")
Back to Journals » Biologics: Targets and Therapy » Volume 16
Progress in Biological Therapies for Adult-Onset Still’s Disease
Authors Galozzi P , Bindoli S, Doria A, Sfriso P
Received 3 January 2022
Accepted for publication 6 April 2022
Published 21 April 2022 Volume 2022:16 Pages 21—34
DOI https://doi.org/10.2147/BTT.S290329
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Doris Benbrook
Paola Galozzi, Sara Bindoli, Andrea Doria, Paolo Sfriso
Rheumatology Unit, Department of Medicine DIMED, University of Padova, Padova, Italy
Correspondence: Paola Galozzi, Rheumatology Unit, Department of Medicine DIMED, University of Padova, via Giustiniani, 2, Padova, 35128, Italy, Tel +39 049 821 8654, Email [email protected]
Abstract: Adult-onset Still’s disease (AOSD) is a rare multifactorial autoinflammatory disorder of unknown etiology, characterized by an excessive release of cytokines triggered by dysregulated inflammation and articular and systemic manifestations. The clinical spectrum of AOSD ranges from self-limiting forms with mild symptoms to life-threatening cases and presents clinical and biological similarities with the juvenile form (sJIA). Nowadays, the advances in biologic agents no longer limit the treatment to NSAIDs, glucocorticoids, or conventional synthetic DMARDs. The blockade of IL-1 and IL-6 is effective in the treatment of systemic and articular inflammation of AOSD patients; however, novel compounds with different properties and targets are now available and others are being studied. In this review, starting from the pathogenesis of AOSD, we summarized the current and emerging biological therapies, possible effective agents for achieving AOSD control and remission.
Keywords: biologics, AOSD, IL-1 inhibitors, IL-6 inhibitors, small molecules, new treatment
Introduction
Adult-onset Still’s disease (AOSD) is a rare inflammatory disorder of unknown etiology, with an estimated prevalence of 1 in 100,000 and an annual incidence of 0.16–0.40 per 100,000 depending on the population studied.1 The overwhelming majority of cases present between the ages of 16–35 with a slight female predominance, however 10% of cases present after 50 years of age.2
AOSD has a heterogeneous clinical presentation. The main clinical features (spiking fever, arthritis, skin rash, and hyperferritinemia), as well as other minor features (sore throat, lymphadenopathy, hepatosplenomegaly, thrombocytosis, serositis, myalgia, neutrophilic leukocytosis, inflammatory anemia), characterized the disease.3
During the diagnostic process, Yamaguchi et al 4 and Fautrel et al5 classification criteria for AOSD are actually used in clinical practice, although primarily designed for research. They include exclusion criteria such as infections, malignancies, and other autoimmune diseases with high sensitivity and specificity; so, they should be used only after a wide diagnostic framework.6 Historically, AOSD phenotypes have been described based on the different evolution courses over time (monogenic, polycyclic, or chronic aspects) and on the type of symptoms (prominent systemic features or chronic arthritis).7 This dichotomous classification distinguishes the systemic subtype characterized by high fever, skin rash, and higher risk to develop life-threatening complications, to the articular subtype with predominant joint pain and arthritis. These two phenotypes could also be predicted by some factors: high fever, hepatitis, and elevated serum levels of C-reactive protein and ferritin better associate with the systemic AOSD, while female gender, steroid dependence, and low ferritin level associate with the chronic AOSD.1 In light of novel evidence about AOSD pathogenesis and the use of biological treatments, a different cytokine profile has been observed associated with distinct AOSD manifestations. The systemic subset, indeed, presents an excessive level of interleukin (IL)-1β and IL-18, while the chronic articular AOSD is principally driven by IL-6 and tumor necrosis factor (TNF)-α.8 Several chemokines, including CXC-chemokine ligand 10 (CXCL10), CXCL13, and macrophage migration inhibitory factor (MIF)), the proteins S100 A8/A9 and A12 and the advanced glycation end products (AGEs) and their receptors (RAGE) have potential as new diagnostic biomarkers for AOSD, upon confirmation with larger patient groups.9
Pathogenesis, Hyperinflammation, and Clinical Complications
Although the exact mechanisms involved in the pathogenesis of AOSD are unknown, significant progresses have been made to confirm the homology between AOSD and the systemic juvenile idiopathic arthritis (sJIA). Both conditions encompass many common clinical and biological findings as well as the disease onset and the clinical course result as undistinguishable from each other, suggesting a continuum of a single disease entity.10
The AOSD pathophysiology remains unclear. However, it is thought that factors including an imbalance in innate and adaptive immunity and an increase of inflammatory cytokines may contribute to disease development.1 Aberrant IL-1 and IL-6 production, indeed, would favor the development of pathogenic adaptive T helper 17 (Th17) cell-mediated responses. Several infectious triggers, including viruses (Parvovirus B19, Epstein Barr virus, Cytomegalovirus) and bacteria (Yersinia and Mycoplasma), as well as genetic factors related to the HLA (HLA-Bw35, -B17, -B18, -B35, -DR2, -DR4, -DR5, -DQ1, DRw6, -DRB1, and -DQB1) are also believed to trigger the AOSD onset.11
The fundamental characteristic of AOSD pathogenesis is the neutrophil activation, responsible for the induction and regulation of inflammation. Neutrophil extracellular traps (NETs) play a pivotal role in the intense activation of macrophages and in stimulating the overproduction of several pro-inflammatory cytokines.12 Specific Toll-like receptors (TLRs) and damage-associated molecular patterns (DAMPs) are reported to initiate and maintain the inflammatory response through the activation of NLRP3 inflammasomes, leading to caspase activation and overproduction of active IL-1β and IL-18.13 These proinflammatory cytokines, in turn, lead to amplifying the inflammatory cascade through the exuberant production of downstream mediators, such as IL-6, IL-8, IL-17, TNF-α, and interferon (IFN)-γ, a so-called cytokine storm (Figure 1). Also, the inadequate resolution of the hyperinflammatory status can play a role in sustaining the cytokine storm. Similar to AOSD, excessive and uncontrolled systemic inflammation has been recognized as a hallmark of COVID-19, the current severe acute respiratory syndrome caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) virus.14 This suggests a common link of the cytokine storm in both diseases’ pathogenesis, although a recent study suggests differences in cytokine and soluble mediator profiles.15 The levels of IL-6 and IL-10 are reported higher in severe COVID-19, while the expression of serum ferritin dramatically increased in AOSD. Interestingly, a misdirected immune response against SARS-CoV-2 virus may have led to the development of AOSD, according to a recent case report.16
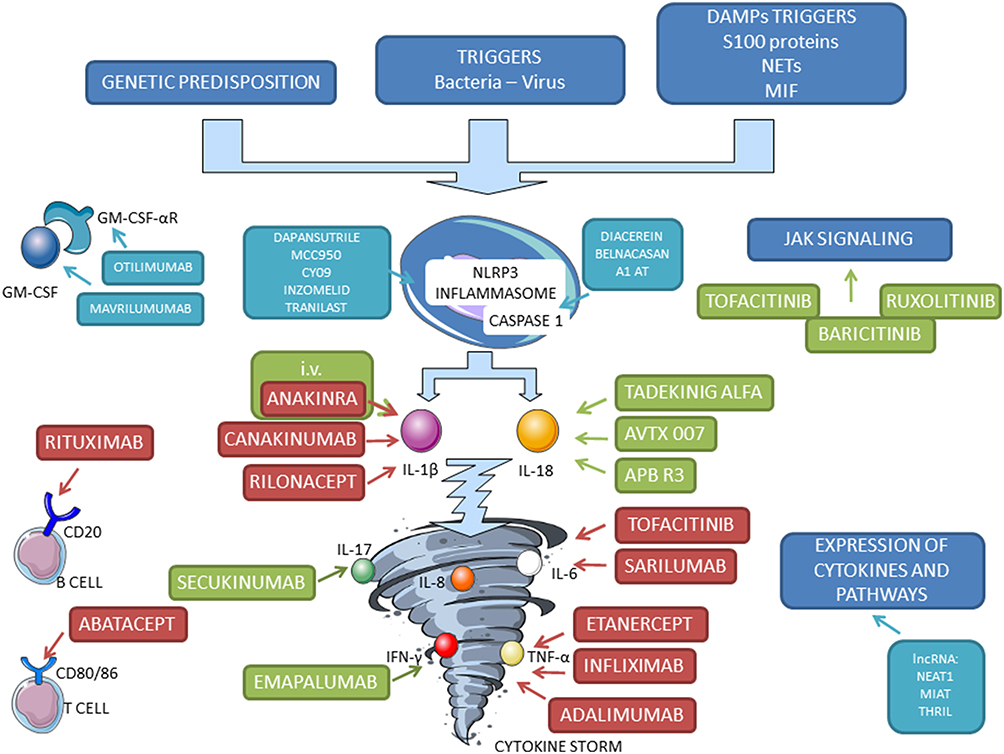 |
Figure 1 Scheme of pathogenesis of AOSD and identification of targets for old (red), new (green), and proposed (light blue) biological therapy. |
The insufficient control of inflammatory activity in AOSD patients is even associated with a risk of serious and different complications. Secondary hemophagocytic lymphohistiocytosis (HLH) or macrophage activation syndrome (MAS) is one of the most severe and life-threatening complications in predisposed AOSD patients.17 Continuous high fever, lymphadenopathy, hepatosplenomegaly, and remarkably elevated levels of serum ferritin and IL-18 contribute to the clinical work-up of MAS patients. Moreover, MAS in AOSD entailed a greater risk of aggressive disease symptoms, difficulties in the response to treatment, and a mortality rate of 20–40%.18 However, there is evidence that cytokine blockade, such as IL-1 and IL-18 inhibitions, might confer additional advantages in the treatment of MAS.19,20 Other complications of AOSD included myocarditis, pericarditis, pulmonary hypertension, multiple organ failure and relapses.21 Clinicians should be aware of these complications, because early recognition and prompt management can significantly reduce morbidity (and mortality). A recent and exhaustive review by Mitrovic and Fautrel21 extensively describes the supportive measures and the immunomodulatory drugs useful to control these specific organ involvements in AOSD.
