")
Back to Journals » The Application of Clinical Genetics » Volume 15
Prominent Mutation of Intron 22 Inversion in Sporadic Hemophilia: Is It Worth the Antenatal Screening?
Authors Sasanakul W, Chuansumrit A , Sirachainan N , Kadegasem P
Received 23 February 2022
Accepted for publication 10 May 2022
Published 19 May 2022 Volume 2022:15 Pages 49—54
DOI https://doi.org/10.2147/TACG.S363132
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Martin Maurer
Werasak Sasanakul, Ampaiwan Chuansumrit, Nongnuch Sirachainan, Praguywan Kadegasem
Department of Pediatrics, Faculty of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok, Thailand
Correspondence: Ampaiwan Chuansumrit, Department of Pediatrics, Faculty of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok, 10400, Thailand, Tel +66 2 2011749, Fax +66 2 2011748, Email [email protected]
Background: Adequate replacement for patients with hemophilia is costly, especially in countries with limited resources.
Objective: Factor VIII gene mutations among Thai patients with hemophilia A were analyzed for the most common mutation. The cost-effectiveness of finding one female without family history of hemophilia possessing the most common factor VIII mutation was compared with the cost of treating one patient with hemophilia.
Methods: In all, 109 unrelated patients with hemophilia A, defined as sporadic cases (n=58) and hereditary cases (n=51), were enrolled for genotypic analysis.
Results: Intron 22 inversion was prominently found in 34 sporadic (58.6%) and 27 hereditary (51.9%) cases. The screening for intron 22 inversion among females without family history of hemophilia at antenatal care has been optionally suggested. A female with a positive result will undergo further prenatal diagnosis of hemophilia in her male offspring. On the contrary, a female with a negative test result remains at risk to have a hemophiliac son caused by other factor VIII gene mutations not included in the screening but the risk is not as high as intron 22 inversion. Although the screening of factor VIII mutation among females without family history of hemophilia is against the current practice, it has been initiated due to the inadequate treatment provided to patients with hemophilia in countries with limited resources. The study calculated approximately one female with intron 22 inversion would exist among 17,064 females without family history of hemophilia. The cost of screening (194,870 USD) was much less than that of treating one patient with hemophilia from birth to 40 years of age by the current regimen (378,000 USD).
Conclusion: Implementing antenatal screening of intron 22 inversion among females without family history of hemophilia is optionally suggested, especially in economically less-developed countries with inadequate treatment service for patients with hemophilia.
Keywords: intron 22 inversion, sporadic, hemophilia, hemophilia A, antenatal screening, female, cost-effectiveness
Introduction
Hemophilia A is one of the most common hereditary severe bleeding disorders, affecting 1 in 10,000 population globally.1 The condition is inherited by the X-linked recessive pattern mainly found among males while females act as carriers. The severity is defined by the levels of deficient factor VIII clotting activity (FVIII:C) as severe (<1 IU/dL), moderate (1–5 IU/dL) and mild hemophilia (>5–<40 IU/dL).2 Patients with severe degree hemophilia A exhibit frequent bleeding at the joints and muscles spontaneously, while moderate and mild degrees are related to minor and more severe trauma, respectively. However, some patients with moderate degree may exhibit similar bleeding manifestations as those with severe degree.3 Effective management involves providing adequate prophylaxis with either factor or nonfactor replacement therapy,4–7 which is costly for economically less-developed countries. Unfortunately, inadequate replacement therapy will lead to high mortality and morbidity rates. Some succumb to severe bleeding episodes during childhood and they seldom reach adulthood.8 Therefore, prevention through effective carrier detection among females in a hemophilia family followed by prenatal diagnosis constitutes an essential tool. In fact, a number of patients with hemophilia involve sporadic cases without family history of hemophilia.9 The new mutation occurs among patients or mothers themselves.
The study aimed to retrospectively analyze the causative factor VIII gene mutations among Thai patients with hemophilia A as well as their mothers to determine whether it would be cost-effective to screen for the commonly found mutation among females without family history of hemophilia at the antenatal setting.
Materials and Methods
A retrospective analysis was conducted among patients with hemophilia A attending the Division of Hematology, Department of Pediatrics, Faculty of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok, Thailand from 2016 to 2020. The study was approved by the Faculty Ethics Committee, Faculty of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok. The study was conducted following the Declaration of Helsinki and written informed consent was obtained from patients and parents.
