Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 17
Prohibitin Protects Pulmonary Microvascular Endothelial Cells Against Cigarette Smoke Extract-Induced Cell Apoptosis and Inflammation
Authors Peng Y ![]() , Cheng W
, Cheng W ![]() , Duan J
, Duan J ![]() , Zhao Y
, Zhao Y ![]() , Zhou Z, Zhou A, Deng M
, Zhou Z, Zhou A, Deng M ![]() , Peng H
, Peng H ![]() , Ouyang R
, Ouyang R ![]() , Chen Y, Chen P
, Chen Y, Chen P ![]()
Received 29 October 2021
Accepted for publication 12 March 2022
Published 29 March 2022 Volume 2022:17 Pages 653—665
DOI https://doi.org/10.2147/COPD.S345058
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Richard Russell
Yating Peng,1– 3 Wei Cheng,1– 3 Jiaxi Duan,1– 3 Yiyang Zhao,1– 3 Zijing Zhou,1– 3 Aiyuan Zhou,1– 3 Minhua Deng,1– 4 Hong Peng,1– 3 Ruoyun Ouyang,1– 3 Yan Chen,1– 3 Ping Chen1– 3
1Department of Pulmonary and Critical Care Medicine, Second Xiang Ya Hospital, Central South University, Changsha, Hunan, 410011, People’s Republic of China; 2Institute of Respiratory Disease, Central South University, Changsha, Hunan, 410011, People’s Republic of China; 3Hunan Centre for Diagnosis and Treatment of Respiratory Disease, Changsha, Hunan, 410011, People’s Republic of China; 4Department of Respiratory, PLA Rocket Force Characteristic Medical Center, Beijing, 100088, People’s Republic of China
Correspondence: Ping Chen, Email [email protected]
Background: Prohibitin has been identified to play roles in cell survival and apoptosis. Here, this study aimed to clarify the role of prohibitin in cigarette smoke extract (CSE)-induced endothelial cell apoptosis.
Methods: The protein level of prohibitin was assessed by Western blot in lung tissues from emphysema and control mice. CSE-induced human pulmonary microvascular endothelial cells (hPMECs) were applied to mimic smoke-related cell apoptosis in vitro. Prohibitin was overexpressed in hPMECs with or without CSE. Mitochondrial function was analyzed by JC-1 staining and ATP assay kits. Oxidative stress was assessed by flow cytometry, fluorescence staining and immunocytochemistry. Apoptosis was analyzed by flow cytometry, Western blot and caspase-3 activity assays. In addition, the expression of inflammatory markers was assessed by Western blot and real-time polymerase chain reaction (PCR). The secretion of inflammatory cytokines was measured by ELISA.
Results: Prohibitin was downregulated in emphysema mouse tissues compared with control experiments. Consistently, CSE inhibited both the protein and RNA levels of prohibitin in hPMECs in a dose-dependent manner. Gain-of-function experiments indicated that CSE induced collapse of mitochondrial membrane potential (MMP) and loss of ATP, while prohibitin improved mitochondrial function. CSE induced robust ROS production and oxidative DNA damage, while prohibitin decreased this damage. Upregulation of prohibitin protected the apoptosis of hPMECs from CSE. Overexpression of prohibitin significantly reduced the levels of the main proinflammatory cytokines. Finally, prohibitin inhibited nuclear factor-kappa B (NF-κB) p65 accumulation and IκBα degradation induced by CSE.
Conclusion: The current findings suggest that CSE-mediated mitochondrial dysfunction, oxidative stress, cell apoptosis and inflammation in hPMECs were reduced by overexpression of prohibitin. We identified prohibitin as a novel regulator of endothelial cell apoptosis and survival in the context of CSE exposure.
Keywords: prohibitin, hPMECs, CSE, apoptosis, mitochondrial, inflammation
Introduction
Chronic obstructive pulmonary disease (COPD) is a progressive destructive lung disease with persistent chronic inflammation characterized by airflow limitation and severe respiratory failure.1 Cigarette smoke (CS) exposure is identified as a major risk factor for COPD. As COPD is frequently exacerbated and often associated with severe complications, the clinical prognosis of COPD patients remains poor.2 Early research into the pathogenesis of COPD focused on the contributions of injury to the extracellular matrix and pulmonary epithelial cells. There is a critical interdependence between alveolar epithelial and microvascular endothelial cells (ECs) in the maintenance of airspace structure, and loss of ECs within the lung directly contributes to emphysematous remodeling.3,4 More recently, CS-induced endothelial apoptosis has been linked to pulmonary lesions in COPD (especially emphysema) and systemic comorbidities, including atherosclerosis, pulmonary hypertension, and chronic renal injury.4 In addition to apoptosis, persistent inflammation, protease–antiprotease imbalance, and oxidative stress in ECs also contribute to COPD.5–7 Chronic smoking exposes the respiratory tree and lungs to reactive oxygen species (ROS), resulting in oxidative stress and injury. Long-term exposure to CS increases ROS levels, decreases total antioxidant capacity, and interferes with DNA repair capacity, which eventually induces oxidative DNA damage.8,9 Simultaneously, compared with control subjects, COPD patients experience an overall heightened state of systemic inflammation reflected by biomarkers, such as cytokines and nitric oxide, which further increase with exacerbations.8
Prohibitin is a highly conserved and pleiotropic protein that is ubiquitously expressed in various compartments of eukaryotes, including the mitochondria, nucleus and plasma membrane. The main function of prohibitin in primary mammalian cells is to regulate nuclear transcription and mitochondrial integrity. Multiple prohibitin1 (PHB) and prohibitin2 subunits form ring-like prohibitin complexes in the inner mitochondrial membrane (IMM) and maintain the mitochondrial cristae structure. Therefore, it is implicated in various cellular functions, including cell apoptosis, autophagy, senescence, and tumor suppression.10 Several studies have reported that ectopic expression of prohibitin protects cells and disease models from oxidative stress injury by regulating mitochondrial function.10,11 In COPD and non-COPD smokers, prohibitin protein and mRNA expression in lung tissue was significantly decreased compared to that in nonsmokers, and prohibitin expression levels were associated with the degree of airway obstruction.12 In ECs, the loss of prohibitin results in a block in electron transport at complex I and increased mitochondrial ROS generation and cellular senescence that culminates in the loss of several endothelial functions.13 However, whether prohibitin can protect pulmonary lung parenchyma cells from cigarette smoke extract (CSE)-induced cytotoxicity is not well understood.
In this study, we first investigated whether CSE treatment can induce changes in prohibitin expression in vivo and in vitro and second investigated whether prohibitin can protect hPMECs from CSE-induced EC death and inflammation. These results may provide us with a novel and potential therapeutic strategy for COPD.