Conventional Treatments
The treatment of AOSD should be addressed to the different variety and severity of symptoms manifested. Clearly, the control of systemic inflammation and the prevention of MAS remain the main therapeutical purposes. For all severe AOSD-related complications, high-dose corticosteroids and supportive measures remain the first-line treatment. In case of inadequate response, combination with IL1 or IL-6 blockers is adequate.21 Nowadays, the diffusion of new biological targeted therapies has extremely ameliorated the management of AOSD, as many approaches are now available and suitable for the diverse clinical manifestations and disease course.
Non-Steroid Anti-Inflammatory Drugs (NSAIDs), Glucocorticoids, and Conventional Synthetic Disease-Modifying Antirheumatic Drugs (csDMARDs)
NSAIDs represent the first line therapy, especially when systemic manifestations are almost absent.22 However, nearly 20% of the patients present a self-limited disease and for most of them NSAIDs fail in control of the disease activity.23 Nevertheless, NSAIDs should be used as a supportive treatment during the diagnostic process. Indeed, high dose indomethacin (150–250 mg/day) or ibuprofen 800 mg up to 4-times a day may control some of the inflammatory manifestations satisfactorily.1,23 Of course, possible side-effects related to the prolonged employment of NSAIDs should be considered, such as gastrointestinal bleeding and for older patients the risk of renal or hepatic insufficiency.24–26
Glucocorticoids represent an efficient choice in approximately 80% of the patients at the initial dose of 0.5–1 mg/kg/day of methylprednisolone.27 The response achievement is usually prompt and sustained on both articular and systemic manifestations. However, prolonged therapy with steroids should be avoided because of the risk of side-effects and adverse reactions including hypertension, diabetes, tachycardia, the possibility of early-onset osteoporosis, the occurrence of ecchymosis, and weight gain.23 On the other hand, it should be considered that a quick reduction of steroid daily-intake may lead to disease relapses, thus the decrease of the posology should be done gradually. Sometimes, methylprednisolone can be insufficient in controlling the most severe manifestations, especially in those presenting with refractory forms or in patients who present with MAS features. In those cases, other types of glucocorticoids such as dexamethasone should be considered, in the same way as it is employed in the HLH-2004 protocol for the treatment of primary HLH.28 However, a current defined posology for dexamethasone in severe AOSD has not been defined yet, although the clinical decision should rely on a careful examination of the clinical manifestations and on the evaluation of serum biomarkers.
Regarding csDMARDS, they are typically considered glucocorticoid-sparing drugs and are usually added to glucocorticoids or NSAIDs to obtain an adequate disease control. Nowadays, their use is quite limited since biological DMARDs are preferred as second line therapy. Methotrexate and Cyclosporine A were the most frequently employed, especially for the treatment of the chronic articular pattern and for MAS, respectively, while data with azathioprine or leflunomide come from small case series or case reports and are scarce.1
Biological Agents
It was reported in different observational studies that 17–32% of AOSD patients can be resistant to both first-line corticosteroids and second-line csDMARDs.23,29–31 Indeed, the systemic inflammation present in AOSD subjects depends on the increase of pivotal pro-inflammatory cytokines, such as IL-1, IL-6, IL-18, IL-17, and TNFα, which correlate with disease activity.32 For this reason, nowadays, targeted biological treatments have emerged to be effective approaches for the management of AOSD, especially for the refractory form steroid-dependent or when severe manifestations occur.23 However, there is no systematic criteria to select the correct drugs nor indications for biologics treatment suspension once clinical remission is achieved. In patients with systemic AOSD, response to IL-1 inhibitors must be expected within hours or days. It has been suggested that a cautious tapering of the drug could be attempted if clinical remission is maintained for at least 6–12 months.33
IL-1 Inhibitors
Anti IL-1 agents undoubtedly represent the milestone for the treatment of AOSD. To date, three IL-1 inhibitors are available for AOSD. IL-1 inhibitors have an overall satisfactory safety profile in AOSD. In terms of the risk of infection, treatment with anti-IL-1 agents seems to have an acceptable safety profile. Although the inhibition of IL-1 remains the gold standard in systemic AOSD and in the refractory forms,34 other approaches should be considered.
Anakinra
Anakinra, a non-glycosylated form of human IL-1 receptor antagonist (IL-1Ra), acts as a pure receptor antagonist binding to the IL-1 receptor (IL-1RI) and preventing the activation of this receptor by either IL-1β or IL-1α.35 Anakinra was firstly approved in 2001 for the treatment of rheumatoid arthritis, but the first employment in AOSD dates back to 2005 when the first cases were described.36–38 As several studies report, it seems that anakinra is more effective if administered early in the disease course and it was proved to be more useful in patients with highly active systemic AOSD than in those with isolated chronic arthritis.39 The confirmed effectiveness of anakinra was assessed in diverse observational studies by now.40–44 Ortiz-Sanjuan et al45 reported nearly 40 patients who experienced a marked reduction of the frequency of joint manifestations (from 87.8% at baseline to 41.5% after 12 months), of cutaneous rash (from 58.5% to 7.3%), fever (from 78% to 14.6%), lymphadenopathy (from 26.8% to 4.9%), and ferritin serum levels (from 63.4% to 36.6%) after 1year of therapy with anakinra. In addition, a general decrease of steroid daily intake was noticed (p<0.01). Similar results were achieved in a larger cohort of 136 consecutive AOSD patients that, after anakinra, exhibited a significant reduction of Pouchot’s score from the baseline (p<0.0001) until the last follow-up period (12 months), with no major safety concerns.46 Anakinra is usually administered at the dosage of 100 mg/day but given the short half-life (6–8 hours) in cases of insufficient response it can be increased to 200 mg, split into twice-daily administrations.1 Treatment with anakinra has been associated with frequent injection-site reactions and occasionally severe cases of hepatotoxicity, reversible after treatment withdrawal.44
Canakinumab
Canakinumab is a fully human monoclonal antibody against IL-1β with a half-life of 26 days, which makes it possible to administer it at the dosage of 150 mg or 300 mg every 4–8 weeks. In 2016, canakinumab was approved by the European Medicines Agency (EMA) and the US Food and Drug Administration (FDA) for a license extension to treat both SJIA and AOSD, based on the concept of the Still’s disease continuum.11 In this context, in 2018, Feist et al47 described the efficacy and the safety of canakinumab in a subgroup of young adolescents with SJIA aged ≥16 years which were representative of an adult population. The phenotypic dichotomy of AOSD with the prevalence of systemic symptoms rather than polyarthritis reflects on the diverse possible therapeutical approaches. In this context, the CONSIDER trial (a multicenter, randomized, double-blind, placebo-controlled trial), which aimed to employ canakinumab in AOSD subjects with active joint involvement, did not achieve the primary endpoint (p=0.18) defined as the change in disease activity score (ΔDAS28>1.2).48,49 These results pave the way to new therapeutical strategies, which should be tailored on each patient according to the clinical phenotype presented.