Methods
Factor VIII Clotting Activity (FVIII:C)
The patients’ whole blood was mixed with 3.2% sodium citrate at the ratio of 9:1, immediately placed on ice and centrifuged at 1,800 g for 15 min to obtain platelet poor plasma. The FVIII:C was assayed within 4 hours or in plasma stored at –70℃ for less than 6 months before testing. The FVIII:C assay was based on one-stage activated partial thromboplastin time10 using the ACL 200 Coagulometer with commercial factor VIII deficient plasma purchased from Instrumentation Laboratories, Lexington, MA, USA.
DNA Extraction
DNA was extracted from 10 mL of EDTA blood using an in-house salting-out method,11 modified from a commercial kit. Briefly, 500 µL of buffy coat was mixed in equal volumes of 10 mM Tris-HCl, 320 mM sucrose, 5 mM MgCl2, 1% Triton X-100, pH 8.8 and spun at 20,000 g for 5 min to obtain the pellets. Then 340 µL of 400 mM Tris-HCl, 60 mM EDTA, 150 mM NaCl, 1% SDS, pH 8.0 and 100 µL of 3 M sodium acetate were added, vigorously mixed and incubated at 65℃ for 20 min. The DNA was extracted by adding 580 µL of the mixture of chloroform and isoamyl alcohol at the ratio of 24:1, gently mixed by tilting the tube up and down for 20 min and spinning at 20,000 g to obtain the supernatant. Finally, 880 µL of cold absolute ethanol was added to precipitate the DNA strands. After spinning at 20,000 g for 10 min, the DNA pellets were obtained, and were cleaned by adding 500 µL of 70% ethanol and spun at 20,000 g. The obtained DNA pellets were dissolved with sterile water to create the DNA solution.
Mutation Detection
Intron 22 inversion was the initial mutation detected using the inverse-shifting PCR technique by modifying the previously reported method.12 Five primers of 1U (CCTTTCAACTCCATCTCCAT), 2U (ACGTGTCTTTTGGAGAAGTC), 3U (CTCACATTGTGTTCTTGTAGTC), ID (ACATACGGTTTAGTCACAAGT) and ED (TCCAGTCACTTAGGCTCAG) were also obtained from the related report.12 On day 1, the patient’s genomic DNA (2 µg) was digested with 20 units of restriction enzyme Bcl 1 at 50℃ overnight in a volume of 50 µL. On day 2, the digested DNA was isolated using 100 µL of phenol and chloroform mixture. After vigorous mixing by tilting the tube up and down 60 times, the sample was spun at 9000 g for 10 min to obtain the supernatant. The DNA was extracted by adding the mixture of chloroform and iso-amyl alcohol as the mentioned procedure to obtain 116 µL of DNA solution. On day 3, DNA fragments were circularized with 1 unit of T4 ligase enzyme and 10x buffer of 13 µL in a total of 130 µL at 15℃ overnight. On day 4, the circularized DNA was isolated using the same method as on day 2 to obtain the final DNA solution of 36 µL. On day 5, PCR was performed on each 10 µL of circularized DNA as a diagnostic test with 0.6 µM of each primer of 1 U, 2 U, 3 U and ID and a complementary test with 0.6 µM of each primer of 1 U, 2 U, 3 U and ED. An additional PCR standard reagent included 1 unit of Taq, 0.2 mM of dNTPs, 1.5 mM MgCl2, buffer and sterile water to reach a total volume of 25 µL. Thermocycling involved 30 cycles of denaturation at 95℃ for 30 sec, annealing at 55℃ for 30 sec and extension at 72℃ for 30 sec; cycling was preceded by 95℃ for 5 min, followed by 10 min at 72℃. The inverse-shifting PCR products were run on 1.5% agarose gel electrophoresis with 50 base pairs (bp) as the ladder marker and stained with 1% ethidium bromide.
Each diagnostic and complementary test showed 3 different bands of 487, 333 and 385 bp; and 559, 457 and 405 bp, respectively. The interpretation of male and female with wild type and intron 22 inversion was based on the visualized bands. The male and female with wild type had 487, 457 and 405 bp. The male with distal type of intron 22 inversion had 333, 559 and 457 bp while the female carrier had 487,333, 559,457 and 405 bp. The male with proximal type of intron 22 inversion had 385, 559 and 405 bp while the female carrier had 487, 385, 559, 457 and 405 bp. The male with duplication of intron 22 and female carrier had 487, 559,457 and 405 bp. Lastly, the male with deletion of intron 22 had 333 and 457 bp or 385 and 405 bp while the female carrier had 487,333, 457 and 405 bp or 487,385,457 and 405 bp. For patients without intron 22 inversion, all exons were amplified and sequenced subsequently.13 Finally, intron 1 inversion was searched for in patients without causative mutation using a similar procedure as intron 22 inversion with different primers.12
Statistical Analysis
The Chi-square test was used for discrete data and the level of significance was set at p <0.05.
