboj/19.13.3159
109. Oda K, Matsuoka Y, Funahashi A, Kitano H. A comprehensive pathway map of epidermal growth factor receptor signaling. Mol Syst Biol. 2005;1:
110. Sunpaweravong P, Sunpaweravong S, Puttawibul P, et al. Epidermal growth factor receptor and cyclin D1 are independently amplified and overexpressed in esophageal squamous cell carcinoma. J Cancer Res Clin Oncol. 2005;131(2):111–119. doi:10.1007/s00432-004-0610-7
111. Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol. 1995;19(3):183–232.
112. Pignochino Y, Sarotto I, Peraldo-Neia C, et al. Targeting EGFR/HER2 pathways enhances the antiproliferative effect of gemcitabine in biliary tract and gallbladder carcinomas. BMC Cancer. 2010;10:631. doi:10.1186/1471-2407-10-631
113. Nakazawa K, Dobashi Y, Suzuki S, Fujii H, Takeda Y, Ooi A. Amplification and overexpression of c-erbB-2, epidermal growth factor receptor, and c-met in biliary tract cancers. J Pathol. 2005;206(3):356–365. doi:10.1002/path.1779
114. Yoshikawa D, Ojima H, Iwasaki M, et al. Clinicopathological and prognostic significance of EGFR, VEGF, and HER2 expression in cholangiocarcinoma. Br J Cancer. 2008;98(2):418–425. doi:10.1038/sj.bjc.6604129
115. Leone F, Cavalloni G, Pignochino Y, et al. Somatic mutations of epidermal growth factor receptor in bile duct and gallbladder carcinoma. Clin Cancer Res. 2006;12(6):1680–1685. doi:10.1158/1078-0432.CCR-05-1692
116. Wiedmann M, Feisthammel J, Blüthner T, et al. Novel targeted approaches to treating biliary tract cancer: the dual epidermal growth factor receptor and ErbB-2 tyrosine kinase inhibitor NVP-AEE788 is more efficient than the epidermal growth factor receptor inhibitors gefitinib and erlotinib. Anticancer Drugs. 2006;17(7):783–795. doi:10.1097/01.cad.0000217433.48870.37
117. Kiguchi K, Ruffino L, Kawamoto T, Ajiki T, Digiovanni J. Chemopreventive and therapeutic efficacy of orally active tyrosine kinase inhibitors in a transgenic mouse model of gallbladder carcinoma. Clin Cancer Res. 2005;11(15):5572–5580. doi:10.1158/1078-0432.CCR-04-2603
118. Philip PA, Mahoney MR, Allmer C, et al. Phase II study of erlotinib in patients with advanced biliary cancer. J Clin Oncol. 2006;24(19):3069–3074. doi:10.1200/JCO.2005.05.3579
119. Lee J, Park SH, Chang HM, et al. Gemcitabine and oxaliplatin with or without erlotinib in advanced biliary-tract cancer: a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2012;13(2):181–188. doi:10.1016/S1470-2045(11)70301-1
120. Gruenberger B, Schueller J, Heubrandtner U, et al. Cetuximab, gemcitabine, and oxaliplatin in patients with unresectable advanced or metastatic biliary tract cancer: a phase 2 study. Lancet Oncol. 2010;11(12):1142–1148. doi:10.1016/S1470-2045(10)70247-3
121. Malka D, Cervera P, Foulon S, et al. Gemcitabine and oxaliplatin with or without cetuximab in advanced biliary-tract cancer (BINGO): a randomised, open-label, non-comparative phase 2 trial. Lancet Oncol. 2014;15(8):819–828. doi:10.1016/S1470-2045(14)70212-8
122. Van Cutsem E, Köhne CH, Hitre E, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360(14):1408–1417. doi:10.1056/NEJMoa0805019
