Back to Journals » Degenerative Neurological and Neuromuscular Disease » Volume 9
Progressive Multifocal Leukoencephalopathy: Current Insights
Authors Kartau M, Sipilä JOT ![]() , Auvinen E, Palomäki M, Verkkoniemi-Ahola A
, Auvinen E, Palomäki M, Verkkoniemi-Ahola A
Received 29 August 2019
Accepted for publication 14 November 2019
Published 2 December 2019 Volume 2019:9 Pages 109—121
DOI https://doi.org/10.2147/DNND.S203405
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Müller
Marge Kartau,1 Jussi OT Sipilä,2–4 Eeva Auvinen,5 Maarit Palomäki,6 Auli Verkkoniemi-Ahola1
1Clinical Neurosciences, Neurology, Helsinki University Hospital and Helsinki University, Helsinki, Finland; 2Department of Neurology, Siun Sote, North Carelia Central Hospital, Joensuu, Finland; 3Division of Clinical Neurosciences, Turku University Hospital, Turku, Finland; 4Clinical Neurosciences, University of Turku, Turku, Finland; 5Department of Virology, University of Helsinki and Helsinki University Hospital, Helsinki, Finland; 6Neuroradiology, HUS Medical Imaging Center, Helsinki, Finland
Correspondence: Marge Kartau
Clinical Neurosciences, Neurology, Helsinki University Hospital and Helsinki University, Helsinki, Finland
Email [email protected]
Abstract: Cases of PML should be evaluated according to predisposing factors, as these subgroups differ by incidence rate, clinical course, and prognosis. The three most significant groups at risk of PML are patients with hematological malignancies mostly previously treated with immunotherapies but also untreated, patients with HIV infection, and patients using monoclonal antibody (mAb) treatments. Epidemiological data is scarce and partly conflicting, but the distribution of the subgroups appears to have changed. While there is no specific anti-JCPyV treatment, restoration of the immune function is the most effective approach to PML treatment. Research is warranted to determine whether immune checkpoint inhibitors could benefit certain PML subgroups. There are no systematic national or international records of PML diagnoses or a risk stratification algorithm, except for MS patients receiving natalizumab (NTZ). These are needed to improve PML risk assessment and to tailor better prevention strategies.
Keywords: progressive multifocal leukoencephalopathy, JC polyomavirus, monoclonal antibodies, HIV, multiple sclerosis, disease modifying therapies
Introduction
Progressive multifocal leukoencephalopathy (PML) is an opportunistic viral infection of the central nervous system (CNS) first described in 1958 by Åström and colleagues.1 PML, named after its pathological features, is a progressive multifocal disease of white matter, which can now be thought of as the classical form of PML. The disease is caused by the JC polyomavirus (JCPyV). The prerequisite for PML is profound suppression of cell-mediated immunity, whether associated with diseases, such as HIV or lymphoproliferative malignancies, or treatment with immunosuppressive or immunomodulatory therapies (multiple sclerosis or rheumatoid arthritis) or both (systemic lupus erythematosus). Epidemiological data for different PML subgroups is scarce and partly conflicting because there are no systematic national or international records of PML diagnoses and only a few population-based studies of PML incidence. During the 1980’s and 1990’s with the emergence of HIV in humans, PML was the most important opportunistic infection of the CNS in these patients and most PML cases occurred in this group. In recent times, PML has been increasingly diagnosed in patients treated with biological therapies such as mAbs which deplete lymphocytes or impede leukocytes trafficking into the CNS. Interest in PML increased in 2005 when its association with the multiple sclerosis (MS) drug natalizumab (NTZ) was discovered2 and MS patients have become an important population at possible risk of PML.3 NTZ provides a good example how the risk-benefit profile must be determined at the individual level. PML risk also affects certain other MS therapies to a smaller degree. Other mAbs, as well as chemotherapy needed to treat patients with malignant diseases or after solid organ (SOT) or hematopoietic stem cell transplantation (HSCT) are also associated with an increased risk of PML.4
PML diagnosis is based upon distinguishing neurological features, characteristic changes in brain magnetic resonance imaging (MRI), and the presence of JCPyV DNA in cerebrospinal fluid (CSF). Currently reconstitution of the immune system gives the best prognosis for this condition.
As the etiology of PML has changed, so have its diagnosis and management. The nature of immunosuppression underlying PML determines the prognosis, treatment, and the risk of PML immune reconstruction inflammatory syndrome (PML-IRIS). Detection of PML at an early stage, when the disease is asymptomatic and restricted, is associated with a better outcome and higher survival rate.5,6
JCPyV Infection
JCPyV is a member of the Polyomaviridae family and has a 5 kb double-stranded circular DNA genome. The initial route of infection is thought to be ingestion or respiratory inhalation. The virus is then transported into kidney epithelial cells, bone marrow and spleen, where it establishes life-long persistence. JCPyV also persists in the lymphocytes.
Primary asymptomatic infection usually occurs in childhood, but adult infections are also possible. Primary infection is caused by the so called “archetype” virus, where the non-coding control region (NCCR) has a certain block structure. Occasionally, in immunosuppressed but also in healthy individuals, asymptomatic reactivation of JCPyV may take place, and the virus is excreted in the urine. Upon active viral replication in immunosuppressed individuals rearrangements in the viral genome may emerge, which mainly affect the NCCR but occasionally also the VP1 viral capsid protein. The archetype virus does not replicate efficiently in the brain, whereas the so called neurotropic variants harboring NCCR rearrangements can actively replicate in glial cells. Mutations in VP1 may additionally favor virus tropism for alternative cell populations, increasing the risk of PML. Mutations within the large T antigen and agnoprotein genes have also been reported in both PML7,8 and non-PML patients.9
Although most primary infections take place in childhood, the development of PML in childhood is extremely uncommon. JCPyV seroprevalence increases with age and reaches 90% in adults with occasional JCPyV shedding in urine within 19–27% of individuals.10 Each year 3% or less of the seronegative population becomes infected.11 The rarity of PML despite the widespread prevalence of JCPyV implies robust barriers to the development of the disease. Cell-mediated immunity is crucial for controlling JCPyV, as reflected by the high rates of the disease in advanced HIV infection, especially when the CD4+ lymphocyte count is below 100 cells/mm3. However, B cells and CD34+ progenitors also play roles in the pathophysiology, acting as viral reservoirs, and as a vector for viral dissemination in the CNS.12 B-cell depletion disrupts CD4- and CD8-positive T-cells homeostasis. Plasma cells regulate inflammatory T-cells activity via the immunocheckpoin pathways, thereby protecting the brain from excessive immune-mediated damage during active JCPyV infection.13 The role of anti-JCPyV antibodies is not yet completely understood. As more than half of PML patients are seropositive before the onset of PML, humoral immune responses seem insufficient to protect the patient from developing PML. Altogether higher antibody levels have been detected in patients before PML diagnosis as compared to patients who did not develop PML.14 Increase in anti-JCPyV antibody levels in NTZ treated patients prior to or coinciding with PML diagnosis has been suggested in some studies,15,16 possibly associated with virus reactivation. Other studies report stable high anti-JCPyV antibody levels prior to PML, although the authors considered the possibility that an increase in antibody levels may have been hidden by antibody assay saturation.17
PML is not the only disorder caused by JCPyV. Nephropathy with or without PML has been observed in renal allograft recipients.18 JCPyV can also infect meningeal and choroid plexus cells causing JCPyV meningitis (JCVM).19 There are reported cases of granule cell neuronopathy (JCVGCN) of the cerebellum.20 Fulminant JCPyV encephalopathy (JCE), involving cortical pyramidal neurons, is characterized by infection and lysis of cortical gray matter.21 JCPyV has been found in the brain of otherwise healthy individuals22 and therefore the presence of the virus is insufficient to make a diagnosis of PML.