Results
A total of 109 unrelated patients with hemophilia A (severe 88, moderate 20, mild 1) were enrolled in the study. They were defined as sporadic cases without family history (n=58) and hereditary cases from known hemophilia families (n=51). The causative factor VIII gene mutations included intron 22 inversion and other mutations including frame-shift, missense, nonsense, deletion and splice site mutations designated as non-intron 22 inversion, as shown in Table 1. The proportion of patients with intron 22 inversion among sporadic cases (34/58=58.6%) was slightly higher than those with hereditary cases (27/51=52.9%), but no statistically significant difference was found (p=0.09). The distal type of intron 22 inversion was more commonly found compared with those with proximal type but no duplication or deletion of intron 22 was found in the current study. However, deletion of 1 to 2 other exons up to a large deletion of 22 exons was found in a few patients.
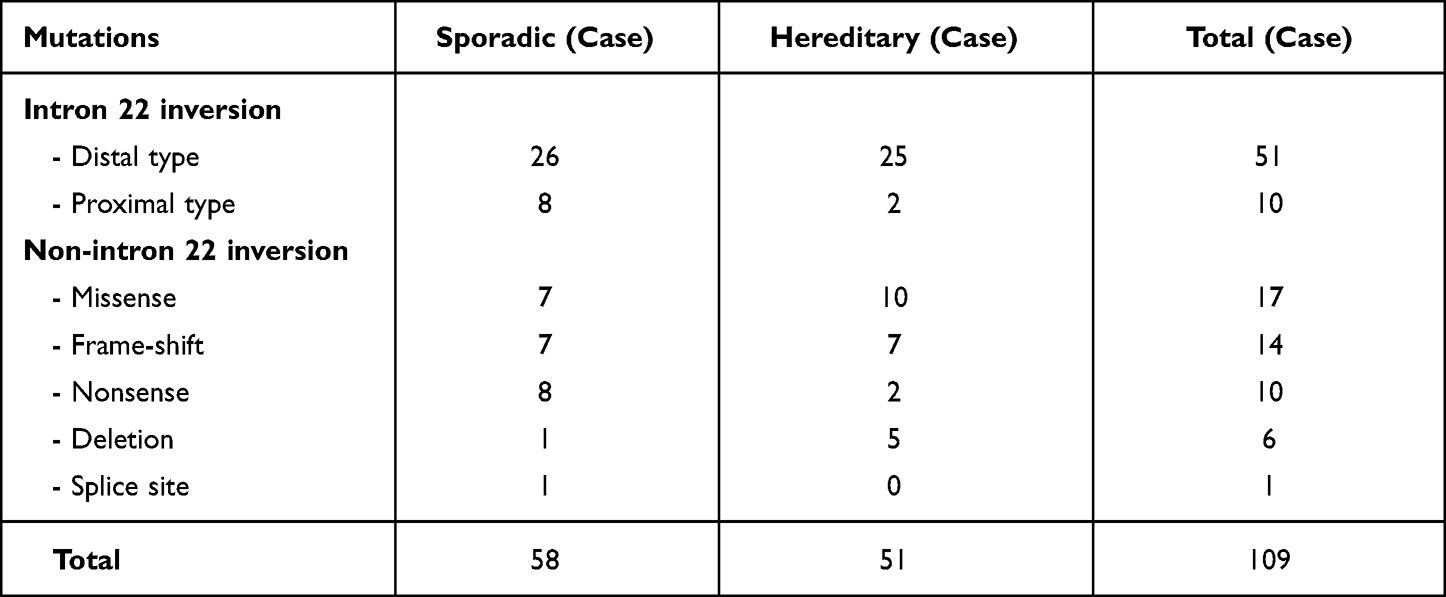 |
Table 1 Mutations on the Factor VIII Gene Among Studied Patients with Hemophilia A |
Only 86 mothers (86/109=78.9%), 50 from sporadic cases and 36 from hereditary cases, were available for the mutation analysis (54 intron 22 inversion, 32 non-intron 22 inversion), while 23 mothers were missing. The results revealed that seven mothers from sporadic cases (7/50=14.0%) had no mutation (Table 2). The proportion of mothers without intron 22 inversion (1/33=3%) was significantly lower than those without non-intron 22 inversion (6/17=35.3%), with a p-value of 0.001. One patient with the distal type of intron 22 inversion showed a moderate degree, with FVIII:C at 1.0 IU/dL, while all 6 patients with non-intron 22 inversion received a diagnosis of severe degree.
