")
Back to Journals » Journal of Inflammation Research » Volume 16
Progress in the Treatment of High Altitude Cerebral Edema: Targeting REDOX Homeostasis
Authors Li Y, Li C, Luo T, Yue T, Xiao W, Yang L, Zhang Z, Han F, Long P , Hu Y
Received 4 April 2023
Accepted for publication 15 June 2023
Published 23 June 2023 Volume 2023:16 Pages 2645—2660
DOI https://doi.org/10.2147/JIR.S415695
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Yubo Li,1,2,* Chengming Li,1,2,* Tao Luo,3,* Tian Yue,4 Wenjing Xiao,5 Ling Yang,1,2 Zaiyuan Zhang,6 Fei Han,3,* Pan Long,3 Yonghe Hu6
1School of Clinical Medicine, Chengdu University of TCM, Chengdu, People’s Republic of China; 2Basic Medical Laboratory, The General Hospital of Western Theater Command, Chengdu, People’s Republic of China; 3Department of Ophthalmology, The General Hospital of Western Theater Command, Chengdu, People’s Republic of China; 4School of Life Science and Engineering, Southwest Jiaotong University, Chengdu, People’s Republic of China; 5Department of Pharmacy, The General Hospital of Western Theater Command, Chengdu, People’s Republic of China; 6College of Medicine, Southwest Jiaotong University, Chengdu, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Pan Long, Department of Ophthalmology, The General Hospital of Western Theater Command, Rongdu Avenue #270, Chengdu, People’s Republic of China, Tel +86-181-9125-6132, Email [email protected] Yonghe Hu, College of Medicine, Southwest Jiaotong University, No. 111, North First section of the Second Ring Road, Chengdu, People’s Republic of China, Tel +86-138-8059-6789, Email [email protected]
Abstract: With the increasing of altitude activities from low-altitude people, the study of high altitude cerebral edema (HACE) has been revived. HACE is a severe acute mountain sickness associated with exposure to hypobaric hypoxia at high altitude, often characterized by disturbance of consciousness and ataxia. As for the pathogenesis of HACE, previous studies suggested that it might be related to the disorder of cerebral blood flow, the destruction of blood-brain barrier and the injury of brain parenchyma cells caused by inflammatory factors. In recent years, studies have confirmed that the imbalance of REDOX homeostasis is also involved in the pathogenesis of HACE, which mainly leads to abnormal activation of microglia and destruction of tight junction of vascular endothelial cells through the excessive production of mitochondrial-related reactive oxygen species. Therefore, this review summarizes the role of REDOX homeostasis and the potential of the treatment of REDOX homeostasis in HACE, which is of great significance to expand the understanding of the pathogenesis of HACE. Moreover, it will also be helpful to further study the possible therapy of HACE related to the key link of REDOX homeostasis.
Keywords: high altitude cerebral edema, hypobaric hypoxia, REDOX homeostasis, mitochondria dysfunction, microglia
Introduction
High altitude (intermediate altitude: 1500–2500 meters; high altitude: 2500–3500 meters; very high altitude: 3500–5500 meters; extreme altitude: >5500 meters) is characterized by low pressure, low oxygen, low temperature, low humidity and high ultraviolet radiation.1 People who live in plain areas for a long time entering high altitude areas (>2500 meters) will inevitably suffer from acute and chronic altitude sickness to varying degrees due to different body adaptability to the environment.2,3 Proper high altitude response (increased respiratory rate, heart rate, metabolic rate, etc.) is beneficial for the body to quickly adapt to hypoxic environment. However, if poorly adapted, a variety of acute and chronic altitude diseases may occur, such as high altitude brain edema (HACE), high altitude pulmonary edema (HAPE) and high altitude heart disease (HAHD). Specifically, the brain is the body’s High oxygen consumption organs that relies primarily on glucose and ketone bodies for energy.4 Therefore, it is extremely sensitive to the hypobaric hypoxia environments at high altitudes.5
HACE typically occurs in areas with altitudes ranging from 4500m to 5500m, is characterized by symptoms such as headache, ataxia, and altered mental status.6,7 Though the incidence is low, it is a rapidly progressive and fatal disease. Without timely and effective treatments, patients can progress to coma or even death within 24 hours.8 However, current treatment options for HACE are limited. The main reason is that its mechanism is complicated, involving a series of pathological changes such as cerebral blood flow disturbance, disruption of the blood-brain barrier (BBB), and brain parenchymal cell damage induced by inflammatory factors in a low-pressure hypoxic environment.9,10 Recently, studies have found that REDOX homeostasis plays an important role in the occurrence, development and outcome of HACE.2,11,12
REDOX homeostasis refers to the dynamic balance between oxidation and reduction in the body, which belongs to the inherent self-defense mechanism of cells and plays a crucial role in maintaining the normal life activities of cell.13 Profound REDOX imbalances are associated with a wide range of diseases, such as virus infections,14,15 tumors16–18 and cardio-cerebrovascular diseases.19–23 Studies found that exposure to low-pressure and hypoxic platforms leads to alterations in a range of metabolic reactions and cellular pathways that are closely related to the maintenance of REDOX homeostasis.10,24–27 Therefore, we focus on the regulation of REDOX homeostasis to explore the pathological mechanism and treatment progress of HACE.
Basic Overview of HACE
Epidemiology and Pathological Mechanism
Unacclimatized individuals who rapidly ascend to heights of more than 2500 meters are at risk of developing one or more acute high altitude illness (HAI), mainly including acute mountain sickness (AMS), HACE and HAPE.3 AMS and HAPE can occur in susceptible individuals at altitudes as low as 2500 meters, while HACE usually occurs at higher elevations.3 Specifically, HACE mostly occurs in the stage of rapidly entering high altitude, and the altitude of 4500~5500 meters is the high incidence area.6 According to previous studies, the main risk factors of HACE mainly include rapid ascent,28 fatigue, cardiopulmonary diseases (such as organic heart diseases, upper respiratory tract infections),29 and neurological diseases (such as Parkinson’s disease, epilepsy).30 Although HACE morbidity and mortality rates have decreased in recent years, the number of HACE patients cannot be underestimated due to increased highland activity. Therefore, it is still necessary to further study the pathological mechanism and prevention and treatment of HACE.
Previous studies have suggested that the main causes of HACE are cerebral hemodynamic disorders (increased cerebral blood flow and velocity) caused by hypobaric hypoxia, destruction of blood-brain barrier and abnormal inflammatory factors. The disturbance of cerebral hemodynamics may disrupt the tight junctions between endothelial cells of cerebral arteries and lead to cerebral vasogenic edema caused by BBB damage.31 Moreover, the hypoxic environment can cause mitochondrial dysfunction, leading to intracellular edema. In addition, neurons and glial cells release inflammatory cytokines such as interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), and interleukin-6 (IL-6) under hypoxia conditions at high altitude.32–35 These inflammatory factors can increase cerebral vascular permeability and result in cerebral edema.36 HACE and HAPE are both acute high-altitude illnesses and share some pathophysiological mechanisms. Pulmonary edema in HAPE patients may cause pulmonary arterial hypertension, leading to increased right heart load, decreased cardiac output and cerebral perfusion pressure, resulting in cerebral hypoxia, cerebral edema, and even HACE. Clinical observations have found that HACE patients often have dual edema in the brain and lungs, indicating a close relationship between the brain and lungs in acute mountain sickness.37–39 Therefore, we believe that the exploration of the pathological mechanisms, prevention, and treatment of high-altitude cerebral edema can be linked with high-altitude pulmonary edema, rather than just focusing on the lesions of a single organ or tissue. Oxidative stress injury has been proven to be ubiquitous in the pathogenesis of HACE and HAPE and has become a new research hotspot in recent years.2,40,41 In the high altitude hypoxia environment, the body produces a large number of free radicals and oxidants, while hypoxia and ischemia lead to the lack of intracellular reductants, resulting in the imbalance of REDOX homeostasis in the body.12,42,43 This REDOX homeostasis imbalance can cause a variety of pathophysiological reactions, including abnormal intracellular metabolism, inflammation, cell apoptosis and so on, resulting in the occurrence of HACE.2,44
Clinical Manifestations and Diagnostic Methods
HACE is an acute and severe HAI caused by dysfunction of central nervous system in the hypobaric hypoxia environment at high altitude. In terms of clinical symptoms, HACE also has common manifestations of AMS, such as headache, anorexia, nausea, vomiting, dizziness, and fatigue.45 In addition, its more prominent feature is abnormal manifestations of the nervous system, mainly including severe headache, altered states of consciousness, ataxia (instability of gait).46 If not treated quickly, HACE can cause epilepsy, coma and even death. Physical signs can present as limb dysfunction, pyramidal tract syndrome, or meningeal irritation, and approximately half of patients with HACE have papilledema, retinal hemorrhage, urinary retention or urinary incontinence.6