Neuropathology
Pathologic features in PML include demyelination with the presence of foamy macrophages, relative preservation of axons, and astrogliosis, sometimes with atypical astrocytic nuclei and opale oligodendroglial nuclei. These oligodendroglial nuclei are filled with virus particles when viewed by electron microscopy, and the nuclei are consistently positive in immunohistochemistry using the monoclonal antibody to JCPyV, and in JCPyV in-situ hybridization (ISH). Neurons in the adjacent cortex can also become infected by the virus. Neurons in the adjacent cortex can also become infected by the virus.23
Epidemiology of PML
Population-based studies on PML epidemiology are scarce and long-term overall incidence trends are largely unknown (Table 1). Most papers report incidence in specific patient populations because PML was rare among patients not infected with HIV up to the mid-2000s,24 also the risk has decreased among HIV-patients after introduction of highly active antiretroviral therapy (HAART).25 A recent population-based Swedish study reported that after two decades of stable PML incidence of 0.026/100,000 person-years, the incidence has increased to 0.11 in 2011–2013, apparently related to the use of mAb therapies.26 Finnish population-based registry study investigating the period 2004–2014 reported an overall lower PML incidence of 0.072/100,000 person-years with no temporal incidence trend despite the increasing use of mAbs at the national level.27 In the most recent study from southern Finland in 2004–2016, we reported a PML-incidence of 0.12/100,000 person-years with no sign of increase.28 In this study the data was obtained from clinical records and the HIV Quality Registry. The discrepancy in incidence trends may be explained by differences in national circumstances and research methods. Nevertheless, they indicate that more studies from different regions and with in-depth analysis of PML causes are needed. Moreover, national PML surveillance and recording protocols would be helpful.
 |
Table 1 Incidence Rate of PML in Different Populations |
Underlying Diseases
Hematologic and Oncologic Conditions
PML was originally described in untreated patients with chronic lymphocytic leukemia (CLL) and Hodgkin’s disease.1 Currently patients with malignancies are the largest PML population,26,27 see Figure 1. PML can still occur in untreated patients but is more commonly associated with immunosuppressive or immunomodulatory treatments.29 Increased use of mAbs and HSCT correlate with rising PML incidence among patients with hematologic malignancies.30 Hematologic conditions associated with PML include CLL, Hodgkin’s disease, non-Hodgkin’s lymphoma, acute myeloid leukemia, acute lymphoblastic leukemia, Waldeström macroglobulinemia, multiple myeloma, polycythemia vera, and mycosis fungoides.28,29,31 Major PML risk factors are uncontrolled Hodgkin’s disease, treatment with purine analogues, and treatments related to HSCT.32 The prognosis is on the same level as in HIV-PML, but worse than in PML associated with autoimmune disorders.26,27
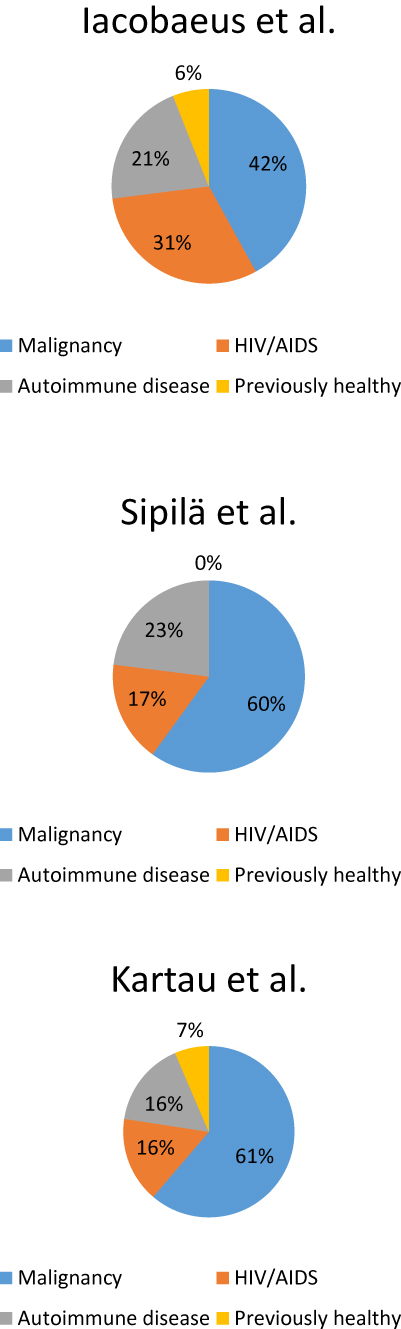 |
Figure 1 The distributions of PML etiologies. |
HIV Infection
Previously HIV was the underlying condition in approximately 80% of PML cases33 but in addition to improving survival, the HAART has reduced the incidence of HIV-PML34 (Table 1). Indeed, HIV’s share as the predisposing disease has been 16–31% in the recent Nordic studies (Figure 1). The median survival time of HIV-PML patients in Denmark increased from 0.4 years in 1995–1996 to 1.8 years in 1997–200635 and a recent study from Spain reported a 1-year survival of 55% (79% when CD4+ count was >100 cells/mm3 at diagnosis) and a 50% 3-year survival.25 Similarly, the recent 1-year survival rate of HIV-PML patients was 75% in Paris, France.36 In the recent Swedish study, HIV-PML was correlated with higher mortality compared to PML associated with autoimmune disease or a malignancy26 whereas the recent Finnish study reported similar survival in HIV-PML or a malignancy (66% at 1 year), albeit poorer than in PML associated with a connective tissue disorder.27 CD4+ lymphocyte counts of <200 cells/µl are a known PML risk factor in HIV patients although patients with CD4+ counts above that and/or receiving HAART may also be affected.37
Organ Transplantations
PML is a known complication associated with bone marrow transplants and SOT. The risk of PML among heart or lung transplant recipients is comparable to the PML risk associated with NTZ treatment for MS (Table 1).2,38 The most important risk factor for PML is immunosuppressive medication to prevent graft rejection.38 Median time from transplantation to onset of PML is 27 months in solid organ vs 11 months in bone marrow recipients. The fatality rate is high, around 84%, and the 1-year survival rate is 56%.38
PML has been described in patients with either autologous or allogenic stem cell transplantation.39,40 PML risk is present due to invasive conditioning regimens and prolonged immune suppression. Symptoms typically develop more than 1 month after HSCT and have been reported as late as several years post-transplantation. In the transplantation population (both SOT and HSCT), PML is mostly associated with fludarabine and RTX therapy.41
Autoimmune Diseases (Other Than MS)
The number of PML cases connected to autoimmune disease has been increasing since 2006.42 PML is usually ascribed to the drugs used to treat these diseases. The incidence rates are shown in Table 1.
