Back to Journals » ImmunoTargets and Therapy » Volume 15
Progress on Potential Therapeutic Targets for Sepsis-Related Cardiac Dysfunction: From Basic Research to Clinical Translation
Authors Gao Y, Dai R, Kong C, Niu Z ![]() , Yuan L, Liu X
, Yuan L, Liu X ![]()
Received 11 November 2025
Accepted for publication 17 February 2026
Published 21 March 2026 Volume 2026:15 580700
DOI https://doi.org/10.2147/ITT.S580700
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Michael Shurin
Yan Gao, Rui Dai, Chang Kong, Zejun Niu, Li Yuan, Xiaojie Liu
Department of Anesthesiology, The Affiliated Hospital of Qingdao University, Qingdao, People’s Republic of China
Correspondence: Xiaojie Liu, Email [email protected]
Abstract: Sepsis-related cardiac dysfunction (SRCD) is a severe complication of sepsis with complex pathophysiology involving dysregulated inflammatory responses, immune cell dysfunction, and multiple forms of programmed cell death. The review addresses the core pathophysiological mechanisms of SRCD, the central role of immune cell crosstalk (particularly between macrophages and neutrophils), and the evaluation of multi-target therapeutic strategies with an emphasis on natural compounds. We highlight promising interventions, including ginsenosides and melatonin, which exert multi-target protective effects. Future research should integrate insights across these themes to accelerate the development of precise therapies and improve patient survival and quality of life.
Keywords: sepsis-related cardiac dysfunction, macrophage, NETosis, natural compounds, ferroptosis, immune–metabolic dysfunction
Introduction
Sepsis is defined as life-threatening organ dysfunction caused by a dysregulated host response to infection, and septic shock is classified as a subtype of sepsis.1–5 Sepsis-related cardiac dysfunction (SRCD) is a common and fatal complication during the progression of sepsis, with a complex pathophysiological mechanism.2–4,6 Cardiac dysfunction is involved in 70% to 90% of sepsis-related deaths.1 Myocardial injury is among the common organ injuries in sepsis, accelerating its progression; approximately 50% of sepsis patients in the intensive care unit (ICU) develop myocardial dysfunction,1 making it a significant factor contributing to multiple organ dysfunction syndrome (MODS) and poor prognosis in sepsis patients.7 Its characteristics include left and/or right ventricular systolic and/or diastolic dysfunction and the elevation of myocardial injury biomarkers.
Sepsis can lead to a dysregulated cardiac response, and increase levels of myocardial depressant factors, oxidative imbalance, mitochondrial damage, and pyroptosis. By suppressing cardiac energy metabolism and inducing cardiomyocyte death, these changes ultimately lead to cardiac dysfunction.7–10 However, these complex pathogenic factors limit the therapeutic value of basic and clinical research.11 It is urgent to develop targeted, highly effective treatments with minimal side effects to address sepsis-induced myocardial injury.
To clearly present the pathophysiological mechanisms and therapeutic strategies related to SRCD, this review is structured around three aspects: first, the core mechanisms underlying SRCD were outlined. Second, the dysregulated crosstalk between innate immune cells—particularly the “NETs-macrophage pyroptosis” positive feedback loop as a central amplifier of myocardial injury was focused. Third, promising therapeutic strategies were evaluated, with emphasis on the multi-target potential of natural compounds and the translational barriers that must be overcome. This framework aims to bridge fundamental discovery and therapeutic innovation, offering a structured pathway from understanding SRCD pathophysiology to develop potential interventions.
Research Progress on the Molecular Mechanisms and Therapeutic Strategies of SRCD
Pathophysiological Mechanisms of Sepsis-Related Cardiac Dysfunction (SRCD)
The pathophysiological mechanisms of SRCD involve a complex multilevel, multipath way regulatory network centered on the uncontrolled inflammatory response and cell damage triggered by pathogen-associated molecular patterns (PAMPs) that activate innate immune signaling pathways.12,13 The Toll-like receptor (TLR) system acts as a key recognition node, transducing signals through adapter proteins such as myeloid differentiation primary response 88 (MyD88) and TRIF.14 It not only promotes nuclear factor kappa B (NF-κB) pathway activation, driving the massive release of inflammatory cytokines (including TNF-α, IL-6, and IL-1β) but also activates stress kinases such as JNK, collectively leading to cardiomyocyte damage.15 Recent studies have shown that novel positive regulators such as OLFM4 (olfactomedin 4), key genes involved in sepsis, can increase NF-κB activity; knocking down OLFM4 expression effectively decreases the LPS-induced release of inflammatory cytokines (TNF-α, IL-6, and IL-1β) in cardiomyocytes and inhibits apoptosis, thereby exerting a protective effect.16 Conversely, endogenous factors such as IL-15 (a proinflammatory cytokine in sepsis) partially inhibit the NF-κB signaling pathway; this regulation suppresses pro-apoptotic proteins (cleaved caspase-3 and Bax) and enhances the anti-apoptotic protein Bcl-2, ultimately mitigating cardiomyocyte apoptosis.17 Additionally, multiple EGF-like domain 9 (MEGF9) regulates energy metabolism and oxidative stress by activating AMPK signaling pathway. Functional experiments have confirmed that MEGF9 overexpression significantly alleviates LPS-induced myocardial inflammation, oxidative stress damage, and cardiac dysfunction, whereas silencing MEGF9 expression exacerbates these pathological changes.18 The GAS6/AXL signaling pathway plays a key role in compensatory protection by inhibiting NLRP3 inflammasome activation and mitigating mitochondrial dysfunction, endoplasmic reticulum stress, and apoptosis.19 Moreover, the cellular senescence-associated protein p16INK4a was shown to exacerbate NLRP3 inflammasome activation and oxidative stress by inhibiting the PI3K/Akt pathway, and its deficiency has a cardioprotective effect.20
These studies not only elucidated the molecular mechanisms of SRCD but also revealed multidimensional intervention strategies targeting the TLR/NF-κB axis, inflammasome activation, and endogenous protective pathways, providing an important theoretical basis for the development of novel treatments for septic myocardial injury.
The Roles of Immune Cells in SRCD and Research Progress on Therapeutic Strategies
The pathophysiological mechanism of SRCD is complex and involves multilevel changes resulting from the imbalance of immune homeostasis and its cascade-damaging effects on cardiomyocytes. Among them, macrophages and neutrophils, as crucial components of the innate immune system, not only participate in inflammation, oxidative stress, and cell death through unique mechanisms but also collectively form an effector network that promotes cardiac dysfunction through complex intercellular communication. Specifically, macrophages can polarize into proinflammatory M1 or anti-inflammatory M2 phenotypes, regulating the local inflammatory environment.21,22 On the other hand, neutrophils can release neutrophil extracellular traps (NETs) through processes such as NETosis, exacerbating tissue damage and promoting immunothrombosis.23–25 In-depth analysis of the regulatory mechanisms of key events such as macrophage polarization and neutrophil NETosis and their interactions is crucial for revealing the pathogenesis of SRCD and developing precise immunomodulatory strategies.
Macrophages
As core executors of innate immunity, macrophages play a key role in SRCD through their phenotypic polarization. Upon pathogen invasion, macrophages polarize into the classically activated M1 phenotype, highly expressing markers such as iNOS, CD86, CD11b, and Cox2 and releasing large amounts of proinflammatory cytokines such as TNF-α and IL-1β, directly driving oxidative stress, mitochondrial dysfunction, and apoptosis. Conversely, M2 macrophages exert anti-inflammatory and tissue repair functions.21,26,27 Research has shown that this polarization process is precisely regulated, and various intervention strategies exert therapeutic effects by modulating macrophage polarization: ginsenoside Rc promotes M2 polarization by inhibiting the STAT3/FoxO3a pathway and upregulating the deacetylase Sirt1;28 myeloid cell-derived angiotensin-converting enzyme (ACE2) promotes M2 polarization by generating Ang1–7 and activating MasR to inhibit NF-κB and STAT1 signalling;29 and losartan effectively promotes macrophage shift towards the M2 phenotype by regulating NF-κB and MAPK signalling.30 Furthermore, atractylenolide I targeting the PARP1/NLRP3 axis,31 Hmgcs2 activating the PI3K/Akt pathway via Src,32 isosteviol sodium (STV-Na) significantly regulating glutathione metabolism, purine metabolism, and glycerophospholipid metabolism pathways, and mediating systemic metabolic reprogramming;33 growth differentiation factor 3 (GDF3) inhibiting NLRP3 inflammasome expression dependent on the Smad2/3 signalling pathway34 and trimetazidine (TMZ) effectively inhibiting LPS-induced proinflammatory responses and oxidative stress in macrophages via Sirt1-mediated regulation of the AMPK/Nrf2/HO-1 and PPARα pathways,35 all of which reveal the diversity and therapeutic potential of regulating macrophage polarization.