123. Chen JS, Hsu C, Chiang NJ, et al. A KRAS mutation status-stratified randomized phase II trial of gemcitabine and oxaliplatin alone or in combination with cetuximab in advanced biliary tract cancer. Ann Oncol. 2015;26(5):943–949. doi:10.1093/annonc/mdv035
124. Leone F, Marino D, Cereda S, et al. Panitumumab in combination with gemcitabine and oxaliplatin does not prolong survival in wild-type KRAS advanced biliary tract cancer: a randomized phase 2 trial (Vecti-BIL study). Cancer. 2016;122(4):574–581. doi:10.1002/cncr.29778
125. Vogel A, Kasper S, Bitzer M, et al. PICCA study: panitumumab in combination with cisplatin/gemcitabine chemotherapy in KRAS wild-type patients with biliary cancer-a randomised biomarker-driven clinical phase II AIO study. Eur J Cancer. 2018;92:11–19. doi:10.1016/j.ejca.2017.12.028
126. Yamashita-Kashima Y, Yoshimura Y, Fujimura T, et al. Molecular targeting of HER2-overexpressing biliary tract cancer cells with trastuzumab emtansine, an antibody-cytotoxic drug conjugate. Cancer Chemother Pharmacol. 2019;83(4):659–671. doi:10.1007/s00280-019-03768-8
127. Nam AR, Kim JW, Cha Y, et al. Therapeutic implication of HER2 in advanced biliary tract cancer. Oncotarget. 2016;7(36):58007–58021. doi:10.18632/oncotarget.11157
128. Javle M, Churi C, Kang HC, et al. HER2/neu-directed therapy for biliary tract cancer. J Hematol Oncol. 2015;8:58. doi:10.1186/s13045-015-0155-z
129. Kawamoto T, Ishige K, Thomas M, et al. Overexpression and gene amplification of EGFR, HER2, and HER3 in biliary tract carcinomas, and the possibility for therapy with the HER2-targeting antibody pertuzumab. J Gastroenterol. 2015;50(4):467–479. doi:10.1007/s00535-014-0984-5
130. Yarlagadda B, Kamatham V, Ritter A, Shahjehan F, Kasi PM. Trastuzumab and pertuzumab in circulating tumor DNA ERBB2-amplified HER2-positive refractory cholangiocarcinoma. NPJ Precis Oncol. 2019;3:19. doi:10.1038/s41698-019-0091-4
131. Czink E, Heining C, Weber TF, et al. [Durable remission under dual HER2 blockade with Trastuzumab and Pertuzumab in a patient with metastatic gallbladder cancer]. Z Gastroenterol. 2016;54(5):426–430. doi:10.1055/s-0042-103498
132. Ramanathan RK, Belani CP, Singh DA, et al. A phase II study of lapatinib in patients with advanced biliary tree and hepatocellular cancer. Cancer Chemother Pharmacol. 2009;64(4):777–783. doi:10.1007/s00280-009-0927-7
133. Peck J, Wei L, Zalupski M, O’Neil B, Villalona Calero M, Bekaii-Saab T. HER2/neu may not be an interesting target in biliary cancers: results of an early phase II study with lapatinib. Oncology. 2012;82(3):175–179. doi:10.1159/000336488
134. Moehler M, Maderer A, Ehrlich A, et al. Safety and efficacy of afatinib as add-on to standard therapy of gemcitabine/cisplatin in chemotherapy-naive patients with advanced biliary tract cancer: an open-label, phase I trial with an extensive biomarker program. BMC Cancer. 2019;19(1):55. doi:10.1186/s12885-018-5223-7
135. Profiling differential responses to Pan-HER inhibition. Cancer Discov. 2017;7(6):Of12. doi:10.1158/2159-8290.CD-NB2017-052