Materials and Methods
Cell Culture
The hPMECs, purchased from Science Cell (Cat NO: CP3000), were used in this study. The cells were grown in endothelial culture medium containing 10% fetal bovine serum (FBS), and were routinely maintained at 37°C in a water-saturated atmosphere with 5% CO2. hPMECs at 60–80% confluence were exposed to medium or CSE, approximately total of 10 million passage 3–4 hPMECs were used in this study.
Preparation of CSE
CSE was prepared as previously reported with modification.14 Briefly, half cigarette without filter was burned for 30 sec and the smoke passed through 10 mL of ECs medium using a vacuum pump. This 100% CSE was adjusted to a pH of 7.4 and filtered through a 0.22 µm filter (Millipore), and the CSE was diluted to the concentration of 0%, 1%, 1.5%, 2% and 2.5% in each and added to ECs within 30 minutes of preparation. Each batch was finished by the same investigator and in the same environment. Cigarettes of a domestic brand were obtained from Changde Tobacco (Changde, Hunan, China).
Overexpression of Prohibitin Plasmid Vectors
Briefly, the human PHB cDNA was subcloned into the multiple cloning site of the shuttle plasmid pAdTrack-CMV. The purified recombinant plasmids were linearized and co-electroporated with pAdEasy-1 adenoviral backbone vector into Escherichia coli BJ5183. The complete adenovectors (Ad-PHB) and empty adenovectors (Ad) were packaged by transfecting 293 cells, where viral particles were further amplified, purified, and titered.
Western Blot Assay
The lung tissues were homogenized and hPMECs were harvested and lysed with lysis buffer for 30 min on ice. After centrifugation for 30 min at 4°C, the supernatants were collected. The protein concentration of lung tissues or hPMECs was determined by a bicinchoninic acid (BCA) protein assay kit (Pierce, USA). Equal amounts of protein were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, electrotransferred to polyvinylidene difluoride (PVDF) membranes. Membrane was blocked with 5% BSA and incubated with the primary antibodies, followed by HRP-conjugated goat anti-rabbit secondary antibodies. Human prohibitin, mtTFA, NF-κB p65, IκB-α (Cell Signaling Technology), GAPDH (Bioworld) and histone H3 (Bioworld) and mouse prohibitin, p65 (Proteintech), GAPDH (Bioworld) antibody was used in this study.
Isolation of RNA and Real-Time Polymerase Chain Reaction (PCR)
The total RNA was extracted using TRIzol reagent and Total RNA was then reverse-transcribed using the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific). Subsequently, real-time PCR was performed using a StepOne Plus real-time PCR system (Life Technologies, Carlsbad, CA, USA). The sequences of all primers used were:
 |
|
Detection of MMP
The hPMECs were washed with cold PBS and centrifuged at 1500rpm for 5min. 5×105 cells were suspended in 0.5 mL cell culture medium and dyed by 0.5mL JC-1 dyeing fluid. Cells were incubated at 37°C for 20 minutes. After the incubation, cells were centrifuged at 1200 rpm for 3 min, and supernatants were discarded. Cells were washed by JC-1 dyeing buffer 2 times, suspended by 0.5 mL JC-1 dyeing buffer and analyzed by flow cytometry.
ATP Contents Assay
Cellular ATP contents were assessed by ATP Assay kit (Beyotime, China). Briefly, cells were rinsed and lysed using ATP lysis buffer on ice. Samples were collected and centrifuged at 12,000 rpm for 10 min at 4°C to acquire supernatant. ATP detection working dilution were added and luminescence activity was measured immediately using luminometer. Standard curve of ATP measure was made in each assay. Subsequently, the intracellular ATP contents were normalized by the protein contents in each sample.
ROS Assay
The activity of ROS within the cell was determined using a standard 2’,7’-dichlorofluorescin diacetate (DCFDA) assay (Beyotime, China). Cells were incubated with DCFDA dye at 37°C for 20 min and washed three times with DMEM. Then, ROS were detected by fluorescence microscopy (Olympus, Japan) and flow cytometry (Beckham, USA).
Immunocytochemistry Staining
hPMECs on 6-chamber slides were treated and fixed with 4% paraformaldehyde at 4°C for 15 min and then permeabilised with 0.2% Triton X-100 in PBS at room temperature (RT) for 15 min. Cells were then incubated with primary antibodies against mouse 8-hydroxy-2’-deoxyguanosine (8-OHdG) (1:200) (ab48508, 1:200 dilution; Abcam, Cambridge, United Kingdom) at 4°C overnight. Subsequently, 100 µL/well working solution of goat anti-mouse secondary antibodies (1:300) was added and incubated at room temperature for 90 min. Then cells were stained with avidin-biotin-peroxidase complex and visualised with DAB. The stained slides were photographed using an inverted microscope (Olympus, Tokyo, Japan) at 200 × magnification.
Detection of Apoptosis
An Annexin V FITC Apoptosis Detection Kit II (BD Biosciences, San Jose, CA) was used to measure cell apoptosis. Briefly, hPMECs were collected and incubated with 5 μL Annexin V and 5 μL propidium iodide for 15 min in the dark at 25°C. The number of apoptotic cells was determined by flow cytometry. Initial gating for total hPMECs (P1) in 100,000–200,000 acquired events was performed. This gating percentage was 96–97%. Among P1 gated cells, hPMECs with positive or negative annexin V-propidium iodide staining were distinguished.
Caspase-3 Activity Assay
Caspase-3 activity was measured using caspase-3 activity assay kit (Beyotime, China) following the manufacturer’s protocol. Briefly, cells were resuspended in lysis buffer and centrifuged. Ac-DEVD-pNA and reaction buffer was added into cell lysis. After incubation at 37°C for 2h, absorbance was read on microplate reader at 405nm.
Enzyme Linked Immunosorbent Assay (ELISA)
Supernatants were collected and stored at 80°C. The levels of TNF-α, IL-6 and IL-1β were quantified by ELISA according to the manufacturer’s instructions (Boster, Wuhan, China) with a detection limits of 1000pg/mL, 300pg/mL and 300pg/mL.
Emphysema Mouse Model
Six 6-week-old male C57BL/6 J mice (Wt: 19.0 ± 21.0 g) were randomly enrolled in this study and randomly divided into two groups: CS exposure group and control group. The studies described here were approved by the Animal Research Ethics Board of Central South University (Chang Sha, Hunan, China) in accordance with the guidelines of the Chinese Council on Animal Care.