Of all rheumatologic conditions, systemic lupus erythematosus (SLE) has the highest PML risk.43 Most SLE patients who develop PML have been subjected to, or are under immunosuppressant therapy. While the main cause of PML in SLE patients is therapy-related, it is possible that SLE may increase PML risk itself. A role of lymphopenia has been suggested.44
Other PML associated rheumatic diseases are vasculitis, most notably Wegener granulomatosis, poly- and dermatomyositis, scleroderma and rheumatoid arthritis (RA). There are reports of PML in patients with Sjögren syndrome and sarcoidosis with no prior immunomodulatory therapy, but with considerable lymphopenia.45 Rheumatological drugs that may cause PML are rituximab, methotrexate, leflunomide, and infliximab.46 In the recent nationwide studies, PML associated with an autoimmune disorder had a more favorable prognosis than PML associated with HIV or malignancy.26,27
Idiopathic Immune Deficiency Syndromes
PML has been described in patients with several types of primary immunodeficiency disorders (PIDs). PIDs are rare inherited disorders, characterized by a wide range of syndromic or genetic defects, mostly with combined B and T cell impairment. Idiopathic CD4+ T lymphocytopenia is the most commonly reported PID behind PML.47 Many PML cases are secondary to immunosuppressive treatments such as prolonged steroid therapy, RTX, azathioprine, or anti-TNF-α used to treat autoimmune, inflammatory, or proliferative PID complications.48 According to a study of 11 PID-PML patients, all patients but one died after a median of 8 months following PML diagnosis.48
PML in the Setting of Minimal or No Immunodeficiency
Some cases of PML have been reported with less overt immunodeficiency, such as chronic kidney and liver diseases, type 2 diabetes mellitus, dementia.28,49 There is one histologically proven PML case each concerning a pregnant woman, and a woman prior to pregnancy without any known underlying immunodeficiency.50,51 There is no epidemiological data suggesting that pregnant MS patients or healthy women have a higher incidence of PML.
PML in the Setting of mAb Therapy and Other Immunomodulating Drugs
Monoclonal antibody therapeutics have dramatically improved the therapy of chronic inflammatory diseases and cancers.52 Some mAbs repress the immune system for long periods, and predispose the patient to opportunistic infections. In addition to NTZ, other mAbs associated with PML are efalizumab and RTX. Efalizumab was withdrawn from the market in 2009 due to its association with PML. The incidence of PML due to RTX treatment is estimated to be 1 case per 32,000.4 There is an increased risk of PML in patients with SLE (1 case per 4000 individuals) and RA (1 case per 25,000 individuals) treated with rituximab (RTX).53 Many other mAbs have also been reported as risk factors of PML (Table 2).54 Patients with PML related to mAb treatment may have different clinical picture compared to classic PML. Once PML is identified, the mAb must be discontinued without delay and removed by plasmapheresis.
 |
Table 2 Drugs Associated with PML |
In addition to mAb, numerous other drugs are associated with PML (Table 2).54,55 These drugs have differences in the incidence rate of PML, the nature of the underlying diseases being treated, and the time from starting treatment to the development of PML.
Risk of PML is associated with several disease modifying therapies (DMTs) for MS (Table 3).56–60 The mode of action of DMTs plays a role in causing PML.61 The best studied DMT associated to PML is NTZ,61 which is a mAb that inhibits the transmembrane leukocyte receptor α-4 integrin. It prevents leukocyte adhesion to vessel walls and subsequent migration across the blood–brain barrier, resulting in immunosuppression within the CNS. The more the CNS compartment is immunocompromised, the greater the risk of PML. NTZ may also inhibit the retention of lymphocytes in bone marrow and spleen, thus leading to an increase of JCPyV-infected peripheral leukocytes and to a possible increase of the peripheral JCPyV load.62 As of September 3, 2019 there have been 825 confirmed PML cases (822 MS, 3 Crohn’s disease) according to latest Tysabry Safety Update information. The overall global incidence of PML in NTZ-treated patients is 4.08 per 1000 patients (95% CI 3.80 to 4.36 per 1000 patients). The duration of NTZ dosing prior to PML diagnosis ranged from 8 to 148 doses, and the mean duration of NTZ dosing at time of PML diagnosis was approximately 51 months (Tysabri Safety Update 09/2019, Biogen data on file). The mortality rate in NTZ-PML is 12–24%.63,64
 |
Table 3 PML Risk Under MS DMTs |
Major risk factors for NTZ-PML are the presence of serum antibodies to JCPyV, history of prior immunosuppression, and a length of NTZ treatment greater than 2 years. A recently acknowledged risk factor is a patient age over 50 years.65 In elderly patients, risk of lymphopenia is higher because of age-related immunosenescence, characterized by lower levels and functionality of lymphocytes. Lower anti-JCPyV antibody levels, or index values, are associated with lower PML risk in patients without previous immunosuppressant treatment group.66 For anti-JCPyV antibody-negative patients, estimated PML risk is less than 0.07 per 1000 patients. The PML risk is small at antibody index values of 0.9 or less, and increases with index values greater than 1.5 in people treated with NTZ for more than 2 years. The cumulative risk of PML begins to increase after two years of treatment and the increase remains constant from year three to year six. In anti-JCPyV antibody-positive patients, the risk of PML over 6 years is 2.7% in patients with previous immunosuppressant use and 1.7% in those without.66
Adjusted odds ratios of PML associated with DMTs in 2015–2017 were reported as follows: NTZ 115.72, fingolimod 4.98, RTX 3.22 and DMF 1.77.67 There is a potential risk of PML with ocrelizumab use.68 There are seven carry-over cases of PML with ocrelizumab and five reports suggestive of carry-over PML, though not meeting AAN diagnostic criteria.56,69 All five cases had MRI findings compatible with PML. In addition a confounded case of ocrelizumab monotherapy associated PML of 78-year old MS patient is reported.70 The incidence rate of PML with fingolimod is 3.12 per 100,000 patient-years.58 DMF-PML cases are usualy associated with drug-induced prolonged lymphopenia and age of ⩾50 years.57 However, recently a DMF-PML case of a 39-year patient without repeated lymphocyte counts below 800 cells/μL was reported.71 One case of MS patient`s PML has been described reported in association with alemtuzumab.59 Several PML cases have been described in lymphoma patients receiving polychemotherapy including alemtuzumab, but lymphoma is a PML risk factor itself as well.57 A few PML cases have been described in patients treated intravenously with cladribine,72 but there have been no cases of PML with oral cladribine.
Diagnosis of PML
According to the consensus statement from the American Academy of Neurology, PML diagnosis requires clinical, imaging, and virological evidence to make a definite, probable, or possible PML diagnosis.73 The highest level of diagnostic certainty requires histopathologic confirmation, though this is a highly invasive procedure. Therefore, the presence of clinical and/or imaging findings in combination with JCPyV DNA in CSF is also considered diagnostic but in the most challenging cases, brain biopsy may be performed. Brain biopsy has sensitivity of 64–96% and specificity of 100% in PML.74
PML Clinical Features
The clinical presentation of PML is heterogeneous. The symptoms result from brain lesions in corresponding areas.75 Clinical symptoms are not mandatory in the diagnosis of confirmed PML, because the disease may be asymptomatic at the beginning.76 Hemiparesis, ataxia, gait disturbance, visual deficits, and cognitive changes are prevalent with limb weakness being the most common in HIV- and MS-associated PML.75,77 Although rare, brain stem involvement causes the most severe symptoms. The disease is progressive and symptoms worsen over time.