Rilonacept
Among other IL-1 inhibitors, it should be mentioned that Rilonacept (IL-1 trap molecule), a construct of two extracellular chains of the IL-1R complex (IL-1R plus IL1RAP), fused to the Fc portion of human immunoglobulin G1 (IgG1). Rilonacept can bind both IL-1β and IL-1α with high affinity and its half-life is longer than that of anakinra.1 For this reason, it was successfully used at 160 mg/week in some refractory forms with a predominant articular involvement as a steroid-sparing agent.50,51 However, Rilonacept is currently FDA-approved only for the treatment of recurrent pericarditis.
IL-6 Inhibitors
IL-6 is a pro-inflammatory cytokine downstream of IL-1, and it represents a therapeutic target as well for AOSD.
Tocilizumab
Tocilizumab (TCZ) is a humanized anti-IL-6 receptor antibody that recognizes both membrane-bound and soluble forms of IL-6 receptor, specifically blocking IL-6. It is administered subcutaneously at doses of up to 8 mg/kg every 2 weeks.52 The efficacy of TCZ was proved in cases of refractory AOSD, especially when the chronic articular pattern was predominant.53,54 However, the confirmation of its effectiveness on articular manifestation derived from a randomized clinical trial of 2018.55 The same study also reported TCZ as acceptable in terms of safety.
Sarilumab
Sarilumab (anti IL-6Rα) is a fully human anti-IL-6Rα mAb that binds membrane-bound and soluble human IL-6Rα with high affinity.56 So far, the efficacy of sarilumab in AOSD was reported only in one patient who was successfully treated after developing resistance to TCZ.57 The rationale of switching to sarilumab in TCZ-resistant subjects derives from the data obtained from the ASCERTAIN EXTEND trial (NCT01146652) on patients affected by rheumatoid arthritis.58 The hypothesis of using sarilumab in refractory cases relies on the fact that in systemic AOSD the high levels of IL-6 may overwhelm the neutralizing capability of TCZ, therefore the direct inhibition of the IL-6 receptor might help to reduce more thoroughly the pro-inflammatory activity of IL-6. The same trial (NCT01146652) evaluated the safety of sarilumab, showing that the incidence rate of adverse events of special interest was generally stable, without any signal for increased rate over time.
TNF-Alpha Inhibitors
Among TNF-α inhibitors, infliximab, etanercept, and adalimumab were the first biologicals used in single case reports or small series in the early 2000s.59 The first employment of infliximab date to 2001, when it was administered i.v. at a dose of 3–5 mg/kg in three refractory Still’s patients with success.60 Other cases regarding the use of infliximab are reported in the literature.61 Data with etanercept, the recombinant soluble form of the human 75-kDa TNF-receptor fusion protein, derive mostly from a small case series in which an improvement of the articular involvement has been observed after biweekly doses of 25 mg s.c in half of the patients with no occurrence of adverse reactions.62 However, the convincing results with TNF-α inhibitors seen at the start were not confirmed by Fautrel et al,63 who noticed a complete remission of symptoms only in five out of 20 patients treated with either infliximab or etanercept. Finally, data with adalimumab are mixed and scarce64,65 and, for this reason, in AOSD other biological approaches are nowadays preferred to anti-TNF-alpha in terms of efficacy and safety. Indeed, a recent meta-analysis showed that the efficacy of TNFα inhibitors on clinical and laboratory manifestations is significantly lower than that of other biologic treatments and the observed adverse reactions, including serious infections, may lead to TNFα inhibitors discontinuation.66
Other Therapeutical Approaches
Rituximab, a chimeric anti CD-20 monoclonal antibody, is approved for rheumatoid arthritis, but the experience in AOSD (administered at 375 mg/m2 given twice at 4-week intervals) is limited and for selected cases.67,68 Likewise, the employment of abatacept, a co-stimulation modulator that inhibits T-lymphocyte activation by binding to CD80 and CD86 and blocks the interaction with CD28, exhibited mixed and scarce results.69,70 Concerning intravenous immunoglobulin (IVIGs) they were proved to be effective and well-tolerated at the usual dose of 2 g/kg in 2–5 days every month in half of the patients in two open-label studies;71,72 however, they should be employed in selected cases or when life-threatening manifestations occur.
Novel Biologics Treatments
Alongside biologics that have now entered the therapeutic routine of AOSD patients, scientific efforts have produced new compounds with different pharmacokinetic and dynamic properties (Table 1). Oral small molecules such as JAK or new anti-cytokine have been developed, so AOSD patients refractory to a specific biologic drug could be easily switched to an alternative highly effective treatment.
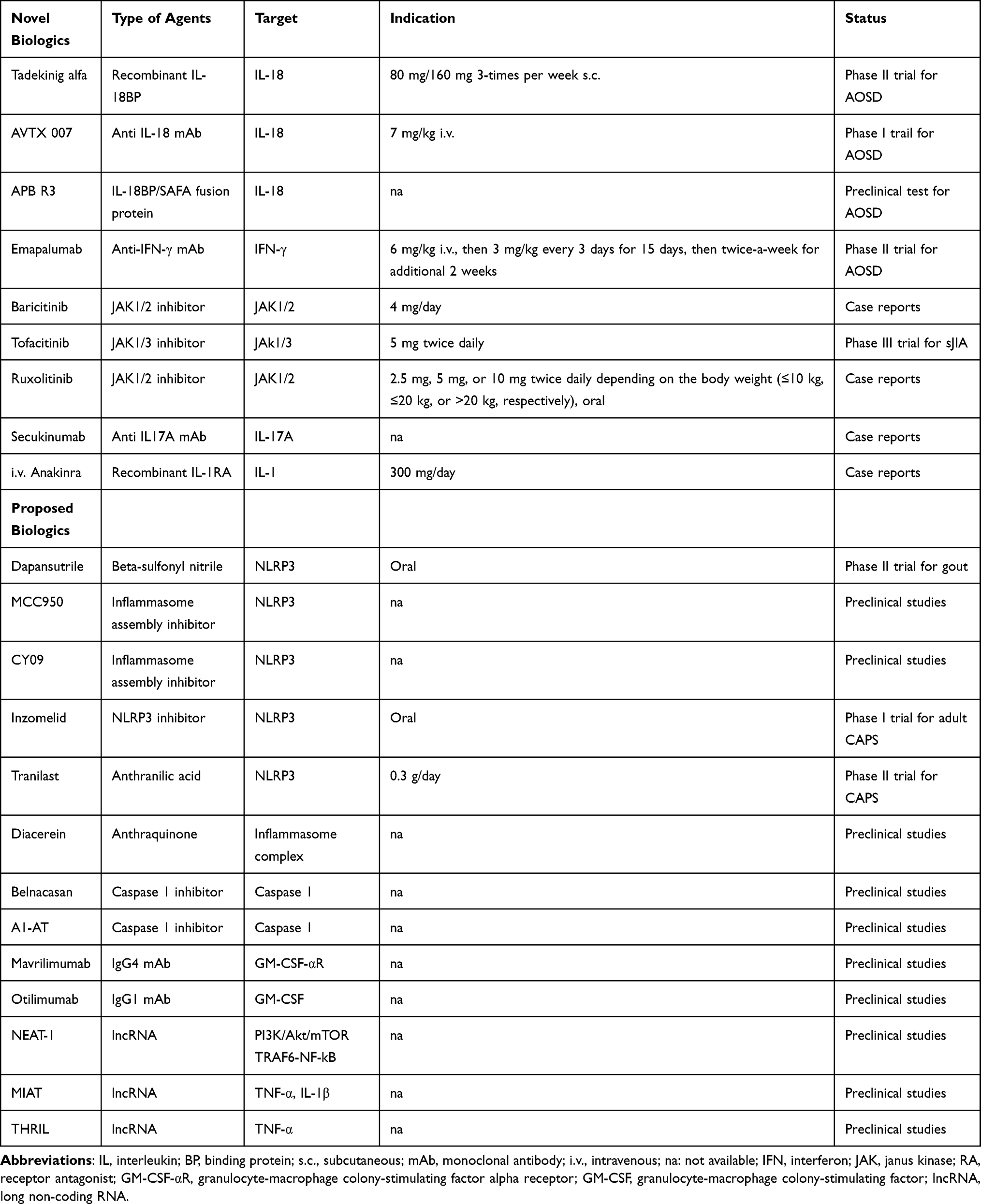 |
Table 1 Novel and Proposed Biologic Treatments for AOSD Patients |
IL-18 Inhibitors
IL-18 is a pro-inflammatory cytokine belonging to the IL-1 superfamily, whose activity is tightly regulated by natural IL-18 binding protein (IL-18BP). As previously described, elevated levels of IL-18 during the active phase of AOSD give it the role of a potential biomarker.14 Actually, three IL-18 inhibitors are under evaluation; however, data on the clinical efficacy of IL-18 blockade is still limited.