At present, the diagnosis of HACE mainly relies on a history of highland activities, the observation of symptoms and signs, as well as a comprehensive analysis of laboratory and imaging tests. In terms of clinical symptoms and signs, the absence of coordination between the trunk and lower or upper limbs (truncal ataxia) is a typical sign.46 Studies showed that ataxia as measured by heel-knee-tibia test, finger-nose test and rapid rotation is the best objective clinical index for the progression of AMS to HACE.47 Laboratory tests such as electrolytes, complete blood counts, blood glucose, ethanol levels, carboxyhemoglobin levels, and toxicological screening can help rule out other diseases with similar clinical symptoms. HACE imaging changes recognized by computed tomography (CT) and magnetic resonance imaging (MRI) are often manifested as vasogenic edema and diffuse microhemorrhage in the white matter area of the splenium of the corpus callosum.48 Hemosiderin deposition in the corpus callosum is often considered to be a highly specific sign of HACE and can be used to distinguish AMS from HACE.49 Other brain diseases related to hypoxia can also be distinguished by the different shapes and locations of lesions in MRI images. For example, hypoxic brain injury, it also presents with angiogenic edema and increased water content in the brain. However, it is generally not accompanied by diffuse microhemorrhage and hemosiderin deposition, and presents with white matter lesions in the cerebral cortex and hippocampus.50 A recent study confirmed that the endpoint of retinal peripapillary capillaries detected by optical coherence tomography angiography can be used as a rapid, non-invasive potential biomarker for AMS, enabling the observation and prediction of changes in the central nervous system microvasculature and the development of HACE.51 In addition, uneven enlargement of the inner diameter of the optic nerve sheath, edema of the optic disc, enlargement of the inner diameter of retinal blood vessels, and leakage of retinal blood vessels may also be the precursor of HACE to a certain extent.52
Progress in Prevention and Treatment of HACE
Because of the low morbidity, rapid progression, and high mortality of HACE, prevention must take precedence over treatment. Pretreatment and slow rise are the most effective means to reduce the incidence of HACE.53 Specifically, intermittent exposure to low or positive pressure anoxia through the use of hypoxic tents, hypoxic chambers, or hypoxic masks pre-acclimates the body to high altitude hypoxia.54 The speed of ascent should be around 600 m/day, and climbers should rest for 1 day for every 600–1200 m of ascent.46 Meanwhile, when reaching more than 3000 meters at high altitude, climbers should increase their sleep height at a rate of no more than 500 meters per day.3 This only applies to those who are at low risk of developing complications and with no previous episodes of altitude sickness. Although it is generally believed that adaptation remains the best strategy for HACE prevention, and prophylactic medication is not recommended for mountain climbers at low HACE risk, prophylactic medication is also necessary for patients at moderate to high risk.54 Acetazolamide (125–250 mg orally, twice daily) is currently the only certified prophylactic drug that helps to adapt to high-altitude conditions and may be useful in patients with a history of altitude sickness.3,55,56
For the treatment of HACE, rapid descent to low altitude is a top priority. During the descent, patients with severe HACE should be given oxygen with a target saturation of 90%. Patients with severe HACE with delayed transport may be treated with a portable hyperbaric chamber to immediately reduce cerebral blood flow, thereby reducing intracranial pressure.3 Medication should be administered when the altitude cannot be reduced immediately or when oxygen therapy is not available due to conditions. At present, there exists several therapeutic ways for HACE, including absolute repose, dehydration therapy, hypothermia, medication and fluid therapy, and oxygen therapy. However, there is no specific medicines for the treatment of HACE, previous clinical experience supports the use of dexamethasone, acetazolamide, and mannitol.3,57 Recently, it has also been suggested that intranasal administration of dexamethasone may be a non-invasive alternative to muscular or intravenous administration. This may be a more convenient and efficient way, but the symptoms of congestion and rhinitis common at high altitudes can limit nasal absorption.58 However, dexamethasone is not easy to get used to, and sudden withdrawal can cause a rebound in symptoms of high altitude disease.59 Nowadays, studies have found that some effective components of traditional Chinese medicine can relieve HACE through antioxidation and reduce oxidative stress in hypobaric and hypoxic environment, such as ginkgo biloba extract,60 salidroside,61 quercetin,62,63 puerarin,64–66 catechin,67 betel nut polyphenols,68 and so on.
The Role of REDOX Imbalance in the Pathogenesis of HACE
REDOX Homeostasis
The photosynthesis of life on earth leads to the accumulation of oxygen in the atmosphere and exposes the human body to an oxygen-rich environment in which oxidation occurs periodically. In order to adapt to this environment, the body has formed a molecular mechanism to regulate and maintain the balance between reduction and oxidation, namely REDOX homeostasis.69 REDOX homeostasis is the most basic driving force of human life activities and the most common metabolic reaction mechanism, but it is a dynamic equilibrium process.70 Maintaining this equilibrium requires the participation of oxidants (oxygen, hydrogen peroxide, superoxide anion, etc.), reductants (glutathione, NADH, NADPH, etc.), redox enzymes (superoxide dismutase, glutathione peroxidase, etc.), and redox buffer system (glutathione-glutamine system, NADH/NAD+, NADPH/NADP+, etc.). Among them, oxygen is the main determinant of cell metabolism and gene expression. Metabolic changes and energy restriction under hypoxia conditions lead to the imbalance of cell REDOX homeostasis.43,71
Reactive oxygen species (ROS) is an important component of intracellular oxides, and its concentration and source may be a critical determinant of redox-mediated signal transduction and cell fate.72 Usually, an appropriate amount of ROS serves as secondary signaling molecules in human body, playing an essential role in regulating energy metabolism and maintaining cell homeostasis. However, excessive amounts of ROS will react non-specifically with biological molecules such as DNA, proteins, unsaturated lipids and carbohydrates, causing oxidative damage such as DNA breakage, protein denaturation and lipid peroxidation, thus inducing cell death (Figure 1).73 Generally, the main source of ROS in human body is mitochondria. Endoplasmic reticulum protein folding and peroxidase activation in peroxisome also promote the production of ROS. In addition, ROS produced by nicotinamide adenine dinucleotide phosphate oxidase is also one of the important sources of endogenous ROS in the cytoplasm.74 Because of the short half-life of most types of ROS, ROS produced from different sources may have different biological functions.
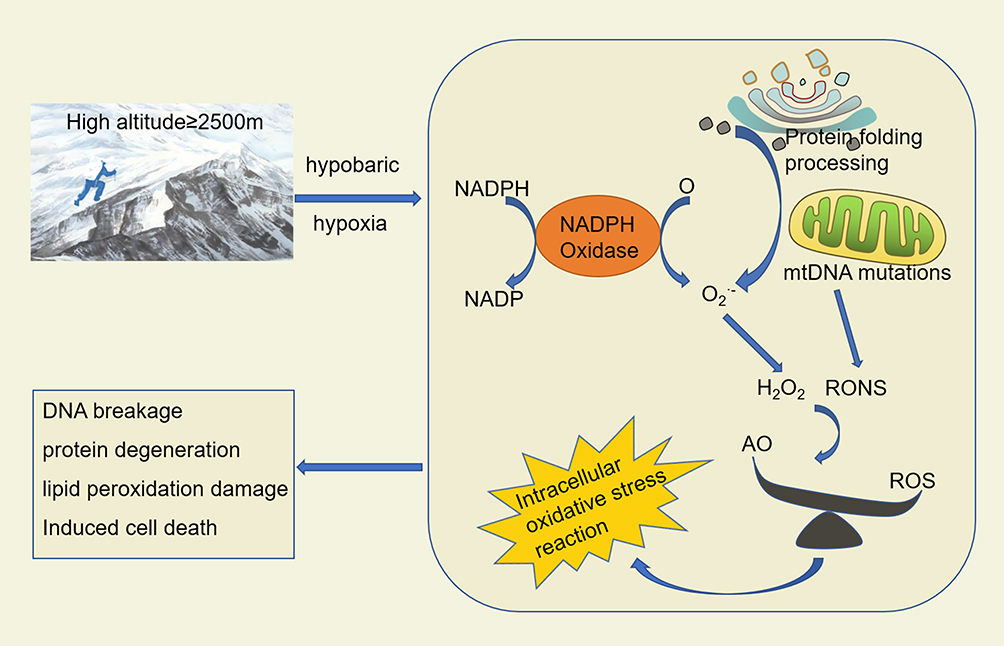 |
Figure 1 Oxidative stress damage of cells induced by hypoxia at high altitude. Abbreviations: RONS, reactive oxygen and nitrogen species; ROS, reactive oxygen species; AO, antioxidant; NADPH, nicotinamide adenine dinucleotide phosphate. |
Under normal circumstances, the production of ROS mediated by nicotinamide adenine dinucleotide phosphate oxidase (NADPH) is a necessary signal for insulin stimulation signal transduction.75 Moreover, the increase of ROS in the oxidative metabolism of mitochondria can lead to a series of diseases such as neurodegenerative diseases, cancer, diabetes and so on.76,77 This is mainly because mitochondria are the main sites of cellular energy metabolism and the center of REDOX reaction.78 When stimulated by hypoxia, inflammation, virus or tumors, large amounts of ROS can induce strong oxidative stress damage.79,80 At the same time, the body itself has a complete antioxidant defense system to protect against oxidative damage caused by ROS, including enzymatic reactions antioxidant defense, non-enzymatic reactions antioxidant defense, and antioxidant molecules. Among them, non-enzymatic reactive antioxidants are the first line of defense against ROS, while subsequent activation of enzymatic antioxidant defenses dominated. Moreover, the antioxidant enzymes produced by the cells mainly include superoxide dismutase, catalase, peroxidase, thioredoxin, glutathione peroxidation and glutathione reductase. These enzymes can be activated successively under the stimulation of ROS, efficiently catalyze the decomposition and metabolism of ROS, and are regulated by NF-E2–related factor 2 (Nrf2), peroxisome proliferators activated receptor-gamma coactivator 1 (PGC-1), p53 and other transcription factors.81
Relationship Between Imbalanced REDOX Homeostasis and HACE
Blood-brain barrier (BBB) plays an important biological role in maintaining the stability of brain tissue and the normal physiological state of central nervous system. REDOX homeostasis in vivo is a critical condition for maintaining the integrity of BBB. However, high altitude hypoxia environment can lead to REDOX imbalance, BBB damage, and induces HACE (Table 1).82,83 Moreover, Table 1 shows the changes of oxidant, reducing agent, REDOX enzyme and REDOX buffer system in the body during the process of HACE. Mitochondria are both REDOX centers and oxygen-sensitive organelles, and their functions are closely related to oxygen levels.73 Under the condition of hypoxia at high altitude, mitochondrial function is inhibited, resulting in abnormal mitochondrial energy metabolism and excessive production of ROS, leading to brain oxidative stress damage and inducing the occurrence of HACE. It can be divided into three aspects: First, oxidative stress leads to abnormal activation of microglia and destruction of tight junctions of vascular endothelial cells through excessive production of mitochondrial-related ROS,56,84,85 leading to massive activation of inflammatory factors and increased BBB permeability,86 thus causing brain edema; Second, mitochondrial dysfunction can also lead to imbalance of intracellular calcium balance, which leads to excessive release of Ca2+ and calcium overload, thus causing cytotoxic edema;87–89 Third, hypoxia inhibits oxidative phosphorylation of cells, resulting in mitochondrial membrane depolarization, depletion of ATP and decrease of cell transport. Then, increased intracellular osmotic pressure may induce a large number of neurons and endothelial cells, leading to destruction of BBB.84,90 Therefore, the role of mitochondrial dysfunction in HACE is essential, and the study of the improvement and protection of mitochondrial function is of great significance for the prevention and treatment of HACE.