There is subtle variation in the presentation of PML as determined by the underlying cause. In HIV-PML, 50–73% of the patients have predominant motor system findings, following cognitive and speech difficulties.26,78 In a recent German study of 142 NTZ-PML patients, 33.8% had cognitive and equally as many had motor symptoms, 26.8% had gait ataxia, 25.3% had speech and language problems, and 16.2% had vision deficits. 7.7% were asymptomatic.79 Seizures may occur in up to one-third of the general PML population and are more frequent with juxtacortical and T1-hyperintense lesions on MRI.21
Brain Imaging
Brain MRI imaging is the most important diagnostic tool for PML diagnosis. Frequent brain scans are recommended for early detection of PML in high-risk patients (those who have three risk factors for NTZ-PML and those who have not had immunosuppressant therapy, but have high serum anti-JCPyV ab index and more than two years of NTZ), because PML lesions in MRI can develop before clinical symptoms.80,81
The typical imaging features of PML include predilection for the frontal and parieto-occipital regions and subcortical location involving U-fibers. Involvement of the overlying cortex has been increasingly reported.82 Corpus callosum83 or deep grey matter may be affected. Posterior fossa involvement has been reported, most commonly involving the cerebellum and middle cerebellar peduncles.84 Lesions have sharp borders toward gray matter contrasting with ill-defined borders toward white matter, and increased signal intensity on T2-weighted and diffusion-weighted images (DWI).85 T2-weighted imaging may demonstrate a microcyst or granular pattern.82 On T1-weighted images PML lesions are hypointense and well-defined, even if small in size. Usually there is no gadolinium enhancement on MRI, but 15% of HIV-PML, and 40% of NTZ-PML patients may exhibit hazy peripheral, sometimes punctate or nodular enhancement.73 Two types of inflammation have been reported in PML: fatal IRIS type and anti-JCPyV immunoreaction type. The latter is associated with good prognosis.86
When mass effect and enhancement are prominent, one should suspect PML-IRIS, particularly when accompanied by clinical deterioration. Enhancement at the time of diagnosis correlates with decreased survival and greater clinical disability compared to those that do not enhance.87
In NTZ-PML, imaging features can be associated with prognosis. Radiologically widespread disease at diagnosis is present in 71% of fatal cases. Asymptomatic cases of PML found on screening MRI have been associated with good clinical outcomes.5,88
Laboratory Diagnostics
Detecting JCPyV DNA in CSF by highly sensitive PCR has great diagnostic value. With new ultrasensitive PCR techniques, the sensitivity is >95% and specificity is >97%.89 Despite the high sensitivity of PCR, a negative test result does not rule out PML,73 because viral loads can be very low (below 100 copies/mL), while most commercial tests are able to detect JCPyV DNA in excess of 200 copies/mL.90 In HIV-infected patients, the viral load in CSF is high and sufficient for diagnosis. HAART decreases viral load and makes laboratory-confirmed diagnosis difficult. In patients with MS or other autoimmune diseases, the viral loads are also low.73 This creates a need to take multiple CSF samples despite a clinically strong suspicion of PML due to initial samples being falsely negative.
Management and Surveillance of PML
There is neither a specific prophylaxis nor an effective anti-JCPyV treatment for PML. A number of drugs such as cytarabine, cidofovir, INF-alpha, mefloquine, mirtazapine, and topotecan were used in clinical trials without benefit.91,92 Currently the approach to treating PML is prompt immune reconstitution without causing IRIS.
In NTZ-PML patients, immune reconstitution is achieved by plasma exchange in order to remove NTZ from peripheral circulation.93 There is one study reporting the benefits of filgrastim (granulocyte-colony stimulating factor) in patients with NTZ-PML.94 In HIV-infected patients, optimization of HAART is the best therapeutic choice.34
In a portion of the PML population, such as patients with hematological malignancies, immune reconstitution cannot be achieved. For these patients, blocking of pathways involving programmed cell death 1 (PD-1), a T-cell surface receptor that keeps the immune system from attacking the body’s own tissues, and its ligand is a new promising treatment option. Pembrolizumab and nivolumab, mAbs that both target PD-1, both seem to reduce JCPy viral load and enhance JCPyV specific cell mediated immune responses by preserving and activating CD4+ and CD8+ cells.95,96 Immune checkpoint inhibitors activate the immune system, and as a side effect they may exacerbate any underlying autoimmune disease. A controlled trial is needed to determine whether PD-1 inhibitors are indeed able to suppress JCPyV in PML patients, and which subgroups would benefit from the treatment. Recently, several new treatment methods, such as systemic cytokine administration (interferon-alpha, interleukin-2, and interleukin-7) and immunization by recombinant human anti-JCPyV VP1 mAbs, JCPyV-specific cytotoxic T lymphocyte therapy, cryopreserved BK virus–specific T cells, and JCPyV capsid protein conjugated with interleukin-7 for PML were reported.91,97,98
The prognosis of PML has evolved over time, and prolonged survival rates are becoming more common in HIV- and NTZ-PML groups. The survival rate in these groups is currently 70–80%.36,99 A low viral load in the CSF, VP1 loop-specific polymorphism, a controlled inflammation mediated by CD4- and CD8-positive T cells, and plasma cells and a regulatory immune system are associated with good PML prognosis.13 Predictors of favorable outcomes in NTZ-PML cases are younger age, lower Expanded Disability Status Scale score prior to diagnosis, shorter diagnostic delay, and more localized brain lesions on MRI at the time of diagnosis.100 PML cases related to HSCT have longer time to diagnosis, better survival rates, and lower mortality rates compared to the cases associated with biological treatments or purine analogues.30
In HIV-PML, complete recovery is not to be expected but one third of the patients may become healthy enough to have no functional disability. Patients with cerebellar lesions have a worse outcome due to gait ataxia and incoordination that precludes independent living.102 Of NTZ-PML survivors, 40% have severe, 47% moderate, and 13% mild disability.101
PML-IRIS
In patients with a reversible cause of immunosuppression, re-establishment of immunocompetence may lead to an excessively overreactive immune response referred to as IRIS. The likelihood of developing PML-IRIS depends on the underlying disease. Approximately 20% of HIV-PML patients and most NTZ-PML patients treated with immunoadsorption develop IRIS.102
IRIS is treated with corticosteroids, which have a profound impact on the virus-specific T-cell response, especially on JCPyV. Therefore, they should not be given before the onset of clinical or radiological signs of IRIS.103 Sometimes prolonged corticosteroid therapy102 and treatment with maraviroc, a CCR5 receptor antagonist, has been reported and may be warranted to prevent overshooting damage by IRIS.104 The observation is not yet confirmed in a large study.