The glass box used for modeling was made by us and had a size of 69 cm × 47 cm × 38 cm, a round hole with a diameter of 1 cm at a density of 1 hole per 100 cm2 on the lid and 1 hole per 250 cm2 on four sides of the box. In the box, a partition with the same size holes at a density of 1 hole per 6 cm2 was placed in the middle of the box to divide it into two parts: the lower part was used for cigarette burning, and the upper part was used for animal exposure to smoke.15 First, six cigarettes were burned at the same time with the smoke lasting for 25 min. Second, the box was opened to let the animals rest for 5 min. Then, the first step was repeated. This process was considered one cycle of CS exposure. Before CS exposure, the animals were acclimatized to the system for 3 d. Mice were exposed for 2 cycles/day, 5 days/week for 4 weeks.16,17 Control animals were exposed to room air alone.
Lung Tissue Morphometry, Apoptosis Assay and Immunohistology
Lung tissue samples were fixed in 4% formaldehyde, cut into 3.5-mm-thick sections, and stained with hematoxylin and eosin (HE). Morphometry was quantified by the values of mean linear intercept (MLI) at a magnification of 100× as previously described.18 MLI was assessed by dividing the length of a line drawn across the section by the total number of intercepts encountered in 36 lines per sample, and 10 random fields per sample were observed by microscopy. The numbers of apoptotic cells in longitudinal lung sections from control and emphysema mice were quantified by In-situ Apoptosis Detection Kit (Nanjing Keygen Biotech, China).
Statistical Analysis
Data are reported as the mean ± SEM. Results were compared by 2-tailed Student’s t-test for 2 groups and two-way ANOVA. SPSS v16.0 (SPSS Inc., Chicago, IL) was used for analysis. Differences were considered statistically significant at P < 0.05.
Results
Increased Apoptosis and Inflammation and Decreased Expression of Prohibitin in an Emphysema Mouse Model
We exposed mice to CSE, and as expected, the MLI values were significantly increased in the emphysema mouse group compared with the control group (Figure 1A and B). Compared to that in the lungs of the control mouse group, apoptotic cells were increased in the emphysema mouse group (Figure 1C and D). In mice exposed to CSE, prohibitin protein levels were significantly decreased (Figure 1E and F). The expression of p65 was further elevated in CSE-exposed mouse lungs compared with control mice (Figure 1E and G).
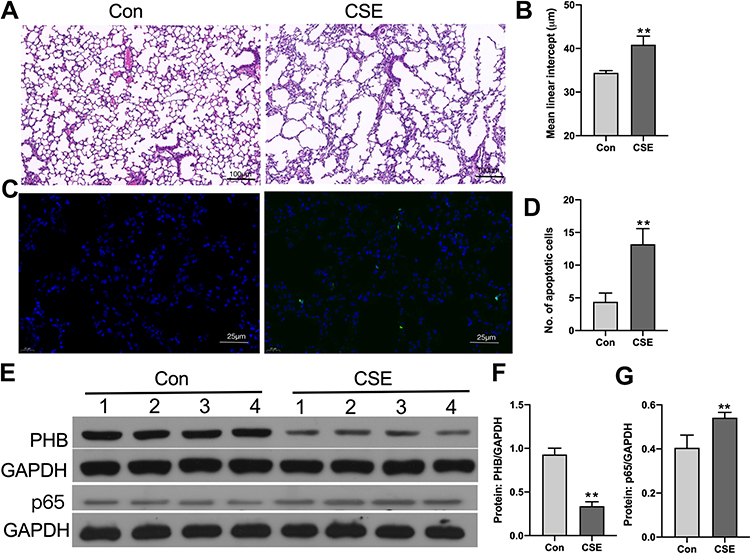 |
Figure 1 Identification of apoptosis and inflammation in the lung in an animal model of COPD. (A) Mouse lungs from the control and CS exposure groups after hematoxylin and eosin staining (10X, scale bar = 100 µm). (B) MLI of mice lungs (Student’s t-tests). (C) TUNEL staining in control and CS-exposed mouse lungs (40X, scale bar = 100 µm). (D) TUNEL-positive apoptotic cells in the control and CS exposure groups. (E) Prohibitin and p65 protein levels were determined by Western blot analysis. (F) Prohibitin protein levels were quantified by densitometry. (G) p65 protein levels were quantified by densitometry. *P < 0.05 vs control. **P < 0.01 vs control. |
Dose-Dependent Downregulation of Prohibitin in hPMECs by CSE
To evaluate whether CSE treatment affects the prohibitin level in vitro, we first treated hPMECs with control medium or 1%, 1.5%, 2% and 2.5% CSE for 12 h. Western blot analysis showed that the protein level of prohibitin was dose-dependently decreased when hPMECs were exposed to control medium or 1%, 1.5%, 2% and 2.5% CSE (Figure 2A and B). Further quantification of prohibitin mRNA levels in hPMECs was performed after 1%, 1.5%, 2% and 2.5% CSE treatment for 12 h. Treatment with CSE resulted in a robust decrease in prohibitin transcription (Figure 2C).
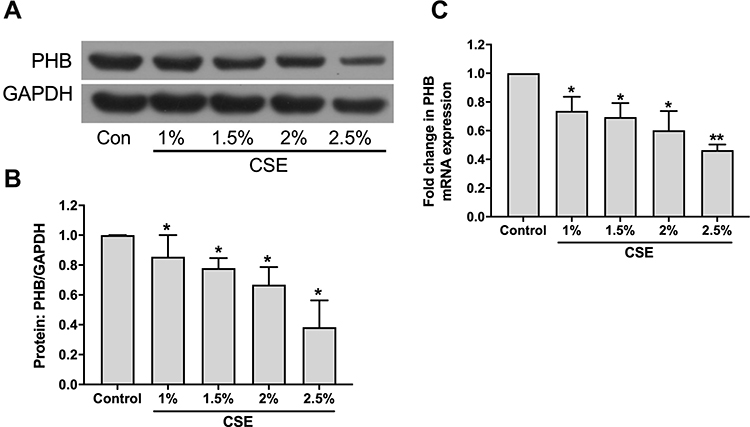 |
Figure 2 CSE treatment affects prohibitin expression of hPMECs. (A) hPMECs were treated with control medium or 1%, 1.5%, 2% and 2.5% CSE for 12 h. Prohibitin protein levels were determined by Western blot analysis. (B) Prohibitin protein levels were quantified by densitometry, and bar graphs represent three independent experiments (unpaired Student’s t-tests). (C) Relative mRNA levels were determined in hPMECs treated with control medium or 1%, 1.5%, 2% and 2.5% CSE for 12 h. *P < 0.05 vs control. **P < 0.01 vs control. |
PHB Suppressed the Collapse of the MMP and Loss of ATP Induced by CSE
Next, we explored the biological relevance of prohibitin upregulation in hPMECs after exposure to CSE. We successfully overexpressed prohibitin by transfecting hPMECs with a prohibitin plasmid. The protein expression of prohibitin was remarkably elevated at 24 and 48 hours after transfection with prohibitin, while transfection with empty vector did not show any change in the levels of prohibitin (Figure 3A). hPMECs transfected with an empty vector or a prohibitin plasmid were challenged with or without 2.5% CSE for 12 h.Prohibitin overexpression improved mitochondrial membrane potential (Figure 3B and C) and defects in ATP levels under CSE treatment (Figure 3D).