Cardiac-resident macrophage subsets, particularly TREM2hi Mac1 cells with unique transcriptional signatures, play an indispensable role in maintaining myocardial homeostasis by actively phagocytosing and clearing dysfunctional mitochondria; impairment of their function directly leads to worsened cardiac function.36 Bioinformatics analysis further revealed key differentially expressed genes (including Socs3, Il1rn, and Ccl7) in septic cardiac macrophages, providing potential new targets for targeted regulation.37 Additionally, recent studies have shown that intercellular communication plays a very important role in SRCD. Macrophage-derived exosomes act as key information carriers, and their biological effects are highly dependent on the activation state of the macrophage. For instance, IL-1β pretreatment enriches macrophage exosomes with miR-146a via the JNK-1/2 pathway, and miR-146a improves cardiomyocyte mitochondrial function by inhibiting MAPK4/Drp-1 signalling.38 Exosomes derived from M2 anti-inflammatory macrophages (M2-exos) deliver miR-24-3p, targeting and inhibiting Tnfsf10 expression, significantly improving cardiac function, reducing cardiomyocyte apoptosis, and lowering serum inflammatory cytokine levels.39 However, TLR9 activation prompts macrophages to secrete exosomes with cardiotoxic effects, exacerbating myocardial mitochondrial oxidative stress and apoptosis, revealing the “double-edged sword” nature of exosome function.40 These findings collectively highlight the regulation of macrophage polarization (M1/M2 balance) and its downstream effects as pivotal intervention points for SRCD.
Neutrophils
In the pathogenesis of SRCD, neutrophils play a dual role; they are both the first line of defence against pathogens and key effector cells that cause myocardial tissue damage.
In a normal immune response, neutrophils are rapidly mobilized to combat infection and clear pathogens by releasing proteases, reactive oxygen species (ROS), and reactive nitrogen species (RNS), which is an important host defence mechanism.41–44 However, in the pathological state of sepsis, the lifespan of neutrophils is abnormally prolonged, and their chemotactic migration ability is impaired, leading to the accumulation of many activated neutrophils in the circulation and myocardial microvasculature.45,46 These accumulated cells are overactivated and produce excessive amounts of ROS, leading to oxidative stress in cardiomyocytes and mitochondrial dysfunction and the release of neutrophil extracellular traps (NETs), which promote inflammation.47 Neutrophils can also abnormally adhere to endothelial cells via high expression of adhesion molecules, obstructing microcirculation, and further activating endothelial cells towards a proinflammatory, procoagulant phenotype through the release of inflammatory mediators, collectively driving SRCD progression.48,49 Furthermore, neutrophils and platelets can directly interact via CD11a; gram-negative bacteria can induce NET formation through TLR4 activation on platelets,50 further exacerbating intravascular inflammation and thrombosis.
The role of neutrophils in SRCD extends far beyond inflammatory cell infiltration; their most notable feature is the release of NETs. NETs are web-like fibrous structures released by activated neutrophils and are characterized by a core of decondensed nuclear chromatin forming DNA fibres 15–17 nm in diameter. These structures not only are important antibacterial weapons but also exhibit significant proinflammatory effects in immune responses.51,52 High concentrations of extracellular DNA can disrupt bacterial membranes by chelating divalent metal ions, but if not timely degraded by nucleases or effectively cleared by phagocytes, they can become signals for tissue damage or programmed cell death, triggering a strong inflammatory response.53,54 Histones (including H1, H2A, H2B, H3, and H4) are the main protein components of NETs, accounting for approximately 70%; within NETs, H2A and H2B are present in high amounts55 and undergo significant posttranslational modifications during NET formation. For instance, serine protease cleavage reduces the molecular weight of histones by 2–5 kDa,43 and acetylation promotes histone separation from DNA through the neutralization of positive charges.56
Peptidylarginine deiminase (PAD)-mediated histone citrullination not only drives chromatin decondensation but also becomes a significant source of autoantibodies in autoimmune diseases such as rheumatoid arthritis (RA).57,58 Furthermore, extracellular histones exhibit antibacterial activity and cytotoxicity and are capable of eliciting strong proinflammatory responses, leading to organ damage and even death.54 PAD4 knockout mice cannot form NETs and exhibit reduced infarct size and improved cardiac function after myocardial ischemia–reperfusion, fully confirming the central role of the PAD4-NET axis in disease progression.59
With respect to therapeutic strategies, studies have shown that using DNase I to degrade extracellular DNA/NETs and using recombinant human ADAMTS13 (rhADAMTS13) to cleave von Willebrand factor (VWF) and inhibit leukocyte recruitment can effectively reduce myocardial infarct size, decrease local tissue NET formation, and improve cardiac systolic function; combined treatment with both drugs has synergistic effects, revealing that simultaneously targeting leukocyte recruitment (VWF pathway) and clearing NETs (extracellular chromatin) is a promising combination strategy.60
Although NET formation is regulated by various mediators,61 lytic NETosis (also known as “suicidal NETosis”) and nonlytic NETosis (also known as “vital NETosis”) are the primary regulatory pathways.25,62,63 The formation of lytic NETosis (“suicidal” or NOX-dependent NETosis) is typically triggered by stimuli such as antibodies, microorganisms, cholesterol crystals, or phorbol myristate acetate (PMA) and is essentially a form of programmed cell death.64 The entire process takes 2–4 hours25,62 and is highly dependent on the production of ROS by the NADPH oxidase (NOX) complex (Figure 1).