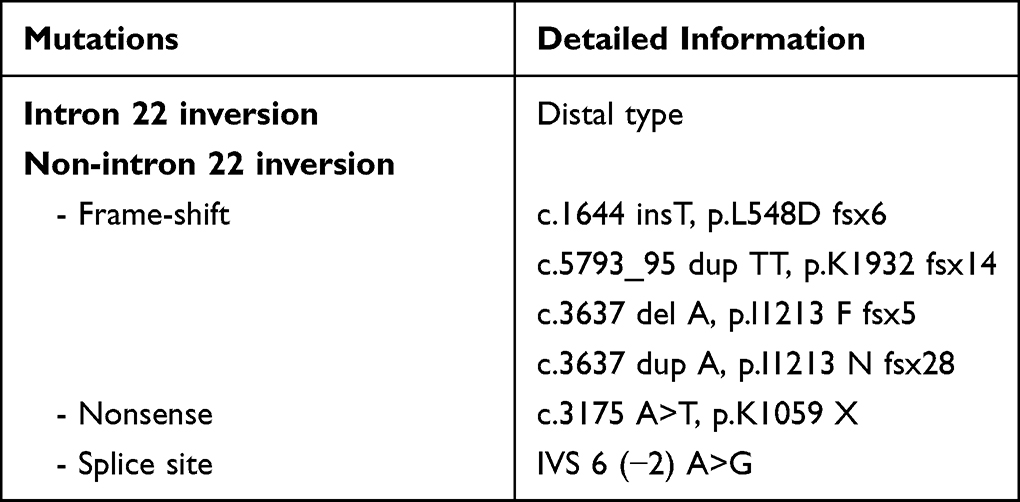 |
Table 2 Seven Mutations in the Sporadic Cases of Hemophilia Whose Mothers Had No Mutation Detected |
The study tabulated the possibility of finding one female possessing intron 22 inversion of 17,064 females without family history of hemophilia from the prevalence of hemophilia A of 1:10,000 and the occurrence of intron 22 inversion among sporadic cases of hemophilia A estimated at 58.6%. The cost of screening among females without family history of hemophilia was compared with the cost of used factor VIII concentrate provided to registered patients with moderate to severe degree hemophilia A by the National Health Security Office in Thailand from birth to 40 years of age, as shown in Table 3. Only episodic treatment and a low dose prophylaxis of 10 to 15 units /kg once to twice weekly have been provided to patients with hemophilia by the fixed budget. The cost of antenatal screening to find one female without family history of hemophilia possessing intron 22 inversion was much less than that of treating one patient with hemophilia. Thai patients with hemophilia are referred by the local provincial hospital in their hometown to hemophilia treatment centers located mostly at regional and university hospitals receiving a fixed budget from the government for laboratory investigation, medication and factor concentrate. The government subsidizes the outpatient and inpatient services; therefore, no service charge for the physician and medical personnel is required.