136. Dokduang H, Jamnongkarn W, Promraksa B, et al. In vitro and in vivo anti-tumor effects of Pan-HER inhibitor varlitinib on cholangiocarcinoma cell lines. Drug Des Devel Ther. 2020;14:2319–2334. doi:10.2147/DDDT.S250061
137. Nam HJ, Kim HP, Yoon YK, et al. The irreversible pan-HER inhibitor PF00299804 alone or combined with gemcitabine has an antitumor effect in biliary tract cancer cell lines. Invest New Drugs. 2012;30(6):2148–2160. doi:10.1007/s10637-011-9782-6
138. Skaper SD. The biology of neurotrophins, signalling pathways, and functional peptide mimetics of neurotrophins and their receptors. CNS Neurol Disord Drug Targets. 2008;7(1):46–62. doi:10.2174/187152708783885174
139. Cocco E, Scaltriti M, Drilon A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol. 2018;15(12):731–747. doi:10.1038/s41571-018-0113-0
140. Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci. 2006;361(1473):1545–1564. doi:10.1098/rstb.2006.1894
141. Nakagawara A. Trk receptor tyrosine kinases: a bridge between cancer and neural development. Cancer Lett. 2001;169(2):107–114. doi:10.1016/S0304-3835(01)00530-4
142. Solomon JP, Linkov I, Rosado A, et al. NTRK fusion detection across multiple assays and 33,997 cases: diagnostic implications and pitfalls. Mod Pathol. 2020;33(1):38–46. doi:10.1038/s41379-019-0324-7
143. Demols A, Perez-Casanova L, Rocq L, et al. 71P NTRK gene fusions in bilio-pancreatic cancers. Ann Oncol. 2020;31:233. doi:10.1016/j.annonc.2020.04.057
144. Lamarca A, Barriuso J, McNamara MG, Valle JW. Molecular targeted therapies: ready for “prime time” in biliary tract cancer. J Hepatol. 2020;73(1):170–185. doi:10.1016/j.jhep.2020.03.007
145. Eguchi M, Eguchi-Ishimae M, Tojo A, et al. Fusion of ETV6 to neurotrophin-3 receptor TRKC in acute myeloid leukemia with t(12;15)(p13;q25). Blood. 1999;93(4):1355–1363. doi:10.1182/blood.V93.4.1355
146. Cook PJ, Thomas R, Kannan R, et al. Somatic chromosomal engineering identifies BCAN-NTRK1 as a potent glioma driver and therapeutic target. Nat Commun. 2017;8:15987. doi:10.1038/ncomms15987
147. Drilon A, Nagasubramanian R, Blake JF, et al. A next-generation TRK kinase inhibitor overcomes acquired resistance to prior TRK kinase inhibition in patients with TRK fusion-positive solid tumors. Cancer Discov. 2017;7(9):963–972. doi:10.1158/2159-8290.CD-17-0507
148. Amatu A, Sartore-Bianchi A, Bencardino K, Pizzutilo EG, Tosi F, Siena S. Tropomyosin receptor kinase (TRK) biology and the role of NTRK gene fusions in cancer. Ann Oncol. 2019;30(Suppl 8):viii5–viii15. doi:10.1093/annonc/mdz383
149. Rolfo C, Ruiz R, Giovannetti E, et al. Entrectinib: a potent new TRK, ROS1, and ALK inhibitor. Expert Opin Investig Drugs. 2015;24(11):1493–1500. doi:10.1517/13543784.2015.1096344
150. Kawamoto M, Ozono K, Oyama Y, Yamasaki A, Oda Y, Onishi H. The novel selective pan-TRK inhibitor ONO-7579 exhibits antitumor efficacy against human gallbladder cancer in vitro. Anticancer Res. 2018;38(4):1979–1986. doi:10.21873/anticanres.12435
151. Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;9(7):517–531.
152. Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;3(6):459–465. doi:10.1038/nrc1097
153. Brown MD, Sacks DB. Protein scaffolds in MAP kinase signalling. Cell Signal. 2009;21(4):462–469. doi:10.1016/j.cellsig.2008.11.013
154. Hazzalin CA, Mahadevan LC. MAPK-regulated transcription: a continuously variable gene switch? Nat Rev Mol Cell Biol. 2002;3(1):30–40. doi:10.1038/nrm715
155. Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26(22):3279–3290. doi:10.1038/sj.onc.1210421
156. Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004;5(11):875–885. doi:10.1038/nrm1498
157. Walter D, Hartmann S, Waidmann O. Update on cholangiocarcinoma: potential impact of genomic studies on clinical management. Z Gastroenterol. 2017;55(6):575–581. doi:10.1055/s-0043-102581
158. Ohashi K, Tstsumi M, Nakajima Y, Nakano H, Konishi Y. Ki-ras point mutations and proliferation activity in biliary tract carcinomas. Br J Cancer. 1996;74(6):930–935. doi:10.1038/bjc.1996.459
159. Voss JS, Holtegaard LM, Kerr SE, et al. Molecular profiling of cholangiocarcinoma shows potential for targeted therapy treatment decisions. Hum Pathol. 2013;44(7):1216–1222. doi:10.1016/j.humpath.2012.11.006
160. Goeppert B, Frauenschuh L, Renner M, et al. BRAF V600E-specific immunohistochemistry reveals low mutation rates in biliary tract cancer and restriction to intrahepatic cholangiocarcinoma. Mod Pathol. 2014;27(7):1028–1034. doi:10.1038/modpathol.2013.206
161. Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med. 2015;373(8):726–736. doi:10.1056/NEJMoa1502309
162. Silkin SV, Startsev SS, Krasnova ME, et al. Complete clinical response of BRAF-mutated cholangiocarcinoma to vemurafenib, panitumumab, and irinotecan. J Gastrointest Cancer. 2016;47(4):502–505. doi:10.1007/s12029-015-9792-2
163. Loaiza-Bonilla A, Clayton E, Furth E, O’Hara M, Morrissette J. Dramatic response to dabrafenib and trametinib combination in a BRAF V600E-mutated cholangiocarcinoma: implementation of a molecular tumour board and next-generation sequencing for personalized medicine. Ecancermedicalscience. 2014;8:479. doi:10.3332/ecancer.2014.479
164. Subbiah V, Lassen U, Élez E, et al. Dabrafenib plus trametinib in patients with BRAF(V600E)-mutated biliary tract cancer (ROAR): a phase 2, open-label, single-arm, multicentre basket trial. Lancet Oncol. 2020;21(9):1234–1243. doi:10.1016/S1470-2045(20)30321-1
165. Lee JK, Capanu M, O’Reilly EM, et al. A phase II study of gemcitabine and cisplatin plus sorafenib in patients with advanced biliary adenocarcinomas. Br J Cancer. 2013;109(4):915–919. doi:10.1038/bjc.2013.432
166. Bekaii-Saab T, Phelps MA, Li X, et al. Multi-institutional phase II study of selumetinib in patients with metastatic biliary cancers. J Clin Oncol. 2011;29(17):2357–2363. doi:10.1200/JCO.2010.33.9473
167. Bridgewater J, Lopes A, Beare S, et al. A phase 1b study of Selumetinib in combination with Cisplatin and Gemcitabine in advanced or metastatic biliary tract cancer: the ABC-04 study. BMC Cancer. 2016;16:153. doi:10.1186/s12885-016-2174-8
168. Ikeda M, Ioka T, Fukutomi A, et al. Efficacy and safety of trametinib in Japanese patients with advanced biliary tract cancers refractory to gemcitabine. Cancer Sci. 2018;109(1):215–224. doi:10.1111/cas.13438
169. Shroff RT, Yarchoan M, O’Connor A, et al. The oral VEGF receptor tyrosine kinase inhibitor pazopanib in combination with the MEK inhibitor trametinib in advanced cholangiocarcinoma. Br J Cancer. 2017;116(11):1402–1407. doi:10.1038/bjc.2017.119