Tadekinig Alfa
As recombinant human IL-18BP, tadekinig alfa binds IL-18 with a high affinity and in turn inhibits the secretion of TNF-α, IFN-γ, and IL-1.73 The most comprehensive study is a Phase II clinical trial showing early clinical and laboratory efficacy independent of dosage (80 mg or 160 mg 3-times per week s.c.) in 23 patients with long-standing multidrug-resistant disease.74 In fact, 50% of patients had previously been treated with csDMARD and with previous biologic agents in about one third of patients. The response rate was nearly 50% with an overall good safety profile. Similar to anakinra, the most frequent adverse event was injection site reaction; mild upper airway infections and arthralgia were also reported. A recent study evaluating the prolonged treatment with tadekining alfa reported a maintained clinical remission and dropped levels of free IL-18 in serum within 2 hours after injection.75
AVTX 007
AVTX 007 (formerly CERC 007, AEVI 007, or MEDI 2338) is a high affinity, fully human anti-IL-18 monoclonal antibody, being developed for the treatment of autoinflammatory diseases, potentially including AOSD, sJIA, and multiple myeloma. Early stage clinical development is underway in the USA (NCT04752371). The Phase I multicenter, open-label study plans to include 12 patients, six of whom will be administered AVTX-007 intravenous (i.v.) at a dose of 7 mg/kg. Based on safety results in the first cohort, six other participants will be administered a dose escalation or reduction of AVTX-007. Safety, tolerability, and efficacy will be evaluated.
APB R3
APB R3 is a long acting recombinant fusion protein composed of IL-18BP fused via a peptide linker to anti-human serum albumin Fab fragment (SAFA). SAFA is a platform technology to produce long-acting therapeutics by modifying antiserum albumin Fab avoiding lysosomal degradation and thereby prolonging the half life of the molecule. As at October 2021, early research of APB R3 is underway for the treatment of adult-onset Still’s disease in South Korea.76
Anti IFN-γ
IL-18 is a potent inducer of IFN-γ which has been found elevated in patients with macrophage activation syndrome complicating sJIA.77 Recent reports20,78 on the efficacy of an IFN-γ antibody (emapalumab) on cases of MAS complicating sJIA has grown great expectations on the potential efficacy of novel therapeutic agent. However, data regarding the effectiveness and safety of the IFN-γ blockade in treating AOSD remain limited.
Emapalumab
Emapalumab is a fully human monoclonal antibody that neutralizes both free- and receptor-bound IFN-γ by inhibiting receptor dimerization and the signaling transduction.79 The efficacy of emapalumab was recently assessed in a clinical trial enrolling 34 young patients with a diagnosis of primary HLH.20 Although the 27 pretreated patients were reported to have worsened or reactivated disease, an unsatisfactory response, or adverse events, only 15% had fever, 70% thrombocytopenia, and 78% hyperferritinemia. However, the study by Locatelli et al20 does not present convincing data for strong efficacy of emapalumab in patients with primary HLH. Recently, Gabr et al80 reported that emapalumab effectively eliminated fever and improved laboratory outcomes of a patient with AOSD complicated by MAS.
JAK Inhibitors
Unlike the inhibition of targeted cytokines, Janus kinase (JAK) inhibitors can block a large variety of proinflammatory molecules through the competitive interaction with JAK region required for downstream JAK/STAT signaling. JAK inhibitors indeed prevent the effect of IL-6, IL-10, IFN-γ, IFN-α, and granulocyte-macrophage colony-stimulating factor (GM-CSF), which are strongly implicated in the AOSD pathogenesis.81 Baricitinib and Ruxolitinib are more selective toward JAK1/2, while tofacitinib is more effective to JAK3.82 Due to the modulating effect on the immune response, JAK inhibitors have a good chance of becoming a very promising treatment in heterogeneous disorders, such as AOSD. However, data regarding the effectiveness and safety of JAK inhibitors in treating AOSD remain limited to case reports.
Baricitinib
Baricitinib, a JAK1/2 inhibitor, was reported by Kacar et al83 as effective in treating two AOSD patients refractory to csDMARDs and other biologics. The combination with anakinra effectively treated a patient with refractory AOSD;84 this observation is partially confirmed by Gillard et al85 in another case. The authors also reported two cases with a concomitant use of baricitinib and steroids but there was no response to therapy at the last follow-up.
Tofacitinib
Tofacitinib, a JAK1/3 inhibitor, has recently been approved by the FDA for polyarticular JIA and is currently under evaluation (5 mg twice daily) in a double-blind trial for sJIA (NTC03000439). At present, only case reports support the use of tofacitinib in Still’s disease complicated by MAS,86 as well as the studies on animal models of HLH.87 Besides, a recent report revealed the successful use of tofacitinib in 14 patients with AOSD.88
Ruxolitinib
The JAK1/2 inhibitor ruxolitinib is known to significantly reduce the proliferation and activation of immune modulating IFN-γ and other cytokines on experimental murine models of HLH.89 Therefore, ruxolitinib may be considered for patients with secondary HLH. Indeed, a pilot study in 12 children with secondary HLH showed a good clinical response after 28 days of oral treatment.90 Partial response was also reported for two Still’s patients with the concurrent use of steroids.85
Anti IL-17
An increase of IL-17 in AOSD has been reported91 and is involved in the recruitment of neutrophils, thus contributing to the maintenance of the inflammatory phenotype.92 Thus, the use of IL-17 inhibitors could be an effective and safe therapeutic option, especially in the chronic patients. However, more evidence and more treated cases are awaited.
Secukinumab
Secukinumab is a human monoclonal antibody that binds to and neutralizes IL-17A. It is currently approved in many countries to treat psoriasis, ankylosing spondylitis, non-radiographic axial spondyloarthritis (SpA), and psoriatic arthritis with a favorable safety profile. Mitrovic et al93 reported a single case of an AOSD patient who achieved complete remission under secukinumab, after loss of efficacy to anakinra and MTX, after SpA onset. This observation could suggest a phenotype shift rather than an overlap between the two diseases. Moreover, one has to keep in mind that the manifestations of SpA should be differentiated from the forms of Still’s disease with chronic articular damage.
A New Strategy for Anti IL-1
The use of anti IL-1 agents is already known to give a fast and robust response in the majority of AOSD cases. Furthermore, both articular and systemic manifestations can respond to this approach, that has shown efficacy in specific organ involvements as well as in other systemic complications of diseases such as hyperinflammation, cytokine storm, and MAS.32 In this context, on a larger series of patients with COVID-19 pneumonia, characterized by systemic hyperinflammation and cytokine storm, Pontali et al94 reported the potential efficacy and safety of the early use of high doses of intravenous (i.v.) anakinra with or without glucocorticoids. We personally treated an AOSD patient with 1 mg/kg of oral prednisone and IL-1 inhibition with i.v. anakinra (100 mg every 8 hours) with clinical improvement in less than 24 hours [personal communication].