 |
Table 3 Comparison Between the Cost of Finding One Female with Intron 22 Inversion of 17,064 Females Without Family History of Hemophilia and That of Factor VIII Concentrate for One Patient with Hemophilia A |
Discussion
Hemophilia is one of the hereditary bleeding disorders involving serious bleeding manifestations. The X-linked recessive inheritance affects males while females act as carriers. However, up to 50% of patients with hemophilia constitute sporadic cases with recent mutations.9 In the current study, intron 22 inversion was shown as the prominent mutation frequently found in both sporadic and hereditary cases of severe and moderate hemophilia A similar to related studies.14–16 In addition, seven mothers from sporadic cases exhibited no mutation similar to their sons. They might have truly noncarrier status or somatic/gonadal mosaicism. In the case of noncarrier status in the mothers, the de novo mutation occurring in sporadic cases of hemophilia themselves was reported at 17.8% (13/73)17 to 27.6% (8/29),18 which was consistent with our current study at 14.0% (7/50). Interestingly, the number of mothers without intron 22 inversion was lower than that without non-intron 22 inversion, similar to the related reported study.18 The intron 22 inversion originates almost exclusively from the male germline19 by an event of homologous recombination between large and inverted repeat sequences. Almost all mothers of hemophiliac sons affected by intron 22 inversion are carriers of the intron 22 inversion.20
In clinical practice, women/girls from a family with hemophilia will receive genetic counseling and laboratory testing whether or not they are carriers of hemophilia. However, in economically less-developed countries, the treatment for patients with hemophilia remains limited. Only episodic treatment and low dose prophylaxis of 10 to 15 units/kg once to twice weekly are provided to Thai registered patients with moderate and severe hemophilia. They have to conduct themselves appropriately to avoid bleeding risk, and refrain from contact sports and high velocity activities. However, breakthrough bleeding was found at 3 to 6 episodes yearly, especially at the target joints. Therefore, antenatal screening for the most commonly found intron 22 inversion of the factor VIII gene among females without family history of hemophilia is optionally suggested. Even though it would be against current practice, it could be cost-effective for the test to be offered in economically constrained countries where affected patients with hemophilia are inadequately treated. Females with positive test results will receive similar care to those from a hemophiliac family receiving a diagnosis of hemophilia carrier status involving genetic counseling and prenatal diagnosis of hemophilia in her male offspring. Upon the prenatal diagnosis of hemophilia, information concerning natural course and available treatment will be provided for their decision of continuing or terminating the pregnancy. The female will benefit from receiving a diagnosis of carrier status of hemophilia before bearing a hemophiliac son. Otherwise, she will have a diagnosis of hemophilia carrier after bearing a hemophiliac son. This represents an effective tool to prevent a new case of hemophilia that should not constitute any social or ethical prejudice. On the contrary, females with negative test results remain at risk to have a hemophiliac son affected by other factor VIII mutations not included in the screening but the risk is not as high as intron 22 inversion. In cases of easy and frequent bruising among their sons, the investigation of congenital bleeding disorders including hemophilia should be determined without delay. Therefore, the comprehensive explanation should be provided to females and family members before screening to reassure that only the most common factor VIII gene mutation of intron 22 inversion will be screened for.
Furthermore, the laboratory technique to determine intron 22 inversion is available using the polymerase chain reaction in facilities of a comprehensive treatment center costing 11.42 USD per test. The EDTA whole blood of 3 mL can be sent by mail without ice to the comprehensive center21,22 for convenient molecular analysis.
The limitation of the current study involved the study center acting as a referral center and receiving a large number of patients with severe hemophilia with fewer cases of moderate hemophilia and only one mild hemophilia case. It would be a selective bias to obtain a higher proportion of patients with intron 22 inversion.
In conclusion, finding one female without family history of hemophilia carrying the hidden risk of intron 22 inversion was cost-effective compared with the cost of treating one patient with hemophilia by calculation in the country with inadequate treatment for patients with hemophilia. Implementing antenatal screening of intron 22 inversion for females without family history of hemophilia is optionally suggested.
Data Sharing Statement
The data from the findings of the study are available from the corresponding author upon reasonable request.
Author Contributions
All authors made a significant contribution to the work reported, whether that was in the conception, study design, execution, acquisition of data, analysis and interpretation, and in all these areas; took part in drafting, revised the article; provided final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agreed to be accountable for all aspects of the work.
Disclosure
The authors state that they have no interests which might be perceived as posing a conflict or bias.