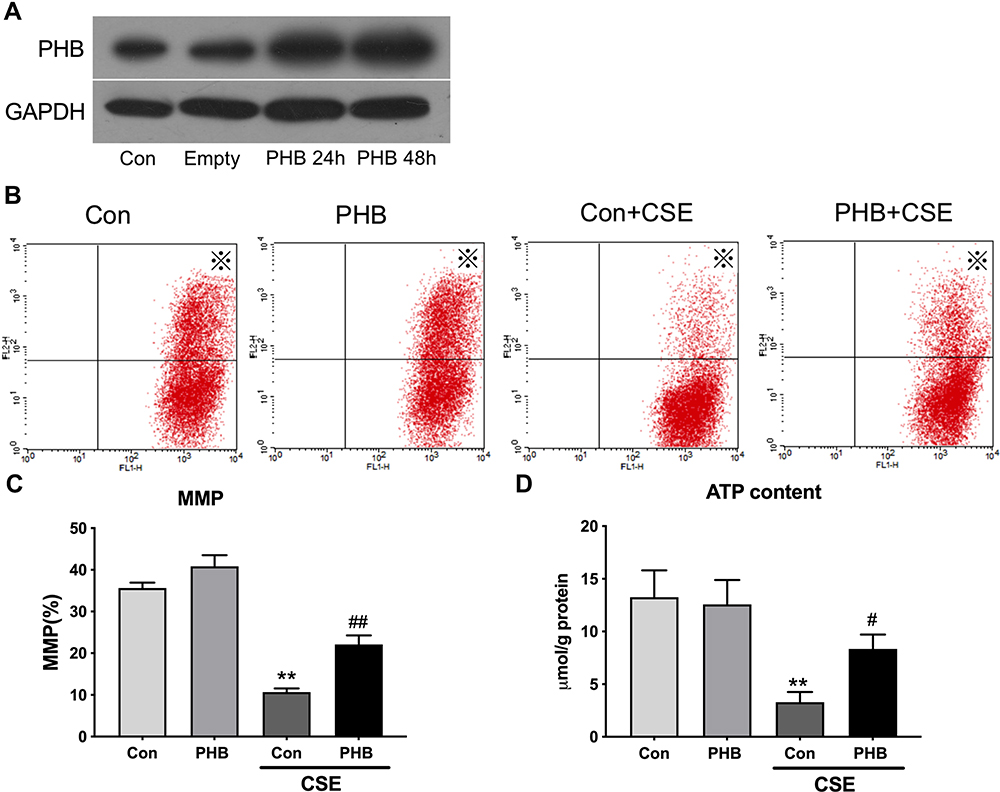 |
Figure 3 Prohibitin modulates changes in the Δψm and ATP content in hPMECs exposed to CSE. hPMECs were transfected with adenoviral PHB constructs for 2 h and cultured for 12 h or 48 h. (A) PHB content in hPMECs was detected by Western blot method a. hPMECs transfected with an empty vector or a prohibitin plasmid were challenged with or without 2.5% CSE for 12 h. (B) Representative cytometry plots of cells incubated with JC-1 probe (labeled with※). (C) Bar graph demonstrates the levels of MMP in different groups. (D) ATP content analysis in hPMECs with the indicated treatments. Bar graphs represent the results from three independent experiments. **P < 0.01 vs empty vector-transfected cells with control medium; ##P < 0.01, #P < 0.05 vs empty vector-transfected cells with CSE. |
Prohibitin Suppressed the Accumulation of Intracellular ROS and Oxidant-Induced DNA Damage Induced by CSE
To determine whether there was a functional consequence of the loss of MMP and ATP, we examined intracellular ROS levels. 12 We revealed that prohibitin overexpression inhibited intracellular ROS production under CSE treatment (Figure 4A, B, Dand E). Moreover, the CSE exposure group showed a remarkable amount of 8-OHdG in both the nucleus and cytoplasm. The results indicated that CSE could seriously damage DNA and that prohibitin overexpression mitigated the oxidative DNA damage associated with it (Figure 4C and F).
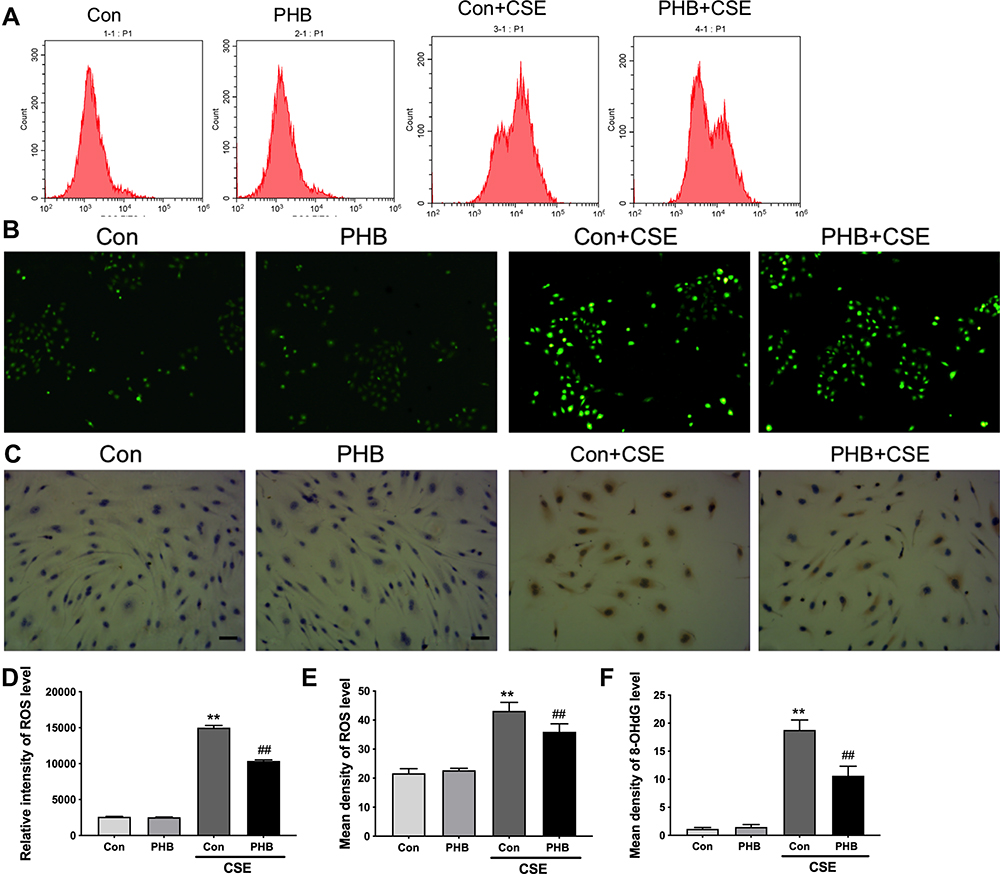 |
Figure 4 Prohibitin suppresses intracellular ROS in hPMECs exposed to CSE. hPMECs transfected with an empty vector or a prohibitin plasmid were challenged with or without 2.5% CSE for 12 h. (A) The level of ROS was determined by flow cytometry. (B) The level of ROS was determined by fluorescence microscopy. Original magnification×200. (C) Representative immunohistochemistry photographs of 8-OHdG expression. Original magnification×400. (D) Quantification of intensity by flow cytometry. (E) Quantification of density by fluorescence microscopy. (F) Quantification of 8-OHdG expression. **P < 0.01 vs empty vector-transfected cells with control medium; ##P < 0.01 vs empty vector-transfected cells with CSE. |
Prohibitin Attenuates Apoptosis Induced by CSE
We previously demonstrated that CSE treatment significantly triggered the apoptotic cascade response in hPMECs.19 After CSE treatment, the early and late apoptosis rates were significantly reduced in the prohibitin overexpression group compared to the control group (Figure 5A and B). CSE induced robust activation of cleaved caspase-3, while prohibitin suppressed this activation (Figure 5C). It has been shown that mtTFA, Bax and Bcl-2 plays an important role in regulating apoptosis. Prohibitin significantly upregulated the protein levels of mtTFA and downregulate the Bax/Bcl-2 ratio in hPMECs induced by CSE (Figure 5D–F). These results suggest the protective role of prohibitin in CSE-induced hPMEC apoptosis.