170. Bendell JC, Javle M, Bekaii-Saab TS, et al. A phase 1 dose-escalation and expansion study of binimetinib (MEK162), a potent and selective oral MEK1/2 inhibitor. Br J Cancer. 2017;116(5):575–583. doi:10.1038/bjc.2017.10
171. Finn RS, Ahn DH, Javle MM, et al. Phase 1b investigation of the MEK inhibitor binimetinib in patients with advanced or metastatic biliary tract cancer. Invest New Drugs. 2018;36(6):1037–1043. doi:10.1007/s10637-018-0600-2
172. Kim JW, Lee KH, Kim JW, et al. Enhanced antitumor effect of binimetinib in combination with capecitabine for biliary tract cancer patients with mutations in the RAS/RAF/MEK/ERK pathway: phase Ib study. Br J Cancer. 2019;121(4):332–339. doi:10.1038/s41416-019-0523-5
173. Lowery MA, Bradley M, Chou JF, et al. Binimetinib plus gemcitabine and cisplatin Phase I/II Trial in patients with advanced biliary cancers. Clin Cancer Res. 2019;25(3):937–945. doi:10.1158/1078-0432.CCR-18-1927
174. Pacold ME, Suire S, Perisic O, et al. Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase gamma. Cell. 2000;103(6):931–943. doi:10.1016/S0092-8674(00)00196-3
175. Tuttle RL, Gill NS, Pugh W, et al. Regulation of pancreatic beta-cell growth and survival by the serine/threonine protein kinase Akt1/PKBalpha. Nat Med. 2001;7(10):1133–1137. doi:10.1038/nm1001-1133
176. Arcaro A, Guerreiro AS. The phosphoinositide 3-kinase pathway in human cancer: genetic alterations and therapeutic implications. Curr Genomics. 2007;8(5):271–306. doi:10.2174/138920207782446160
177. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129(7):1261–1274. doi:10.1016/j.cell.2007.06.009
178. Keniry M, Parsons R. The role of PTEN signaling perturbations in cancer and in targeted therapy. Oncogene. 2008;27(41):5477–5485. doi:10.1038/onc.2008.248
179. Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol. 2009;4:127–150. doi:10.1146/annurev.pathol.4.110807.092311
180. Kim RD, Alberts SR, Peña C, et al. Phase I dose-escalation study of copanlisib in combination with gemcitabine or cisplatin plus gemcitabine in patients with advanced cancer. Br J Cancer. 2018;118(4):462–470. doi:10.1038/bjc.2017.428
181. Jin L, Jin MH, Nam AR, et al. Anti-tumor effects of NVP-BKM120 alone or in combination with MEK162 in biliary tract cancer. Cancer Lett. 2017;411:162–170. doi:10.1016/j.canlet.2017.10.002
182. McRee AJ, Sanoff HK, Carlson C, Ivanova A, O’Neil BH. A phase I trial of mFOLFOX6 combined with the oral PI3K inhibitor BKM120 in patients with advanced refractory solid tumors. Invest New Drugs. 2015;33(6):1225–1231. doi:10.1007/s10637-015-0298-3
183. Bendell JC, Rodon J, Burris HA, et al. Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2012;30(3):282–290. doi:10.1200/JCO.2011.36.1360
184. Hyman DM, Snyder AE, Carvajal RD, et al. Parallel phase Ib studies of two schedules of buparlisib (BKM120) plus carboplatin and paclitaxel (q21 days or q28 days) for patients with advanced solid tumors. Cancer Chemother Pharmacol. 2015;75(4):747–755. doi:10.1007/s00280-015-2693-z
185. Sakamoto Y, Yamagishi S, Tanizawa Y, Tajimi M, Okusaka T, Ojima H. PI3K-mTOR pathway identified as a potential therapeutic target in biliary tract cancer using a newly established patient-derived cell panel assay. Jpn J Clin Oncol. 2018;48(4):396–399. doi:10.1093/jjco/hyy011
186. Corti F, Nichetti F, Raimondi A, et al. Targeting the PI3K/AKT/mTOR pathway in biliary tract cancers: a review of current evidences and future perspectives. Cancer Treat Rev. 2019;72:45–55. doi:10.1016/j.ctrv.2018.11.001