References
1. Iorio A, Stonebraker JS, Chambost H, et al. Establishing the prevalence and prevalence at birth of hemophilia in males: a meta-analytic approach using national registry. Ann Intern Med. 2019;17(8):540–546. doi:10.7326/M19-1208
2. Blanchette VS, Key NS, Ljung LR, Manco-Johnson MJ, van den Berg HM, Srivastava A. Subcommittee on factor VIII, factor IX and rare coagulation disorders of the scientific and standardization committee of the international society on thrombosis and hemostasis. definitions in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost. 2014;12(11):1935–1939. doi:10.1111/jth.12672
3. Di Minno MND, Ambrosino P, Franchini M, Coppola A, Di Minno G. Arthropathy in patients with moderate hemophilia A: a systematic review of the literature. Semin Thromb Hemost. 2013;39(7):723–731. doi:10.1055/s-0033-1354422
4. Nilsson IM, Berntorp E, Lofqvist T, Petersson H. Twenty-five years’ experience of prophylactic treatment in severe haemophilia A and B. J Intern Med. 1992;232(1):25–32. doi:10.1111/j.1365-2796.1992.tb00546.x
5. Berntorp E, Andersson NG. Prophylaxis for hemophilia in the era of extended half-life factor VIII and factor IX products. Semin Thromb Hemost. 2016;42(5):518–525. doi:10.1055/s-0036-1571315
6. Weyand AC, Pipe SW. New therapies for hemophilia. Blood. 2019;133(5):389–398. doi:10.1182/blood-2018-08-872291
7. Shima M, Nogami K, Nagami S, et al. A multicenter, open-label study of emicizumab given every 2 or 4 weeks in children with severe haemophilia A without inhibitors. Haemophilia. 2019;25(6):979–989. doi:10.1111/hae.13848
8. Evatt BL. Demographics of hemophilia in developing countries. Semin Thromb Hemost. 2005;31(5):489–494. doi:10.1055/s-2005-922218
9. Ljung R, Kling S, Sjorin E, Nilsson IM. More than half of the sporadic cases of hemophilia A in Sweden are due to recent mutation. Acta Paediatr Scand. 1991;80(3):343–348. doi:10.1111/j.1651-2227.1991.tb11860.x
10. Hardisty RM, Macpherson JC. A one-stage factor VIII (anti-hemophilic globulin) assay and its use on venous blood and capillary plasma. Thromb Diath Haemorrh. 1962;7:215–228. doi:10.1055/s-0038-1655470
11. Sasanakul W, Chuansumrit A, Kadegasem P, Hathirat P. A comparison of DNA extraction between conventional phenol-chloroform and in-house modified method. Rama Med J. 1997;20:119–124.
12. Rossetti LC, Radic CP, Larripa IB, De Brasi CD. Developing a new generation of tests for genotyping hemophilia-causative rearrangements involving int22h and int1h hotspots in the factor VIII gene. J Thromb Haemost. 2008;6(5):830–836. doi:10.1111/j.1538-7836.2008.02926.x
13. Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74(12):5463–5467. doi:10.1073/pnas.74.12.5463
14. Lakich D, Kazazian HH Jr, Antonarakis SE, Gitschier J. Inversions disrupting the factor VIII gene are a common cause of severe haemophilia A. Nat Genet. 1993;5(3):236–241. doi:10.1038/ng1193-236
15. Antonarakis SE, Rossiter JP, Young M, et al. Factor VIII gene inversion in severe hemophilia A: results of an international consortium study. Blood. 1995;86(6):2106–2112. doi:10.1182/blood.V86.6.2206.bloodjournal8662206
16. Naylor J, Brinke A, Hassock S, Green PM, Giannelli F. Characteristic mRNA abnormality found in half the patients with severe hemophilia A is due to large DNA inversion. Hum Mol Genet. 1993;2(11):1773–1778. doi:10.1093/hmg/2.11.1773
17. Becker J, Schwaab R, Moller-Taube A, et al. Characterization of the factor VIII defect in 147 patients with sporadic hemophilia A: family studies indicate a mutation type-dependent sex ratio of mutation frequencies. Am J Hum Genet. 1996;58(4):657–670.
18. Ljung RC, Sjorin E. Origin of mutation in sporadic cases of haemophilia A. Br J Haematol. 1999;106(4):870–874. doi:10.1111/cge.12709
19. Rossiter JP, Young M, Kimberland ML, et al. Factor VIII gene inversion causing severe hemophilia A originates almost exclusively in male germ cells. Hum Mol Genet. 1994;3(7):1035–1039. doi:10.1093/hmg/3.7.1035
20. Tizzano EF, Domenech M, Baiget M. Inversion of intron 22 in isolated cases of severe hemophilia A. Thromb Haemost. 1995;73(1):6–9. doi:10.1055/s-0038-1651667
21. Sasanakul W, Chuansumrit A, Rurgkhum S, Udomsubpayakul U, Hathirat P. DNA extraction and amplification of 10-day, room-temperature blood samples. J Med Assoc Thai. 1999;82(Suppl 1):S186–S189.
22. Kadegasem P, Rurgkhum S, Sasanakul W, Chuansumrit A. DNA extraction from buffy coat sent by mail without ice. Thai J Hematol Transfu Med. 2001;11(3):167–171.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.