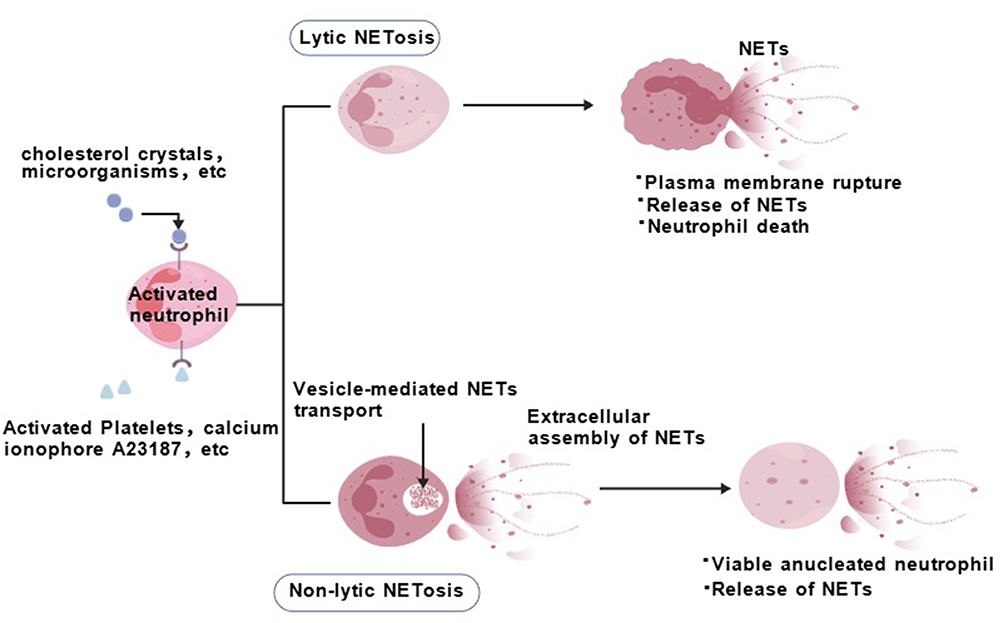 |
Figure 1 Neutrophil extracellular traps (NETs) formation pathways. This figure illustrates two mechanisms of NETs generation. Lytic NETosis: Activated neutrophils form and release NETs via vesicular transport and extracellular assembly, culminating in membrane rupture and cell death. Non-lytic NETosis: Neutrophils release NETs while remaining viable, resulting in anucleate living cells. Both pathways are activated by diverse stimuli and involve intracellular transport and extracellular assembly of NETs. |
On the one hand, ROS burst activates neutrophil elastase (NE), causing its release from azurophilic granules into the cytoplasm; NE binds to F-actin and mediates the degradation of actin filaments, after which it is translocated to the nucleus, where it mediates histone hydrolysis to promote chromatin decondensation.65 On the other hand, ROS promote the activation of peptidylarginine deiminase 4 (PAD4), catalysing the citrullination of histone arginine residues, thereby neutralizing their positive charge and weakening their electrostatic interaction with DNA, collectively promoting chromatin decondensation.65,66 Concurrently, cell cycle-related signals (eg, activation of CDK4/6) and M-phase-like events (eg, lamin phosphorylation and centrosome separation) participate in driving nuclear envelope disintegration. Ultimately, the decondensed chromatin mixes with various granular proteins (eg, MPO and NE) from the cytoplasm and is released extracellularly through the ruptured plasma membrane, leading to cell death.67 Both MPO and NE on chromatin fibres stabilize NETs and ensure their antibacterial properties; complexes of free DNA with MPO or NE are considered specific biomarkers for NETs. Citrullinated histone H3 (citH3), which is associated with NETosis, reflects only PAD4-dependent histone modification. Kenny et al reported that NETosis can occur independently of histone H3 citrullination and that pharmacological inhibition of PAD does not always block NETosis.64
The formation of nonlytic NETosis (vital or NOX-independent NETosis) can occur rapidly (within 5–60 minutes) without causing immediate cell death, which is primarily induced by activated platelets, specific microorganisms (eg, Staphylococcus aureus and Candida albicans), and the calcium ionophore A23187,23,68,69 triggered by receptors such as TLR2, complement C3, or TLR4.51,70–72 This process is independent of NOX-derived ROS but involves the activation of the PAD4 enzyme, causing histone citrullination and chromatin decondensation.73 Cells release mitochondrial DNA (mtDNA) or partial nuclear DNA along with granular proteins directly to the extracellular space via vesicular transport. After NET release, the cells remain metabolically active and retain phagocytic and chemotactic functions, becoming anucleate but functionally active phagocytes.74 Notably, several studies describe a new mode of NET formation involving mitochondrial DNA rather than nuclear DNA. In neutrophils preactivated with IL-8/IFNγ or LPS, a large and rapid release of mtDNA is detected without loss of cell viability. Unlike nonlytic NETosis involving nuclear DNA, mitochondrial NET formation is dependent on ROS, as neutrophils treated with ROS inhibitors cannot release NETs. However, its detailed molecular mechanism remains unclear.51,72 Although the two NET formation pathways differ in terms of inducers, kinetics, and cell fate, they are not completely independent. Modifications such as histone acetylation can regulate the immunoreactivity of NETs; using low concentrations of deacetylase inhibitors can promote NET formation, but when the dose increases to a certain level, NET formation is inhibited.75 Additionally, different neutrophil subsets (eg, high-density vs low-density neutrophils) differ in their ability and ability to form NETs under pathological conditions; low-density neutrophils (LDNs) commonly form NETs and exert immunosuppressive effects in patients with systemic lupus erythematosus (SLE), antiphospholipid syndrome, and pulmonary infections.76–79
NET clearance relies primarily on deoxyribonucleases (DNases) and phagocytosis by macrophages. DNase-I, DNase-II, and DNase-1L3 preferentially degrade free DNA, apoptotic body DNA, and chromatin-bound DNA, respectively. Genetic deficiencies in DNase-I or DNase-1L3 are associated with severe autoimmune diseases such as rheumatoid arthritis and scleroderma. Impaired NET clearance leads to exposure to intracellular antigens, potentially driving autoimmunity. DNase deficiency in mouse models also causes NET-associated thrombosis in multiple organs.59
Crosstalk Between Macrophages and Neutrophils
A self-amplifying inflammatory circuit, driven by the interaction between macrophages and neutrophils, is central to the progression of SRCD.
Recent research has shown that NETosis activation stems not only from direct stimulation by pathogens or inflammatory factors but also from precise regulation by crosstalk between immune cells. For example, Chen et al revealed that NETs induce macrophage pyroptosis via the HMGB1-RAGE-CatB-ASC-Caspase-1 signaling pathway, thereby exacerbating the inflammatory response in sepsis.80 More recent research revealed that macrovesicles released by pyroptotic macrophages are rich in mitochondria that carry GSDMD-N fragments, which are taken up by neutrophils, disrupting their mitochondrial homeostasis, inducing mitochondrial ROS (mtROS) bursts, and subsequently activating GSDMD-N-dependent NETosis.81 These studies reveal a “NET–macrophage pyroptosis–NET” positive feedback loop, resulting in the formation of a self-amplifying inflammatory storm during the pathological process of SRCD (Figure 2). Additionally, a study by Lee et al revealed that in mice, specific depletion of alveolar macrophages leads to excessive recruitment, infiltration, and activation of neutrophils, causing uncontrolled inflammation and tissue damage, ultimately promoting the development of sepsis.82 These findings collectively establish the central role of immune cell crosstalk in the pathogenesis of SRCD, providing a new theoretical framework for understanding the disease’s pathological process.
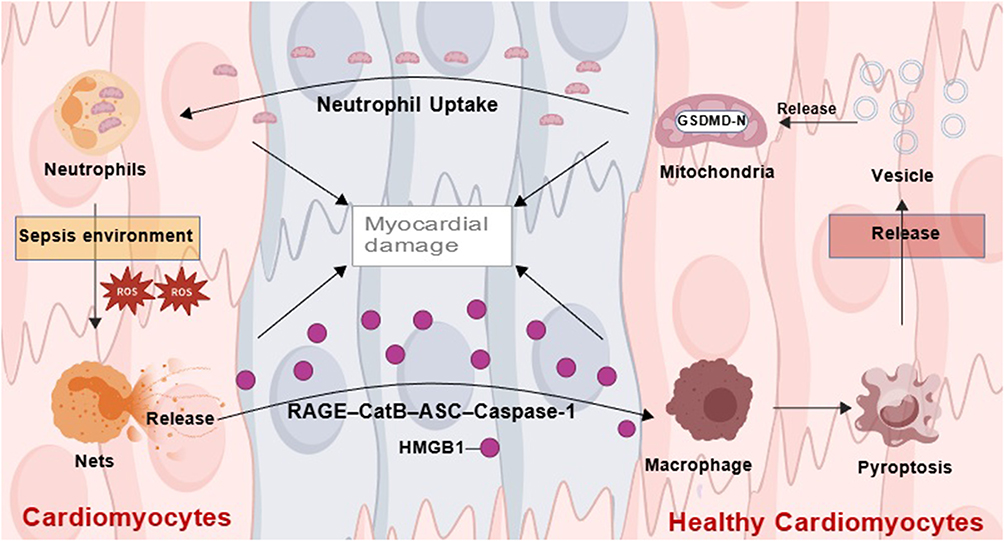 |
Figure 2 The Neutrophil-Macrophage Positive Feedback Loop in Sepsis-Related Cardiac Dysfunction. This figure illustrates the key neutrophil-macrophage positive feedback loop driving sepsis-related cardiac dysfunction. NETs release HMGB1, activating macrophage pyroptosis via the CatB/ASC/Caspase-1 pathway. Pyroptotic macrophages release microvesicles rich in GSDMD-N-tagged mitochondria. Upon uptake by neutrophils, these disrupt mitochondrial homeostasis, leading to mtROS outburst and GSDMD-N-dependent NETosis, which in turn promotes further NETs release. This cycle forms a self-amplifying “NETs → macrophage pyroptosis → NETs” circuit that aggravates cardiac injury in SRCD. |
Non-Coding RNAs
In SRCD, a complex network of non-coding RNAs, particularly long non-coding RNAs (lncRNAs), plays a key regulatory role through mechanisms such as competing endogenous RNA (ceRNA). Some lncRNAs primarily drive inflammatory responses; for example, PVT1 upregulates HMGB1 by sponging miR-29a, exacerbating inflammation,83 while GAS5 activates the NF-κB pathway through the miR-449b/HMGB1 axis, promoting cardiomyocyte injury.84 Others directly regulate cardiomyocyte death: CYTOR modulates XIAP via miR-24 to influence apoptosis;85 MAPKAPK5-AS1 promotes apoptosis and inflammation through the miR-124-3p/E2F3 axis;86 and MCM3AP-AS1 drives cellular inflammation and apoptosis via the miR-28-5p/CASP2 pathway and is highly expressed in the serum of SRCD patients, indicating potential as a biomarker.87 Certain lncRNAs exhibit cardioprotective functions, such as ZFAS1, which upregulates SIRT1 by sponging miR-34b-5p,88 and lncRNA-AABR07066529.3, which alleviates myocardial injury by inhibiting MyD88.89 Additionally, FGD5-AS1 participates in inflammatory regulation through the miR-133a-3p/AQP1 axis,90 whereas MALAT1 exacerbates injury via non-ceRNA mechanisms, such as histone modification.91 Table 1 clearly summarizes the molecular mechanisms, functions, and clinical potential of these key lncRNAs.