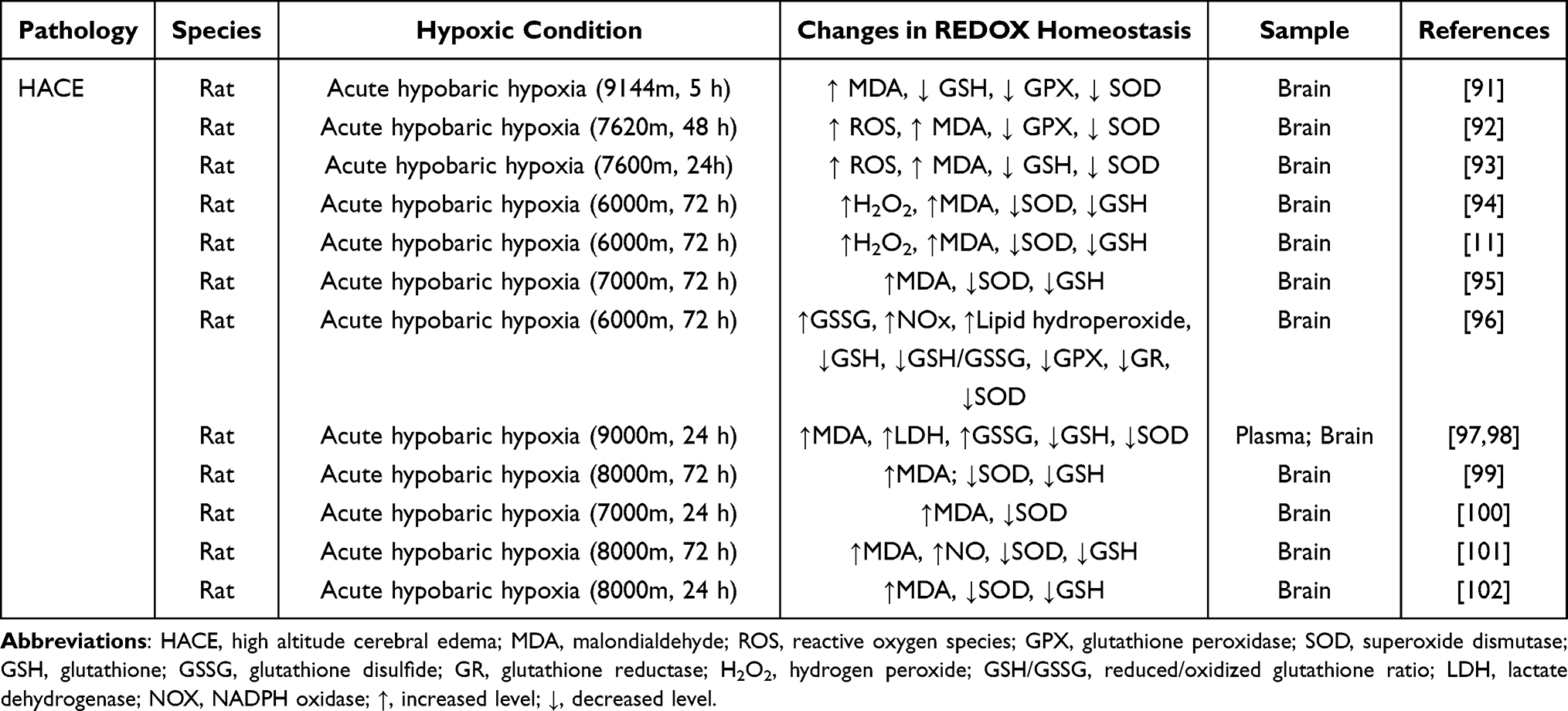 |
Table 1 High Altitude Cerebral Edema and REDOX Imbalance |
Additionally, microglia are macrophages in brain tissue and are the only immune cellular substances in the central nervous system that help maintain the homeostasis in brain tissue. In the altitude hypoxia environment, microglia will produce a large number of free radicals and oxidants, thus causing oxidative stress response. Oxidative stress reaction will lead to the decrease of antioxidant enzyme activity and the enhancement the activity of oxidase in microglia, thus reducing the resistance of microglia to free radicals and oxidants, and further aggravating the degree of oxidative stress.103 At the same time, oxidative stress can also cause inflammation. On the one hand, this inflammatory response promotes the increase of M1-polarized microglia through up-regulation of NRF1,34,35 thus breaking the tight junctions of vascular endothelium and increasing vascular permeability.104,105 On the other hand, by upregulation of Aquaporin-4 (AQP4) (marker of HACE cytotoxic edema), astrocytes swell and release a large amount of cytotoxic compounds, which leads to tissue damage and increased swelling, forming a vicious cycle.106 These pathological changes lead to a breakdown of the blood-brain barrier, swelling of brain tissue, and eventually HACE disease (Figure 2). In addition, oxidative stress response of microglia can also lead to mitochondrial membrane oxidative damage and mitochondrial dysfunction, leading to energy metabolism disorders and cell apoptosis.107 Apoptotic cells will release a large number of intracellular substances, which further cause inflammation and oxidative stress, thus aggravating the degree of HACE.