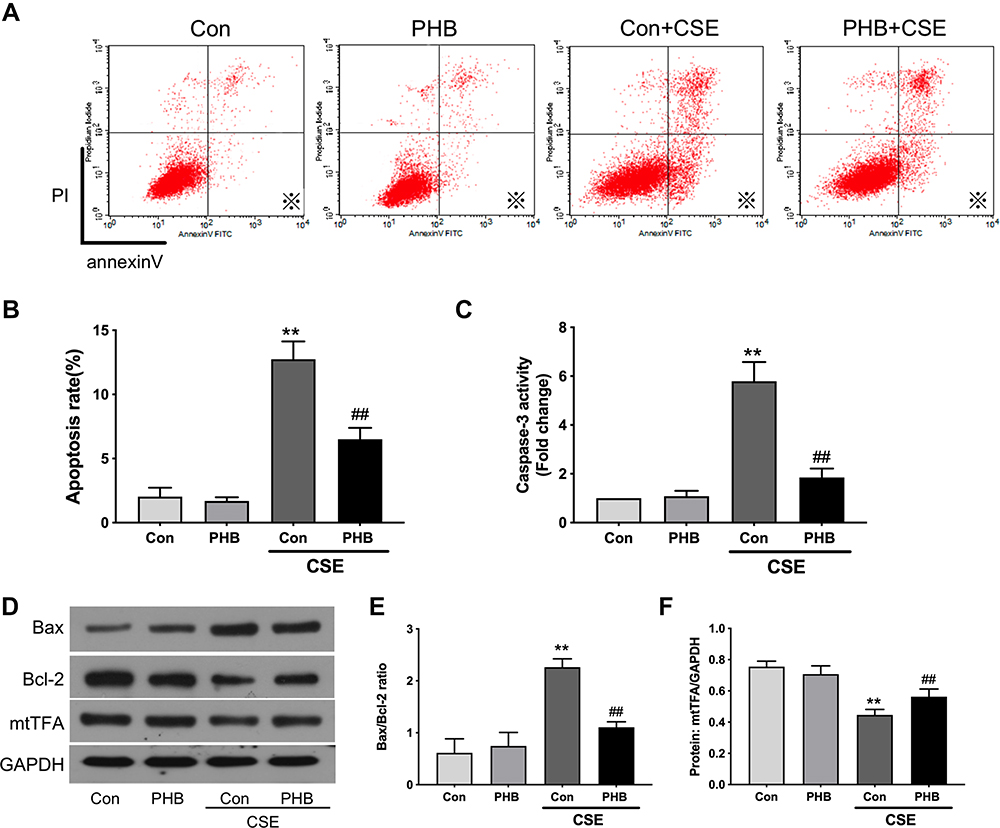 |
Figure 5 Prohibitin alleviates apoptosis of hPMECs exposed to CSE. hPMECs were transfected with an empty vector or prohibitin overexpression plasmid and then treated with or without 2.5% CSE for 12 h. (A) Representative pictures of apoptotic cells by flow cytometry. (B) Quantification of apoptotic cells by flow cytometry (labeled with※). (C) Cell lysates were harvested and immunoblotted for cleaved caspase-3. (D) Levels of mtTFA, Bax, Bcl-2 proteins were determined by Western blot. (E) The levels of Bax/Bcl-2 were quantified. (F) The levels of mtTFA were quantified by densitometry. The results are representative of three independent experiments. **P < 0.01 vs empty vector-transfected cells with control medium; ##P < 0.01 vs empty vector-transfected cells with CSE. |
Prohibitin Prevents Inflammation Induced by CSE
Inflammation is known to participate in the pathogenesis of emphysema,8,20 and proinflammatory cytokines have been reported to play critical roles in the pathogenesis of COPD.20 We demonstrated increased TNF-α, IL-6 and IL-1β contents in culture media (Figure 6A–C) and mRNA expression levels (Figure 6D–F) in hPMECs treated with CSE; however, they were reduced by prohibitin overexpression.