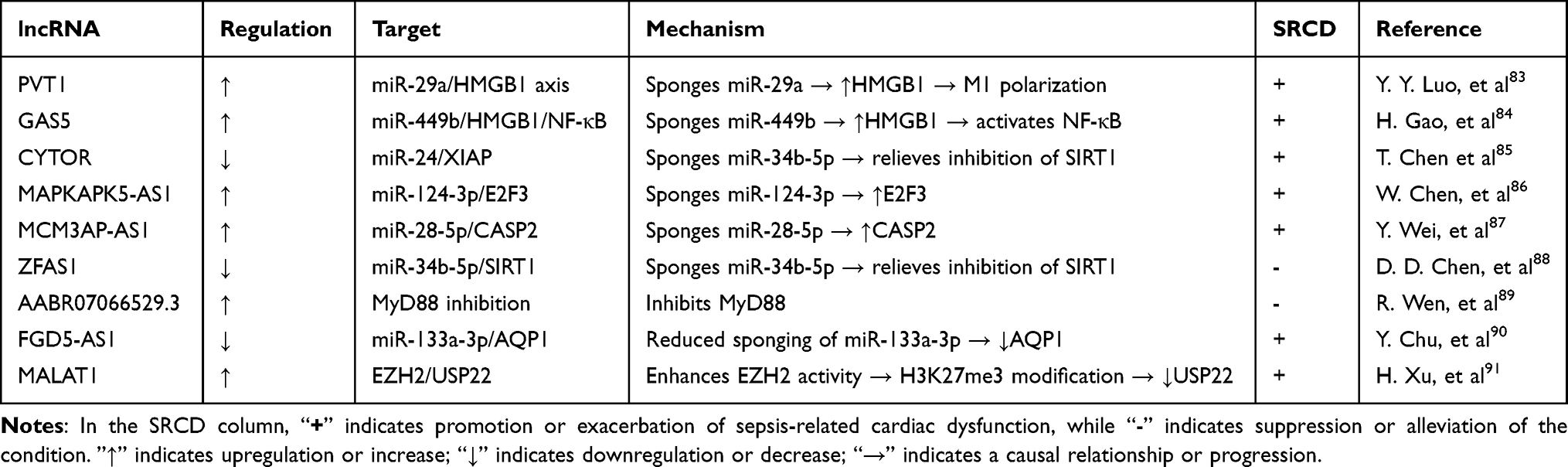 |
Table 1 Regulatory Roles of lncRNAs in Sepsis-Related Cardiac Dysfunction |
Moreover, novel types of cell death, such as ferroptosis, have been confirmed to be involved. Ferroptosis essentially involves the accumulation of lipid peroxides due to inhibited glutathione peroxidase 4 (GPX4) activity.92 The lncRNA Lcn2-204 promotes myocardial iron overload and lipid peroxidation by positively regulating lipocalin-2 protein expression, thereby driving the process of cardiomyocyte ferroptosis.93 These mechanisms provide a solid theoretical foundation and promising intervention targets for the development of new immunomodulatory strategies targeting different pathological phases and multitarget synergy.
Promising Novel Therapeutic Drugs Alleviate SRCD Through Multiple Mechanisms
The pathogenesis of sepsis-related cardiac dysfunction (SRCD) involves immune-inflammatory imbalance and various programmed cell death mechanisms, while multiple natural compounds demonstrate therapeutic potential through multi-target and multi-pathway synergistic effects.94–102 For instance, Sodium octanoate exerts protective effects via anti-inflammatory, antioxidant, and enhanced energy metabolism;94 Ginsenoside-Rg1 synergistically inhibits cardiomyocyte apoptosis and regulates iron homeostasis;95 Melatonin mitigates injury by suppressing ferroptosis and activating AMPK signaling.96,97 Further, other natural compounds exert anti-inflammatory, antioxidant, and anti-cell death activities through their respective mechanisms. Through their respective mechanisms, they collectively exhibit anti-inflammatory, antioxidant, and anti-cell death activities. Table 2 clearly outlines the targets, mechanisms, and therapeutic effects of these compounds in SRCD. These studies provide an important foundation for developing natural compound-based strategies for the prevention and treatment of SRCD.