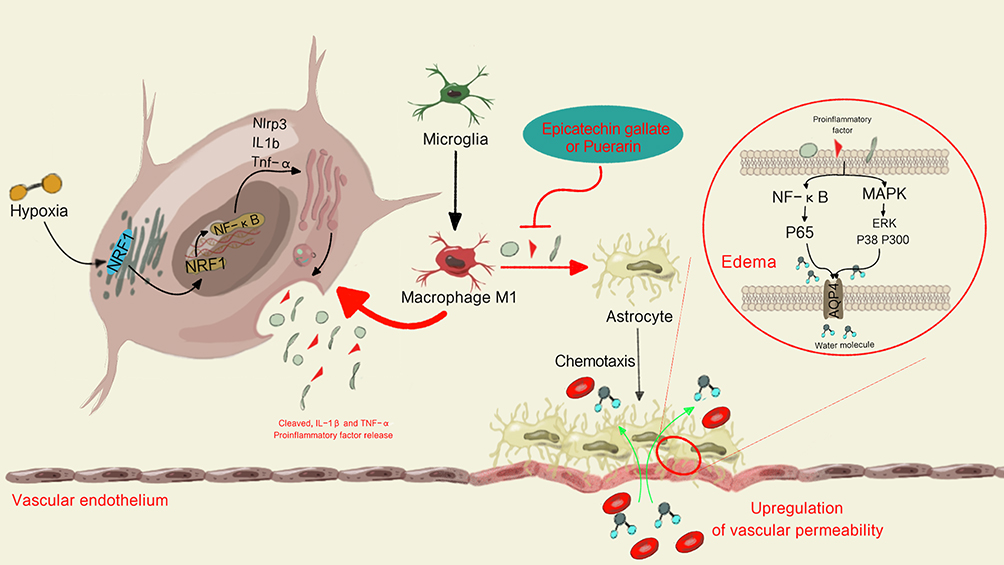 |
Figure 2 HACE mediated by microglia in hypoxia environment at high altitude. Hypoxia-induced systemic inflammation can promote M1 polarization of microglia by up-regulating the expression of Nrf1, releasing a large number of pro-inflammatory factors to activate MAPKs and NF-κB signaling pathways in astrocytes, up-regulating the expression of AQP4, destroying the integrity of BBB, and inducing HACE. |
Study on REDOX Homeostasis in the Treatment of HACE
Related Targets
HACE is mediated by oxidative stress damage caused by the imbalance of REDOX homeostasis in low atmospheric pressure and low oxygen at high altitude. Therefore, maintaining REDOX homeostasis of brain cells is an important method to prevent and treat HACE. As mentioned above, antioxidant enzymes in the body are regulated by several transcription factors. These key transcription factors maintain REDOX homeostasis by regulating transcription levels of antioxidant enzymes and protein content of related enzymes. Among them, Nrf2 is a key regulatory factor of cellular antioxidant response, which can reduce the oxidative stress damage of the body through several ways.108 Specifically, Nrf2 binds to Kelch-like ECH-associated protein 1 (Keap1) in the cytoplasm and is targeted for degradation, thus maintaining a steady-state level under normal physiological conditions. However, some cysteine residues in Keap1 are oxidized to cysteine sulfonic acid at high altitude, which inhibits Keap1’s ability to promote Nrf2 ubiquitination and degradation in the proteasome. As a result, the steady-state level of Nrf2 is elevated, and it dissociates from Keap1 and trans-locates to the nucleus to form a complexes with other proteins that regulate the expression of more than 100 genes involved in cellular stress responses via antioxidant response elements (AREs) or electrophile response elements (EpREs) (Figure 3a).109 On the one hand, Nrf2 can regulate the production of ROS and reactive nitrogen species (RNS) by controlling the transcription of ROS/RNS-producing enzyme.110 On the other hand, Nrf2 can induce the expression of antioxidant enzymes (such as catalase) to remove ROS and improve the antioxidant capacity of the body.111
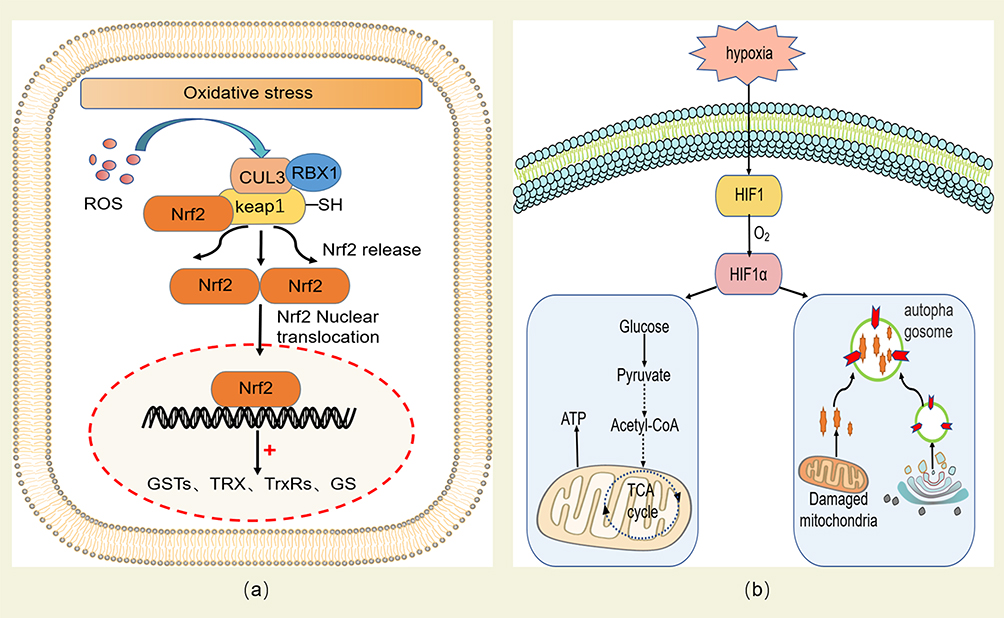 |
Figure 3 (a) Nrf2 activates antioxidant factors to reduce oxidative stress. The oxidation of SH- group in Keap1 inhibits its ability to target Nrf2 degradation. Nrf2 is isolated from Keap1 group and located in the nucleus, where it activates several antioxidant factors, including glutathione S-transferase (GSTs), TRX, thioredoxin reductase (TrxRs) and glutathione synthase (GS), thus maintaining intracellular REDOX homeostasis. (b) HIF-1α activates the hypoxic adaptation response of mitochondria under hypoxic conditions. Under hypoxia, the stability of O2-regulated HIF-1α can regulate the transformation of mitochondria to O2-independent ATP production based on glycolysis, activate mitochondrial autophagy, eliminate severely damaged mitochondria that produce large amounts of ROS, and finally alleviate the oxidative stress damage caused by mitochondria under hypoxic conditions. |
In addition, under hypoxic conditions, mitochondrial hypoxia impairs normal oxidative phosphorylation and electron transport chain function, leading to the production of a large number of ROS, thus promoting the occurrence of HACE. Hypoxia-inducible factor-1 (HIF-1), as a transcription factor that is activated under hypoxic conditions and directly targets several mitochondrial proteins to protect the body from oxidative stress.112,113 HIF-1 is a heterodimer composed of stable HIF-1β and unstable HIF-1α regulated by O2. Among them, HIF-1α is a constitutive expression protein and its expression level is very low under the condition of normal oxygen due to the continuous degradation of HIF-1α by 26S proteasome. However, under hypoxia conditions, HIF-1α can be stabilized by inducing inhibition of HIF-1α hydroxylase, thereby activating mitochondrial adaptation to hypoxia. This adaptive response promotes cell survival by reshaping cellular energy metabolism, including the transition to oxygen-independent glycolytic based ATP production and down-regulation of mitochondrial oxidative phosphorylation to reduce oxygen-dependent energy production.40,97 In addition, HIF-1α also activates mitochondrial autophagy through two typical hypoxia targets BCL2 and adenovirus E1B 19-kDa-interacting protein 3 (BNIP3) and BNIP3-like (BNIP3L), thus eliminating severely damaged mitochondria that produce large amounts of ROS,113 and ultimately alleviating oxidative stress damage caused by mitochondria under hypoxia conditions (Figure 3b).
AQP4 is an aquaporin in the astrocyte foot process, which is an important part of the formation of aquaporin in brain. Microglia can regulate the expression and distribution of AQP4, thus affecting the permeability of brain water channels and water flow velocity. Microglia activate A1 reactive astrocytes by activating the inflammatory pathway of nuclear factor kappa-B (NF-κB) and releasing fission mitochondria in extracellular cells under altitude hypoxia. AQP4 is upregulated in the bottom of astrocytes, leading to increased permeability of brain water channels, making water flow between the brain tissue and the brain easier, causing astrocytes cytotoxic edema and increasing the risk of HACE development.114 A series of inflammatory responses mediated by NF-κB signaling pathway and BBB damage caused by AQP4 accumulation are the main links leading to HACE.115–117 In a word, the study on them is helpful to further understand the pathological mechanism of HACE and provide new ideas and methods for its prevention and treatment.
HACE Therapy Targeting REDOX Homeostasis
The balance of REDOX homeostasis is crucial to the physiological activities of the body, and its imbalance can lead to various diseases. However, it can also be utilized as a way to treat these diseases. For example, hypoxic tumors grow in anoxic environments resulting in dramatic changes in cellular metabolic pathways and molecular mechanisms, leading to an imbalance in cellular REDOX status and lack sensitivity to various antitumor therapies. Moreover, recent studies suggest that intervention in cellular REDOX status via chemical or gene therapy may enhance low-oxygen tumors’ sensitivity to treatment, thereby improving the therapeutic efficacy of tumors.118,119 These findings demonstrate that maintaining cellular REDOX homeostasis may provide new directions and ideas for the treatment of some diseases. It is also feasible to explore the treatment of HACE with REDOX imbalance as the entry point. In this paper, the existing relevant studies are discussed.