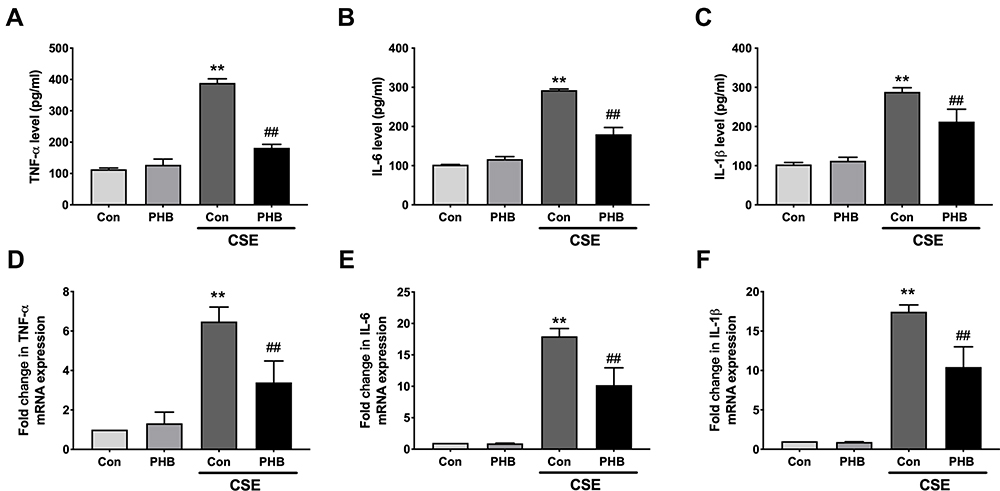 |
Figure 6 Prohibitin reduce transcription and release of pro-inflammatory cytokines in hPMECs exposed to CSE. hPMECs were transfected with an empty vector or prohibitin overexpression plasmid and then treated with or without 2.5% CSE for 12 h. The levels of (A) TNF-α, (B) IL-6 and (C) IL-1β in culture media was measured by ELISA.The levels of (D) TNF-α, (E) IL-6 and (F) IL-1β mRNA expression was determined by RT-PCR. **P < 0.01 vs empty vector-transfected cells with control medium; ##P < 0.01 vs empty vector-transfected cells with CSE. |
Prohibitin Inhibits NF-κB Activation Induced by CSE
Oxidative stress (eg, ROS) activates NF-κB/p65-mediated transcription of proinflammatory mediators through activation of its activating inhibitory kappa B kinase (IKK) in COPD.8 As expected, nuclear NF-κB/p65 was obviously elevated and IκB was reduced by CSE treatment. In contrast, prohibitin reduced the levels of nuclear NF-κB/p65 and inhibited IκBα degradation induced by CSE (Figure 7A–C). Therefore, prohibitin can inhibit the activation of the NF-κB pathway induced by CSE.
 |
Figure 7 Prohibitin inhibits NF-κB pathway induced by CSE in hPMECs. hPMECs were transfected with an empty vector or prohibitin overexpression plasmid and then treated with or without 2.5% CSE for 12 h. (A) Nuclear extracts were subjected to Western blot analysis for NF-κB p65, histone H3 and whole cell lysates were prepared for IκBα and GAPDH was used as loading control. The levels of (B) nuclear NF-κB p65, and (C) IκBα were quantifed by densitometry. **P < 0.01 vs empty vector-transfected cells with control medium; ##P < 0.01, #P < 0.05 vs empty vector-transfected cells with CSE. |
Discussion
A large number of research studies have shown the close relationship between cigarette smoke and the development of COPD.21,22 We and others have demonstrated that cigarette smoke, as a common inducer of COPD, can induce apoptosis, the inflammatory response, and oxidative stress in ECs, which aggravates lung injury.19,23–25
We first identified increased apoptosis and inflammation in our CS-induced emphysema mouse model. We then measured the levels of prohibitin after treatment with CSE. Both the protein and mRNA levels of prohibitin were reduced with CSE challenge. These data are consistent with the conclusion of studies based on decreased expression of prohibitin in lung tissue from COPD patients.12 These studies showed that prohibitin expression decreased in lungs from COPD as a result of CSE pathology. Previous studies have shown a negative effect of oxidative stress, such as hydrogen peroxide (H2O2), on the expression of prohibitin in human renal epithelial cells.26 In agreement with these studies, we discovered that both the protein and mRNA expression of prohibitin were decreased in ECs in a CSE dose-dependent manner, indicating a defect in the transcriptional regulation of prohibitin. However, as microRNAs may directly target prohibitin and phosphorylation of prohibitin plays a pivotal role in cell survival,27,28 a posttranslational deimination of prohibitin under CSE treatment cannot be excluded. In summary, we speculate that the downregulation of prohibitin in the lungs of patients with COPD may be involved in the pathophysiological process of emphysema.
We then tested how prohibitin affected CSE-induced endothelial injury. It is now well established that CS causes mitochondrial dysfunction, and restoration of mitochondrial function has been proven to be beneficial in various models of COPD.29 We observed a protective effect of prohibitin on the decline in ATP levels and MMP in hPMECs under CSE treatment. Prohibitin is a key regulator of mitochondrial quality control in multiple physiological and pathological conditions.30 As demonstrated by other studies, prohibitin prevented mitochondrial fragmentation, preserved mitochondrial respiratory function, and attenuated mitochondrial complex I oxidative degradation in cells exposed to oxidative stress.31 Our data provide evidence that CSE caused mitochondrial depolarization, decreased energy production and prohibitin modulation improved mitochondrial function in hPMECs under CSE treatments.
In addition to mitochondrial function, oxidative damage was also alleviated by prohibitin in CSE-treated hPMECs. Upon mitochondrial damage or dysfunction, mitochondrial pores open and allow the influx of potassium and calcium cations, thereby depolarizing the mitochondrial membrane, which in turn induces ROS production and release. Meanwhile, excessive ROS directly causes the collapse of MMP and depletion of ATP, which later activates a series of signaling pathways that induce apoptosis.32 The oxidative damage in DNA by CSE is either caused directly or through the generation of ROS.26,33,34 Several studies have shown that prohibitin acts as a coactivator for ARE-dependent gene expression and promotes endogenous antioxidant defense components under oxidative stress.26,33,34 Prohibitin interacts with the nicotinamide adenine dinucleotide hydrogen (NADH) dehydrogenase protein complex, which is essential for oxidoreductase activity and DNA repair within cells.35,36 Hence, prohibitin may act as an antioxidative agent by inhibiting CSE-induced oxidative damage and oxidative DNA damage.
It has been confirmed that CSE induces apoptosis via an intrinsic apoptotic pathway, which involves mitochondrial fragmentation and release of cytochrome c, as well as changed expression levels of pro-apoptotic and anti-apoptotic molecules.37,38 mtTFA, an important antiapoptotic agent,39 is responsible for both the transcription and maintenance of mitochondrial DNA40 and protects mtDNA from oxidative stress.41 Our group showed that the expression of mtTFA mRNA was downregulated in the lungs of CODP patients and that the expression of mtTFA was decreased by CSE as a consequence of hypermethylation of the mtTFA promoter.42 In this study, we observed a clear protective effect of prohibitin on the apoptosis of hPMECs insulted by CSE, and the protein level of mtTFA was significantly reversed with prohibitin overexpression. Importantly, the ring-shaped prohibitin complex in mitochondria may regulate mitochondrial dynamics, mitochondrial morphology, the mitochondrial genome, the electron transport chain (ETC), ROS homeostasis, and antiapoptotic proteins, which further prevent mitochondria-mediated apoptosis.10 Nuclear prohibitin colocalizes with many transcription factors and coordinately regulates transcription.10 Thus, prohibitin may stabilize mtTFA in endothelial cells and subsequently prevent apoptosis to cigarette smoke.