Proposal Future Treatments
Although anti-cytokine agents are characterized by a good safety profile that allows long periods of continued treatment, it would be reasonable to believe that the risks may outweigh the benefits once an inactive disease state is achieved. In these regards, new strategies for the treatment of the adult form of Still’s disease are now under consideration.
NLRP3 Inflammasome Inhibitors
Great attention was recently addressed to the blockade of the inflammasome NLRP3 and its components, that are well-known to be involved in autoinflammatory processes. So far, different compounds able to bind NLRP3 and consequently the release of IL-1 have been investigated.
Dapansutrile
Dapansutrile (OLT1177), an orally active beta-sulfonyl nitrile, functions as a direct inhibitor of NLRP3 and it is currently tested in gout.95 The first proof-of-concept, phase II trial on adult gouty patients was recently carried out in The Netherlands and dapansutrile was orally administered at variable dosages, eliciting a significant reduction of joint tenderness and swelling.96 In addition, a dampening of pro-inflammatory cytokines, and in particular IL-6, was noticed. Despite the limitations of this preliminary study, the combination of its efficacy, the good safety profile, and the oral administration make dapansutrile a promising therapeutical option not only for gouty patients, but it is conceivable it could also be employed in AOSD, in particular in those patients having a predominant articular involvement.
Other NLRP3 Inflammasome Complex Inhibitors
By now, other blockers of NLRP3 or its components are under examination. MCC950 directly binds to the NACHT domain and inhibits the inflammasome assembly by hampering ASC oligomerization.97 This compound was proven to reduce IL-1β production in vivo and in mouse models of CAPS.98 A similar mechanism was described for CY09 which affects the NLRP3 ATPase activity hindering the assembly of NLRP3 itself.99 On CAPS there are currently ongoing studies with Inzomelid (NCT04015076), another oral selective NLRP3 inhibitors, and tranilast (NCT03923140), an anthranilic acid able to bind the NACHT domain;99 diacerein, an anthraquinone compound, may downregulate both NLRP3/caspase-1/IL-1β either the IL-6/pSTAT3 axis,100 while belnacasan (VX-765) and A1-AT target directly the caspase I component blocking the IL-1β release.95,101 Despite the studies on these inflammasome inhibitors currently being at an early stage, it is conceivable they could have future employment in Still’s disease.
Granulocyte-Macrophage Colony-Stimulating Factor Inhibitors
Another target that has been investigated in recent times is GM-CSF.102 GM-CSF can upregulate neutrophils and macrophages, can induce the formation of NETs, and in turn enhances the production of pro-inflammatory cytokines by binding to its receptor.
Mavrilimumab and Otilimab
In this context, mavrilimumab (CAM-3001), an IgG4 monoclonal antibody (mAB), and otilimab (MOR-103), an IgG1 mAB, can bind the GM-CSF-alpha receptor and GM-CSF, respectively, preventing the release of pro-inflammatory cytokines. Both otilimab and mavrilimumab were proved to be effective and safe in rheumatoid arthritis patients, providing a significant reduction of DAS28-CRP in the clinical trials carried out by now.103–105 In addition, mavrilimumab was recently employed for the treatment of severe COVID-19 pneumonia, relying on the fact that the inhibition of GM-CSF could curb the hyperinflammation provoked by the virus.106 For this reason, it is conceivable these inhibitors could be used for treating both the chronic articular pattern and the systemic form of Still’s disease.
Long Non-Coding RNAs
Recently, the expression signature of long non-coding RNAs (lncRNAs) in AOSD patients was investigated to understand whether they could correlate with the disease phenotype. LncRNAs are known to be central regulators of the immune responses107–110 and their expression is associated to certain pathways or cytokines that contribute to AOSD pathogenesis. For example, NEAT-1 (nuclear enriched abundant transcript 1) levels, detected in serum samples of AOSD patients, strongly correlate with the expression of other lncRNAs after cyclosporine or anti IL-6, while they were not observed before starting the treatments. NEAT-1 can upregulate the PI3K/Akt/mTOR and TRAF6-NfkB axis and in turn regulates the production of IL-6, IL-17, and TNF- α. Similarly, MIAT (myocardial infarction associated transcript) was described as a suppressor on TNF-α and IL-1β, while, on the other hand, THRIL (TNF-alpha and hnRNPL-related immunoregulatory lincRNA) was found to upregulate TNF-α; in AOSD indeed, high levels of MIAT and a low expression of THRIL were observed in comparison to healthy controls.111
Taken together these results suggest that, according to the different type of lncRNA signature detected, it is possible to understand the axis, or the group of cytokines mostly involved in AOSD pathogenesis and, thus, managing properly the patients contributing to the treat-to target strategy.
Conclusions
Recent advances in biologic drug development have had a major impact on AOSD patients in terms of improved quality-of-life and coping strategies. Specific inhibition of IL-1 and IL-6 is now considered a safe and effective therapy to better control the disease. As reported in this work, there are currently a number of biological agents, possible effective targets for achieving AOSD control and remission. The early and targeted blockade of the inflammation is critical for treatment, and early biological therapies in an appropriate window-of-opportunity is expected in the near future.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Sfriso P, Bindoli S, Galozzi P. Adult-onset still’s disease: molecular pathophysiology and therapeutic advances. Drugs. 2018;78(12):1187–1195. doi:10.1007/s40265-018-0956-9
2. Mollaeian A, Chen J, Chan NN, et al. Adult onset Still’s disease in the elderly: a case-based literature review. BMC Rheumatol. 2021;5(1):12. doi:10.1186/s41927-021-00183-6
3. Efthimiou P, Kontzias A, Hur P, et al. Adult-onset Still’s disease in focus: clinical manifestations, diagnosis, treatment, and unmet needs in the era of targeted therapies. Semin Arthritis Rheum. 2021;51(7):858–874. doi:10.1016/j.semarthrit.2021.06.004
4. Yamaguchi M, Ohta A, Tsunematsu T, et al. Preliminary criteria for classification of adult Still’s disease. J Rheumatol. 1992;19(3):424–430.
5. Fautrel B, Le Moël G, Saint-Marcoux B, et al. Diagnostic value of ferritin and glycosylated ferritin in adult onset Still’s disease. J Rheumatol. 2001;28(2):322–329.
6. Lebrun D, Mestrallet S, Dehoux M, et al. Validation of the Fautrel classification criteria for adult-onset Still’s disease. Semin Arthritis Rheum. 2018;47(4):578–585. doi:10.1016/j.semarthrit.2017.07.005
7. Maria AT, Le Quellec A, Jorgensen C, et al. Adult onset Still’s disease (AOSD) in the era of biologic therapies: dichotomous view for cytokine and clinical expressions. Autoimmun Rev. 2014;13(11):1149–1159. doi:10.1016/j.autrev.2014.08.032
8. Inoue N, Shimizu M, Tsunoda S, et al. Cytokine profile in adult-onset Still’s disease: comparison with systemic juvenile idiopathic arthritis. Clin Immunol. 2016;169:8–13. doi:10.1016/j.clim.2016.05.010
9. Mitrovic S, Fautrel B. New markers for adult-onset Still’s disease. Joint Bone Spine. 2018;85(3):285–293. doi:10.1016/j.jbspin.2017.05.011
10. Nirmala N, Brachat A, Feist E, et al. Gene-expression analysis of adult-onset Still’s disease and systemic juvenile idiopathic arthritis is consistent with a continuum of a single disease entity. Pediatr Rheumatol Online J. 2015;13:50. doi:10.1186/s12969-015-0047-3
11. Fautrel B. Adult-onset Still disease. Best Pract Res Clin Rheumatol. 2008;22(5):773–792. doi:10.1016/j.berh.2008.08.006
12. Hu Q, Shi H, Zeng T, et al. Increased neutrophil extracellular traps activate NLRP3 and inflammatory macrophages in adult-onset Still’s disease. Arthritis Res Ther. 2019;21(1):9. doi:10.1186/s13075-018-1800-z
13. Jung JY, Kim JW, Suh CH, et al. Roles of interactions between toll-like receptors and their endogenous ligands in the pathogenesis of systemic juvenile idiopathic arthritis and adult-onset still’s disease. Front Immunol. 2020;11:583513. doi:10.3389/fimmu.2020.583513
14. Tang Y, Liu J, Zhang D, et al. Cytokine storm in COVID-19: the current evidence and treatment strategies. Front Immunol. 2020;11:1708. doi:10.3389/fimmu.2020.01708
15. Meng J, Ma Y, Jia J, et al. Cytokine storm in coronavirus disease 2019 and adult-onset still’s disease: similarities and differences. Front Immunol. 2021;11:603389. doi:10.3389/fimmu.2020.603389
16. Bamidis AD, Koehler P, Di Cristanziano V, et al. First manifestation of adult-onset Still’s disease after COVID-19. Lancet Rheumatol. 2021;3(5):e319–e321. doi:10.1016/S2665-9913(21)00072-2
17. Wang R, Li T, Ye S, et al. Macrophage activation syndrome associated with adult-onset Still’s disease: a multicenter retrospective analysis. Clin Rheumatol. 2020;39(8):2379–2386. doi:10.1007/s10067-020-04949-0
18. Yang XP, Wang M, Li TF, et al. Predictive factors and prognosis of macrophage activation syndrome associated with adult-onset Still’s disease. Clin Exp Rheumatol. 2019;Suppl 121(6):83–88.