Non-Drug Therapy
Hyperbaric oxygen is one of the main ways to treat HACE. It can rapidly increase the partial pressure of blood oxygen in patients with HACE and correct the state of cerebral ischemia and hypoxia. Studies have shown that hyperbaric oxygen preconditioning can increase the activity of antioxidant enzymes and the ability of scavenging lactic acid by inducing the expression of heat shock protein 70, so as to reduce the oxidative stress response of mouse brain in hypoxic environment and prevent the occurrence of HACE.60,96 In addition, intermittent hypoxia exposure has also been shown to reduce the incidence of HACE through anti-oxidation and anti-inflammation,25,120 which may be related to multiple modifications and functional improvements of mitochondrial DNA sequences under long-term / chronic hypoxia.90
Drug Therapy
Anti-inflammatory agents and antioxidants are the first choice for treating the imbalance of antioxidant homeostasis caused by oxidative stress and inflammation. Glucocorticoids have long been used for the treatment of HACE and have good anti-inflammatory effects. However, it may cause adverse reactions such as hyperglycemia, indigestion, and withdrawal symptoms, and there are contraindications for peptic ulcer, breastfeeding and pregnancy. Therefore, in recent years, there has been increasing attention to anti-inflammatory and antioxidant substances derived from a number of natural substances, especially traditional Chinese herbal medicines. Natural antioxidants such as ginkgolide B,121 quercetin,62 tetrahydrocurcumin,100 areca polyphenols,68 salidroside,97,122 phenylethanol glycoside99 and potentilla anserine L. polysaccharide101 can reduce oxidative stress and inflammation in a hypobaric and hypoxic environment by enhancing the activity of antioxidant enzymes and inhibiting NF-κB inflammatory signaling, so as to prevent or treat HACE. In addition, endogenous Exendin-4 and ganglioside GM1 are also thought to alleviate HACE by inhibiting inflammation and oxidative stress (Table 2). Sun et al95 found that Exendin-4 effectively protected the integrity of BBB by decreasing the expression of IL-6, TNF-α and NF-κB, increasing the expression of VEGF to promoting angiogenesis. Ganglioside GM1 can down-regulate the levels of pro-inflammatory cytokines IL-1β, TNF-α and IL-6 in serum and brain tissue by activating PI3K/AKT-Nrf2 pathway, thus playing a therapeutic intervention role in HACE.93
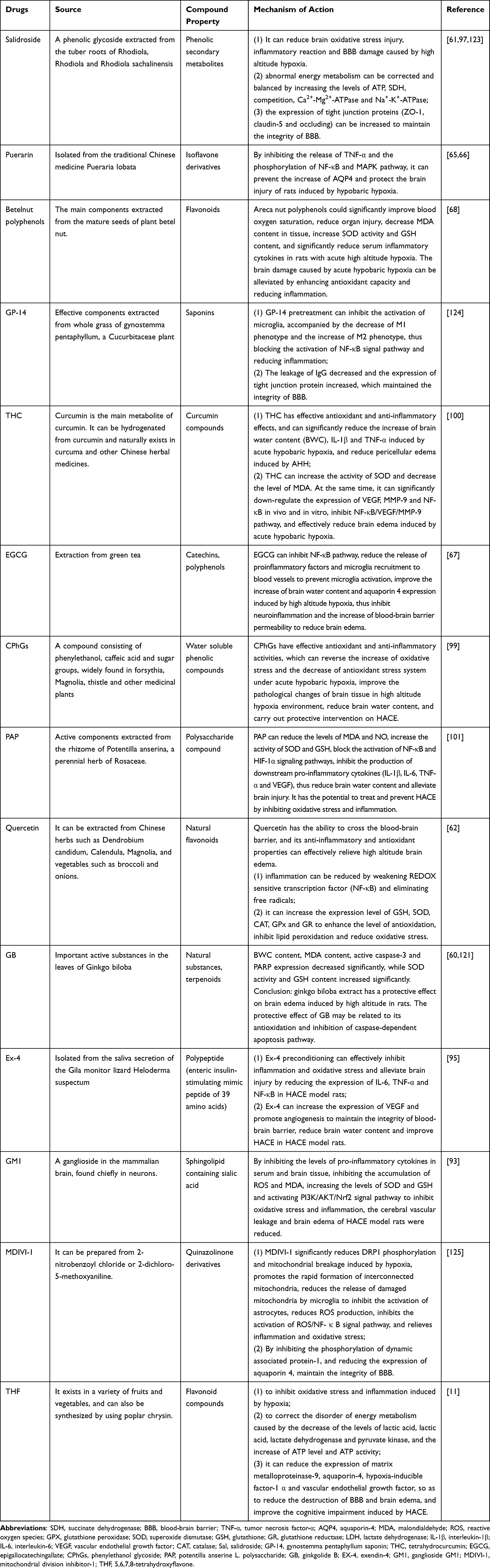 |
Table 2 The Drug Treatment of HACE |
HIF-1α protects mitochondria from hypoxia by regulating vascular metabolism, inflammatory response, apoptosis and cell metabolism. Therefore, the study of mitochondrial functional stability and HIF-1α activation may provide new ideas for the prevention and treatment of HACE. Studies have shown that cobalt chloride,126 taurine127 and salidroside97 can maintain high levels of heme oxygenase-1 and reduce ROS-induced lipid and protein oxidation through HIF-1α signaling pathway, thus protecting the integrity of mitochondrial structure and function and reducing oxidative stress damage. Mitochondrial kinetic dysfunction can directly promote the increase of mitochondrial ROS production. Studies have shown that mitochondrial division inhibitor-1 (MDIVI-1) has a protective effect on brain edema in mice induced by simulated high altitude exposure.125 On the one hand, MDIVI-1 reduces the secretion of IL-6 and TNF-α by inhibiting ROS/NF-κB signal pathway. On the other hand, MDIVI-1 may reduce the activation of astrocytes by reducing microglia release to damaged mitochondria, thus reducing the expression of AQP4 to relieve brain edema (Figure 4a).
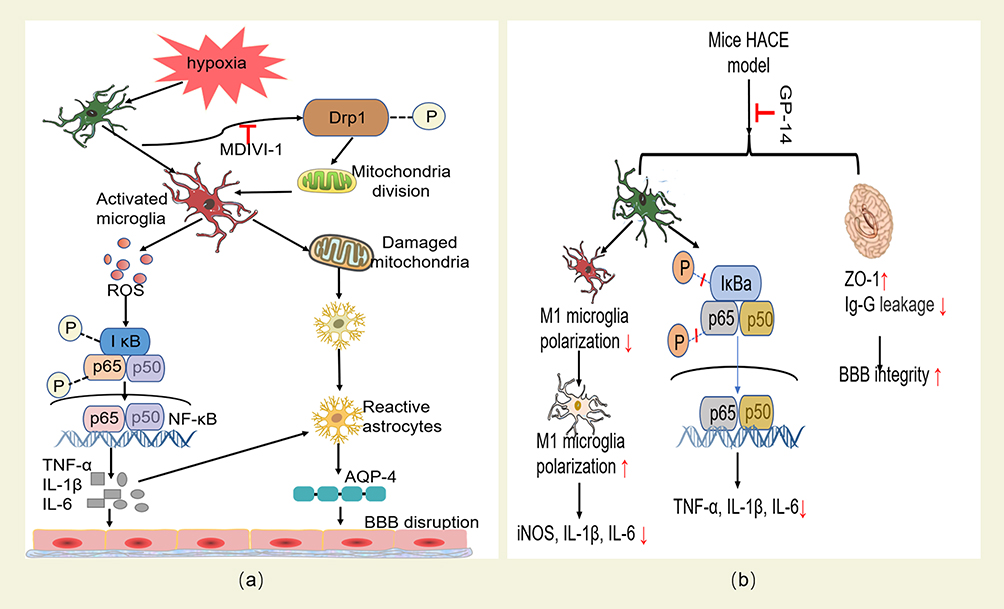 |
Figure 4 (a) The protective effect of MDIVI-1 on HACE and its mechanism. MIDIVI1 inhibits Drp1-mediated mitochondrial division under hypoxia, reduces the release of ROS and blocks the activation of ROS/NF-κB inflammatory pathway, protects the integrity of mitochondria released by microglia under hypoxia, and reduces the production of reactive astrocytes and the expression of AQP4, and finally alleviates inflammatory reaction and oxidative stress damage, and protects the integrity of BBB. (b) The protective effect of GP-14 on HACE and its mechanism. GP-14 can reduce the level of inflammation by inhibiting the expression of pro-inflammatory cytokines regulated by IκB-α and p65 phosphorylation. It also regulates the polarization of microglia M1/M2 by inhibiting the polarization of microglia to M1 phenotype and promoting microglia polarization to anti-inflammatory M2 phenotype; At the same time, it also reduces IgG leakage and increases the expression level of tight junction scaffold protein ZO-1, thus protecting the integrity of BBB and reducing the occurrence of HACE. |
Studies128,129 confirmed that the up-regulated expression of AQP4 in brain tissue of HACE patients was positively correlated with the damage degree of BBB. Therefore, inhibiting the overactivation of microglia and down-regulating the expression level of AQP4 may be a new way to prevent and treat HACE. Current studies have found that puerarin,65 catechin67 and gynostemma pentaphyllum saponins (GP-14)124 (Figure 4b) can inhibit the activation of microglia to astrocytes by inactivating NF-κB inflammatory signal pathway. Therefore, it can realize the protection effects on HACE by blocking the effect of AQP4 on BBB permeability.