Inflammation was reported as a significant factor contributing to the destruction of pulmonary tissue in COPD.25 NF-κB p65 nuclear translocation subsequently regulates the transcription of many inflammatory cytokines. Cigarette smoke-mediated oxidative stress activates NF-κB-dependent transcription of proinflammatory mediators.6,22,24,30 In this study, we determined that prohibitin inhibited the translocation of NF-κB from the cytoplasm to the nucleus and degradation of IκBα under CSE treatment. Meanwhile, the transcription and release of proinflammatory cytokines (TNF-α, IL-6 and IL-1β) in hPMECs induced by CES was also decreased by prohibitin. These results indicate that the NF-κB pathway is essential in the anti-inflammatory effects of prohibitin against CSE.
There are several open questions worthy of future study. First, we did not explore the phosphorylation or other post transcriptional modification states of prohibitin, since these epigenetic changes are far more important than relative expression for cell function. Second, it is interesting to explore whether there is a protein–protein interaction between mtTFA and prohibitin. Third, whether changed NF-κB and mtTFA signaling could influence the protective effect of prohibitin on hPMECs under CSE is unclear.
Conclusion
In summary, prohibitin expression was downregulated significantly in the lung tissues of the emphysema mouse model. Prohibitin protected hPMECs from CSE-induced apoptosis, inflammation, oxidative stress and mitochondrial dysfunction, highlighting the contribution of prohibitin to the maintenance of EC homeostasis under CSE.
Abbreviations
CSE, cigarette smoke extract; hPMECs, human pulmonary microvascular endothelial cells; ROS, reactive oxygen species; mtTFA, mitochondrial transcription factor A; IMM, inner mitochondrial membrane; TNF-ɑ, tumor necrosis factor alpha; COPD, chronic obstructive pulmonary disease; Ad, adenovectors; BCA, bicinchoninic acid; PVDF, polyvinylidene difluoride; MMP, mitochondrial membrane potential; ELISA, enzyme linked immunosorbent assay; HE, hematoxylin and eosin; MLI, mean linear intercept; H2O2, hydrogen peroxide; NADH, nicotinamide adenine dinucleotide hydrogen; NF-κB, nuclear factor-kappa B.
Data Sharing Statement
The data used to support the findings of this study are available from the corresponding author upon request.
Ethical Approval and Consent to Participate
Ethical approval was approved by the Institutional Animal Care and Use Committee (IACUC), The second Xiangya Hospital, Central South University (NO.2021sydw0072).
Acknowledgments
This project supported by the National Key Clinical Specialist Construction Projects ((2012)No. 650), the National Natural Science Foundation of China (NSFC, Grants 81770046), (NSFC, Grants 81970044) and Xiangya Mingyi grant (2013).
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that there is no conflict of interest regarding the publication of this paper.
References
1. Vogelmeier CF, Criner GJ, Martinez FJ, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease 2017 report. GOLD executive summary. Am J Respir Crit Care Med. 2017;195(5):557–582. doi:10.1164/rccm.201701-0218PP
2. Criner RN, Han MK. COPD care in the 21st century: a public health priority. Respir Care. 2018;63(5):591. doi:10.4187/respcare.06276
3. Goel K, Beatman EL, Egersdorf N, et al. Sphingosine 1 Phosphate (S1P) receptor 1 is decreased in human lung microvascular endothelial cells of smokers and mediates S1P effect on autophagy. Cells. 2021;10(5):1200. doi:10.3390/cells10051200
4. Polverino F, Celli BR, Owen CA. COPD as an endothelial disorder: endothelial injury linking lesions in the lungs and other organs?(2017 Grover Conference Series). Pulm Circ. 2018;8(1):2045894018758528. doi:10.1177/2045894018758528
5. Fischer BM, Voynow JA, Ghio AJ. COPD: balancing oxidants and antioxidants. Int J Chron Obstruct Pulmon Dis. 2015;2(10):261–276. doi:10.2147/COPD.S42414
6. Cornwell WD, Kim V, Song C, et al. Pathogenesis of inflammation and repair in advanced COPD. Semin Respir Crit Care Med. 2010;31(03):257–266.
7. Fischer BM, Pavlisko E, Voynow JA. Pathogenic triad in COPD: oxidative stress, protease–antiprotease imbalance, and inflammation. Int J Chron Obstruct Pulmon Dis. 2011;6:413. doi:10.2147/COPD.S10770
8. Rahman I, Adcock IM. Oxidative stress and redox regulation of lung inflammation in COPD. Eur Respir J. 2006;28(1):219–242. doi:10.1183/09031936.06.00053805
9. Chen Z, Wang D, Liu X, et al. Oxidative DNA damage is involved in cigarette smoke-induced lung injury in rats. Environ Health Prev Med. 2015;20(5):318–324. doi:10.1007/s12199-015-0469-z
10. Peng YT, Chen P, Ouyang RY, et al. Multifaceted role of prohibitin in cell survival and apoptosis. Apoptosis. 2015;20(9):1135–1149. doi:10.1007/s10495-015-1143-z
11. Signorile A, Sgaramella G, Bellomo F, et al. Prohibitins: a critical role in mitochondrial functions and implication in diseases. Cells. 2019;8(1):71. doi:10.3390/cells8010071
12. Soulitzis N, Neofytou E, Psarrou M, et al. Downregulation of lung mitochondrial prohibitin in COPD. Respir Med. 2012;106(7):954–961. doi:10.1016/j.rmed.2012.03.019
13. Schleicher M, Shepherd BR, Suarez Y, et al. Prohibitin-1 maintains the angiogenic capacity of endothelial cells by regulating mitochondrial function and senescence. J Cell Biol. 2008;180(1):101–112. doi:10.1083/jcb.200706072
14. Shi Z, Chen Y, Pei Y, et al. The role of cyclooxygenase-2 in the protection against apoptosis in vascular endothelial cells induced by cigarette smoking. J Thorac Dis. 2017;9(1):30. doi:10.21037/jtd.2017.01.23
15. He ZH, Chen P, Chen Y, et al. Comparison between cigarette smoke-induced emphysema and cigarette smoke extract-induced emphysema. Tob Induc Dis. 2015;13:6. doi:10.1186/s12971-015-0033-z