PML-IRIS is a potentially disabling and fatal condition with a mortality rate of 28%. Fatal PML-IRIS is characterized by excessive cytotoxic CD8-positive T-cell infiltration in the brain. In PML with controlled inflammation, both T- and B-cell lineages appear cooperatively.105
However, several studies have not been able to demonstrate significant differences in survival rates between PML with and without IRIS.106
PML Prevention Strategies for MS Patients
The optimal PML mitigation strategy is prevention and appropriate surveillance. PML is associated with several drugs, but so far, a risk stratification algorithm has only been established for NTZ (Table 3). Prior to starting NTZ, patients must be risk-stratified for the presence of serum JCPyV antibodies. Antibody testing is recommended for all NTZ-treated JCPyV antibody negative patients every six months, and in the event of seroconversion their therapy should be changed. MRI screening every 3–4 months is recommended for patients who receive NTZ for ≥18 months while JCPyV seropositive. A yearly brain MRI is recommended for patients at low risk of PML (JCPyV seronegative). MRI screening every 3–4 months up to 12 months is suggested when switching from NTZ to fingolimod, DMF or alemtuzumab.80 In a recent study by Ryerson et al, for anti-JCPyV antibody-positive MS patients, extended interval dosing of NTZ was associated with significantly lower PML risk than standard interval dosing.107
The European Medicines Agency (EMA) recommends that a complete blood count should be performed before starting treatment with DMF, and every 3 months during treatment. A baseline brain MRI should be available as a reference. If during treatment the lymphocyte counts drop to low levels for more than 6 months, the doctor should consider discontinuation of DMF.107 Similar EMA guidelines exist for fingolimod. Before introducing fingolimod treatment, a baseline MRI scan should be available as a reference. If PML is suspected, MRI should be performed immediately and treatment with fingolimod should be suspended until PML has been excluded.109
Long-term safety risks of sequential multiple therapies are still unknown. Currently, succession of therapies for patients with relapsing-remitting MS is determined by disease activity, patient-, and drug-related factors. The risk–benefit ratio of DMTs should be regularly re-evaluated as it changes with duration of therapy. Neurologists must remain vigilant for signs and symptoms of PML when switching from MS therapies associated with PML to a new MS therapy. PML should be carefully excluded by clinical-, imaging, and CSF exams, especially when switching from NTZ or fingolimod to long-lasting B-cell depleting therapy.
Safety protocols for PML exist for the treatment of MS patients. Other patients, such as cancer and rheumatic patients, currently have no such protocols in place.
Conclusion
Many different subgroups of PML, based on the underlying diseases have been identified. These subgroups of PML have different incidences and prevalences, as well as different clinical courses and prognoses. While often associated with a bad prognosis, in some cases PML may become inactive, leaving patients with permanent neurological deficits. The mainstay of PML treatment is immune reconstitution including discontinuation of the inciting agent, and with some medications plasma exchange, although blocking of PD-1 and its ligand, as well as direct antiviral therapeutics are an active area of investigation. Diagnosis is challenging and may need repeated clinical judgement, CSF testing, and MRI.
There is no systematic national or international record of PML diagnoses, and no risk stratification algorithm except for NTZ. The long-term risk of multiple successive immunomodulatory and immunosuppressive therapies is unknown. Global database and recording systems were developed by the company producing NTZ in order to follow up the emergence of PML and other adverse events during NTZ treatment. Surveillance registries as well as analysis of real-world data to improve the assessment of PML risk and to tailor prevention strategies for other PML subgroups are urgently required as well. In addition to neurologists, physicians from other fields must be aware of PML risk associated with mAb and other drugs. Current information concerning drugs causing PML is mostly provided by pharmaceutical companies. Large nationwide studies on the incidence and prevalence of PML with respect to the underlying diseases and treatments would provide evidence that is more conclusive.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Åström KE, Mancall EL, Richardson EP
2. Zaheer F, Berger JR. Treatment-related progressive multifocal leukoencephalopathy: current understanding and future steps. Ther Adv Drug Saf. 2012;3:5. doi:10.1177/2042098612453849
3. Anton R, Haas M, Arlett P, et al. Drug-induced progressive multifocal leukoencephalopathy in multiple sclerosis: European regulators’ perspective. Clin Pharmacol Ther. 2017;102:283–289. doi:10.1002/cpt.v102.2
4. Bohra C, Sokol L, Dalia S. Progressive multifocal leukoencephalopathy and monoclonal antibodies: a review. Cancer Control. 2017;24:4. doi:10.1177/1073274817729901
5. Lindå H, von Heijne A. Presymptomatic diagnosis with MRI and adequate treatment ameliorate the outcome after natalizumab-associated progressive multifocal leukoencephalopathy. Front Neurol. 2013. doi:10.3389/fneur.2013.00011
6. Dahlhaus S, Hoepner R, Chan A, et al. Disease course and outcome of 15 monocentrically treated natalizumab-associated progressive multifocal leukoencephalopathy patients. J Neurol Neurosurg Psychiatry. 2013;84:10. doi:10.1136/jnnp-2013-304897
7. Dang X, Wüthrich C, Gordon J, Sawa H, Koralnik IJ. JC virus encephalopathy is associated with a novel agnoprotein-deletion JCV variant. PLoS One. 2012;7:4. doi:10.1371/journal.pone.0035793
8. Takahashi K, Sekizuka T, Fukumoto H, et al. Deep-sequence identification and role in virus replication of a JC virus quasispecies in patients with progressive multifocal leukoencephalopathy. J Virol. 2016;91:1.
9. Seppälä HM, Helanterä IT, Laine PKS, et al. Archetype JC Polyomavirus (JCPyV) Prevails in a Rare Case of JCPyV nephropathy and in stable renal transplant recipients with JCPyV viruria. J Infect Dis. 2017;216:8. doi:10.1093/infdis/jix435
10. Bellizzi A, Nardis C, Anzivino E, et al. Human polyomavirus JC reactivation and pathogenetic mechanisms of progressive multifocal leukoencephalopathy and cancer in the era of monoclonal antibody therapies. J Neurovirol. 2012;18:1. doi:10.1007/s13365-012-0080-7
11. Auer M, Hegen H, Sellner J, et al. Conversion and reversion of anti‐John Cunningham virus antibody serostatus: a prospective study. Brain Behav. 2019;9:7.
12. Durali D, de Goër de Herve MG, Gasnault J, Taoufik Y. B cells and progressive multifocal leukoencephalopathy: search for the missing link. Front Immunol. 2015;6. doi:10.3389/fimmu.2015.00241
13. Sanjo N, Nose Y, Shishido-Hara Y, et al. A controlled inflammation and a regulatory immune system are associated with more favorable prognosis of progressive multifocal leukoencephalopathy. J Neurol. 2019;266:2. doi:10.1007/s00415-018-9140-0
14. Trampe AK, Hemmelmann C, Stroet A, et al. Anti-JC virus antibodies in a large German natalizumab-treated multiple sclerosis cohort. Neurology. 2012;78:22. doi:10.1212/WNL.0b013e3182583022
15. Warnke C, Ramanujam R, Plavina T, et al. Changes to anti-JCV antibody levels in a Swedish national MS cohort. J Neurol Neurosurg Psychiatry. 2013;84:11. doi:10.1136/jnnp-2012-304332
16. Outteryck O, Zéphir H, Salleron J, et al. JC-virus seroconversion in multiple sclerosis patients receiving natalizumab. Mult Scler. 2014;20:7. doi:10.1177/1352458513505353
17. Vennegoor A, van Rossum JA, Polman CH, Wattjes MP, Killestein J. Longitudinal JCV serology in multiple sclerosis patients preceding natalizumab-associated progressive multifocal leukoencephalopathy. Mult Scler. 2015;21:12. doi:10.1177/1352458514567728
18. Delbue S, Ferraresso M, Ghio L, et al. A review on JC virus infection in kidney transplant recipients. Clin Dev Immunol. 2013;2013:1–7. doi:10.1155/2013/926391
19. Corbridge SM, Rice RC, Bean LA, et al. JC virus infection of meningeal and choroid plexus cells in patients with progressive multifocal leukoencephalopathy. J Neurovirol. 2019;25:520–524. doi:10.1007/s13365-019-00753-y
20. Tan CS, Koralnik IJ. Beyond progressive multifocal leukoencephalopathy: expanded pathogenesis of JC virus infection in the central nervous system. Lancet Neurol. 2010;9:4.