Conclusions and Future Directions
REDOX homeostasis is a delicate balance between the oxidation and reduction systems. At high altitudes, this balance is disrupted, leading to oxidative stress and inflammation, and mediating the onset of HACE. We reviewed the changes in REDOX homeostasis during HACE pathogenesis, in which the activation and expression levels of Nrf2, HIF-1α and AQP4 are key factors. A number of potential therapeutic agents have been explored for these targets. However, their mechanisms and efficacy remain unclear due to the universality of the REDOX system and complexity of HACE. Therefore, it is necessary to further explore these signaling molecules and pathways to guide the targeted regulation of complex underlying pathophysiology and thus improving the therapeutic level of HACE.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was financially supported by grants from the National Nature Science Foundation of China (No.82001484), the Science Foundation of The General Hospital of Western Theater Command (No.2021-XZYG-B33), and the Science Foundation of Sichuan Science and Technology Department (No.2023JDRC0099).
Disclosure
All authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Márquez MF. The heart at high altitude. Trends Cardiovasc Med. 2022;2022:e15.
2. Pena E, El Alam S, Siques P, Brito J. Oxidative stress and diseases associated with high-altitude exposure. Antioxidants. 2022;11(2):267.
3. Luks AM, Auerbach PS, Freer L, et al. Wilderness medical society clinical practice guidelines for the prevention and treatment of acute altitude illness: 2019 update. Wilderness Environ Med. 2019;30(4s):S3–s18.
4. Biller A, Badde S, Heckel A, et al. Exposure to 16 h of normobaric hypoxia induces ionic edema in the healthy brain. Nat Commun. 2021;12(1):5987.
5. Chen Y, He Y, Zhao S, He X, Xue D, Xia Y. Hypoxic/ischemic inflammation, microRNAs and δ-opioid receptors: hypoxia/ischemia-sensitive versus-insensitive organs. Front Aging Neurosci. 2022;14:847374.
6. Jensen JD, Vincent AL. High Altitude Cerebral Edema. Treasure Island (FL): StatPearls Publishing; 2022.
7. Zelmanovich R, Pierre K, Felisma P, Cole D, Goldman M, Lucke-Wold B. High altitude cerebral edema: improving treatment options. Biologics. 2022;2(1):81–91.
8. Sawicka M, Szymczak RK. A fatal case of high-altitude cerebral oedema on a climbing expedition to Karakoram. Travel Med Infect Dis. 2022;2022:102493.
9. Hao GS, Fan QL, Hu QZ, Hou Q. Research progress on the mechanism of cerebral blood flow regulation in hypoxia environment at plateau. Bioengineered. 2022;13(3):6353–6358.
10. Murray AJ, Montgomery HE, Feelisch M, Grocott MPW, Martin DS. Metabolic adjustment to high-altitude hypoxia: from genetic signals to physiological implications. Biochem Soc Trans. 2018;46(3):599–607.
11. Jing L, Wu N, Zhang J, Da Q, Ma H. Protective effect of 5,6,7,8-Tetrahydroxyflavone on high altitude cerebral edema in rats. Eur J Pharmacol. 2022;928:175121.
12. Mrakic-Sposta S, Montorsi M, Porcelli S, et al. Effects of prolonged exposure to hypobaric hypoxia on oxidative stress: overwintering in antarctic concordia station. Oxid Med Cell Longev. 2022;2022:4430032.
13. Chiang S, Braidy N, Maleki S, Lal S, Richardson DR, Huang ML. Mechanisms of impaired mitochondrial homeostasis and NAD(+) metabolism in a model of mitochondrial heart disease exhibiting redox active iron accumulation. Redox Biol. 2021;46:102038.
14. Checconi P, De Angelis M, Marcocci ME, et al. Redox-modulating agents in the treatment of viral infections. Int J Mol Sci. 2020;21(11):154.
15. Fraternale A, Zara C, De Angelis M, et al. Intracellular redox-modulated pathways as targets for effective approaches in the treatment of viral infection. Int J Mol Sci. 2021;22(7):e245.
16. Arslanbaeva LR, Santoro MM. Adaptive redox homeostasis in cutaneous melanoma. Redox Biol. 2020;37:101753.
17. Li H, Fu X, Yao F, Tian T, Wang C, Yang A. MTHFD1L-mediated redox homeostasis promotes tumor progression in tongue squamous cell carcinoma. Front Oncol. 2019;9:1278.
18. Kubli SP, Bassi C, Roux C. AhR controls redox homeostasis and shapes the tumor microenvironment in BRCA1-associated breast cancer. Proc Natl Acad Sci U S A. 2019;116(9):3604–3613.
19. Shao D, Oka S, Brady CD, Haendeler J, Eaton P, Sadoshima J. Redox modification of cell signaling in the cardiovascular system. J Mol Cell Cardiol. 2012;52(3):550–558.
20. He X, Liu J, Zang WJ. Mitochondrial homeostasis and redox status in cardiovascular diseases: protective role of the vagal system. Free Radic Biol Med. 2022;178:369–379.
21. Paspalj D, Nikic P, Savic M, et al. Redox status in acute ischemic stroke: correlation with clinical outcome. Mol Cell Biochem. 2015;406(1–2):75–81.
22. Eleftheriadou D, Kesidou D, Moura F, Felli E, Song W. Redox-responsive nanobiomaterials-based therapeutics for neurodegenerative diseases. Small. 2020;16(43):e1907308.
23. Akanji MA, Rotimi DE, Elebiyo TC, Awakan OJ, Adeyemi OS. Redox homeostasis and prospects for therapeutic targeting in neurodegenerative disorders. Oxid Med Cell Longev. 2021;2021:9971885.
24. Cao XF, Bai ZZ, Ma L, Ma S, Ge RL. Metabolic alterations of qinghai-tibet plateau pikas in adaptation to high altitude. High Alt Med Biol. 2017;18(3):219–225.
25. Gangwar A, Paul S, Ahmad Y, Bhargava K. Intermittent hypoxia modulates redox homeostasis, lipid metabolism associated inflammatory processes and redox post-translational modifications: benefits at high altitude. Sci Rep. 2020;10(1):7899.
26. Ni Q, Shao Y, Wang YZ, Jing YH, Zhang YC. Impact of high altitude on the hepatic fatty acid oxidation and synthesis in rats. Biochem Biophys Res Commun. 2014;446(2):574–579.
27. Gao WX, Wu G, Gao YQ. Pathophysiological changes in mitochondria of mammalian exposed to hypoxia at high altitude. Chin j Appl Physiol. 2014;30(6):502–505.
28. Canouï-Poitrine F, Veerabudun K, Larmignat P, Letournel M, Bastuji-Garin S, Richalet JP. Risk prediction score for severe high altitude illness: a cohort study. PLoS One. 2014;9(7):e100642.
29. Higgins JP, Tuttle T, Higgins JA. Altitude and the heart: is going high safe for your cardiac patient? Am Heart J. 2010;159(1):25–32.
30. Falla M, Giardini G, Angelini C. Recommendations for traveling to altitude with neurological disorders. J Central Nervous Syst Dis. 2021;13:11795735211053448.
31. Xue Y, Wang X, Wan B, et al. Caveolin-1 accelerates hypoxia-induced endothelial dysfunction in high-altitude cerebral edema. Cell Commun Signal. 2022;20(1):160.
32. Pu X, Li F, Lin X, Wang R, Chen Z. Oxidative stress and expression of inflammatory factors in lung tissue of acute mountain sickness rats. Mol Med Rep. 2022;25(2):85.
33. Liu B, Chen J, Zhang L, et al. IL-10 dysregulation in acute mountain sickness revealed by transcriptome analysis. Front Immunol. 2017;8:628.
34. Zhou Y, Huang X, Zhao T, et al. Hypoxia augments LPS-induced inflammation and triggers high altitude cerebral edema in mice. Brain Behav Immun. 2017;64:266–275.
35. Wang X, Chen G, Wan B, et al. NRF1-mediated microglial activation triggers high-altitude cerebral edema. J Mol Cell Biol. 2022;22:121.
36. Song TT, Bi YH, Gao YQ, et al. Systemic pro-inflammatory response facilitates the development of cerebral edema during short hypoxia. J Neuroinflammation. 2016;13(1):63.
37. Bajinka O, Simbilyabo L, Tan Y, Jabang J, Saleem SA. Lung-brain axis. Crit Rev Microbiol. 2022;48(3):257–269.
38. Li C, Chen W, Lin F, et al. Functional two-way crosstalk between brain and lung: the brain-lung axis. Cell Mol Neurobiol. 2022;2022:1–13.
39. Riech S, Kallenberg K, Moerer O, et al. The pattern of brain microhemorrhages after severe lung failure resembles the one seen in high-altitude cerebral edema. Crit Care Med. 2015;43(9):e386–9.
40. Sharma S, Singh Y, Sandhir R, et al. Mitochondrial DNA mutations contribute to high altitude pulmonary edema via increased oxidative stress and metabolic reprogramming during hypobaric hypoxia. Biochimica et Biophysica Acta Bioenergetics. 2021;1862(8):148431.
41. Mishra A, Ali Z, Vibhuti A, et al. CYBA and GSTP1 variants associate with oxidative stress under hypobaric hypoxia as observed in high-altitude pulmonary oedema. Clin Sci. 2012;122(6):299–309.