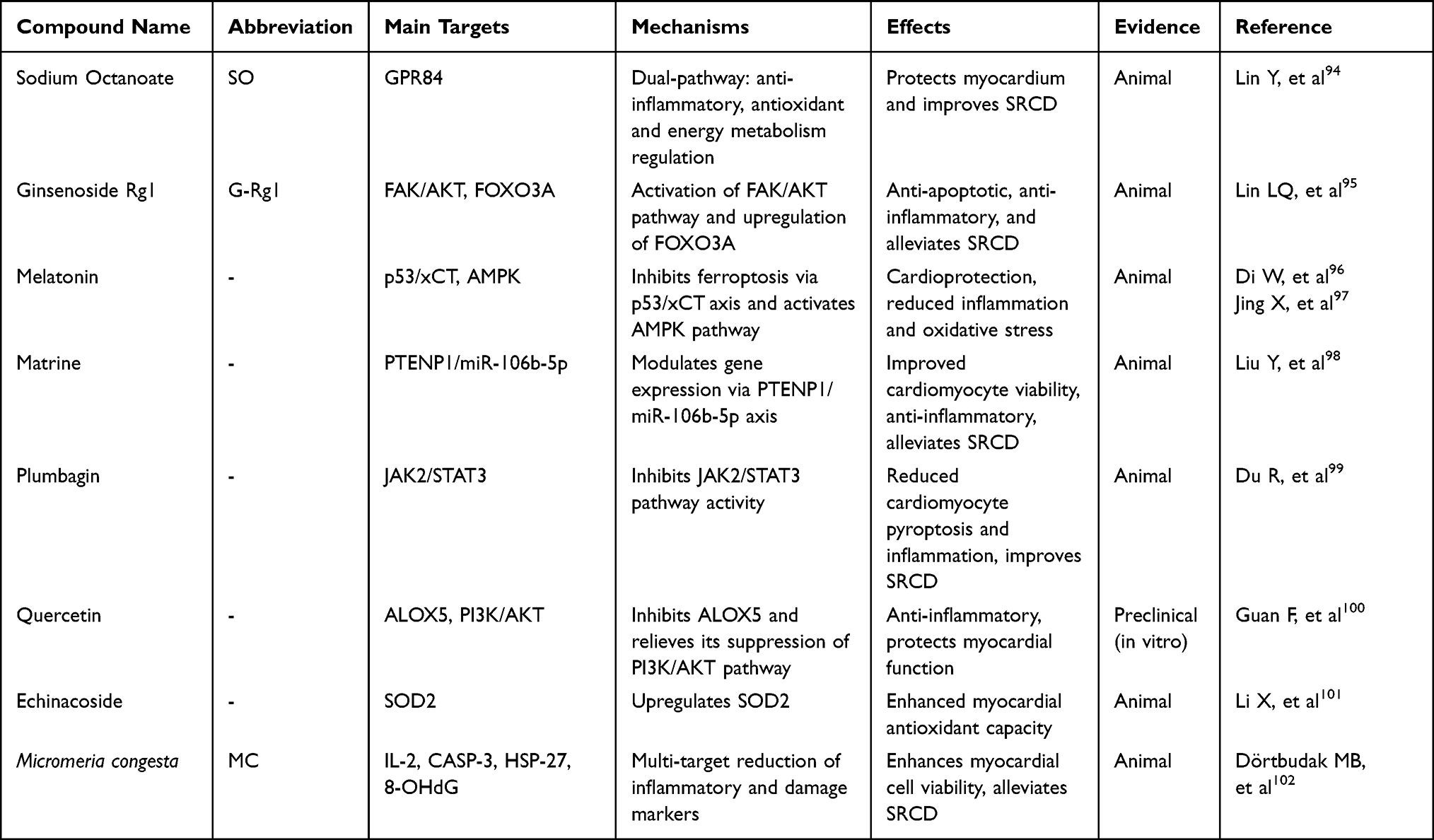 |
Table 2 Summary of Natural Compound Mechanisms in Sepsis-Related Cardiac Dysfunction |
Summary
Summary of Key Mechanisms and Therapeutic Strategies
Sepsis-related cardiac dysfunction (SRCD) is a highly fatal complication of sepsis, with a complex pathogenesis involving systemic inflammatory dysregulation, immune cell dysfunction, and various programmed cell death pathways. This review outlines the expanding scope of SRCD research—from classical signaling pathways such as TLR/NF-κB and the NLRP3 inflammasome, to novel regulators like OLFM4 and MEGF9, as well as injury-related factors such as p16INK4a. It particularly highlights the crosstalk between macrophages and neutrophils, emphasizing the central role of the “NETs–macrophage pyroptosis” positive feedback loop initiated by neutrophil extracellular traps (NETs) in SRCD pathogenesis. Additionally, emerging regulatory mediators such as exosomes, non-coding RNAs, and ferroptosis further exacerbate the pathological progression of SRCD.
In terms of therapeutic strategies, research focus has shifted from single-target anti-inflammatory interventions toward multi-target synergistic regulation. Several natural compounds have shown promising cardioprotective effects in preclinical studies, primarily through multiple mechanisms including immunomodulation, inhibition of cell death, and improvement of energy metabolism. The field is currently at a critical stage transitioning from basic mechanistic exploration to clinical translation. Future progress will depend on the following directions: clarifying how immune cells communicate, developing drugs that target molecules such as MEGF9, GAS6/AXL, and specific lncRNAs, and creating combination treatments suited to different stages of the disease. The goal is to improve outcomes for patients, ultimately aiming to improve clinical outcomes for patients.
Current Limitations and Challenges
Despite these advances, the translation of SRCD research into effective therapies continues to encounter significant translational hurdles. Firstly, most pharmacological studies are reliant on animal models, which fail to fully recapitulate the complexity and heterogeneity of human sepsis, contributing to a high rate of failure in clinical translation. Secondly, the efficacy of many interventions in animal models is critically time-dependent, with optimal outcomes typically observed when administered prior to sepsis induction—a scenario that is often suboptimal or unfeasible in the unpredictable clinical setting. Moreover, the optimal therapeutic window following the onset of sepsis remains poorly defined. Thirdly, promising natural compounds face considerable pharmacological challenges, including low bioavailability, a lack of human pharmacokinetic data, and difficulties in the identification of active components and standardization of extracts. Finally, clinical validation is constrained by the predominant use of all-cause mortality as a primary endpoint.103 There is a notable scarcity of dedicated clinical trials for SRCD that utilize direct and sensitive measures of cardiac function improvement as key outcomes.
This formidable translational dilemma is reflected in clinical guidelines. For example, the Japanese Clinical Practice Guidelines for Management of Sepsis and Septic Shock 2024 (J-SSCG 2024) acknowledges the high incidence and poor prognosis of SRCD in septic shock, noting that while mechanical circulatory support may be considered in refractory cases, evidence remains insufficient and indications are not standardized.104 Beyond recommendations for general supportive care of septic shock—such as fluid resuscitation and vasopressor use—the guideline does not provide specific, evidence-based recommendations for pharmacological or management strategies targeting the pathological process of SRCD. This profoundly illustrates the discrepancy between the current mechanistic understanding of SRCD and established, actionable clinical management protocols, further highlighting the urgent need for targeted clinical research.
Future Perspectives
In summary, the deepening understanding of SRCD mechanisms continues to generate novel therapeutic concepts. Future efforts should focus on integrating multi-omics data, utilizing advanced preclinical models that better mimic human disease, and, most critically, conducting well-designed clinical trials. By bridging these gaps, we can aspire to translate these promising basic research findings into reliable clinical strategies capable of improving patient outcomes.
Acknowledgments
The authors would like to express their gratitude to Mengmeng Zhang from Qingdao Municipal Hospital who finished the experimental pathological staining with Created with BioGDP.com.
Funding
The study was supported by Natural Science Foundation of Shandong Province (ZR2022QH217).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Lin Y, Xu Y, Zhang Z. Sepsis-Induced Myocardial Dysfunction (SIMD): the pathophysiological mechanisms and therapeutic strategies targeting mitochondria. Inflammation. 2020;43(4):1184–13. doi:10.1007/s10753-020-01233-w
2. Nong Y, Wei X, Yu D. Inflammatory mechanisms and intervention strategies for sepsis-induced myocardial dysfunction. Immun Inflamm Dis. 2023;11(5):e860. doi:10.1002/iid3.860
3. Wen R, Zhang TN, Yang N. Recent research on pyroptosis in sepsis-induced myocardial depression. Zhongguo Dang Dai Er Ke Za Zhi. 2024;26(7):774–781. doi:10.7499/j.issn.1008-8830.2312039
4. Yu YY, Wang R, Chen GQ, et al. Mechanisms and targeted therapeutic strategies in sepsis-induced myocardial dysfunction: the role of NLRP3 Inflammasome-Mediated inflammation. J Inflamm Res. 2025;18:8875–8897. doi:10.2147/JIR.S521655
5. Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315(8):801–810. doi:10.1001/jama.2016.0287
6. Frencken JF, Donker DW, Spitoni C, et al. Myocardial injury in patients with sepsis and its association with long-term outcome. Circ Cardiovasc Qual Outcomes. 2018;11(2):e004040. doi:10.1161/CIRCOUTCOMES.117.004040
7. Lv X, Wang H. Pathophysiology of sepsis-induced myocardial dysfunction. Mil Med Res. 2016;3:30. doi:10.1186/s40779-016-0099-9
8. Carbone F, Liberale L, Preda A, Schindler TH, Montecucco F. Septic cardiomyopathy: from pathophysiology to the clinical setting. Cells. 2022;11(18):2833. doi:10.3390/cells11182833
9. Martin L, Derwall M, Thiemermann C, Schürholz T. Heart in sepsis: molecular mechanisms, diagnosis and therapy of septic cardiomyopathy. Anaesthesist. 2017;66(7):479–490. doi:10.1007/s00101-017-0329-x
10. Wang C, Yuan W, Hu A, et al. Dexmedetomidine alleviated sepsis‑induced myocardial ferroptosis and septic heart injury. Mol Med Rep. 2020;22(1):175–184. doi:10.3892/mmr.2020.11114
11. Rhodes A, Evans LE, Alhazzani W, et al. Surviving sepsis campaign: International guidelines for management of sepsis and septic shock: 2016. Crit Care Med. 2017;45(3):486–552. doi:10.1097/CCM.0000000000002255
12. Poveda-Jaramillo R. Heart dysfunction in sepsis. J Cardiothorac Vasc Anesth. 2021;35(1):298–309. doi:10.1053/j.jvca.2020.07.026
13. Zhang G, Dong D, Wan X, Zhang Y. Cardiomyocyte death in sepsis: mechanisms and regulation (Review). Mol Med Rep. 2022;26(2). doi:10.3892/mmr.2022.12773
14. Zhu Y, Zhang M, Ouyang M, Zhou D, Li L. Roles of MyD88 and TRIF in cardiac dysfunction during sepsis. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. 2017;29(8):684–688. doi:10.3760/cma.j.issn.2095-4352.2017.08.003
15. Chang C, Hu L, Sun S, et al. Regulatory role of the TLR4/JNK signaling pathway in sepsis‑induced myocardial dysfunction. Mol Med Rep. 2021;23(5). doi:10.3892/mmr.2021.11973.