21. Khoury MN, Alsop DC, Agnihotri SP, et al. Hyperintense cortical signal on magnetic resonance imaging reflects focal leukocortical encephalitis and seizure risk in progressive multifocal leukoencephalopathy. Ann Neurol. 2014;75:5. doi:10.1002/ana.v75.5
22. White FA, Ishaq M, Stoner GL, Frisque RJ. JC virus DNA is present in many human brain samples from patients without progressive multifocal leukoencephalopathy. J Virol. 1992;66:10.
23. Wüthrich C, Koralnik IJ. Frequent infection of cortical neurons by JC virus in patients with progressive multifocal leukoencephalopathy. J Neuropathol Exp Neurol. 2012;71:1. doi:10.1097/NEN.0b013e31823ede59
24. Amend KL, Turnbull B, Foskett N, Napalkov P, Kurth T, Seeger J. Incidence of progressive multifocal leukoencephalopathy in patients without HIV. Neurology. 2010;12:75.
25. Casado JL, Corral I, García J, et al. Continued declining incidence and improved survival of progressive multifocal leukoencephalopathy in HIV/AIDS patients in the current era. Eur J Clin Microbiol Infect Dis. 2014;33:2. doi:10.1007/s10096-013-1941-6
26. Iacobaeus E, Burkill S, Bahmanyar S, et al. The national incidence of PML in Sweden, 1988–2013. Neurology. 2018;90:e498–e506. doi:10.1212/WNL.0000000000004926
27. Sipilä JOT, Soilu-Hänninen M, Rautava P, Kytö V. Progressive multifocal leukoencephalopathy in Finland: a cross-sectional registry study. J Neurol. 2019;266:2. doi:10.1007/s00415-018-09167-y
28. Kartau M, Verkkoniemi-Ahola A, Paetau A, et al. The incidence and predisposing factors of John Cunningham virus-induced progressive multifocal leukoencephalopathy in Southern Finland: a population-based study. Open Forum Infect Dis. 2019;6:2. doi:10.1093/ofid/ofz024
29. Neil EC, DeAngelis LM. Progressive multifocal leukoencephalopathy and hematologic malignancies: a single cancer center retrospective review. Blood Adv. 2017;1:23. doi:10.1182/bloodadvances.2017008201
30. Adrianzen Herrera D, Ayyappan S, Jasra S, et al. Characteristics and outcomes of progressive multifocal leukoencephalopathy in hematologic malignancies and stem cell transplant – a case series. Leuk Lymphoma. 2019;60:2. doi:10.1080/10428194.2018.1474523
31. Carson KR, Newsome SD, Kim EJ, et al. Progressive multifocal leukoencephalopathy associated with brentuximab vedotin therapy: a report of 5 cases from the Southern Network on Adverse Reactions (SONAR) project. Cancer. 2014;120:16. doi:10.1002/cncr.28712
32. García-Suárez J, de Miguel D, Krsnik I, Bañas H, Arribas I, Burgaleta C. Changes in the natural history of progressive multifocal leukoencephalopathy in HIV-negative lymphoproliferative disorders: impact of novel therapies. Am J Hematol. 2005;80:4. doi:10.1002/ajh.20492
33. Weber T. Progressive multifocal leukoencephalopathy. Neurol Clin. 2008;26:3. doi:10.1016/j.ncl.2008.03.007
34. d’Arminio Monforte A, Cinque P, Mocroft A, et al. Changing incidence of central nervous system diseases in the EuroSIDA cohort. Ann Neurol. 2004;55:3. doi:10.1002/ana.10788
35. Engsig FN, Hansen AB, Omland LH, et al. Incidence, clinical presentation, and outcome of progressive multifocal leukoencephalopathy in HIV-infected patients during the highly active antiretroviral therapy era: a nationwide cohort study. J Infect Dis. 2009;199:1. doi:10.1086/595299
36. Gasnault J, Costagliola D, Hendel-Chavez H, et al. Improved survival of HIV-1-infected patients with progressive multifocal leukoencephalopathy receiving early 5-drug combination antiretroviral therapy. PLoS One. 2011;6:6. doi:10.1371/journal.pone.0020967
37. Falcó V, Olmo M, del Saz SV, et al. Influence of HAART on the clinical course of HIV-1-infected patients with progressive multifocal leukoencephalopathy: results of an observational multicenter study. J Acquir Immune Defic Syndr. 2008;49:1. doi:10.1097/QAI.0b013e31817bec64
38. Mateen FJ, Muralidharan R, Carone M, et al. Progressive multifocal leukoencephalopathy in transplant recipients. Ann Neurol. 2011;70:2. doi:10.1002/ana.v70.2
39. Kharfan-Dabaja MA, Ayala E, Greene J. Two cases of progressive multifocal leukoencephalopathy after allogeneic hematopoietic cell transplantation and a review of the literature. Bone Marrow Transplant. 2007;39(2):101–117.
40. Steurer M, Clausen J, Gotwald T. Progressive multifocal leukoencephalopathy after allogeneic stem cell transplantation and posttransplantation rituximab. Transplantation. 2003;76:435–436. doi:10.1097/01.TP.0000078897.11633.5F
41. Pruitt AA, Graus F, Rosenfeld MR. Neurological complications of transplantation. Neurohospitalist. 2013;3:1.
42. Clavel G, Moulignier A, Semerano L. Progressive multifocal leukoencephalopathy and rheumatoid arthritis treatments. Joint Bone Spine. 2017;84:6. doi:10.1016/j.jbspin.2017.03.002
43. Molloy ES, Calabrese LH. Progressive multifocal leukoencephalopathy: a national estimate of frequency in systemic lupus erythematosus and other rheumatic diseases. Arthritis Rheum. 2009;60. doi:10.1002/art.24966
44. Brandao M, Damasio J, Marinho A, et al. Systemic lupus erythematosus, progressive multifocal leukoencephalopathy, and T-CD4+ lymphopenia. Clin Rev Allergy Immunol. 2012;43:302–307. doi:10.1007/s12016-012-8327-x
45. Jamilloux Y, Néel A, Lecouffe-Desprets M, et al. Progressive multifocal leukoencephalopathy in patients with sarcoidosis. Neurology. 2014;82:15. doi:10.1212/WNL.0000000000000318
46. Palazzo E, Yahia SA. Progressive multifocal leukoencephalopathy in autoimmune diseases. Joint Bone Spine. 2012;79:351–355. doi:10.1016/j.jbspin.2011.11.002
47. Bag AK, Curé JK, Chapman PR, Roberson GH, Shah R. JC virus infection of the brain. AJNR Am J Neuroradiol. 2010;31:9. doi:10.3174/ajnr.A2035
48. Hadjadj J, Guffroy A, Delavaud C, et al. Progressive multifocal leukoencephalopathy in primary immunodeficiencies. J Clin Immunol. 2019;39:1. doi:10.1007/s10875-018-0578-8
49. Gheuens S, Pierone G, Peeters P, Koralnik IJ. Progressive multifocal leukoencephalopathy in individuals with minimal or occult immunosuppressio. J Neurol Neurosurg Psychiatry. 2010;81:3. doi:10.1136/jnnp.2009.187666
50. Rosas MJ, Simões-Ribeiro F, An SF, Sousa N. Progressive multifocal leukoencephalopathy: unusual MRI findings and prolonged survival in a pregnant woman. Neurology. 1999;52:3. doi:10.1212/WNL.52.3.657
51. Stockhammer G, Poewe W, Wissel J, Kiechl U, Maier H, Felber S. Progressive multifocal leukoencephalopathy presenting with an isolated focal movement disorder. Mov Disord. 2000;15:5. doi:10.1002/1531-8257(200009)15:5<>1.0.CO;2-S
52. Shepard HM, Phillips GL, Thanos CD, Feldmann M. Developments in therapy with monoclonal antibodies and related proteins. Clin Med. 2017;17:3. doi:10.7861/clinmedicine.17-3-220
53. Clifford DB, Ances B, Costello C, et al. Rituximab-associated progressive multifocal leukoencephalopathy in rheumatoid arthritis. Arch Neurol. 2011;68:9. doi:10.1001/archneurol.2011.103
54. Yukitake M. Drug‐induced progressive multifocal leukoencephalopathy in multiple sclerosis: A comprehensive review. Clin Exp Neuroimmunol. 2018;9:37–47. doi:10.1111/cen3.2018.9.issue-S1
55. Raisch DW, Rafi JA, Chen C, Bennett CL. Detection of cases of progressive multifocal leukoencephalopathy associated with new biologicals and targeted cancer therapies from the FDA’s adverse event reporting system. Expert Opin Drug Saf. 2016;15:8. doi:10.1080/14740338.2016.1198775
56. Genentech. [Online]. September 30, 2019. Available from: https://www.ocrelizumabinfo.com/content/dam/gene/ocrelizumabinfo/pdfs/progressive-multifocal-leukoencephalopathy.pdf.