16. Jobse BN, Rhem RG, Wang IQ, Counter WB, Stämpfli MR, Labiris NR. Detection of lung dysfunction using ventilation and perfusion SPECT in a mouse model of chronic cigarette smoke exposure. J Nucl Med. 2013;54(4):616–623. doi:10.2967/jnumed.112.111419
17. He S, Li L, Sun S, Zeng Z, Lu J, Xie L. A novel murine chronic obstructive pulmonary disease model and the pathogenic role of microRNA-21. Front Physiol. 2018;9:503. doi:10.3389/fphys.2018.00503
18. Zeng H, Li T, He X, et al. Oxidative stress mediates the apoptosis and epigenetic modification of the Bcl-2 promoter via DNMT1 in a cigarette smoke-induced emphysema model. Respir Res. 2020;21(1):1–14. doi:10.1186/s12931-020-01495-w
19. Zong D, Li J, Cai S, et al. Notch1 regulates endothelial apoptosis via the ERK pathway in chronic obstructive pulmonary disease. Am J Physiol Cell Physiol. 2018;315(3):C330–C340. doi:10.1152/ajpcell.00182.2017
20. Caramori G, Adcock IM, Di Stefano A, et al. Cytokine inhibition in the treatment of COPD. Int J Chron Obstruct Pulmon Dis. 2014;9:397. doi:10.2147/COPD.S42544
21. Eisner MD, Anthonisen N, Coultas D, et al. An official American Thoracic Society public policy statement: novel risk factors and the global burden of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;182(5):693–718. doi:10.1164/rccm.200811-1757ST
22. Pauwels NS, Bracke KR, Dupont LL, et al. Role of IL-1α and the Nlrp3/caspase-1/IL-1β axis in cigarette smoke-induced pulmonary inflammation and COPD. Eur Respir J. 2011;38(5):1019–1028. doi:10.1183/09031936.00158110
23. Long YJ, Liu XP, Chen SS, et al. miR-34a is involved in CSE-induced apoptosis of human pulmonary microvascular endothelial cells by targeting Notch-1 receptor protein. Respir Res. 2018;19(1):21. doi:10.1186/s12931-018-0722-2
24. Comer DM, Kidney JC, Ennis M, Elborn JS. Airway epithelial cell apoptosis and inflammation in COPD, smokers and nonsmokers. Eur Respir J. 2013;41(5):1058–1067. doi:10.1183/09031936.00063112
25. Gong J, Zhao H, Liu T, et al. Cigarette smoke reduces fatty acid catabolism, leading to apoptosis in lung endothelial cells: implication for pathogenesis of COPD. Front Pharmacol. 2019;10:941. doi:10.3389/fphar.2019.00941
26. Ye J, Li J, Xia R, et al. Prohibitin protects proximal tubule epithelial cells against oxidative injury through mitochondrial pathways. Free Radic Res. 2015;49(11):1393–1403. doi:10.3109/10715762.2015.1075654
27. Kang T, Lu W, Xu W, et al. MicroRNA-27 (miR-27) targets prohibitin and impairs adipocyte differentiation and mitochondrial function in human adipose-derived stem cells. J Biol Chem. 2013;288(48):34394–34402. doi:10.1074/jbc.M113.514372
28. Chiu CF, Ho MY, Peng JM, et al. Raf activation by Ras and promotion of cellular metastasis require phosphorylation of prohibitin in the raft domain of the plasma membrane. Oncogene. 2013;32(6):777. doi:10.1038/onc.2012.86
29. Maremanda KP, Sundar IK, Rahman I. Role of inner mitochondrial protein OPA1 in mitochondrial dysfunction by tobacco smoking and in the pathogenesis of COPD. Redox Biol. 2021;45:102055. doi:10.1016/j.redox.2021.102055
30. Rajendrasozhan S, Yang SR, Edirisinghe I, et al. Deacetylases and NF-κB in redox regulation of cigarette smoke-induced lung inflammation: epigenetics in pathogenesis of COPD. Antioxid Redox Signal. 2008;10(4):799–812. doi:10.1089/ars.2007.1938
31. Anderson CJ, Kahl A, Qian L, et al. Prohibitin is a positive modulator of mitochondrial function in PC 12 cells under oxidative stress. J Neurochem. 2018;146(3):235–250. doi:10.1111/jnc.14472
32. Mazat JP, Devin A, Ransac S. Modelling mitochondrial ROS production by the respiratory chain. Cell Mol Life Sci. 2020;77(3):455–465. doi:10.1007/s00018-019-03381-1
33. Yang H, Li TWH, Zhou Y, et al. Activation of a novel c-Myc-miR27-prohibitin 1 circuitry in cholestatic liver injury inhibits glutathione synthesis in mice. Antioxid Redox Signal. 2015;22(3):259–274. doi:10.1089/ars.2014.6027
34. Kathiria AS, Butcher MA, Hansen JM, et al. Nrf2 is not required for epithelial prohibitin-dependent attenuation of experimental colitis. Am J Physiol Gastrointest Liver Physiol. 2013;304(10):G885–G896. doi:10.1152/ajpgi.00327.2012
35. Neofytou E, Tzortzaki EG, Chatziantoniou A, et al. DNA damage due to oxidative stress in chronic obstructive pulmonary disease (COPD). Int J Mol Sci. 2012;13(12):16853–16864. doi:10.3390/ijms131216853
36. Fouquerel E, Sobol RW. ARTD1 (PARP1) activation and NAD+ in DNA repair and cell death. DNA Repair (Amst). 2014;23:27–32. doi:10.1016/j.dnarep.2014.09.004
37. Zeng H, Shi Z, Kong X, et al. Involvement of B-cell CLL/lymphoma 2 promoter methylation in cigarette smoke extract-induced emphysema. Exp Biol Med. 2016;241(8):808–816. doi:10.1177/1535370216635759
38. Xuan L, Shi J, Yao C, et al. Vam3, a resveratrol dimer, inhibits cigarette smoke-induced cell apoptosis in lungs by improving mitochondrial function. Acta Pharmacol Sin. 2014;35(6):779. doi:10.1038/aps.2014.17
39. Wang J, Silva JP, Gustafsson CM, et al. Increased in vivo apoptosis in cells lacking mitochondrial DNA gene expression. Proc Natl Acad Sci. 2001;98(7):4038–4043. doi:10.1073/pnas.061038798
40. Ekstrand M, Larsson NG. Breeding and genotyping of Tfam conditional knockout mice. Methods Mol Biol. 2002;197:391–400. doi:10.1385/1-59259-284-8:391
41. Dai XG, Li T, Huang WB, et al. Upregulation of mitochondrial transcription factor a promotes the repairment of renal tubular epithelial cells in sepsis by inhibiting reactive oxygen species-mediated toll-like receptor 4/p38MAPK signaling. Pathobiology. 2019;86:1–11.
42. Peng H, Guo T, Chen Z, et al. Hypermethylation of mitochondrial transcription factor A induced by cigarette smoke is associated with chronic obstructive pulmonary disease. Exp Lung Res. 2019;45(3–4):101–111. doi:10.1080/01902148.2018.1556748
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.