16. Chen H, Liu S, Fang G. Knockdown of OLFM4 protects cardiomyocytes from sepsis by inhibiting apoptosis and inflammatory responses. Allergol Immunopathol (Madr). 2024;52(5):15–20. doi:10.15586/aei.v52i5.1145
17. He C, Yu Y, Wang F, Li W, Ni H, Xiang M. Pretreatment with interleukin-15 attenuates inflammation and apoptosis by inhibiting NF-κB signaling in sepsis-induced myocardial dysfunction. Eur J Histochem. 2024;68(2). doi:10.4081/ejh.2024.4019
18. Jin Z, Li X, Liu H, et al. MEGF9 prevents lipopolysaccharide-induced cardiac dysfunction through activating AMPK pathway. Redox Rep. 2025;30(1):2435252. doi:10.1080/13510002.2024.2435252
19. Ji T, Liu Q, Yu L, et al. GAS6 attenuates sepsis-induced cardiac dysfunction through NLRP3 inflammasome-dependent mechanism. Free Radic Biol Med. 2024;210:195–211. doi:10.1016/j.freeradbiomed.2023.11.007
20. Li B, Wang K, Wang X, et al. p16(INK4a) aggravated sepsis-associated cardiac injury by inhibiting the PI3K/AKT pathway and inducing redox imbalance. J Cardiovasc Transl Res. 2025;18(2):375–391. doi:10.1007/s12265-024-10588-6
21. Boutilier AJ, Elsawa SF. Macrophage polarization states in the tumor microenvironment. Int J Mol Sci. 2021;22(13):6995. doi:10.3390/ijms22136995
22. Cutolo M, Campitiello R, Gotelli E, Soldano S. The role of M1/M2 macrophage polarization in rheumatoid arthritis synovitis. Front Immunol. 2022;13:867260. doi:10.3389/fimmu.2022.867260
23. Demkow U. Molecular mechanisms of Neutrophil Extracellular Trap (NETs) degradation. Int J Mol Sci. 2023;24(5):4896. doi:10.3390/ijms24054896
24. Döring Y, Libby P, Soehnlein O. Neutrophil extracellular traps participate in cardiovascular diseases: recent experimental and clinical insights. Circ Res. 2020;126(9):1228–1241. doi:10.1161/CIRCRESAHA.120.315931
25. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. 2018;18(2):134–147. doi:10.1038/nri.2017.105
26. Funes SC, Rios M, Escobar-Vera J, Kalergis AM. Implications of macrophage polarization in autoimmunity. Immunology. 2018;154(2):186–195. doi:10.1111/imm.12910
27. Luo M, Zhao F, Cheng H, Su M, Wang Y. Macrophage polarization: an important role in inflammatory diseases. Front Immunol. 2024;15:1352946. doi:10.3389/fimmu.2024.1352946
28. Jinzhong Wang MS, Jian Fu MS. STAT3/FoxO3a/Sirt1 pathway inhibition by ginsenoside Rc ameliorates cardiomyocyte damage in septic cardiomyopathy by altering macrophage polarization. J Mol Histol. 2025;56(3):148. doi:10.1007/s10735-025-10417-3
29. Xiao X, Li JX, Li HH, Teng F. ACE2 alleviates sepsis-induced cardiomyopathy through inhibiting M1 macrophage via NF-κB/STAT1 signals. Cell Biol Toxicol. 2024;40(1):82. doi:10.1007/s10565-024-09923-z
30. Chen XS, Wang SH, Liu CY, et al. Losartan attenuates sepsis-induced cardiomyopathy by regulating macrophage polarization via TLR4-mediated NF-κB and MAPK signaling. Pharmacol Res. 2022;185:106473. doi:10.1016/j.phrs.2022.106473
31. Wang D, Lin Z, Zhou Y, et al. Atractylenolide I ameliorates sepsis-induced cardiomyocyte injury by inhibiting macrophage polarization through the modulation of the PARP1/NLRP3 signaling pathway. Tissue Cell. 2024;89:102424. doi:10.1016/j.tice.2024.102424
32. Zou XZ, Hao JF, Hou MX. Hmgcs2 regulates M2 polarization of macrophages to repair myocardial injury induced by sepsis. Aging. 2023;15(15):7794–7810. doi:10.18632/aging.204944
33. Wang S, Tan KS, Beng H, et al. Protective effect of isosteviol sodium against LPS-induced multiple organ injury by regulating of glycerophospholipid metabolism and reducing macrophage-driven inflammation. Pharmacol Res. 2021;172:105781. doi:10.1016/j.phrs.2021.105781
34. Wang L, Li Y, Wang X, et al. GDF3 protects mice against sepsis-induced cardiac dysfunction and mortality by suppression of macrophage pro-inflammatory phenotype. Cells. 2020;9(1).