57. Klotz L, Havla J, Schwab N, et al. Risks and risk management in modern multiple sclerosis immunotherapeutic treatment. Ther Adv Neurol Disord. 2019;12:175628641983657. doi:10.1177/1756286419836571
58. Berger JR, Cree BA, Greenberg B, et al. Progressive multifocal leukoencephalopathy after fingolimod treatment. Neurology. 2018;90:e1815–e1821. doi:10.1212/WNL.0000000000005529
59. Gerevini S, Capra R, Bertoli D, Sottini A, Imberti L. Immune profiling of a patient with alemtuzumab-associated progressive multifocal leukoencephalopathy. Mult Scler. 2019;25:8. doi:10.1177/1352458519832259
60. Lehmann HC, Krüger K, Fink GR, Schroeter M. Progressive multifocal leukoencephalopathy after interferon beta-1a monotherapy. J Neurol. 2015;262:3. doi:10.1007/s00415-014-7620-4
61. Berger JR. Classifying PML risk with disease modifying therapies. Mult Scler Relat Disord. 2017;12:59–63. doi:10.1016/j.msard.2017.01.006
62. von Andrian UH, Engelhardt B. Alpha4 integrins as therapeutic targets in autoimmune disease. N Engl J Med. 2003;348:1. doi:10.1056/NEJMe020157
63. Maas RP, Muller-Hansma AH, Esselink RA, et al. Drug-associated progressive multifocal leukoencephalopathy: a clinical, radiological, and cerebrospinal fluid analysis of 326 cases. J Neurol. 2016;263:10. doi:10.1007/s00415-016-8217-x
64. Prosperini L, de Rossi N, Scarpazza C, et al. Natalizumab-related progressive multifocal leukoencephalopathy in multiple sclerosis: findings from an Italian independent registry. PLoS One. 2016;11:12. doi:10.1371/journal.pone.0168376
65. Prosperini L, Scarpazza C, Imberti L, Cordioli C, De Rossi N, Capra R. Age as a risk factor for early onset of natalizumab-related progressive multifocal leukoencephalopath. J Neurovirol. 2017;23:5. doi:10.1007/s13365-017-0561-9
66. Ho PR, Koendgen H, Campbell N, Haddock B, Richman S, Chang I. Risk of natalizumab-associated progressive multifocal leukoencephalopathy in patients with multiple sclerosis: a retrospective analysis of data from four clinical studies. Lancet Neurol. 2017;16:11. doi:10.1016/S1474-4422(17)30282-X
67. Oshima Y, Tanimoto T, Yuji K, Tojo A. Drug-associated progressive multifocal leukoencephalopathy in multiple sclerosis patients. Mult Scler. 2019;25:8. doi:10.1177/1352458518786075
68. Hughes S. PML reported in patient receiving ocrelizumab. S.L. Available from: https://www.medscape.com/viewarticle/880654. Medscape. 2017.
69. Clifford DB, Gass A, Richert N, et al. Cases reported as progressive multifocal leukoencephalopathy in ocrelizumab-treated patients with multiple sclerosis.
70. ocrelizumabinfo home page. F. Hoffmann La-Roche Ltd. [Online]. 2019. Available from: https://www.ocrelizumabinfo.global.
71. Diebold M, Altersberger V, Décard BF, Kappos L, Derfuss T, Lorscheider J. A case of progressive multifocal leukoencephalopathy under dimethyl fumarate treatment without severe lymphopenia or immunosenescence. Mult Scler. 2019;25:12. doi:10.1177/1352458519852100
72. Alstadhaug KB, Fykse Halstensen R, Odeh F. Progressive multifocal leukoencephalopathy in a patient with systemic mastocytosis treated with cladribine. J Clin Virol. 2017;88:17–20. doi:10.1016/j.jcv.2016.12.005
73. Berger JR, Aksamit AJ, Clifford DB, et al. PML diagnostic criteria: consensus statement from the AAN neuroinfectious disease section. Neurology. 2013;80:1430–1438. doi:10.1212/WNL.0b013e31828c2fa1
74. Tan SC, Koralnik IJ. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases (Eighth Edition). SL: Elsevier Health Sciences; 2015.
75. Williamson EML, Berger JR. Diagnosis and treatment of progressive multifocal leukoencephalopathy associated with multiple sclerosis therapies. Neurotherapeutics. 2017;14:961–973. doi:10.1007/s13311-017-0570-7
76. Scarpazza C. Early diagnosis of PML: results from the Italian cohort. [Online]. October 27, 2017. Available from: http://onlinelibrary.ectrims-congress.eu/ectrims/2017/ACTRIMS-ECTRIMS2017/200861/cristina.scarpazza.early.diagnosis.of.pml.results.from.the.italian.cohort.html.
77. Berger JR. Progressive multifocal leukoencephalopathy. Handb Clin Neurol. 2014;123.
78. Berger JR, Pall L, Lanska D. Progressive multifocal leukoencephalopathy in patients with HIV infection. J Neurovirol. 1998;4:1. doi:10.3109/13550289809113482
79. Blankenbach K, Schwab N, Hofner B, Adams O, Keller-Stanislawski B, Warnke C. Natalizumab-associated progressive multifocal leukoencephalopathy in Germany. Neurology. 2019;92:19. doi:10.1212/WNL.0000000000007451
80. Wattjes MP, Rovira À, Miller D, et al. Evidence-based guidelines: MAGNIMS consensus guidelines on the use of MRI in multiple sclerosis–establishing disease prognosis and monitoring patients. Nat Rev Neurol. 2015;11:10.
81. McGuigan C, Craner M, Guadagno J, et al. Stratification and monitoring of natalizumab-associated progressive multifocal leukoencephalopathy risk: recommendations from an expert group. J Neurol Neurosurg Psychiatry. 2016;87(2):117–125.