42. Ugalde JM, Aller I, Kudrjasova L, et al. Endoplasmic reticulum oxidoreductin provides resilience against reductive stress and hypoxic conditions by mediating luminal redox dynamics. Plant Cell. 2022;2022:147.
43. Sasidharan R, Schippers JHM, Schmidt RR. Redox and low-oxygen stress: signal integration and interplay. Plant Physiol. 2021;186(1):66–78.
44. Mallet RT, Burtscher J, Pialoux V, et al. Molecular mechanisms of high-altitude acclimatization. Int J Mol Sci. 2023;24(2):221.
45. Hackett PH, Roach RC. High-altitude illness. N Engl J Med. 2001;345(2):107–114.
46. Savioli G, Ceresa IF, Gori G, et al. Pathophysiology and therapy of high-altitude sickness: practical approach in emergency and critical care. J Clin Med. 2022;11(14):26.
47. Wu T, Ding S, Liu J, et al. Ataxia: an early indicator in high altitude cerebral edema. High Alt Med Biol. 2006;7(4):275–280.
48. Hackett PH, Yarnell PR, Hill R, Reynard K, Heit J, McCormick J. High-altitude cerebral edema evaluated with magnetic resonance imaging: clinical correlation and pathophysiology. JAMA. 1998;280(22):1920–1925.
49. Bailey DM, Bärtsch P, Knauth M, Baumgartner RW. Emerging concepts in acute mountain sickness and high-altitude cerebral edema: from the molecular to the morphological. Cell Mol Life Sci. 2009;66(22):3583–3594.
50. Li N, Chen K, Bai J, et al. Tibetan medicine Duoxuekang ameliorates hypobaric hypoxia-induced brain injury in mice by restoration of cerebrovascular function. J Ethnopharmacol. 2021;270:113629.
51. Xie Y, Yang D, Huang AS, et al. Retinal microvasculature is a potential biomarker for acute mountain sickness. Sci China Life Sci. 2023;2023:187.
52. Willmann G, Gekeler F, Schommer K, Bärtsch P. Update on high altitude cerebral edema including recent work on the eye. High Alt Med Biol. 2014;15(2):112–122.
53. Molano Franco D, Nieto Estrada VH, Gonzalez Garay AG, Martí-Carvajal AJ, Arevalo-Rodriguez I. Interventions for preventing high altitude illness: part 3. Miscellaneous and non-pharmacological interventions. Cochrane Database Sys Rev. 2019;4(4):Cd013315.
54. Davis C, Hackett P. Advances in the prevention and treatment of high altitude illness. Emerg Med Clin North Am. 2017;35(2):241–260.
55. Clark ST, Sheraton M. EMS High-Altitude Field Prophylaxis and Treatment. Treasure Island (FL): StatPearls Publishing; 2022.
56. Gao D, Wang Y, Zhang R, Zhang Y. Efficacy of Acetazolamide for the prophylaxis of acute mountain sickness: a systematic review, meta-analysis, and trial sequential analysis of randomized clinical trials. Ann Thorac Med. 2021;16(4):337–346.
57. Murata T, Suzuki N, Yamawaki H, et al. Dexamethasone prevents impairment of endothelium-dependent relaxation in arteries cultured with fetal bovine serum. Eur J Pharmacol. 2005;515(1–3):134–141.
58. Szymczak RK, Sawicka M. Can intranasal delivery of dexamethasone facilitate the management of severe altitude disease? J Travel Med. 2023;2023:78.
59. Johnson NJ, Luks AM. High-altitude medicine. Med Clin North Am. 2016;100(2):357–369.
60. Luo QQ, Yang JX, Zhang XM. The protective effects of ginkgolide B and hypoxic preconditioning against acute hypoxia injury in mice. Chin J Appl Physiol. 2009;25(3):362–365.
61. Jiang S, Fan F, Yang L, et al. Salidroside attenuates high altitude hypobaric hypoxia-induced brain injury in mice via inhibiting NF-κB/NLRP3 pathway. Eur J Pharmacol. 2022;925:175015.
62. Patir H, Sarada SK, Singh S, Mathew T, Singh B, Bansal A. Quercetin as a prophylactic measure against high altitude cerebral edema. Free Radic Biol Med. 2012;53(4):659–668.
63. Dong YS, Wang JL, Feng DY, et al. Protective effect of quercetin against oxidative stress and brain edema in an experimental rat model of subarachnoid hemorrhage. Int J Med Sci. 2014;11(3):282–290.
64. Li X, Cheng Z, Chen X, Yang D, Li H, Deng Y. Purpurogallin improves neurological functions of cerebral ischemia and reperfusion mice by inhibiting endoplasmic reticulum stress and neuroinflammation. Int Immunopharmacol. 2022;111:109057.
65. Wang C, Yan M, Jiang H, et al. Mechanism of aquaporin 4 (AQP 4) up-regulation in rat cerebral edema under hypobaric hypoxia and the preventative effect of puerarin. Life Sci. 2018;193:270–281.
66. Wang C, Yan M, Jiang H, et al. Protective effects of puerarin on acute lung and cerebrum injury induced by hypobaric hypoxia via the regulation of aquaporin (AQP) via NF-κB signaling pathway. Int Immunopharmacol. 2016;40:300–309.
67. Chen G, Cheng K, Niu Y, Zhu L, Wang X. (-)-Epicatechin gallate prevents inflammatory response in hypoxia-activated microglia and cerebral edema by inhibiting NF-κB signaling. Arch Biochem Biophys. 2022;2022:109393.
68. Huo Y, Zhao A, Song J, Li J, Wang R. 槟榔多酚对急进高原大鼠具有抗缺氧作用 [Betelnut polyphenols provide protection against high-altitude hypoxia in rats]. J Southern Med Univ. 2021;41(5):671–678. Chinese.
69. Le Gal K, Schmidt EE, Sayin VI. Cellular redox homeostasis. Antioxidants. 2021;10(9):24.
70. Wang X, Li S, Liu Y, Ma C. Redox regulated peroxisome homeostasis. Redox Biol. 2015;4:104–108.
71. Solaini G, Baracca A, Lenaz G, Sgarbi G. Hypoxia and mitochondrial oxidative metabolism. Biochim Biophys Acta. 2010;1797:1171–7.
72. Lee IG, Lee BJ. How bacterial redox sensors transmit redox signals via structural changes. Antioxidants. 2021;10(4):224.
73. Wu R, Li S, Hudlikar R, et al. Redox signaling, mitochondrial metabolism, epigenetics and redox active phytochemicals. Free Radic Biol Med. 2022;179:328–336.
74. Sies H, Jones DP. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol. 2020;21(7):363–383.
75. Santos LR, Rebelato E, Graciano MF, Abdulkader F, Curi R, Carpinelli AR. Oleic acid modulates metabolic substrate channeling during glucose-stimulated insulin secretion via NAD(P)H oxidase. Endocrinology. 2011;152(10):3614–3621.
76. Shadel GS, Horvath TL. Mitochondrial ROS signaling in organismal homeostasis. Cell. 2015;163(3):560–569.
77. Yee C, Yang W, Hekimi S. The intrinsic apoptosis pathway mediates the pro-longevity response to mitochondrial ROS in C. elegans. Cell. 2014;157(4):897–909.
78. Ježek P. Mitochondrial redox regulations and redox biology of mitochondria. Antioxidants. 2021;10(12):21.
79. Ciccarese F, Ciminale V. Escaping death: mitochondrial redox homeostasis in cancer cells. Front Oncol. 2017;7:117.
80. Wang CH, Wei YH. Roles of mitochondrial sirtuins in mitochondrial function, redox homeostasis, insulin resistance and type 2 diabetes. Int J Mol Sci. 2020;21(15):54.
81. Van Aken O. Mitochondrial redox systems as central hubs in plant metabolism and signaling. Plant Physiol. 2021;186(1):36–52.
82. Baneke A. What role does the blood brain barrier play in acute mountain sickness? Travel Med Infect Dis. 2010;8(4):257–262.
83. Huang X, Zhou Y, Zhao T, et al. A method for establishing the high-altitude cerebral edema (HACE) model by acute hypobaric hypoxia in adult mice. J Neurosci Methods. 2015;245:178–181.
84. Farías JG, Herrera EA, Carrasco-Pozo C, et al. Pharmacological models and approaches for pathophysiological conditions associated with hypoxia and oxidative stress. Pharmacol Ther. 2016;158:1–23.
85. Fuhrmann DC, Brüne B. Mitochondrial composition and function under the control of hypoxia. Redox Biol. 2017;12:208–215.
86. Behn C, Araneda OF, Llanos AJ, Celedón G, González G. Hypoxia-related lipid peroxidation: evidences, implications and approaches. Respir Physiol Neurobiol. 2007;158(2–3):143–150.
87. Wilson MH, Newman S, Imray CH. The cerebral effects of ascent to high altitudes. Lancet Neurol. 2009;8(2):175–191.
88. Bogdanova A, Petrushanko IY, Hernansanz-Agustín P, Martínez-Ruiz A. ”Oxygen Sensing” by Na,K-ATPase: these miraculous thiols. Front Physiol. 2016;7:314.