35. Chen J, Lai J, Yang L, et al. Trimetazidine prevents macrophage-mediated septic myocardial dysfunction via activation of the histone deacetylase sirtuin 1. Br J Pharmacol. 2016;173(3):545–561. doi:10.1111/bph.13386
36. Zhang K, Wang Y, Chen S, et al. TREM2(hi) resident macrophages protect the septic heart by maintaining cardiomyocyte homeostasis. Nat Metab. 2023;5(1):129–146. doi:10.1038/s42255-022-00715-5
37. Hu DX, Chen SS, Yu Y, Hu LL, Liu L, Yu LL. Bioinformatics analysis and key gene verification of sepsis myocardial macrophage microarray data based on GEO database. Zhonghua Xin Xue Guan Bing Za Zhi. 2023;51(7):759–768. doi:10.3760/cma.j.cn112148-20230522-00295
38. Ma C, Yang Z, Wang J, et al. Interleukin-1β-stimulated macrophage-derived exosomes improve myocardial injury in sepsis via regulation of mitochondrial homeostasis: experimental research. Int J Surg. 2025;111(1):283–301. doi:10.1097/JS9.0000000000001915
39. Sun X, Liu Y, Wang J, Zhang M, Wang M. Cardioprotection of M2 macrophages-derived exosomal microRNA-24-3p/Tnfsf10 axis against myocardial injury after sepsis. Mol Immunol. 2022;141:309–317. doi:10.1016/j.molimm.2021.11.003
40. Li X, Luo J, Li Y, et al. Macrophage-derived exosomes in TLR9(-/-) mice ameliorate sepsis-induced mitochondrial oxidative stress and apoptosis in cardiomyocytes. Oxid Med Cell Longev. 2022;2022:5719974. doi:10.1155/2022/5719974
41. Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. 2006;6(3):173–182. doi:10.1038/nri1785
42. Furze RC, Rankin SM. Neutrophil mobilization and clearance in the bone marrow. Immunology. 2008;125(3):281–288. doi:10.1111/j.1365-2567.2008.02950.x
43. Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191(3):677–691. doi:10.1083/jcb.201006052
44. Metzler KD, Fuchs TA, Nauseef WM, et al. Myeloperoxidase is required for neutrophil extracellular trap formation: implications for innate immunity. Blood. 2011;117(3):953–959. doi:10.1182/blood-2010-06-290171
45. Sônego F, Castanheira FV, Ferreira RG, et al. Paradoxical roles of the neutrophil in sepsis: protective and deleterious. Front Immunol. 2016;7:155. doi:10.3389/fimmu.2016.00155
46. Milot E, Fotouhi-Ardakani N, Filep JG. Myeloid nuclear differentiation antigen, neutrophil apoptosis and sepsis. Front Immunol. 2012;3:397. doi:10.3389/fimmu.2012.00397
47. Yipp BG, Kubes P. NETosis: how vital is it? Blood. 2013;122(16):2784–2794. doi:10.1182/blood-2013-04-457671
48. Aldabbous L, Abdul-Salam V, McKinnon T, et al. Neutrophil extracellular traps promote angiogenesis: evidence from vascular pathology in pulmonary hypertension. Arterioscler Thromb Vasc Biol. 2016;36(10):2078–2087. doi:10.1161/ATVBAHA.116.307634
49. Ma Y, Yang X, Chatterjee V, Meegan JE, Beard RS, Yuan SY. Role of neutrophil extracellular traps and vesicles in regulating vascular endothelial permeability. Front Immunol. 2019;10:1037. doi:10.3389/fimmu.2019.01037
50. Clark SR, Ma AC, Tavener SA, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13(4):463–469. doi:10.1038/nm1565
51. Herre M, Cedervall J, Mackman N, Olsson AK. Neutrophil extracellular traps in the pathology of cancer and other inflammatory diseases. Physiol Rev. 2023;103(1):277–312. doi:10.1152/physrev.00062.2021
52. Zhang H, Wang Y, Qu M, et al. Neutrophil, neutrophil extracellular traps and endothelial cell dysfunction in sepsis. Clin Transl Med. 2023;13(1):e1170. doi:10.1002/ctm2.1170
53. Jiménez-Alcázar M, Rangaswamy C, Panda R, et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science. 2017;358(6367):1202–1206. doi:10.1126/science.aam8897
54. Morales-Primo AU, Becker I, Zamora-Chimal J. Neutrophil extracellular trap-associated molecules: a review on their immunophysiological and inflammatory roles. Int Rev Immunol. 2022;41(2):253–274. doi:10.1080/08830185.2021.1921174
55. Richards CM, McRae SA, Ranger AL, Klegeris A. Extracellular histones as damage-associated molecular patterns in neuroinflammatory responses. Rev Neurosci. 2023;34(5):533–558. doi:10.1515/revneuro-2022-0091
56. Hamam HJ, Khan MA, Palaniyar N. Histone acetylation promotes neutrophil extracellular trap formation. Biomolecules. 2019;9(1):32. doi:10.3390/biom9010032
57. Spengler J, Lugonja B, Ytterberg AJ, et al. Release of active peptidyl arginine deiminases by neutrophils can explain production of extracellular citrullinated autoantigens in rheumatoid arthritis synovial fluid. Arthritis Rheumatol. 2015;67(12):3135–3145. doi:10.1002/art.39313
58. Sánchez-Tirado E, Agüí L, Sánchez-Paniagua M, et al. Serum autoantibody biomarkers for management of rheumatoid arthritis disease. Biosensors. 2023;13(3). doi:10.3390/bios13030381.
59. Natorska J, Ząbczyk M, Undas A. Neutrophil extracellular traps (NETs) in cardiovascular diseases: from molecular mechanisms to therapeutic interventions. Kardiol Pol. 2023;81(12):1205–1216. doi:10.33963/v.kp.98520
60. Savchenko AS, Borissoff JI, Martinod K, et al. VWF-mediated leukocyte recruitment with chromatin decondensation by PAD4 increases myocardial ischemia/reperfusion injury in mice. Blood. 2014;123(1):141–148. doi:10.1182/blood-2013-07-514992
61. Kaltenmeier C, Simmons RL, Tohme S, Yazdani HO. Neutrophil Extracellular Traps (NETs) in cancer metastasis. Cancers. 2021;13(23):6131. doi:10.3390/cancers13236131
62. Aubé FA, Bidias A, Pépin G. Who and how, DNA sensors in NETs-driven inflammation. Front Immunol. 2023;14:1190177. doi:10.3389/fimmu.2023.1190177
63. Byrd AS, O’Brien XM, Johnson CM, Lavigne LM, Reichner JS. An extracellular matrix-based mechanism of rapid neutrophil extracellular trap formation in response to Candida albicans. J Immunol. 2013;190(8):4136–4148. doi:10.4049/jimmunol.1202671
64. Kenny EF, Herzig A, Krüger R, et al. Diverse stimuli engage different neutrophil extracellular trap pathways. Elife. 2017;6.
65. Hidalgo A, Libby P, Soehnlein O, Aramburu IV, Papayannopoulos V, Silvestre-Roig C. Neutrophil extracellular traps: from physiology to pathology. Cardiovasc Res. 2022;118(13):2737–2753. doi:10.1093/cvr/cvab329
66. Guo W, Gong Q, Zong X, et al. GPR109A controls neutrophil extracellular traps formation and improve early sepsis by regulating ROS/PAD4/Cit-H3 signal axis. Exp Hematol Oncol. 2023;12(1):15. doi:10.1186/s40164-023-00376-4
67. Amulic B, Knackstedt SL, Abu Abed U, et al. Cell-cycle proteins control production of neutrophil extracellular traps. Dev Cell. 2017;43(4):449–62.e5. doi:10.1016/j.devcel.2017.10.013
68. Douda DN, Khan MA, Grasemann H, Palaniyar N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc Natl Acad Sci U S A. 2015;112(9):2817–2822. doi:10.1073/pnas.1414055112
69. Lee KH, Kronbichler A, Park DD, et al. Neutrophil extracellular traps (NETs) in autoimmune diseases: a comprehensive review. Autoimmun Rev. 2017;16(11):1160–1173. doi:10.1016/j.autrev.2017.09.012
70. Tabrizi ZA, Khosrojerdi A, Aslani S, et al. Multi-facets of neutrophil extracellular trap in infectious diseases: moving beyond immunity. Microb Pathog. 2021;158:105066. doi:10.1016/j.micpath.2021.105066
71. Cristinziano L, Modestino L, Antonelli A, et al. Neutrophil extracellular traps in cancer. Semin Cancer Biol. 2022;79:91–104. doi:10.1016/j.semcancer.2021.07.011
72. Vorobjeva NV, Chernyak BV. NETosis: molecular mechanisms, role in physiology and pathology. Biochemistry. 2020;85(10):1178–1190. doi:10.1134/S0006297920100065
73. Leshner M, Wang S, Lewis C, et al. PAD4 mediated histone hypercitrullination induces heterochromatin decondensation and chromatin unfolding to form neutrophil extracellular trap-like structures. Front Immunol. 2012;3:307. doi:10.3389/fimmu.2012.00307
74. Jayaseelan VP, Paramasivam A. Emerging role of NET inhibitors in cardiovascular diseases. Hypertens Res. 2020;43(12):1459–1461. doi:10.1038/s41440-020-0527-9
75. Hamam HJ, Palaniyar N. Histone deacetylase inhibitors dose-dependently switch neutrophil death from NETosis to apoptosis. Biomolecules. 2019;9(5):184. doi:10.3390/biom9050184
76. Tay SH, Celhar T, Fairhurst AM. Low-density neutrophils in systemic lupus erythematosus. Arthritis Rheumatol. 2020;72(10):1587–1595. doi:10.1002/art.41395
77. Mehdipour P, Marhon SA, Ettayebi I, et al. Epigenetic therapy induces transcription of inverted SINEs and ADAR1 dependency. Nature. 2020;588(7836):169–173. doi:10.1038/s41586-020-2844-1
78. Mauracher LM, Krall M, Roiß J, et al. Neutrophil subpopulations and their activation potential in patients with antiphospholipid syndrome and healthy individuals. Rheumatology. 2021;60(4):1687–1699. doi:10.1093/rheumatology/keaa532
79. Rankin AN, Hendrix SV, Naik SK, Stallings CL. Exploring the role of low-density neutrophils during Mycobacterium tuberculosis infection. Front Cell Infect Microbiol. 2022;12:901590. doi:10.3389/fcimb.2022.901590
80. Chen L, Zhao Y, Lai D, et al. Neutrophil extracellular traps promote macrophage pyroptosis in sepsis. Cell Death Dis. 2018;9(6):597. doi:10.1038/s41419-018-0538-5
81. Kuang L, Wu Y, Shu J, Yang J, Zhou H, Huang X. Pyroptotic macrophage-derived microvesicles accelerate formation of neutrophil extracellular traps via GSDMD-N-expressing mitochondrial transfer during sepsis. Int J Biol Sci. 2024;20(2):733–750. doi:10.7150/ijbs.87646
82. Lee HH, Aslanyan L, Vidyasagar A, et al. Depletion of alveolar macrophages increases pulmonary neutrophil infiltration, tissue damage, and sepsis in a murine model of Acinetobacter baumannii pneumonia. Infect Immun. 2020;88(7). doi:10.1128/IAI.00128-20.