19. Sönmez HE, Demir S, Bilginer Y, Özen S. Anakinra treatment in macrophage activation syndrome: a single center experience and systemic review of literature. Clin Rheumatol. 2018;37(12):3329–3335. doi:10.1007/s10067-018-4095-1
20. Locatelli F, Jordan MB, Allen C, et al. Emapalumab in children with primary hemophagocytic lymphohistiocytosis. N Engl J Med. 2020;382(19):1811–1822. doi:10.1056/NEJMoa1911326
21. Mitrovic S, Fautrel B. Complications of adult-onset Still’s disease and their management. Expert Rev Clin Immunol. 2018;14(5):351–365. doi:10.1080/1744666X.2018.1465821
22. Franchini S, Dagna L, Salvo F, et al. Efficacy of traditional and biologic agents in different clinical phenotypes of adult-onset Still’s disease. Arthritis Rheum. 2010;62(8):2530–2535. doi:10.1002/art.27532
23. Cavalli G, Farina N, Campochiaro C, et al. Current treatment options and safety considerations when treating adult-onset Still’s disease. Expert Opin Drug Saf. 2020;19(12):1549–1558. doi:10.1080/14740338.2020.1839411
24. O’Connor N, Dargan PI, Jones AL. Hepatocellular damage from non-steroidal anti-inflammatory drugs. QJM. 2003;96(11):787–791. doi:10.1093/qjmed/hcg138
25. Savage RL, Moller PW, Ballantyne CL, et al. Variation in the risk of peptic ulcer complications with nonsteroidal antiinflammatory drug therapy. Arthritis Rheum. 1993;36(1):84–90. doi:10.1002/art.1780360114
26. Clive DM, Stoff JS. Renal syndromes associated with nonsteroidal antiinflammatory drugs. N Engl J Med. 1984;310(9):563–572. doi:10.1056/NEJM198403013100905
27. Gerfaud-Valentin M, Maucort-Boulch D, Hot A, et al. Adult-onset still disease: manifestations, treatment, outcome, and prognostic factors in 57 patients. Medicine. 2014;93(2):91–99. doi:10.1097/MD.0000000000000021
28. Henter JI, Samuelsson-Horne A, Aricò M, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100(7):2367–2373. doi:10.1182/blood-2002-01-0172
29. Kalyoncu U, Solmaz D, Emmungil H, et al. Response rate of initial conventional treatments, disease course, and related factors of patients with adult-onset Still’s disease: data from a large multicenter cohort. J Autoimmun. 2016;69:59–63. doi:10.1016/j.jaut.2016.02.010
30. Sfriso P, Priori R, Valesini G, et al. Adult-onset Still’s disease: an Italian multicentre retrospective observational study of manifestations and treatments in 245 patients. Clin Rheumatol. 2016;35(7):1683–1689. doi:10.1007/s10067-016-3308-8
31. Liu Z, Lv X, Tang G. Clinical features and prognosis of adult-onset Still’s disease: 75 cases from China. Int J Clin Exp Med. 2015;8(7):16634–16639.
32. Feist E, Mitrovic S, Fautrel B. Mechanisms, biomarkers and targets for adult-onset Still’s disease. Nat Rev Rheumatol. 2018;14(10):603–618. doi:10.1038/s41584-018-0081-x
33. Govoni M, Bortoluzzi A, Rossi D, Modena V. How I treat patients with adult onset Still’s disease in clinical practice. Autoimmun Rev. 2017;16(10):1016–1023. doi:10.1016/j.autrev.2017.07.017
34. Laskari K, Tektonidou PMG, Katsiari PC, et al. Outcome of refractory to conventional and/or biologic treatment adult Still’s disease following canakinumab treatment: countrywide data in 50 patients. Semin Arthritis Rheum. 2021;51(1):137–143. doi:10.1016/j.semarthrit.2020.10.011
35. Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. 2011;117(14):3720–3732. doi:10.1182/blood-2010-07-273417
36. Rudinskaya A, Trock DH. Successful treatment of a patient with refractory Adult-onset Still disease with anakinra. J Clin Rheumatol. 2003;9(5):330–332. doi:10.1097/01.rhu.0000089966.48691.7f
37. Fitzgerald AA, Leclercq SA, Yan A, et al. Rapid responses to anakinra in patients with refractory adult-onset Still’s disease. Arthritis Rheum. 2005;52:1794–1803. doi:10.1002/art.21061
38. Kötter I, Wacker A, Koch S, et al. Anakinra in patients with treatment-resistant adult-onset Still’s disease: four case reports with serial cytokine measurements and a review of the literature. Semin Arthritis Rheum. 2007;37(3):189–197. doi:10.1016/j.semarthrit.2007.04.002
39. Guignard S, Dien G, Dougados M. Severe systemic inflammatory response syndrome in a patient with adult onset Still’s disease treated with the anti-IL1 drug anakinra: a case report. Clin Exp Rheumatol. 2007;25(5):758–759.
40. Nordström D, Knight A, Luukkainen R, et al. Beneficial effect of interleukin 1 inhibition with anakinra in adult-onset Still’s disease. An open, randomized, multicenter study. J Rheumatol. 2012;39:2008–2011. doi:10.3899/jrheum.111549
41. Lequerré T, Quartier P, Rosellini D, et al. Interleukin-1 receptor antagonist (anakinra) treatment in patients with systemic-onset juvenile idiopathic arthritis or adult onset Still disease: preliminary experience in France. Ann Rheum Dis. 2008;67(3):302–308. doi:10.1136/ard.2007.076034
42. Laskari K, Tzioufas AG, Moutsopoulos HM. Efficacy and long-term follow-up of IL-1R inhibitor anakinra in adults with Still’s disease: a case-series study. Arthritis Res Ther. 2011;13(3):R91. doi:10.1186/ar3366
43. Giampietro C, Ridene M, Lequerre T, et al. Anakinra in adult-onset still’s disease: long-term treatment in patients resistant to conventional therapy. Arthritis Care Res. 2013;65(5):822–826. doi:10.1002/acr.21901
44. Hong D, Yang Z, Han S, et al. Interleukin 1 inhibition with anakinra in adult-onset still disease: a meta-analysis of its efficacy and safety. Drug Des Devel Ther. 2014;8:2345–2357. doi:10.2147/DDDT.S73428
45. Ortiz-Sanjuán F, Blanco R, Riancho-Zarrabeitia L, et al. Efficacy of anakinra in refractory adult-onset still’s disease multicenter study of 41 patients and literature review. Med. 2015;94(39):e34.