82. Yousry TA, Pelletier D, Cadavid D. Magnetic resonance imaging pattern in natalizumab-associated progressive multifocal leukoencephalopathy. Ann Neurol. 2012;72:779–787. doi:10.1002/ana.v72.5
83. Shah R, Bag AK, Chapman PR, et al. Imaging manifestations of progressive multifocal leukoencephalopathy. Clin Radiol. 2010;65:431–439. doi:10.1016/j.crad.2010.03.001
84. Scholten P, Kralt P, Jacobs B. Posterior fossa progressive multifocal leukoencephalopathy: first presentation of an unknown autoimmune disease. BMJ Case Rep. 2017;
85. Hodel J, Outteryck O, Dubron C, et al. Asymptomatic progressive multifocal leukoencephalopathy associated with natalizumab: diagnostic precision with MR imaging. Radiology. 2016;278(3):863–872.
86. Sanjo N, Kina S, Shishido-Hara Y, et al. Progressive multifocal leukoencephalopathy with balanced CD4/CD8 T-cell infiltration and good response to mefloquine treatment. Intern Med. 2016;55:2. doi:10.2169/internalmedicine.55.6051
87. Tan IL, McArthur JC, Clifford DB, Major EO, Nath A. Immune reconstitution inflammatory syndrome in natalizumab-associated PML. Neurology. 2011;77:11. doi:10.1212/WNL.0b013e31822e55e7
88. Phan-Ba R, Lommers E, Tshibanda L, et al. MRI preclinical detection and asymptomatic course of a progressive multifocal leucoencephalopathy (PML) under natalizumab therapy. J Neurol Neurosurg Psychiatry. 2012;83:224–226. doi:10.1136/jnnp-2011-300511
89. Adang L, Berger J. Progressive multifocal leukoencephalopathy. F1000Res. 2015;4. doi:10.12688/f1000research.7071.1
90. Clifford DB, DeLuca A, Simpson DM, Arendt G, Giovannoni G, Nath A. Natalizumab-associated progressive multifocal leukoencephalopathy in patients with multiple sclerosis: lessons from 28 cases. Lancet Neurol. 2010;9:438–446. doi:10.1016/S1474-4422(10)70028-4
91. Pavlovic D, Patera AC, Nyberg F, Gerber M, Liu M. Progressive multifocal leukeoncephalopathy consortium. Progressive multifocal leukoencephalopathy: current treatment options and future perspectives. Ther Adv Neurol Disord. 2015;8:6. doi:10.1177/1756285615602832
92. Jamilloux Y, Kerever S, Ferry T, Broussolle C, Honnorat J, Sève P. Treatment of progressive multifocal leukoencephalopathy with mirtazapine. Clin Drug Investig. 2016;36:10. doi:10.1007/s40261-016-0433-8
93. Khatri BO, Man S, Giovannoni G, et al. Effect of plasma exchange in accelerating natalizumab clearance and restoring leukocyte function. Neurology. 2009;72:5. doi:10.1212/01.wnl.0000341766.59028.9d
94. Stefoski D, Balabanov R, Waheed R, Ko M, Koralnik IJ, Sierra Morales F. Treatment of natalizumab-associated PML with filgrastim. Ann Clin Transl Neurol. 2019;6:5. doi:10.1002/acn3.2019.6.issue-5
95. Cortese I, Muranski P, Enose-Akahata Y, et al. Pembrolizumab treatment for progressive multifocal leukoencephalopathy. N Engl J Med. 2019;380:1597–1605. doi:10.1056/NEJMoa1815039
96. Walter O, Treiner E, Bonneville F, et al. Treatment of progressive multifocal leukoencephalopathy with nivolumab. N Engl J Med. 2019;380:17. doi:10.1056/NEJMc1816198
97. Grimm J, Martin R, Combauzier B Recombinant human antibodies for therapy and prevention of polyomavirus-related diseases. Patent WO2014/102399 Al 2014.
98. Muftuoglu M, Olson A, Marin D, et al. Allogeneic BK virus–specific T cells for progressive multifocal leukoencephalopathy. N Engl J Med. 2018;379:15. doi:10.1056/NEJMoa1801540
99. Vermersch PD, Kappos LM, Gold RM, et al. Effective therapy for PML by achieving immune reconstitution has resulted in survival as 70–80% in both HIV and natalizumab associated PML. Neurology. 2011;76:20.
100. Vermersch P, Kappos L, Gold R, et al. Clinical outcomes of natalizumab-associated progressive multifocal leukoencephalopathy. Neurology. 2011;76:20. doi:10.1212/WNL.0b013e31821a446b
101. Lima MA, Bernal-Cano F, Clifford DB, Gandhi RT, Koralnik IJ. Clinical outcome of long-term survivors of progressive multifocal leukoencephalopathy. J Neurol Neurosurg Psychiatry. 2010;81:11. doi:10.1136/jnnp.2009.179002
102. Tan K, Roda R, Ostrow L, McArthur J, Nath A. PML-IRIS in patients with HIV infection: clinical manifestations and treatment with steroids. Neurology. 2009;72:17. doi:10.1212/01.wnl.0000343510.08643.74
103. Antoniol C, Jilek S, Schluep M, et al. Impairment of JCV-specific T-cell response by corticotherapy: effect on PML-IRIS management? Neurology. 2012;79:23. doi:10.1212/WNL.0b013e3182768983
104. Giacomini PS, Rozenberg A, Metz I, et al. Maraviroc and JC virus-associated immune reconstitution inflammatory syndrome. N Engl J Med. 2014;370:5. doi:10.1056/NEJMc1304828
105. Nishigori R, Warabi Y, Shishido-Hara Y, et al. Inflammatory cerebellar PML with a CD4/CD8 ratio of 2.9 showed a favorable prognosis in a patient with rheumatoid arthritis: a case report. Intern Med. 2019;58:3323–3329. doi:10.2169/internalmedicine.3038-19
106. Fournier A, Martin-Blondel G, Lechapt-Zalcman E, et al. Immune reconstitution inflammatory syndrome unmasking or worsening AIDS-related progressive multifocal leukoencephalopathy: a literature review. Front Immunol. 2017;8. doi:10.3389/fimmu.2017.00577
107. Ryerson LZ, Foley J, Chang I, et al. Risk of natalizumab-associated PML in patientswith MS is reduced with extended interval dosing. Neurology. 2019;93:15. doi:10.1212/WNL.0000000000007938
108. European Medicines Agency. Updated recommendations to minimise the risk of the rare brain infection PML with tecfidera. [Online]. 2015. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Press_release/2015/10/WC500196017.pdf.
109. New recommendations to minimise risks of the rare brain infection PML and a type of skin cancer with gilenya. [Online]. 2015. Available from: https://www.ema.europa.eu/en/documents/press-release/new-recommendations-minimise-risks-rare-brain-infection-pml-type-skin-cancer-gilenya_en.pdf.
110. Tuccori M, Focosi D, Blandizzi C, Del Tacca M, Petrini M. Re: rituximab maintenance for the treatment of patients with follicular lymphoma: systematic review and meta-analysis of randomized trials. J Natl Cancer Inst. 2009;101:18. doi:10.1093/jnci/djp256
111. Bower JH, Hammack JE, McDonnell SK, Tefferi A. The neurologic complications of B-cell chronic lymphocytic leukemia. Neurology. 1997;48:2. doi:10.1212/WNL.48.2.407
112. van Rijswijk RE, Verbeek J, Haanen C, Dekker AW, van Daal WA, van Peperzeel HA. Major complications and causes of death in patients treated for hodgkin’s disease. J Clin Oncol. 1987;5:10. doi:10.1200/JCO.1987.5.10.1624
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.