89. Oechmichen M, Meissner C. Cerebral hypoxia and ischemia: the forensic point of view: a review. J Forensic Sci. 2006;51(4):880–887.
90. Luo Y, Yang X, Gao Y. Mitochondrial DNA response to high altitude: a new perspective on high-altitude adaptation. Mitochondrial DNA. 2013;24(4):313–319.
91. Purushothaman J, Suryakumar G, Shukla D, et al. Modulatory effects of seabuckthorn (Hippophae rhamnoides L.) in hypobaric hypoxia induced cerebral vascular injury. Brain Res Bull. 2008;77(5):246–252.
92. Himadri P, Kumari SS, Chitharanjan M, Dhananjay S. Role of oxidative stress and inflammation in hypoxia-induced cerebral edema: a molecular approach. High Alt Med Biol. 2010;11(3):231–244.
93. Gong G, Yin L, Yuan L, et al. Ganglioside GM1 protects against high altitude cerebral edema in rats by suppressing the oxidative stress and inflammatory response via the PI3K/AKT-Nrf2 pathway. Mol Immunol. 2018;95:91–98.
94. Jing L, Wu N, He L, Shao J, Ma H. Establishment of an experimental rat model of high altitude cerebral edema by hypobaric hypoxia combined with temperature fluctuation. Brain Res Bull. 2020;165:253–262.
95. Sun ZL, Jiang XF, Cheng YC, et al. Exendin-4 inhibits high-altitude cerebral edema by protecting against neurobiological dysfunction. Neural Regeneration Res. 2018;13(4):653–663.
96. Lin H, Chang CP, Lin HJ, Lin MT, Tsai CC. Attenuating brain edema, hippocampal oxidative stress, and cognitive dysfunction in rats using hyperbaric oxygen preconditioning during simulated high-altitude exposure. J Trauma Acute Care Surg. 2012;72(5):1220–1227.
97. Wang X, Hou Y, Li Q, et al. Rhodiola crenulata attenuates apoptosis and mitochondrial energy metabolism disorder in rats with hypobaric hypoxia-induced brain injury by regulating the HIF-1α/microRNA 210/ISCU1/2(COX10) signaling pathway. J Ethnopharmacol. 2019;241:111801.
98. Hou Y, Wang X, Chen X, et al. Establishment and evaluation of a simulated high‑altitude hypoxic brain injury model in SD rats. Mol Med Rep. 2019;19(4):2758–2766.
99. Luan F, Li M, Han K, et al. Phenylethanoid glycosides of Phlomis younghusbandii Mukerjee ameliorate acute hypobaric hypoxia-induced brain impairment in rats. Mol Immunol. 2019;108:81–88.
100. Pan Y, Zhang Y, Yuan J, et al. Tetrahydrocurcumin mitigates acute hypobaric hypoxia-induced cerebral oedema and inflammation through the NF-κB/VEGF/MMP-9 pathway. Phytother Res. 2020;34(11):2963–2977.
101. Shi J, Wang J, Zhang J, et al. Polysaccharide extracted from Potentilla anserina L ameliorate acute hypobaric hypoxia-induced brain impairment in rats. Phytother Res. 2020;34(9):2397–2407.
102. Tormos KV, Chandel NS. Inter-connection between mitochondria and HIFs. J Cell Mol Med. 2010;14(4):795–804.
103. Zemtsova I, Görg B, Keitel V, Bidmon HJ, Schrör K, Häussinger D. Microglia activation in hepatic encephalopathy in rats and humans. Hepatology. 2011;54(1):204–215.
104. Haruwaka K, Ikegami A, Tachibana Y, et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat Commun. 2019;10(1):5816.
105. Kang R, Gamdzyk M, Lenahan C, Tang J, Tan S, Zhang JH. The dual role of microglia in blood-brain barrier dysfunction after stroke. Curr Neuropharmacol. 2020;18(12):1237–1249.
106. Thrane AS, Rappold PM, Fujita T, et al. Critical role of aquaporin-4 (AQP4) in astrocytic Ca2+ signaling events elicited by cerebral edema. Proc Natl Acad Sci U S A. 2011;108(2):846–851.
107. Chausse B, Lewen A, Poschet G, Kann O. Selective inhibition of mitochondrial respiratory complexes controls the transition of microglia into a neurotoxic phenotype in situ. Brain Behav Immun. 2020;88:802–814.
108. Kang TC. Nuclear factor-erythroid 2-related factor 2 (Nrf2) and mitochondrial dynamics/mitophagy in neurological diseases. Antioxidants. 2020. 9:617.
109. Liu S, Pi J, Zhang Q. Signal amplification in the KEAP1-NRF2-ARE antioxidant response pathway. Redox Biol. 2022;54:102389.
110. Strom J, Xu B, Tian X, Chen QM. Nrf2 protects mitochondrial decay by oxidative stress. FASEB J. 2016;30(1):66–80.
111. Wang L, Zhang X, Xiong X, et al. Nrf2 regulates oxidative stress and its role in cerebral ischemic stroke. Antioxidants. 2022;11(12):57.
112. Li HS, Zhou YN, Li L, et al. HIF-1α protects against oxidative stress by directly targeting mitochondria. Redox Biol. 2019;25:101109.
113. Cyran AM, Zhitkovich A. HIF1, HSF1, and NRF2: oxidant-responsive trio raising cellular defenses and engaging immune system. Chem Res Toxicol. 2022;35(10):1690–1700.
114. Simka M, Latacz P, Czaja J. Possible role of glymphatic system of the brain in the pathogenesis of high-altitude cerebral edema. High Alt Med Biol. 2018;19(4):394–397.
115. Clément T, Rodriguez-Grande B, Badaut J. Aquaporins in brain edema. J Neurosci Res. 2020;98(1):9–18.
116. Jayakumar AR, Tong XY, Ruiz-Cordero R, et al. Activation of NF-κB mediates astrocyte swelling and brain edema in traumatic brain injury. J Neurotrauma. 2014;31(14):1249–1257.
117. Stokum JA, Kurland DB, Gerzanich V, Simard JM. Mechanisms of astrocyte-mediated cerebral edema. Neurochem Res. 2015;40(2):317–328.
118. Li Y, Zhao P, Gong T, et al. Redox dyshomeostasis strategy for hypoxic tumor therapy based on DNAzyme-loaded electrophilic ZIFs. Angewandte Chemie. 2020;59(50):22537–22543.
119. Li Y, Gong T, Gao H, et al. ZIF-based nanoparticles combine X-ray-induced nitrosative stress with autophagy management for hypoxic prostate cancer therapy. Angewandte Chemie. 2021;60(28):15472–15481.
120. González-Candia A, Candia AA, Paz A, et al. Cardioprotective antioxidant and anti-inflammatory mechanisms induced by intermittent hypobaric hypoxia. Antioxidants. 2022;11(6):59.
121. Botao Y, Ma J, Xiao W, et al. Protective effect of ginkgolide B on high altitude cerebral edema of rats. High Alt Med Biol. 2013;14(1):61–64.
122. Xie N, Fan F, Jiang S, et al. Rhodiola crenulate alleviates hypobaric hypoxia-induced brain injury via adjusting NF-κB/NLRP3-mediated inflammation. Phytomedicine. 2022;103:154240.
123. Yan X, Liu J, Zhu M, et al. Salidroside orchestrates metabolic reprogramming by regulating the Hif-1α signalling pathway in acute mountain sickness. Pharm Biol. 2021;59(1):1540–1550.
124. Geng Y, Yang J, Cheng X, et al. A bioactive gypenoside (GP-14) alleviates neuroinflammation and blood brain barrier (BBB) disruption by inhibiting the NF-κB signaling pathway in a mouse high-altitude cerebral edema (HACE) model. Int Immunopharmacol. 2022;107:108675.
125. Lu Y, Chang P, Ding W, et al. Pharmacological inhibition of mitochondrial division attenuates simulated high-altitude exposure-induced cerebral edema in mice: involvement of inhibition of the NF-κB signaling pathway in glial cells. Eur J Pharmacol. 2022;929:175137.
126. Shrivastava K, Shukla D, Bansal A, Sairam M, Banerjee PK, Ilavazhagan G. Neuroprotective effect of cobalt chloride on hypobaric hypoxia-induced oxidative stress. Neurochem Int. 2008;52(3):368–375.
127. Hovsepyan LM, Zakaryan GV, Melkonyan MM, Zakaryan AV. [The effects of taurine on oxidative processes in brain edema]. Zhurnal nevrologii i psikhiatrii imeni S S Korsakova. 2015;115(5):64–67. Russian.
128. Ding Y, Liu J, Xu Y, Dong X, Shao B. Evolutionary adaptation of aquaporin-4 in Yak (Bos grunniens) brain to high-altitude hypoxia of Qinghai-Tibetan plateau. High Alt Med Biol. 2020;21(2):167–175.
129. Fukuda AM, Badaut J. Aquaporin 4: a player in cerebral edema and neuroinflammation. J Neuroinflammation. 2012;9:279.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.