83. Luo YY, Yang ZQ, Lin XF, et al. Knockdown of lncRNA PVT1 attenuated macrophage M1 polarization and relieved sepsis induced myocardial injury via miR-29a/HMGB1 axis. Cytokine. 2021;143:155509. doi:10.1016/j.cyto.2021.155509
84. Gao H, Ma H, Gao M, Chen A, Zha S, Yan J. Long non-coding RNA GAS5 aggravates myocardial depression in mice with sepsis via the microRNA-449b/HMGB1 axis and the NF-κB signaling pathway. Biosci Rep. 2021;41(4). doi:10.1042/BSR20201738
85. Chen T, Zhu C, Ye C. LncRNA CYTOR attenuates sepsis-induced myocardial injury via regulating miR-24/XIAP. Cell Biochem Funct. 2020;38(7):976–985. doi:10.1002/cbf.3524
86. Chen W, Gao G, Yan M, Yu M, Shi K, Yang P. Long noncoding RNA MAPKAPK5-AS1 promoted lipopolysaccharide-induced inflammatory damage in the myocardium by sponging microRNA-124-3p/E2F3. Mol Med. 2021;27(1):131. doi:10.1186/s10020-021-00385-1
87. Wei Y, Bai C, Xu S, Cui M, Wang R, Wu M. Diagnostic and predictive value of LncRNA MCM3AP-AS1 in sepsis and its regulatory role in sepsis-induced myocardial dysfunction. Cardiovasc Toxicol. 2024;24(10):1125–1138. doi:10.1007/s12012-024-09903-z
88. Chen DD, Wang HW, Cai XJ. Long non-coding RNA ZFAS1 alleviates sepsis-induced myocardial injury via target miR-34b-5p/SIRT1. Innate Immun. 2021;27(5):377–387. doi:10.1177/17534259211034221
89. Wen R, Zhang TN, Zhang T, et al. A novel long noncoding RNA-lncRNA-AABR07066529.3 alleviates inflammation, apoptosis, and pyroptosis by inhibiting MyD88 in lipopolysaccharide-induced myocardial depression. FASEB J. 2023;37(8):e23063. doi:10.1096/fj.202201680R
90. Chu Y, Wang X, Yu N, Li Y, Kan J. Long non‑coding RNA FGD5‑AS1/microRNA‑133a‑3p upregulates aquaporin 1 to decrease the inflammatory response in LPS‑induced sepsis. Mol Med Rep. 2021;24(5). doi:10.3892/mmr.2021.12424
91. Xu H, Ye W, Shi B. LncRNA MALAT1 regulates USP22 expression through EZH2-mediated H3K27me3 modification to accentuate sepsis-induced myocardial dysfunction. Cardiovasc Toxicol. 2022;22(9):813–830. doi:10.1007/s12012-022-09758-2
92. Wang Z, Wu C, Yin D, Dou K. Ferroptosis: mechanism and role in diabetes-related cardiovascular diseases. Cardiovasc Diabetol. 2025;24(1):60. doi:10.1186/s12933-025-02614-x
93. Huang Y, Li L, Li Y, et al. Knockdown of LncRNA Lcn2-204 alleviates sepsis-induced myocardial injury by regulation of iron overload and ferroptosis. J Mol Cell Cardiol. 2024;192:79–93. doi:10.1016/j.yjmcc.2024.05.007
94. Lin Y, Zhang W, Jiang X, et al. Sodium octanoate mediates GPR84-dependent and independent protection against sepsis-induced myocardial dysfunction. Biomed Pharmacother. 2024;180:117455. doi:10.1016/j.biopha.2024.117455
95. Lin LQ, Mao FK, Lin J, Guo L, Yuan WR, Wang BY. Ginsenoside Rg1 induces ferroptosis by regulating the focal adhesion kinase/protein kinase B-forkhead box O3A signaling pathway and alleviates sepsis-induced myocardial damage. J Physiol Pharmacol. 2024;75(4). doi:10.26402/jpp.2024.4.04
96. Di W, Jin Z, Lei W, et al. Protection of melatonin treatment and combination with traditional antibiotics against septic myocardial injury. Cell Mol Biol Lett. 2023;28(1):35. doi:10.1186/s11658-022-00415-8
97. Jing X, Chen Z, Zhang M, et al. Melatonin mitigates the lipopolysaccharide-induced myocardial injury in rats by blocking the p53/xCT pathway-mediated ferroptosis. Naunyn Schmiedebergs Arch Pharmacol. 2025;398(2):1653–1663. doi:10.1007/s00210-024-03367-2
98. Liu Y, Liu L, Zhang J. Protective role of matrine in sepsis-associated cardiac dysfunction through regulating the lncRNA PTENP1/miR-106b-5p axis. Biomed Pharmacother. 2021;134:111112. doi:10.1016/j.biopha.2020.111112
99. Du R, Yun Q, Wang Y, et al. [Plumbagin protect against sepsis-induced myocardial injury in mice by inhibiting the JAK2/STAT3 signaling pathway to reduce cardiomyocyte pyroptosis]. Nan Fang Yi Ke Da Xue Xue Bao. 2024;44(11):2209–2219. doi:10.12122/j.issn.1673-4254.2024.11.18
100. Guan F, Du H, Li J, Ren H, Dong A. Quercetin alleviates LPS-stimulated myocardial injury through regulating ALOX5/PI3K/AKT pathway in sepsis. Cardiovasc Toxicol. 2024;24(10):1116–1124. doi:10.1007/s12012-024-09901-1
101. Li X, Zhang Z, Zhang X, et al. Echinacoside prevents sepsis-induced myocardial damage via targeting SOD2. J Med Food. 2024;27(2):123–133. doi:10.1089/jmf.2023.K.0222
102. Dörtbudak MB, Demircioğlu M, Kapucuk FS. Micromeria congesta alleviates LPS-induced inflammation, apoptosis, oxidative stress and DNA damage in rat heart and kidneys. Vet Med Sci. 2025;11(2):e70264. doi:10.1002/vms3.70264
103. Zou S, Bai J, Liang C, Yang J, Xiao J. Plasmocytoid dendritic cells in sepsis-associated acute lung injury: a potential therapeutic target. Anesthesiol Perioperative Sci. 2025;3:36. doi:10.1007/s44254-025-00121-6
104. Shime N, Nakada TA, Yatabe T, et al. The Japanese clinical practice guidelines for management of sepsis and septic shock 2024. Acute Med Surg. 2025;12(1):e70037. doi:10.1002/ams2.70037
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.