187. Tanjak P, Thiantanawat A, Watcharasit P, Satayavivad J. Genistein reduces the activation of AKT and EGFR, and the production of IL6 in cholangiocarcinoma cells involving estrogen and estrogen receptors. Int J Oncol. 2018;53(1):177–188. doi:10.3892/ijo.2018.4375
188. Wilson JM, Kunnimalaiyaan S, Kunnimalaiyaan M, Gamblin TC. Inhibition of the AKT pathway in cholangiocarcinoma by MK2206 reduces cellular viability via induction of apoptosis. Cancer Cell Int. 2015;15(1):13. doi:10.1186/s12935-015-0161-9
189. Ahn DH, Li J, Wei L, et al. Results of an abbreviated phase-II study with the Akt Inhibitor MK-2206 in patients with advanced biliary cancer. Sci Rep. 2015;5:12122. doi:10.1038/srep12122
190. Verzoni E, Pusceddu S, Buzzoni R, et al. Safety profile and treatment response of everolimus in different solid tumors: an observational study. Future Oncol. 2014;10(9):1611–1617. doi:10.2217/fon.14.31
191. Costello BA, Borad MJ, Qi Y, et al. Phase I trial of everolimus, gemcitabine and cisplatin in patients with solid tumors. Invest New Drugs. 2014;32(4):710–716. doi:10.1007/s10637-014-0096-3
192. Buzzoni R, Pusceddu S, Bajetta E, et al. Activity and safety of RAD001 (everolimus) in patients affected by biliary tract cancer progressing after prior chemotherapy: a phase II ITMO study. Ann Oncol. 2014;25(8):1597–1603. doi:10.1093/annonc/mdu175
193. Kim ST, Lee J, Park SH, et al. Prospective phase II trial of everolimus in PIK3CA amplification/mutation and/or PTEN loss patients with advanced solid tumors refractory to standard therapy. BMC Cancer. 2017;17(1):211. doi:10.1186/s12885-017-3196-6
194. Lau DK, Tay RY, Yeung YH, et al. Phase II study of everolimus (RAD001) monotherapy as first-line treatment in advanced biliary tract cancer with biomarker exploration: the RADiChol Study. Br J Cancer. 2018;118(7):966–971. doi:10.1038/s41416-018-0021-1
195. Rizell M, Andersson M, Cahlin C, Hafström L, Olausson M, Lindnér P. Effects of the mTOR inhibitor sirolimus in patients with hepatocellular and cholangiocellular cancer. Int J Clin Oncol. 2008;13(1):66–70. doi:10.1007/s10147-007-0733-3
196. Jung KS, Lee J, Park SH, et al. Pilot study of sirolimus in patients with PIK3CA mutant/amplified refractory solid cancer. Mol Clin Oncol. 2017;7(1):27–31. doi:10.3892/mco.2017.1272
197. Zhang KS, Zhou Q, Wang YF, Liang LJ. Inhibition of Wnt signaling induces cell apoptosis and suppresses cell proliferation in cholangiocarcinoma cells. Oncol Rep. 2013;30(3):1430–1438. doi:10.3892/or.2013.2560
198. Shinada K, Tsukiyama T, Sho T, Okumura F, Asaka M, Hatakeyama S. RNF43 interacts with NEDL1 and regulates p53-mediated transcription. Biochem Biophys Res Commun. 2011;404(1):143–147. doi:10.1016/j.bbrc.2010.11.082
199. Loregger A, Grandl M, Mejías-Luque R, et al. The E3 ligase RNF43 inhibits Wnt signaling downstream of mutated β-catenin by sequestering TCF4 to the nuclear membrane. Sci Signal. 2015;8(393):ra90. doi:10.1126/scisignal.aac6757
200. Ong CK, Subimerb C, Pairojkul C, et al. Exome sequencing of liver fluke-associated cholangiocarcinoma. Nat Genet. 2012;44(6):690–693. doi:10.1038/ng.2273
201. Rizzo A, Ricci AD, Tavolari S, Brandi G. Circulating tumor DNA in biliary tract cancer: current evidence and future perspectives. Cancer Genomics Proteomics. 2020;17(5):441–452. doi:10.21873/cgp.20203
202. Ettrich TJ, Schwerdel D, Dolnik A, et al. Genotyping of circulating tumor DNA in cholangiocarcinoma reveals diagnostic and prognostic information. Sci Rep. 2019;9(1):13261. doi:10.1038/s41598-019-49860-0
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.