46. Colafrancesco S, Priori R, Valesini G, et al. Response to interleukin-1 inhibitors in 140 Italian patients with adult-onset still’s disease: a multicentre retrospective observational study. Front Pharmacol. 2017;8:369. doi:10.3389/fphar.2017.00369
47. Feist E, Quartier P, Fautrel B, et al. Efficacy and safety of canakinumab in patients with Still’s disease: exposure-response analysis of pooled systemic juvenile idiopathic arthritis data by age groups. Clin Exp Rheumatol. 2018;36(4):668–675.
48. Kedor C, Listing J, Zernicke J, et al. Canakinumab for treatment of adult-onset still’s disease to achieve reduction of arthritic manifestation (CONSIDER): Phase II, randomised, double-blind, placebo-controlled, multicentre, investigator-initiated trial. Ann Rheum Dis. 2020;79(8):1090–1097. doi:10.1136/annrheumdis-2020-217155
49. Cota-Arce JM, Cota J, De León-nava MA, et al. Efficacy and safety of canakinumab in the treatment of adult-onset Still’s disease: a systematic review. Semin Arthritis Rheum. 2021;51(6):1282–1290. doi:10.1016/j.semarthrit.2021.08.007
50. Henderson C, Wilson M, Pham T-H, et al. Safety and efficacy of IL-1 Trap in resistant adult onset Still’s disease: 24 month follow-up of open label treatment and biomarkers of response.
51. Petryna O, Cush JJ, Efthimiou P. IL-1 Trap rilonacept in refractory adult onset Still’s disease. Ann Rheum Dis. 2012;71(12):2056–2057. doi:10.1136/annrheumdis-2012-201409
52. Ortiz-Sanjuán F, Blanco R, Calvo-Rio V, et al. Efficacy of tocilizumab in conventional treatment-refractory adult-onset Still’s disease: multicenter retrospective open-label study of thirty-four patients. Arthritis Rheumatol. 2014;66(6):1659–1665. doi:10.1002/art.38398
53. Cipriani P, Ruscitti P, Carubbi F, et al. Tocilizumab for the treatment of adult-onset Still’s disease: results from a case series. Clin Rheumatol. 2014;33(1):49–55. doi:10.1007/s10067-013-2381-5
54. Puéchal X, De Bandt M, Berthelot JM, et al. Tocilizumab in refractory adult still’s disease. Arthritis Care Res. 2011;63(1):155–159. doi:10.1002/acr.20319
55. Kaneko Y, Kameda H, Ikeda K, et al. Tocilizumab in patients with adult-onset still’s disease refractory to glucocorticoid treatment: a randomised, double-blind, placebo-controlled Phase III trial. Ann Rheum Dis. 2018;77(12):1720–1729. doi:10.1136/annrheumdis-2018-213920
56. Lamb YN, Deeks ED, Lamb YN, Deeks ED. Sarilumab: a review in moderate to severe rheumatoid arthritis. Drugs. 2018;78(9):929–940. doi:10.1007/s40265-018-0929-z
57. Simeni Njonnou SR, Soyfoo MS, Vandergheynst FA. Efficacy of sarilumab in adult-onset Still’s disease as a corticosteroid-sparing agent. Rheumatology. 2019;58(10):1878–1879. doi:10.1093/rheumatology/kez154
58. Emery P, van Hoogstraten H, Jayawardena S, et al. Efficacy of sarilumab in patients with rheumatoid arthritis who previously received sarilumab or tocilizumab [abstract]. Arthritis Rheumatol. 2017;69(suppl 10):key075–446.
59. Al-Homood IA. Biologic treatments for adult-onset Still’s disease. Rheumatology. 2014;53(1):32–38. doi:10.1093/rheumatology/ket250
60. Cavagna L, Caporali R, Epis O, et al. Infliximab in the treatment of adult Still’s disease refractory to conventional therapy. Clin Exp Rheumatol. 2001;19(3):329–332.
61. Kraetsch HG, Antoni C, Kalden JR, et al. Successful treatment of a small cohort of patients with adult onset of Still’s disease with infliximab: first experiences. Ann Rheum Dis. 2001;60 Suppl 3(Suppl 3):iii55–iii57. doi:10.1136/ard.60.90003.iii55
62. Husni ME, Maier AL, Mease PJ, et al. Etanercept in the treatment of adult patients with Still’s disease. Arthritis Rheum. 2002;46(5):1171–1176. doi:10.1002/art.10231
63. Fautrel B, Sibilia J, Mariette X, et al. Tumour necrosis factor α blocking agents in refractory adult Still’s disease: an observational study of 20 cases. Ann Rheum Dis. 2005;64(2):262–266. doi:10.1136/ard.2004.024026
64. Benucci M, Li GF, Del Rosso A, et al. Adalimumab (anti-TNF-alpha) therapy to improve the clinical course of adult-onset Still’s disease: the first case report. Clin Exp Rheumatol. 2005;23(5):733.
65. Rech J, Ronneberger M, Englbrecht M, et al. Successful treatment of adult-onset Still’s disease refractory to TNF and IL-1 blockade by IL-6 receptor blockade. Ann Rheum Dis. 2011;70(2):390–392. doi:10.1136/ard.2010.129403
66. Zhou S, Qiao J, Bai J, et al. Biological therapy of traditional therapy-resistant adult-onset Still’s disease: an evidence-based review. Ther Clin Risk Manag. 2018;14:167–171. doi:10.2147/TCRM.S155488
67. Ahmadi-Simab K, Lamprecht P, Jankowiak C, Gross WL. Successful treatment of refractory adult onset Still’s disease with rituximab. Ann Rheum Dis. 2006;65(8):1117–1118. doi:10.1136/ard.2005.047621
68. Bartoloni E, Alunno A, Luccioli F, et al. Successful treatment of refractory adult-onset Still’s disease with anti-CD20 monoclonal antibody. Clin Exp Rheumatol. 2009;27(5):888–889.
69. Quartuccio L, Maset M, De Vita S. Efficacy of Abatacept in a refractory case of adult-onset Still’s disease. Clin Exp Rheumatol. 2010;28(2):265–267.
70. Ostrowski RA, Tehrani R, Kadanoff R. Refractory adult-onset still disease successfully treated with Abatacept. J Clin Rheumatol. 2011;17(6):315–317. doi:10.1097/RHU.0b013e31822c53ad
71. Permal S, Wechsler B, Cabane J, et al. Treatment of Still’s disease in adults with intravenous immunoglobulins. Rev Med Interne. 1995;16(4):250–254. French. doi:10.1016/0248-8663(96)80703-X
72. Vignes S, Wechsler B, Amoura Z, et al. Intravenous immunoglobulin in adult Still’s disease refractory to non-steroidal anti-inflammatory drugs. Clin Exp Rheumatol. 1998;16(3):295–298.
73. Dinarello CA, Novick D, Kim S, et al. Interleukin-18 and IL-18 binding protein. Front Immunol. 2013;4:289. doi:10.3389/fimmu.2013.00289
74. Gabay C, Fautrel B, Rech J, et al. Open-label, multicentre, dose-escalating phase II clinical trial on the safety and efficacy of tadekinig alfa (IL-18BP) in adult-onset Still’s disease. Ann Rheum Dis. 2018;77(6):840–847. doi:10.1136/annrheumdis-2017-212608
75. Kiltz U, Kiefer D, Braun J, et al. Prolonged treatment with Tadekinig alfa in adult-onset Still’s disease. Ann Rheum Dis. 2020;79(1):e10. doi:10.1136/annrheumdis-2018-214496
76. Adisinsight Drug Profile: APB R3. Available from: https://adisinsight.springer.com/drugs/800065974.
77. Bracaglia C, de Graaf K, Pires Marafon D, et al. Elevated circulating levels of interferon-γ and interferon-γ-induced chemokines characterise patients with macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. Ann Rheum Dis. 2017;76(1):166–172. doi:10.1136/annrheumdis-2015-209020
78. Benedetti FD, Brogan P, Grom A, et al. Emapalumab, an interferon gamma (IFN-Y)-blocking monoclonal antibody, in patients with macrophage activation syndrome (MAS) complicating systemic juvenile idiopathic arthritis (SJIA). [abstract]. Ann Rheum Dis. 2019;78(suppl 4):178.
79. Hatterer E, Richard F, Malinge P, et al. Investigating the novel mechanism of action for NI-0501, a human interferon gamma monoclonal antibody. Cytokine. 2012;59(3):570. doi:10.1016/j.cyto.2012.06.257
80. Gabr JB, Liu E, Mian S, et al. Successful treatment of secondary macrophage activation syndrome with emapalumab in a patient with newly diagnosed adult-onset Still’s disease: case report and review of the literature. Ann Transl Med. 2020;8(14):887. doi:10.21037/atm-20-3127
81. Schwartz DM, Kanno Y, Villarino A, et al. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discov. 2017;17(1):78. doi:10.1038/nrd.2017.267
82. Clark JD, Flanagan ME, Telliez JB. Discovery and development of Janus kinase (JAK) inhibitors for inflammatory diseases. J Med Chem. 2014;57(12):5023–5038. doi:10.1021/jm401490p
83. Kacar M, Fitton J, Gough AK, et al. Mixed results with baricitinib in biological-resistant adult-onset Still’s disease and undifferentiated systemic autoinflammatory disease. RMD Open. 2020;6(2):e001246. doi:10.1136/rmdopen-2020-001246
84. Ladhari C, Jorgensen C, Pers Y-M. Treatment of refractory adult onset Still’s disease with combination anakinra and baricitinib therapy. Rheumatology. 2019;58(4):736–737. doi:10.1093/rheumatology/key414
85. Gillard L, Mitrovic S, Reumaux H, et al. AB0772 Jak inhibitors in refractory adult and childhood onset still’s disease. Ann Rheum Dis. 2021;80(Suppl.10):1412–1413. doi:10.1136/annrheumdis-2021-eular.2210
86. Honda M, Moriyama M, Kondo M, et al. Tofacitinib-induced remission in refractory adult-onset Still’s disease complicated by macrophage activation syndrome. Scand J Rheumatol. 2020;49(4):336–338. doi:10.1080/03009742.2020.1729405
87. Maschalidi S, Sepulveda F-E, Garrigue A, et al. Therapeutic effect of JAK1/2 blockade on the manifestations of hemophagocytic lymphohistiocytosis in mice. Blood. 2016;128(1):60–71. doi:10.1182/blood-2016-02-700013
88. Hu Q, Wang M, Jia J, et al. Tofacitinib in refractory adult-onset Still’s disease: 14 cases from a single centre in China. Ann Rheum Dis. 2020;79(6):842–844. doi:10.1136/annrheumdis-2019-216699
89. Das R, Guan P, Sprague L, et al. Janus kinase inhibition lessens inflammation and ameliorates disease in murine models of hemophagocytic lymphohistiocytosis. Blood. 2016;127(13):1666–1675. doi:10.1182/blood-2015-12-684399
90. Zhang Q, Wei A, Ma HH, et al. A pilot study of ruxolitinib as a front-line therapy for 12 children with secondary hemophagocytic lymphohistiocytosis. Haematologica. 2021;106(7):1892–1901. doi:10.3324/haematol.2020.253781
91. Jamilloux Y, Gerfaud-Valentin M, Martinon F, Belot A, Henry T, Sève P. Pathogenesis of adult-onset Still’s disease: new insights from the juvenile counterpart. Immunol Res. 2015;61(1–2):53–62. doi:10.1007/s12026-014-8561-9
92. Cua D-J, Tato C-M. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol. 2010;10(7):479–489. doi:10.1038/nri2800
93. Mitrovic S, Hassold N, Kamissoko A, et al. Adult-onset Still’s disease or systemic-onset juvenile idiopathic arthritis and spondyloarthritis: overlapping syndrome or phenotype shift? Rheumatology;2021. keab726. doi:10.1093/rheumatology/keab726
94. Pontali E, Volpi S, Signori A, et al. Efficacy of early anti-inflammatory treatment with high doses of intravenous anakinra with or without glucocorticoids in patients with severe COVID-19 pneumonia. J Allergy Clin Immunol. 2021;147:1217–1225. doi:10.1016/j.jaci.2021.01.024
95. Oliviero F, Bindoli S, Scanu A, et al. Autoinflammatory mechanisms in crystal-induced arthritis. Front Med. 2020;7:166. doi:10.3389/fmed.2020.00166
96. Klück V, Jansen TLTA, Janssen M, et al. Dapansutrile, an oral selective NLRP3 inflammasome inhibitor, for treatment of gout flares: an open-label, dose-adaptive, proof-of-concept, phase 2a trial. Lancet Rheumatol. 2020;2(5):e270–e280. doi:10.1016/S2665-9913(20)30065-5
97. Coll RC, Robertson AAB, Chae JJ, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21(3):248–255. doi:10.1038/nm.3806
98. Vande Walle L, Stowe IB, Šácha P, et al. MCC950/CRID3 potently targets the NACHT domain of wildtype NLRP3 but not disease-associated mutants for inflammasome inhibition. PLoS Biol. 2019;17(9):e3000354. doi:10.1371/journal.pbio.3000354
99. Jiang H, He H, Chen Y, et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J Exp Med. 2017;214(11):3219–3238. doi:10.1084/jem.20171419
100. Fouad AA, Abdel-Aziz AM, Hamouda AAH. Diacerein downregulates NLRP3/Caspase-1/IL-1β and IL-6/STAT3 pathways of inflammation and apoptosis in a rat model of cadmium testicular toxicity. Biol Trace Elem Res. 2020;195(2):499–505. doi:10.1007/s12011-019-01865-6
101. Xu S, Li X, Liu Y, et al. Inflammasome inhibitors: promising therapeutic approaches against cancer. J Hematol Oncol. 2019;12(1):64. doi:10.1186/s13045-019-0755-0
102. Ma Y, Meng J, Jia J, et al. Current and emerging biological therapy in adult-onset Still’s disease. Rheumatology. 2021;60(9):3986–4000. doi:10.1093/rheumatology/keab485
103. Behrens F, Tak PP, Østergaard M, et al. MOR103, a human monoclonal antibody to granulocyte - Macrophage colony-stimulating factor, in the treatment of patients with moderate rheumatoid arthritis: results of a phase Ib/IIa randomised, double-blind, placebo-controlled, dose-escalation trial. Ann Rheum Dis. 2015;74(6):1058–1064. doi:10.1136/annrheumdis-2013-204816
104. Burmester GR, McInnes IB, Kremer JM, et al. Mavrilimumab, a fully human granulocyte-macrophage colony-stimulating factor receptor α monoclonal antibody: long-term safety and efficacy in patients with rheumatoid arthritis. Arthritis Rheumatol. 2018;70(5):679–689. doi:10.1002/art.40420
105. Weinblatt ME, McInnes IB, Kremer JM, et al. A randomized Phase IIb study of mavrilimumab and golimumab in rheumatoid arthritis. Arthritis Rheumatol. 2018;70(1):49–59. doi:10.1002/art.40323
106. De Luca G, Cavalli G, Campochiaro C, et al. GM-CSF blockade with mavrilimumab in severe COVID-19 pneumonia and systemic hyperinflammation: a single-centre, prospective cohort study. Lancet Rheumatol. 2020;2(8):e465–e473. doi:10.1016/S2665-9913(20)30170-3
107. Carpenter S, Aiello D, Atianand MK, et al. A long noncoding RNA mediates both activation and repression of immune response genes. Science. 2013;341(6147):789–792. doi:10.1126/science.1240925
108. Mathy NW, Chen XM. Long non-coding RNAs (lncRNAs) and their transcriptional control of inflammatory responses. J Biol Chem. 2017;292(30):12375–12382. doi:10.1074/jbc.R116.760884
109. Atianand MK, Fitzgerald KA. Long non-coding RNAs and control of gene expression in the immune system. Trends Mol Med. 2014;20(11):623–631. doi:10.1016/j.molmed.2014.09.002
110. Hadjicharalambous MR, Lindsay MA. Long non-coding RNAs and the innate immune response. Noncoding RNA. 2019;5(2):34.
111. Yang CA, Chen PK, Lan JL, et al. Expression signature of inflammation-associated long non-coding RNAs in adult-onset Still’s disease. Clin Exp Rheumatol. 2021;39 Suppl 132(5):67–74.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.